Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain
 Utpal S. Bhalala
Utpal S. Bhalala Raymond C. Koehler
Raymond C. Koehler Sujatha Kannan
Sujatha Kannan- 1Department of Anesthesiology, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2Department of Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Hypoxic-ischemic (HI) injury to developing brain results from birth asphyxia in neonates and from cardiac arrest in infants and children. It is associated with varying degrees of neurologic sequelae, depending upon the severity and length of HI. Global HI triggers a series of cellular and biochemical pathways that lead to neuronal injury. One of the key cellular pathways of neuronal injury is inflammation. The inflammatory cascade comprises activation and migration of microglia – the so-called “brain macrophages,” infiltration of peripheral macrophages into the brain, and release of cytotoxic and proinflammatory cytokines. In this article, we review the inflammatory and immune mechanisms of secondary neuronal injury after global HI injury to developing brain. Specifically, we highlight the current literature on microglial activation in relation to neuronal injury, proinflammatory and anti-inflammatory/restorative pathways, the role of peripheral immune cells, and the potential use of immunomodulators as neuroprotective compounds.
Introduction
Regions of the developing brain become vulnerable to hypoxic-ischemic (HI) as the neurons develop operating voltage-dependent ion channels, neurotransmitter receptors, synaptic connections, and increased mitochondria to supply the ATP for the consequent increase in energy demand (1). Global HI injury in developing brain can result from a variety of clinical conditions, including perinatal asphyxia and cardiac arrest in infants and older children. Although rates of survival to hospital discharge from in-hospital cardiac arrest in children has improved significantly over the last decade, neurologic outcomes remain poor (2).
A cascade of cellular and biochemical responses to the initial HI insult can lead to secondary neuronal injury after reoxygenation. One of the crucial but understudied mechanisms of secondary neuronal injury after global HI is inflammation (3). It is characterized by activation of microglia, the innate immune cells of brain, migration of peripheral macrophages; release of proinflammatory cytokines and chemokines, and phagocytosis of injured and uninjured neurons. Some evidence suggests that blocking the inflammatory reaction promotes neuroprotection and has potential for use in the clinical treatment of ischemic brain injury (4–8).
Therapeutic hypothermia has been shown to protect the brain after cardiac arrest in adults and after HI in term newborns (9–11); it is currently undergoing a multicenter trial for children after cardiac arrest (12). Although therapeutic hypothermia reduces mortality and improves early neurologic outcome after HI injury in neonates, significant neurologic deficits and learning disability persist into childhood (13, 14). Cell death initiated before the onset of hypothermia, or possibly after rewarming, will likely recruit inflammatory processes that can contribute to the low efficacy of hypothermia in newborns who experience the most severe HI insult. In a multicenter, randomized trial of induced hypothermia for neonatal hypoxic-ischemic encephalopathy (HIE), a rapid and dramatic rise in levels of proinflammatory cytokines immediately after HI injury was not amenable to induced hypothermia (15). Adjunct treatment targeted to inflammation and immune dysregulation may help improve the overall efficacy of therapeutic hypothermia (16).
In this article, we review current understanding about the inflammatory and immune mechanisms involved in acute HI injury of developing brain and the neuroprotective agents that can curtail inflammation and immune dysregulation. New anti-inflammatory targets continue to be identified and constitute an important area for translational medicine (17–22). Overall, the prospects for safe neuroprotective therapies to improve outcome after acute HI brain injury remain promising.
HI Neuronal Injury in Developing Brain
A wealth of information on HI brain injury and repair is available through translational adult stroke research. The immature brain differs from the adult in its capability to use metabolic fuels, vulnerability to glutamate excitotoxicity, and oxidative stress (1). There is an evidence to suggest that age could have a significant effect on response to cytokines and hence neuroinflammation after exposure to lipopolysaccharide (LPS) and/or HI (23). Since the mechanisms of injury and strategies for repair often are very different in the immature as compared with the adult brain, our pediatric community and researchers need to focus on HI brain injury and repair in the developing brain (24, 25).
Global HI of brain initiates a cascade of excitotoxicity and oxidative damage that in turn causes microvascular injury, blood–brain barrier (BBB) dysfunction, and postischemic inflammation. These events all exacerbate the initial injury and can lead to permanent cerebral damage. The modes of secondary neuronal loss after HI are apoptosis, autophagy, programed necrosis, and unregulated necrosis, which arise from cell swelling and bursting of nuclear and cell membranes (26). Apoptosis is more prominent in the neonatal brain than in the adult brain after HI insult (27–29). Also, selective neuronal vulnerability has been observed in developing brain whereby neuronal injury secondary to HI is present predominantly in regions that function in sensorimotor integration and movement control. The regions in the developing brain, which are vulnerable to HI are sensory-motor cortex, basal ganglia, thalamus, and hippocampus (30). Considerable research has been targeted toward different pathways of secondary neuronal injury and therapies to counter one or more such pathways.
Inflammation and Immune Dysregulation after Acute Insult to the Brain
For many years, the brain was considered an immune-privileged organ, but advances in neuroimmunology have challenged this dogma and helped to expand our understanding of the immune processes that occur in healthy and diseased brain (31). It is well accepted that the brain and immune system are engaged in bidirectional crosstalk. Microglia, the resident innate immune cells of the brain, elicit inflammatory responses under neuropathologic conditions such as perinatal HI encephalopathy, infection, and traumatic brain injury, as well as in autoimmune and neurodegenerative disorders (32). Also, growing evidence indicates that, like in peripheral organs, inflammatory cells play a crucial role in remodeling and repair after an acute insult to the brain. The neuroinflammatory response after acute HI brain injury is characterized by activation of microglia; migration of peripheral macrophages, monocytes, and neutrophils; and release of cytokines and chemokines by the inflammatory cells (33).
Microglia – Innate Immune Cells of Brain
Microglia are resident macrophages of brain and are known to actively remove cellular debris during normal development and under pathologic conditions. They were first identified as innate cells of the central nervous system (CNS), distinct from neurons and other glia, by Nissl in 1899. Many years later, in the early part of the 20th century, del Rio Hortega confirmed this distinction using silver carbonate staining methods (34) and proposed that these cells were primarily of hematopoietic origin. Microglia constitute 10–15% of the total glia within the brain (35) and are present throughout the brain. Some areas are more heavily populated than others, and white matter generally contains fewer microglial cells than does gray matter (36). Microglia are highly ramified cells, and under non-pathologic conditions, they have a small cell body with long and densely branched processes.
Role of Microglia in Developing Brain
Although microglia are highly active during pathologic conditions, they are also active under physiologic conditions as they survey the microenvironment and the status of neurons and participate in housekeeping and remodeling within the CNS (37). In developing brain, highly active neurogenesis forms large numbers of neurons every minute, vastly exceeding the ultimate requirements of an adult. Consequently, many neurons undergo apoptosis during early infancy and childhood (38). Microglia have a functional role in the phagocytosis of cell debris and in the release of trophic factors in developing brain. They also are involved with synaptic pruning in the developing brain, thereby influencing its maturation (37, 39–42).
Role of Microglia in HI Injury of Developing Brain
Microglia fulfill a variety of tasks after HI of the brain. They engulf cellular debris, lipids, and apoptotic cells. They also promote cytotoxicity through release of proteases and proinflammatory cytokines, activation of respiratory burst, and N-methyl-d-aspartate (NMDA)-mediated excitotoxicity. These mechanisms not only scavenge the HI damaged neurons but they can also affect viable neurons. Much of our current knowledge of inflammation, particularly microglial activation, after HI injury of developing brain comes from the Vannucci model, in which HI is induced in postnatal rats and mice by exposure to low oxygen and carotid ligation (43, 44).
Time Course of Microglial Activation after HI
Hypoxia–ischemia and intra-cerebral administration of excitotoxins such as NMDA result in a fast and robust microglial reaction in the developing brain (45). One of the earliest studies of microglial activation after HI brain injury in a rat model of developing brain described microglial activation within 2 h after HI injury (6). In another study, microglia exhibited a time-dependent, differential upregulation of MHC and CD4/CD8 immunomolecules from day 1 to day 28 post-injury (46).
Whereas early neuronal cell death undoubtedly activates microglia, a key question is whether activated microglia contribute to delayed cell death of other neurons. Co-culture studies of microglia and oxygen-glucose-deprived neurons have shown that stressed neurons activate microglia, which release proinflammatory cytokines. These cytokines then induce neuronal damage (47). However, uncertainty persists about the extent to which neuronal and non-neuronal cells communicate in vivo as they do in vitro, especially under pathologic conditions involving multiple cell types. Nevertheless, the concept that the proinflammatory cytokines released by active microglia contribute to ongoing secondary neuronal injury has gained support from the neuroprotective effects seen with therapies targeting microglial activation (4–8, 17–22).
Little is known about the morphologic and functional changes that microglia undergo over time after acute, global, HI developmental brain injury, or about the regional distribution of active microglia in relation to selective neuronal vulnerability. Also, the duration of the proinflammatory response beyond the period of acute injury is not clearly defined. Acute inflammation can also be shifted to a chronic inflammatory state and/or adversely affect brain development (48).
Activated microglia play an equally important role in the restorative and reparative processes after neuronal injury (49). Neuroprotective therapies targeted toward microglia could potentially be a double-edged sword if used without appropriate information on time course of the proinflammatory and restorative responses after acute, global HI.
Microglial Activation and White-Matter Injury after HI
Apart from secondary neuronal injury after HI, microglia have been found to play a crucial role in oligodendrocyte injury and disturbance in myelination through a cytokine-mediated mechanism in neonatal hypoxic rats (50). Likewise, overproduction of local cytokines by activated microglia has been reported to induce axonal injury after hypoxia in developing rats (51). Microglia contribute to white mater injury in immature, developing brain (52). In studies of perinatal brain injury, intrauterine inflammation has shown to cause white-matter injury through microglial activation (53). Perinatal HI injury in preterm brain is associated with a T-helper-type immune response (54). Perinatal inflammation, which triggers neuroinflammation is also believed to predispose the immature brain to HI injury (55, 56).
Microglial Activation and BBB Integrity after HI
Under normal conditions, the BBB is important for maintaining micro-environmental homeostasis in the brain and its so-called immune-privileged status by preventing the entry of T lymphocytes (57, 58). In a combined in vivo and in vitro study of the relationship between microglial activation and BBB under HI conditions, increased BBB disruption was associated with activated microglia. However, this association was inhibited by the anti-inflammatory action of minocycline, which is known to inhibit the release of matrix metalloproteinase (MMP)-9 and breakdown of collagen and laminin in the vascular basement membrane (59).
Microglial Proliferation and Migration after Injury
Microglia have a remarkable ability to multiply and migrate in response to neurologic injury (60, 61). Microglial proliferation has been implicated in the onset and/or progression of ischemic brain injury (62). Active microglia express receptors for a variety of molecules, such as interleukin (IL)-3, IL-6, and granulocyte-macrophage colony-stimulating factor, which play an important role in microglial proliferation (63). In vivo studies of the cell proliferation markers Ki67 and bromodeoxyuridine have confirmed that microglia can proliferate in a developing brain environment (64). A rapid increase in number of microglia at the site of injury is related to influx of peripheral monocytes and movement of innate microglia from other parts of the brain (65).
Role of Astrocytes
Astrocytes express a wide variety of receptors of innate immunity (66). In response to HI, there is reactive astrogliosis with release of MMPs, which degrade BBB and facilitate entry of peripherally derived immune cells (67, 68). Through toll-like receptors, astrocytes, on one hand promote inflammation and on the other end, facilitate tissue repair (66, 69). In response to ischemia, astrocytes not only potentiate excitotoxicity through inducible nitric oxide synthase (iNOS) but also release a myriad of cytokines, many of which have dual proinflammatory and anti-inflammatory effects (70–74).
Role of Proinflammatory and Anti-Inflammatory Cytokines and Chemokines after HI Injury of Developing Brain
Cytokines and chemokines released by active microglia in response to an acute neurologic insult take part in innate immune response; modulate influx of peripheral immune and inflammatory cells into the brain; contribute to secondary neuronal, oligodendrocyte, and axonal injury; and ultimately promote tissue repair and recovery (75) (Figure 1). In the brain, cytokines and chemokines are expressed on the neurons and glia (76). There is increased chemokine gene expression and release in the developing brain after HI (77). Cytokines and chemokines released by peripheral immune cells contribute to neuroinflammation (78, 79) and their inhibition or deficiency is associated with reduced injury (80–82). TRAIL (tumor necrosis factor-related apoptosis inducing ligand) is expressed primarily on microglia and astrocytes and it has been shown to participate in neonatal brain injury after inflammation and HI (83). Elevated levels of IL-6 and IL-8 in the cerebrospinal fluid of term newborns have been correlated with an increased degree of encephalopathy and poor neurodevelopmental outcome (84). Reactive oxygen species (ROS) and nitrogen metabolites generated within the active microglia induce the release of proinflammatory cytokines. In a study of mixed astroglial/microglial cultures, stimulated microglia produced NO in a time-dependent manner (85). Following HI injury, hydrogen peroxide (H2O2) levels rise significantly (86) and cause extensive damage to iron-rich developing brain (87). In fact, microglial exposure to continuous H2O2 leads to pleiotropic and biphasic effects (88). Since the effects of cytokines are influenced by one another and majority of cytokines have pleiotropic and cell-specific effects, the final effect of individual cytokines is difficult to establish (89–93).
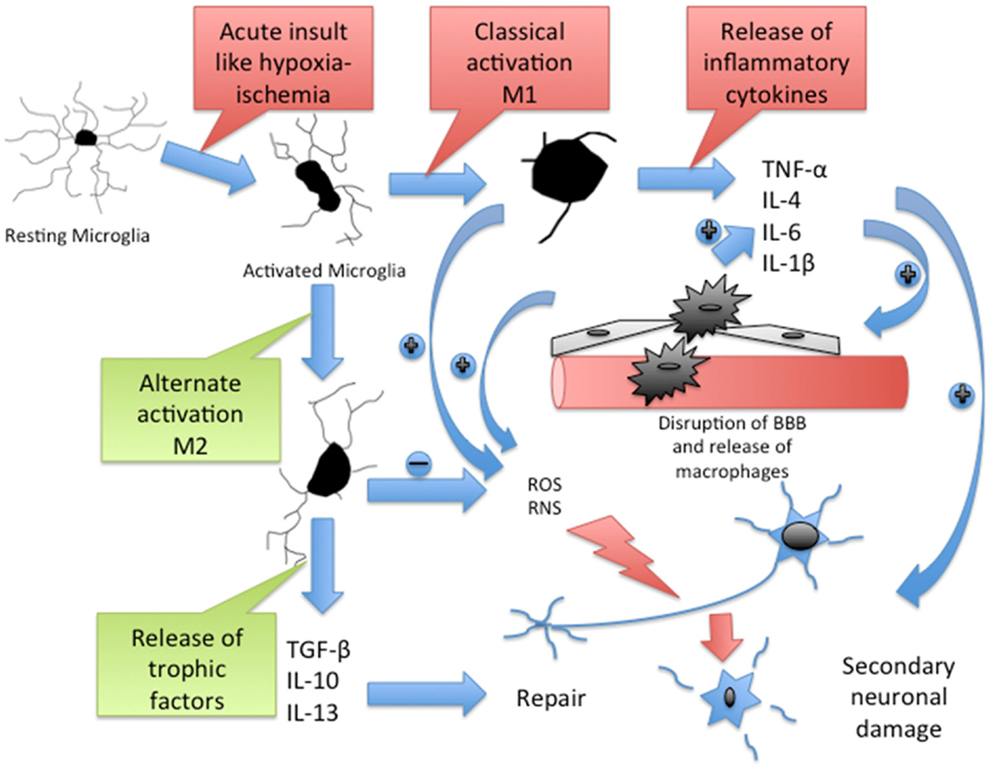
Figure 1. Cascade of inflammatory pathway in brain after acute HI. Resting microglia get activated to M1 type through classical pathway and M2 type through alternate pathway. M1 microglia release inflammatory cytokines, which cause disruption of blood–brain barrier (BBB). The BBB disruption promotes infiltration of macrophages, which further release inflammatory cytokines. Active microglia and macrophages release reactive oxygen species (ROS) and reactive nitrogen species (RNS), which contribute to the secondary neuronal injury. M2 microglia release trophic factors, which contribute to the neuronal repair. TNF-α, a potent proinflammatory cytokine contributes to peripheral immune cell recruitment and proliferation in the brain, neuronal apoptosis, oligodendrocytes, and axonal injury after HI (94). IL-1β blocks oligodendrocyte proliferation (95) and elevates levels of circulating IL-6, another potent cytokine that contributes to early neurologic deterioration after brain ischemia (95, 96). Matrix metalloproteinases (MMPs) disrupt the BBB and allow peripheral leukocyte infiltration (97). Macrophage colony stimulating factor (MCSF), released mainly by macrophages, T cells, B cells, and microglia, induces proliferation, migration, and activation of microglia and regulates the release of proinflammatory cytokines from macrophages (98, 99). MCP-1, a chemokine secreted by active microglia and astrocytes in response to injury (100), mediates the migration of microglia, monocytes, and lymphocytes to the site of injury in the CNS (101, 102).
Microglial Activation after Ischemia – Harmful or Helpful?
Microglia function as CNS macrophages and help clear debris and invading pathogens. When activated in response to a variety of stimuli and triggering events such as HI, proinflammatory and cytotoxic pathways are initiated that can contribute to secondary neuronal injury. Conversely, stimulation of proliferating microglia after cerebral ischemia by MCSF leads to release of insulin-like growth factor (IGF), a neurotrophic factor with neuroprotective properties (103). In a rodent model of focal cerebral ischemia, time-lapse imaging showed that microglia exert neuroprotection by rapidly engulfing apoptotic neurons and motile polymorphonuclear cells (104). In a study of rodent cerebral ischemia, intra-arterial injection of microglia prevented the ischemia-induced decline of brain-derived neurotrophic factor (BDNF) in hippocampus and offered neuroprotection (105). Similarly, in a rodent model of acute neonatal stroke, inhibition of microglia with liposomal clodronate led to elevation of levels of cytokines and chemokines and exacerbation of injury (103). There is growing evidence that microglia activated by injured or dying neurons mediate a decrease in neuronal damage and promote tissue regeneration and repair (106). In response to injury, activated microglia ensheath damaged neurons and remove excitatory input through the displacement of afferent synapses (107). Much of our current understanding of the difference between helpful and harmful microglial phenotypes is derived from literature on acute neuroinflammation after stroke and chronic neuroinflammatory conditions like Alzheimer’s disease (108, 109). The innate immune response is characterized by activation of microglia to an M1 phenotype and the subsequent proinflammatory response followed by resolution, and alternative activation to an M2 phenotype that leads to anti-inflammatory signaling (M2a), the clearance of ROS and reactive nitrogen species (RNS) (M2b), and wound healing (M2c) (110). Depending on the type of insult, the phenotype of microglial activation switches over time from M1 to M2 or vice versa (111). The differences between M1 and M2 microglial phenotypes are shown in Table 1. The M1 phenotype is associated with greater neuronal death than is the alternatively activated M2 phenotype (112); therefore, there is a growing interest in inhibiting the M1 phenotype.
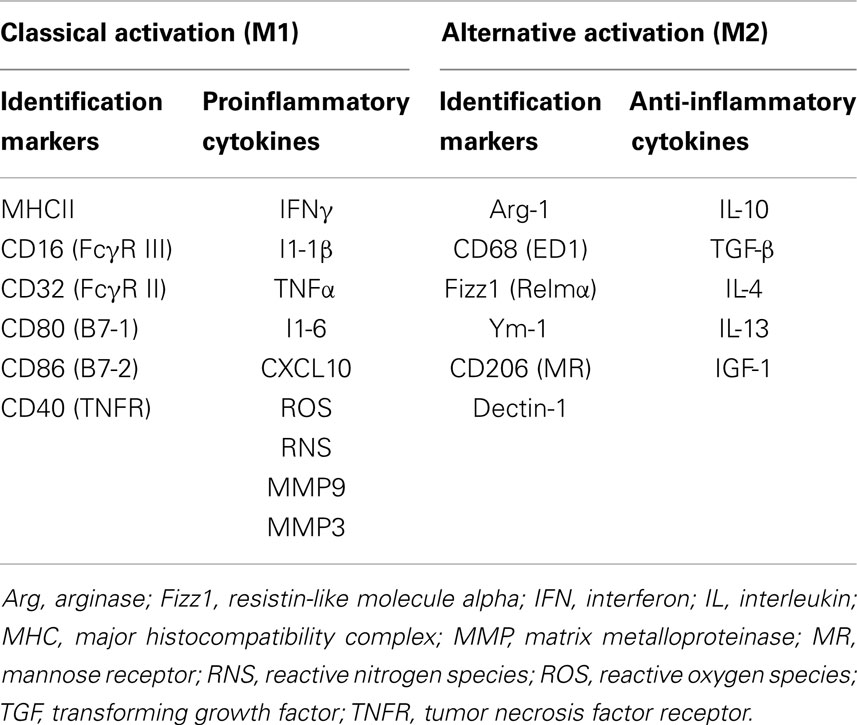
Table 1. The M1 (classical) and M2 (alternate) phenotypes of microglia.
Regulation of Microglia
Microglia are kept under check through neuronal–glial cross talk. Chemokine receptors and corresponding ligands allow interactions between neurons and microglia and control proinflammatory responses of microglia under physiologic conditions (113). CD200/CD200R1, fractalkine (CX3CL1)/CX3CR1, SIRPα/CD47, and heat shock protein 60 (HSP60)/TREM2 are cell–cell interaction molecules that regulate microglia (114–117). After injury, disruption of these interactions from neuronal damage may cause activation of microglia to the proinflammatory M1 phenotype. Modifications of these neuronal–glial regulatory interactions have been shown to attenuate neuronal damage in models of focal cerebral ischemia and chronic neuroinflammation (118–122). Neuronal–glial interactions and their role in secondary neuronal injury after global HI injury in developing brain are understudied and warrant evaluation. Modifications of these regulatory interactions in developing brain after global, HI injury could potentially open new avenues of neuroprotection.
Role of Peripheral Inflammatory and Immune Cells in the Developing Brain after HI
Studies from adult rodent models of focal HI suggest that, in addition to innate inflammation, the peripheral immune system may have a role in the etiology of neuronal damage. More precisely, several studies have shown that acute brain injury from focal ischemia is associated with a massive activation of the peripheral immune system, with rapid mobilization of immune effector cells from the spleen (123, 124). These mobilized effector cells can invade the brain and aggravate the existing injury (123). In a study of preterm sheep in which global HI was induced with the umbilical cord occlusion method, the authors showed mobilization of peripheral immune cells from spleen and reduction of splenic size. These changes were unrelated to splenic HI injury and occurred in parallel with marked injury and functional loss of the preterm brain (125). Yilmaz and co-authors showed that T lymphocytes, but not B lymphocytes, contribute to inflammatory and thrombogenic responses, brain injury, and neurologic deficit associated with experimental stroke in rodents (126). Though studies have shown that activated microglia outnumber the peripherally derived macrophages at the site of infarct (127, 128), inhibition of macrophages has shown to reduce infarct volume (129–131). These reports indicate that infiltrating mononuclear inflammatory cells play a significant role in neuroinflammation and may be necessary for the activation of microglia. Suppression of peripheral immune and inflammatory cells through splenectomy reduces cellular infiltration into the brain and interaction with the activated microglia at the site of ischemic injury, resulting in decreased damage (123). A severe, global HI injury following cardiac arrest would not only induce necrosis and apoptosis in brain but also might exert different effects in peripheral immune organs like spleen, which is influenced by the autonomic nervous system. Therefore, additional studies are warranted to determine the role of the peripheral immune system in whole-body HI (Figure 2).
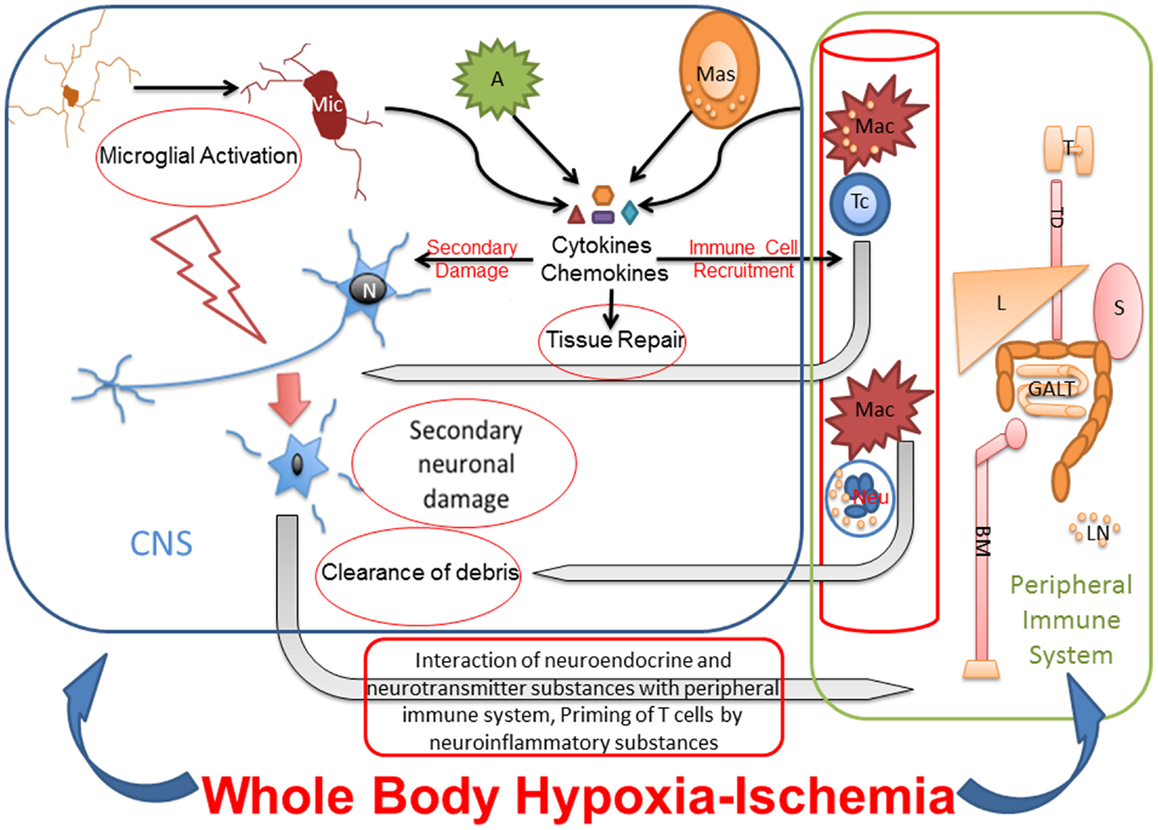
Figure 2. Whole-body hypoxia-ischemia activates central and peripheral immune components. The whole-body HI not only induces neuroinflammation and necrosis and apoptosis in brain but also potentially exerts different effects in peripheral immune organs like spleen, which is influenced by the autonomic nervous system. Microglial activation after global HI leads to secondary neuronal injury. The cytokines and chemokines released by activated microglia, astrocytes, mast cells, and peripheral immune cells cause secondary neuronal damage, degrade BBB for leukocyte recruitment from blood stream, and eventually also contribute to tissue repair. Primed and polarized T cells enter CNS in response to HI, recognize neuronal surface receptors like major histocompatibility complex, and interact with damaged neurons for repair. Macrophages and neutrophils also enter the CNS in response to HI for clearance of debris. Under pathologic conditions, neurotransmitter and neuroinflammatory substances interact with peripheral immune system for priming and activating immunologic pathways for clearance of pathogen and/or debris. Due to cross talk between peripheral and central immune systems, the effects of peripheral immune organs like thymus, liver, spleen, gut associated lymphoid tissues and bone marrow on neuroinflammation, and secondary neuronal injury after whole-body HI need to be studied. Mic, microglia; N, neuron; A, astrocyte; Mas, mast cell; Mac, macrophage; Tc, T cell; Neu, neutrophil; T, thymus; TD, thoracic duct; L, liver; S, spleen; GALT, gut-associated lymphoid tissue; BM, bone marrow.
Anti-Inflammatory and Immunomodulatory Therapies for Neuroprotection after HI Brain Injury
Preconditioning, salvaging, and repair are three main modes of achieving neuroprotection (132).
Hypothermia is now standard of care for term HI encephalopathy, so studies focused on adjunct therapies will be added to that treatment (133). As our understanding has grown regarding the role that inflammatory and immune cells play in the pathophysiology of secondary neuronal injury after HI, so has interest in using anti-inflammatory and immunomodulatory strategies as neuroprotective therapies after brain injury. Such therapies range widely from steroidal and non-steroidal molecules to cannabinoids (CB) and statins.
Cannabinoids
Cannabinoids are diverse chemical compounds, which are either endogenously produced in body (endocannabinoids) or derived from cannabis and related plants (phytocannabinoids) or prepared chemically (synthetic cannabinoids). CB compounds act on cell surface CB receptors to exert a variety of effects, including potent anti-inflammatory and immunomodulatory effects. There are two major subtypes of CB receptors – CB1 and CB2. The CB1 receptors present on neurons produce the psychoactive effects of non-selective CBs like tetrahydrocannabinol (134). CB2 receptors are present on immune cells (135) and are expressed on the surface of activated microglial cells under pathologic conditions (136–138). Under physiologic state, CB2 receptors remain dormant, and they are expressed in the active form on the surface of the immune cells after an acute insult. CB2 receptor agonists such as cannabidiol (CBD) exert potent anti-inflammatory and immunomodulatory actions through a CB2 receptor-G-protein-coupled mechanism (139, 140). Two orphan G-protein-coupled receptors, possibly activated by multiple different cannabinoid ligands, have been recently proposed as novel cannabinoid receptors (141).
Cannabidiol is a major constituent of the cannabis plant, representing up to 40% in plant extracts. CBD has shown neuroprotective effects in adult rodent models of stroke (142–144). CBD studies have also shown significant neuroprotective effects in a piglet model of HI (17–21). CB2 agonists reduce microglial activation, proliferation and migration to the site of injury, and also reduce release of proinflammatory cytokines and chemokines like IL-1β, TNFα, MCP-1, and MIP-1α (20). The neuroprotective effects of CBD are related not only to the anti-inflammatory and immunomodulator effects but also to serotonergic, antiexcitotoxic, antioxidant, adenosine receptor agonist, and antiepileptic effects (18–21). Due to multi-pronged neuroprotective effects, including but not limited to anti-inflammatory and immunomodulatory effects, CB have gained recent interest in research on developmental brain injury. Because CBD is a selective CB2 receptor agonist and lacks CB1-induced psychoactive effects, the potential for its clinical use seems favorable (145). However, certain crucial questions that need to be addressed in preclinical setting before considering CBD for clinical trials are dose–effect relationship, the role of CB2 agonists as adjunct neuroprotective agents with therapeutic hypothermia, and the role of CB2 agonists as neuroprotective agents through peripheral versus central immunomodulation.
Anti-TNF-α
TNF-α is a potent proinflammatory cytokine that plays a key role in neurotoxicity after ischemia. Anti-TNF-α has been shown to be neuroprotective in rodent models of focal cerebral ischemia (146, 147). The challenges with the use of anti-TNF-α have been solubility and brain penetration. Additionally, TNF inhibitors can have harmful side effects, including lymphoma, infections (especially tuberculosis reactivation), congestive heart failure, demyelinating disease, and lupus-like syndrome (148).
IL-1 Antagonists
Recombinant human IL-1 receptor antagonist (rhIL-1ra) has been shown to protect against focal cerebral ischemia in the rat through its actions on microglia (149). A randomized phase II study of rhIL-1ra in acute stroke patients showed that it is safe and well tolerated in acute stroke. In addition, rhIL-1ra exhibited biologic activity that was relevant to the pathophysiology and clinical outcome of ischemic stroke (150).
Minocycline
Minocycline is a tetracycline derivative that has been shown to be safe and effective as an antibiotic and anti-inflammatory drug for treating systemic inflammatory conditions. Minocycline crosses the BBB and has demonstrated neuroprotective qualities in experimental models of post-arrest global HI (7), traumatic brain injury, stroke, spinal cord injury, and neurodegenerative diseases (151). Minocycline is believed to have anti-inflammatory, antiapoptotic, and antioxidant effects. It inhibits microglial activation, T-cell migration, and release of proinflammatory cytokines and chemokines (151). Minocycline administered either immediately before or immediately after a HI insult substantially blocked tissue damage in a rodent model of neonatal HI brain injury (152). In a model of neonatal stroke, minocycline significantly reduced the volume of injury at 24 h but not 7 days after transient MCA occlusion (153). Unfortunately, minocycline has also been shown to have variable and even detrimental effects in different species and models of neurological disorders (151, 154–158). Although its anti-inflammatory actions are likely to contribute to its neuroprotective effects, its contrasting effect in mouse and rat HI models could be related to its reported action on the regulation of prostaglandin pathways (154). Also, long-term minocycline therapy in chronic neuroinflammatory diseases has proved disappointing due to either minimal or no clinical effects, even worse effects, and safety concerns (159–161).
Ibudilast
Ibudilast is mainly a phosphodiesterase (PDE) inhibitor, but it also affects the function of lymphocytes, endothelial cells, and glial cells. In a neuronal-microglial co-culture study, ibudilast suppressed neuronal necrosis that was induced by LPS and interferon-γ activation of microglia (162). Idibulast acts through phosphodiesterase 4 (PDE4) receptors and inhibits release of TNF- α by inflammatory cells and inhibits tyrosine kinase in neutrophils to mitigate inflammation (163). Currently, there are no clinical trials on neuroproctective effects of ibudilast.
Vitamin D
Recent evidence supports the involvement of vitamin D3 in immunologic processes that protect the nervous system (164). In the CNS, the active form of vitamin D – calcitriol – acts as an immunosuppressor. It induces the anti-inflammatory cytokine IL-4 and transforming growth factor and decreases expression of proinflammatory cytokines IL-6, TNF, and MCSF (165–169). Calcitriol decreases expression of MHC class II proteins and cofactor CD4, which play important roles in autoimmune processes in the nervous system (170). Role of vitamin D in modulating inflammation and immune dysregulation and ability to offer neuroprotection in a model of global HI have not yet been well studied.
Steroid and Non-Steroidal Anti-Inflammatory Drugs
Steroid molecules, including sex steroids, inhibit microglia and prevent release of proinflammatory cytokines. Steroid molecules also offer neuroprotection through release of neurotrophic factors from microglia (171). However, we need more basic information regarding the mechanisms by which steroids contribute to neuroprotection before we can predict the conditions in which hormone treatments may have positive outcomes for brain function in human beings. Such knowledge will enable researchers to design the best possible therapeutic approaches. Non-steroidal anti-inflammatory medications have been shown to curtail neuroinflammation associated with Alzheimer’s disease, Parkinson’s disease, and HI brain injury (170, 172).
Statins
Statins have been shown to reduce infarct size in experimental animal models of stroke. Statins attenuate the inflammatory cytokine responses that occur after cerebral ischemia, and their antioxidant properties ameliorate ischemic oxidative stress in the brain. Additionally, statins upregulate endothelial nitric oxide synthase and inhibit iNOS, effects that are potentially neuroprotective (173). Currently, NeuSTART2 (neuroprotection with statin therapy for acute recovery trial phase 2) is an ongoing phase 2 randomized safety trial, in which ischemic stroke patients are randomly assigned to placebo or standard dose lovastatin versus short-term high-dose lovastatin.
Propentofylline
Propentofylline acts by blocking the uptake of adenosine and inhibiting the PDE enzyme system (174, 175). Adenosine is released in response to cell damage after ischemic injury and acts on specific G-protein-coupled receptors on astrocytes, microglia, and infiltrating immune cells to regulate immune system function in the brain. Although the effects of adenosine protect neuronal integrity, adenosine might also aggravate neuronal injury by promoting inflammatory processes (176, 177). A detailed understanding of adenosine receptor function in the brain immune system should help researchers develop novel therapeutic strategies to treat HI-induced brain injury, which is associated with a dysfunctional immune response.
Melatonin
In preclinical models of term brain HI, melatonin markedly decreased microglial activation in association with neuroprotection (178). A study of melatonin with therapeutic hypothermia in a piglet model of HI showed a substantial improvement compared with therapeutic hypothermia alone in preserving brain function measured by amplitude-integrated electroencephalogram, and reduced cell death in selectively vulnerable areas (179). There is an ongoing, prospective, double-blinded, randomized trial of premature newborns of less than 28 weeks’ gestational age for assessing neuroprotective role of melatonin in the United Kingdom (MINT trial, ISRCTN15119574).
Epo
Epo and its receptor (EpoR) are expressed in the developing CNS and are required for normal brain development (180). Epo inhibits early mechanisms of brain injury by its anti-inflammatory, antiexcitotoxic, antioxidant, and antiapoptotic effects on neurons and oligodendrocytes (181–184). Currently, Epo is undergoing a trial of neuroprotection in preterm neonates (PENUT trial – preterm erythropoietin neuroprotection trial).
Xenon
Xenon is an odorless, dense noble gas with anesthetic properties. Xenon’s neuroprotective properties have been demonstrated in cell culture (185), a rodent model of hypoxia-ischemia (186–190) and a neonatal pig model of global hypoxia-ischemia, wherein it can augment hypothermic neuropotection (191, 192). The precise mechanism of neuroprotection and any possible direct effects on inflammation remain to be explored.
Delivery of Anti-Inflammatory and Immunomodulator Neuroprotective Agents Across BBB
Anti-inflammatory and immunomodulator agents like CBD are hydrophobic and, therefore, delivery across BBB becomes an important consideration for optimal neuroprotective effects within a safe dose-range. A novel approach is to use alternative delivery methods like nanoparticles, targeting the inflammatory system. Nanoparticles, such as polyamidoamine dendrimers have been shown to concentrate in activated microglia and astrocytes in the brains of newborn rabbits with cerebral palsy, but not healthy controls. This nanotechnology approach has shown excellent results to deliver dendrimer-bound N-acetyl-l-cysteine (NAC) to microglia to suppress neuroinflammation, using much lower concentrations than are needed with systemic dosing (193–195).
Conclusion
Neuroinflammatory and neuroimmune dysregulation play a key role in secondary neuronal damage after global HI injury in the developing brain. Microglia activated by HI, initiate a cascade of inflammatory reactions that lead to neuronal damage. Indeed, a growing number of anti-inflammatory and immunomodulatory compounds have shown promising neuroprotective effects in preclinical settings. Some of these compounds have even entered clinical trials for adult victims of stroke. However, substantial work is needed to improve our understanding of neuroinflammation after global HI injury in developing brain. The specific issues yet to be determined in relation to inflammatory and immunologic mechanisms after global HI include the temporal, topographic, and gender pattern of neuroinflammation, neuronal–glial regulatory interactions, and their contribution to secondary neuronal injury, the role of the peripheral immune system in potentiating secondary brain injury after multi-organ ischemia, and the development of anti-inflammatory and immunomodulatory compounds as neuroprotective agents after HI injury of developing brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors were partly supported by grants R01 NS060703 (Raymond C. Koehler) and R01 HD069562 (Sujatha Kannan) from the National Institutes of Health.
References
1. Vannucci SJ, Hagberg H. Hypoxia-ischemia in the immature brain. J Exp Biol (2005) 207:3149–54. doi:10.1242/jeb.01064
2. Girotra S, Spertus JA, Li Y, Berg RA, Nadkarni VM, Chan PS, et al. Survival trends in pediatric in-hospital cardiac arrests – an analysis from get with the guidelines-resuscitation. Circ Cardiovasc Qual Outcomes (2013) 6:42–9. doi:10.1161/CIRCOUTCOMES.112.967968
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin (2006) 24:1–21. doi:10.1016/j.ncl.2005.10.004
4. Carty ML, Wixey JA, Colditz PB, Buller KM. Minocycline treatment attenuates HI induced inflammation and white matter injury in the neonatal rat: a comparison of two different dose regimens. Int J Dev Neurosci (2008) 26:477–85. doi:10.1016/j.ijdevneu.2008.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev (2006) 12:105–12. doi:10.1002/mrdd.20102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. McRae A, Gilland E, Bona E, Hagberg H. Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res (1995) 84:245–52. doi:10.1016/0165-3806(94)00177-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Tang M, Alexander H, Clark RS, Kochanek PM, Kagan VE, Bayir H. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab (2010) 30(1):119–29. doi:10.1038/jcbfm.2009.194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Simão F, Matté A, Pagnussat AS, Netto CA, Salbego CG. Resveratrol preconditioning modulates inflammatory response in the rat hippocampus following global cerebral ischemia. Neurochem Int (2012) 61(5):659–65. doi:10.1016/j.neuint.2012.06.009
9. The Hypothermia After Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med (2002) 346:549–56. doi:10.1056/NEJMoa012689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet (2005) 365:663–70. doi:10.1016/S0140-6736(05)17946-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Sarkar S, Barks JD, Donn SM. Whole-body hypothermia for neonates with HI encephalopathy. N Engl J Med (2005) 353:1574–84. doi:10.1056/NEJMcps050929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Moler FW, Silverstein FS, Meert KL, Clark AE, Holubkov R, Browning B, et al. Rationale, timeline, study design, and protocol overview of the therapeutic hypothermia after pediatric cardiac arrest trials. Pediatr Crit Care Med (2013) 14(7):e304–15. doi:10.1097/PCC.0b013e31828a863a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med (2012) 366:2085–92. doi:10.1056/NEJMoa1112066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med (2009) 361:1349–58. doi:10.1056/NEJMoa0900854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Jenkins DD, Rollins LG, Perkel JK, Wagner CL, Katikaneni LP, Bass WT, et al. Serum cytokines in a clinical trial of hypothermia for neonatal hypoxic-ischemic encephalopathy. J Cereb Blood Flow Metab (2012) 32:1888–96. doi:10.1038/jcbfm.2012.83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Cilio MR, Ferriero DM. Synergistic neuroprotective therapies with hypothermia. Semin Fetal Neonatal Med (2010) 15:293–8. doi:10.1016/j.siny.2010.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Fernández-López D, Faustino J, Derugin N, Wendland M, Lizasoain I, Moro MA, et al. Reduced infarct size and accumulation of microglia in rats treated with WIN 55,212-2 after neonatal stroke. Neuroscience (2012) 207:307–15. doi:10.1016/j.neuroscience.2012.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Alvarez FJ, Lafuente H, Rey-Santano MC, Mielgo VE, Gastiasoro E, Rueda M, et al. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in HI newborn piglets. Pediatr Res (2008) 64:653–8. doi:10.1203/PDR.0b013e318186e5dd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lafunte H, Alvarez FJ, Pazos MR, Alvarez A, Rey-Santano MC, Mielgo V, et al. Cannabidiol reduces brain damage and improves functional recovery after acute HI in newborn pigs. Pediatr Res (2011) 70:272–7. doi:10.1038/pr.2011.497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Pazos MR, Mohammed N, Lafuente H, Santos M, Martínez-Pinilla E, Moreno E, et al. Mechanisms of cannabidiol neuroprotection in hypoxic ischemic newborn pigs: role of 5HT1A and CB2 receptors. Neuropharmacology (2013) 71:282–91. doi:10.1016/j.neuropharm.2013.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Castillo A, Tolón MR, Fernández-Ruiz J, Romero J, Martinez-Orgado J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB2 and adenosine receptors. Neurobiol Dis (2010) 37:434–40. doi:10.1016/j.nbd.2009.10.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shen H, Hu X, Liu C, Wang S, Zhang W, Gao H, et al. Ethyl pyruvate protects against HI brain injury via anti-cell death and anti-inflammatory mechanisms. Neurobiol Dis (2010) 37:711–22. doi:10.1016/j.nbd.2009.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Brochu ME, Girard S, Lavoie K, Sébire G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: an experimental study. J Neuroinflammation (2011) 8:55. doi:10.1186/1742-2094-8-55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Ferriero DM. Neonatal brain injury. N Engl J Med (2004) 351(19):1985–95. doi:10.1056/NEJMra041996
25. Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med (2006) 40(3):388–97. doi:10.1016/j.freeradbiomed.2005.08.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal HI. Ann Neurol (2011) 69(5):743–58. doi:10.1002/ana.22419
27. Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after HI in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis (2001) 8:207–19. doi:10.1006/nbdi.2000.0371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Beilharz EJ, Williams CE, Dragunow M, Sirimanne ES, Gluckman PD. Mechanisms of delayed cell death following HI injury in the immature rat: evidence for apoptosis during selective neuronal loss. Brain Res Mol Brain Res (1995) 29:1–14. doi:10.1016/0169-328X(94)00217-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Edwards AD, Yue X, Cox P, Hope PL, Azzopardi DV, Squier MV, et al. Apoptosis in the brains of infants suffering intrauterine cerebral injury. Pediatr Res (1997) 42:684–9. doi:10.1203/00006450-199711000-00022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Martin LJ, Brambrink A, Koehler RC, Traystman RJ. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by HI. J Comp Neurol (1997) 377(2):262–85. doi:10.1002/(SICI)1096-9861(19970113)377:2<262::AID-CNE8>3.0.CO;2-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol (2004) 5(6):575–81. doi:10.1038/ni1078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol (2009) 9:429–39. doi:10.1038/nri2565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Liu F, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin (2013) 34:1121–30. doi:10.1038/aps.2013.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. del Rio Hortega P, Penfield W. Cerebral cicatrix: the reaction of neuroglia and microglia to brain wounds. Bull Johns Hopkins Hosp (1927) 41:278–82.
35. Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience (1992) 48:405–15. doi:10.1016/0306-4522(92)90500-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience (1990) 39(1):151–70. doi:10.1016/0306-4522(90)90229-W
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308:1314–8. doi:10.1126/science.1110647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Pakkenberg B, Pelvig D, Marner L, Bundgaard MJ, Gundersen HJ, Nyengaard JR, et al. Aging and the human neocortex. Exp Gerontol (2003) 38:95–9. doi:10.1016/S0531-5565(02)00151-1
39. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333(6048):1456–8. doi:10.1126/science.1202529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement dependent manner. Neuron (2012) 74:1–15. doi:10.1016/j.neuron.2012.03.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci (2012) 35:369–89. doi:10.1146/annurev-neuro-061010-113810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates developmental CNS synapse elimination. Cell (2007) 131:1164–78. doi:10.1016/j.cell.2007.10.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Rice JE III, Vannucci RC, Brierley JB. The influence of immaturity on HI brain damage in the rat. Ann Neurol (1981) 9(2):131–41. doi:10.1002/ana.410090206
44. Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, et al. Rat model of perinatal HI brain damage. J Neurosci Res (1999) 55:158–63. doi:10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.3.CO;2-T
45. Hagberg H. Glycine and modulation of the NMDA receptor after severe asphyxia. Acta Paediatr (1999) 88:1049–50. doi:10.1111/j.1651-2227.1999.tb00984.x
46. Morioka T, Kalehua AN, Streit WJ. Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia. Acta Neuropathol (1992) 83(2):149–57. doi:10.1007/BF00308474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci (2008) 28(9):2221–30. doi:10.1523/JNEUROSCI.5643-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol (2012) 71(4):444–57. doi:10.1002/ana.22620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain (2009) 132(2):288–95. doi:10.1093/brain/awn109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Deng Y, Lu J, Sivakumar V, Ling EA, Kaur C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol (2008) 18:387–400. doi:10.1111/j.1750-3639.2008.00138.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Deng YY, Lu J, Ling EA, Kaur C. Microglia-derived macrophage colony stimulating factor promotes generation of proinflammatory cytokines by astrocytes in the periventricular white matter in the hypoxic neonatal brain. Brain Pathol (2010) 20:909–25. doi:10.1111/j.1750-3639.2010.00387.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Baburamani AA, Supramaniam VG, Hagberg H, Mallard C. Microglia toxicity in preterm brain injury. Reprod Toxicol (2014) 48:106–12. doi:10.1016/j.reprotox.2014.04.002
53. Saadani-Makki F, Kannan S, Makki M, Muzik O, Janisse J, Romero R, et al. Intrauterine endotoxin administration leads to white matter diffusivity changes in newborn rabbits. J Child Neurol (2009) 24(9):1179–89. doi:10.1177/0883073809338213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Albertsson AM, Bi D, Duan L, Zhang X, Leavenworth JW, Qiao L, et al. The immune response after HI in a mouse model of preterm brain injury. J Neuroinflammation (2014) 11(1):153. doi:10.1186/s12974-014-0153-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Van Steenwinckel J, Schang AL, Sigaut S, Chhor V, Degos V, Hagberg H, et al. Brain damage of the preterm infant: new insights into the role of inflammation. Biochem Soc Trans (2014) 42(2):557–63. doi:10.1042/BST20130284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, et al. Lipopolysaccharide sensitizes neonatal HI brain injury in a MyD88-dependent manner. J Immunol (2009) 183:7471–7. doi:10.4049/jimmunol.0900762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Kaur C, Ling EA. Blood brain barrier in hypoxic ischemic conditions. Curr Neurovasc Res (2008) 5:71–81. doi:10.2174/156720208783565645
58. Brown RC, Mark KS, Egleton RD, Davis TP. Protection against hypoxia-induced blood-brain barrier disruption: changes in intracellular calcium. Am J Physiol Cell Physiol (2004) 286:C1045–52. doi:10.1152/ajpcell.00360.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents improvement by minocycline in vivo and in vitro. Stroke (2006) 37:1087–93. doi:10.1161/01.STR.0000206281.77178.ac
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Ladeby R, Wirenfeldt M, Dalmau I, Gregersen R, García-Ovejero D, Babcock A, et al. Proliferating resident microglia express the stem cell antigen CD34 in response to acute neural injury. Glia (2005) 50:121–31. doi:10.1002/glia.20159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, et al. Microglial cell population dynamics in the injured adult CNS. Brain Res Brain Res Rev (2005) 48:196–206. doi:10.1016/j.brainresrev.2004.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab (2007) 27:1941–53. doi:10.1038/sj.jcbfm.9600495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Sawada M, Suzumura A, Yamamoto H, Marunouchi T. Activation and proliferation of the isolated microglia by colony stimulating factor-1 and possible involvement of protein kinase C. Brain Res (1990) 509:119–24. doi:10.1016/0006-8993(90)90317-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Schöbitz B, de Kloet ER, Sutanto W, Holsboer F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur J Neurosci (1993) 5:1426–35. doi:10.1111/j.1460-9568.1993.tb00210.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Brockhaus J, Moller T, Kettenmann H. Phagocytozing ameboid microglial cells studied in a mouse corpus callosum slice preparation. Glia (1996) 16:81–90. doi:10.1002/(SICI)1098-1136(199601)16:1<81::AID-GLIA9>3.0.CO;2-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol (2007) 28(3):138–45. doi:10.1016/j.it.2007.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Leonardo CC, Eakin AK, Ajmo JM, Collier LA, Pennypacker KR, Strongin AY, et al. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after HI in the neonatal rat. J Neuroinflammation (2008) 5:34. doi:10.1186/1742-2094-5-34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Leonardo CC, Pennypacker KR. Neuroinflammation and MMPs: potential therapeutic targets in neonatal HI injury. J Neuroinflammation (2009) 6:13. doi:10.1186/1742-2094-6-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia (2006) 53:688–95. doi:10.1002/glia.20328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Endoh M, Maiese K, Pulsinelli WA, Wagner JA. Reactive astrocytes express NADPH diaphorase in vivo after transient ischemia. Neurosci Lett (1993) 154:125–8. doi:10.1016/0304-3940(93)90187-P
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Iadecola C, Xu X, Zhang F, el-Fakahany EE, Ross ME, et al. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J Cereb Blood Flow Metab (1995) 15:52–9. doi:10.1038/jcbfm.1995.6
72. Nakashima MN, Yamashita K, Kataoka Y, Yamashita YS, Niwa M. Time course of nitric oxide synthase activity in neuronal, glial, and endothelial cells of rat striatum following focal cerebral ischemia. Cell Mol Neurobiol (1995) 15:341–9. doi:10.1007/BF02089944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Feuerstein GZ, Wang X, Barone FC. The role of cytokines in the neuropathology of stroke and neurotrauma. Neuroimmunomodulation (1998) 5:143–59. doi:10.1159/000026331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Dong Y, Benveniste EN. Immune function of astrocytes. Glia (2001) 36:180–90. doi:10.1002/glia.1107
75. Hanisch UK. Microglia as a source and target of cytokines. Glia (2002) 40:140–55. doi:10.1002/glia.10161
76. Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab (1999) 19:819–34. doi:10.1097/00004647-199908000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Hedtjarn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after HI. J Cereb Blood Flow Metab (2004) 24(12):1333–51. doi:10.1097/01.WCB.0000141559.17620.36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Ferrarese C, Mascarucci P, Zoia C, Cavarretta R, Frigo M, Begni B, et al. Increased cytokine release from peripheral blood cells after acute stroke. J Cereb Blood Flow Metab (1999) 19:1004–9. doi:10.1097/00004647-199909000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, et al. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol (1995) 56:127–34. doi:10.1016/0165-5728(94)00138-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, et al. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol (2002) 125:59–65. doi:10.1016/S0165-5728(02)00033-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab (2002) 22:308–17. doi:10.1097/00004647-200203000-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kumai Y, Ooboshi H, Takada J, Kamouchi M, Kitazono T, Egashira K, et al. Anti-monocyte chemoattractant protein-1 gene therapy protects against focal brain ischemia in hypertensive rats. J Cereb Blood Flow Metab (2004) 24:1359–68. doi:10.1097/01.WCB.0000143534.76388.3C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Kichev A, Rousset CI, Baburamani AA, Levison SW, Wood TL, Gressens P, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling and cell death in the immature CNS after HI and inflammation. J Biol Chem (2014) 289(13):9430–9. doi:10.1074/jbc.M113.512350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Savman K, Blennow M, Gustafson K, Tarkowski E, Hagberg H. Cytokine response in cerebrospinal fluid after birth asphyxia. Pediatr Res (1998) 43(6):746–51. doi:10.1203/00006450-199806000-00006
85. Fullerton HJ, Ditelberg JS, Chen SF, Sarco DP, Chan PH, Epstein CJ, et al. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol (1998) 44:357–64. doi:10.1002/ana.410440311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Ferriero DM. Oxidant mechanisms in neonatal HI. Dev Neurosci (2001) 23:198–202. doi:10.1159/000046143
87. Mischel RE, Kim YS, Sheldon RA, Ferriero DM. Hydrogen peroxide is selectively toxic to immature murine neurons in vitro. Neurosci Lett (1997) 231:17–20. doi:10.1016/S0304-3940(97)00531-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Pathipati P, Müller S, Jiang X, Ferriero D. Phenotype and secretory responses to oxidative stress in microglia. Dev Neurosci (2013) 35:241–54. doi:10.1159/000346159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Maeda Y, Matsumoto M, Hori O, Kuwabara K, Ogawa S, Yan SD, et al. Hypoxia/reoxygenation-mediated induction of astrocyte interleukin 6: a paracrine mechanism potentially enhancing neuron survival. J Exp Med (1994) 180(6):2297–308. doi:10.1084/jem.180.6.2297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Ali C, Nicole O, Docagne F, Lesne S, MacKenzie ET, Nouvelot A, et al. Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitotoxicity in the brain. J Cereb Blood Flow Metab (2000) 20(6):956–66. doi:10.1097/00004647-200006000-00008
91. Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron (1994) 12(1):139–53. doi:10.1016/0896-6273(94)90159-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to HI brain injury in neonatal rats. J Neurosci (2003) 23(28):9459–68.
93. Järlestedt K, Rousset CI, Ståhlberg A. Receptor for complement peptide C3a: a therapeutic target for neonatal HI brain injury. FASEB J (2013) 27(9):3797–804. doi:10.1096/fj.13-230011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Vila N, Castillo J, Dávalos A, Chamorro A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke (2001) 31:2325–9. doi:10.1161/01.STR.31.10.2325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Vela JM, Molina-Holgado E, Arévalo-Martín A, Almazán G, Guaza C. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Mol Cell Neurosci (2002) 20:489–502. doi:10.1006/mcne.2002.1127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Clark WM, Rinker LG, Lessov NS, Hazel K, Eckenstein F. Time course of IL-6 expression in experimental CNS ischemia. Neurol Res (1999) 21:287–92.
97. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med (2011) 17:796–808. doi:10.1038/nm.2399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, et al. Biology and action of colony – stimulating factor-1. Mol Reprod Dev (1997) 46:4–10. doi:10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Hao AJ, Dheen ST, Ling EA. Expression of macrophage colony-stimulating factor and its receptor in microglia activation is linked to teratogen-induced neuronal damage. Neuroscience (2002) 112:889–90. doi:10.1016/S0306-4522(02)00144-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein-1 and other beta-chemokines in murine glial cells. J Neuroimmunol (1995) 60:143–50. doi:10.1016/0165-5728(95)00064-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Taub DD, Proost P, Murphy WJ, Anver M, Longo DL, van Damme J, et al. Monocyte chemotactic protein-1 (MCP-1), -2, and -3 are chemotactic for human T lymphocytes. J Clin Invest (1995) 95:1370–6. doi:10.1172/JCI117788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Gunn MD, Nelken NA, Liao X, Williams LT. Monocyte chemoattractant protein-1 is sufficient for the chemotaxis of monocytes and lymphocytes in transgenic mice but requires an additional stimulus for inflammatory activation. J Immunol (1997) 158:376–83.
103. Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci (2007) 27(10):2596–605. doi:10.1523/JNEUROSCI.5360-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Neumann J, Sauerzweig S, Rönicke R, Gunzer F, Dinkel K, Ullrich O, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci (2008) 28(23):5965–75. doi:10.1523/JNEUROSCI.0060-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Hayashi Y, Tomimatsu Y, Suzuki H, Yamada J, Wu Z, Yao H, et al. The intra-arterial injection of microglia protects hippocampal CA1 neurons against global ischemia-induced functional deficits in rats. Neuroscience (2006) 142:87–96. doi:10.1016/j.neuroscience.2006.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Faustino JV, Wang X, Johnson CE, Klibanov A, Derugin N, Wendland MF, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci (2011) 31(36):12992–3001. doi:10.1523/JNEUROSCI.2102-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia (2002) 40:133–9. doi:10.1002/glia.10154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat (1968) 85:145–57. doi:10.1007/BF00325030
109. Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol (2013) 5(2):73–90.
110. Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) (2012) 60(4):251–66. doi:10.1007/s00005-012-0181-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci (2008) 28:11650–61. doi:10.1523/JNEUROSCI.3024-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke (2012) 43:3063–70. doi:10.1161/STROKEAHA.112.659656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci (2007) 30:596–602. doi:10.1016/j.tins.2007.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Koning N, Swaab DF, Hoek RM, Huitinga I. Distribution of the immune inhibitory molecules CD200 and CD200R in the normal CNS and multiple sclerosis lesions suggests neuron-glia and glia-glia interactions. J Neuropathol Exp Neurol (2009) 68:159–67. doi:10.1097/NEN.0b013e3181964113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab (2008) 28:1707–21. doi:10.1038/jcbfm.2008.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, et al. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol (2005) 174:2004–11. doi:10.4049/jimmunol.174.4.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina Bordignon P, et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem (2009) 110:284–94. doi:10.1111/j.1471-4159.2009.06130.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science (2000) 290:1768–71. doi:10.1126/science.290.5497.1768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci (2006) 9:917–24. doi:10.1038/nn1715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci (2010) 13:411–3. doi:10.1038/nn.2511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci (2011) 31:16327–35. doi:10.1523/JNEUROSCI.3611-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Koning N, Bö L, Hoek RM, Huitinga I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol (2007) 62:504–14. doi:10.1002/ana.21220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Ajmo CT Jr., Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, et al. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res (2008) 86:2227–34. doi:10.1002/jnr.21661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab (2006) 26:654–65. doi:10.1038/sj.jcbfm.9600217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Jellema RK, Lima Passos V, Zwanenburg A, Ophelders DR, De Munter S, Vanderlocht J, et al. Cerebral inflammation and mobilization of the peripheral immune system following global HI in preterm sheep. J Neuroinflammation (2013) 10(13):1–19. doi:10.1186/1742-2094-10-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation (2006) 113:2105–12. doi:10.1161/CIRCULATIONAHA.105.593046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther (2003) 304(1):1–7. doi:10.1124/jpet.102.035048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol (2003) 183:25–33. doi:10.1016/S0014-4886(03)00082-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Chen H, Chopp M, Zhang RL, Bodzin G, Chen Q, Rusche JR, et al. Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann Neurol (1994) 35:458–63. doi:10.1002/ana.410350414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Chopp M, Zhang RL, Chen H, Li Y, Jiang N, Rusche JR. Postischemic administration of an anti-Mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke (1994) 25:869–75. doi:10.1161/01.STR.25.4.869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Zhang ZG, Chopp M, Tang WX, Jiang N, Zhang RL. Postischemic treatment (2-4 h) with anti-CD11b and anti-CD18 monoclonal antibodies are neuroprotective after transient (2 h) focal cerebral ischemia in the rat. Brain Res (1995) 698:79–85. doi:10.1016/0006-8993(95)00830-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Juul SE, Ferriero DM. Pharmacologic neuroprotective strategies in neonatal brain injury. Clin Perinatol (2014) 41(1):119–31. doi:10.1016/j.clp.2013.09.004
133. Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol (2011) 10(4):372–82. doi:10.1016/S1474-4422(11)70016-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev (2006) 58(3):389–462. doi:10.1124/pr.58.3.2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Núñez E, Benito C, Pazos MR, Barbachano A, Fajardo O, González S, et al. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse (2004) 53(4):208–13. doi:10.1002/syn.20050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Carlisle SJ, Marciano-Cabral F, Staab A, Ludwick C, Cabral GA. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int Immunopharmacol (2002) 2:69–82. doi:10.1016/S1567-5769(01)00147-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci (2003) 23:1398–405.
139. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature (1993) 365:61–5. doi:10.1038/365061a0
140. Hayakawa K, Mishima K, Hazekawa M, Sano K, Irie K, Orito K, et al. Cannabidiol potentiates pharmacological effects of δ9-tetrahydrocannabinol via CB1 receptor-dependent mechanism. Brain Res (2008) 1188:157–64. doi:10.1016/j.brainres.2007.09.090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Brown AJ. Novel cannabinoid receptors. Br J Pharmacol (2007) 152(5):567–75. doi:10.1038/sj.bjp.0707481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Braida D, Pegorini S, Arcidiacono MV, Consalez GG, Croci L, Sala M. Post-ischemic treatment with cannabidiol prevents electroencephalographic flattening, hyperlocomotion and neuronal injury in gerbils. Neurosci Lett (2003) 346:61–4. doi:10.1016/S0304-3940(03)00569-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Mishima K, Hayakawa K, Abe K, Ikeda T, Egashira N, Iwasaki K, et al. Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism. Stroke (2003) 36:1077–82.
144. Hayakawa K, Mishima K, Nozako M, Hazekawa M, Irie K, Fujioka M, et al. Delayed treatment with cannabidiol has a cerebroprotective action via a cannabinoid receptor-independent myeloperoxidase-inhibiting mechanism. J Neurochem (2007) 102:1488–96. doi:10.1111/j.1471-4159.2007.04565.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Scuderi C, Filippis DD, Iuvone T, Blasio A, Steardo A, Esposito G. Cannabidiol in medicine: a review of its therapeutic potential in CNS disorders. Phytother Res (2009) 23:597–602. doi:10.1002/ptr.2625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Nawashiro H, Martin D, Hallenbeck JM. Neuroprotective effects of TNF binding protein in focal cerebral ischemia. Brain Res (1997) 778(2):265–71. doi:10.1016/S0006-8993(97)00981-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Lavine SD, Hofman FM, Zlokovic BV. Circulating antibody against tumor necrosis factor-alpha protects rat brain from reperfusion injury. J Cereb Blood Flow Metab (1998) 18:52–8. doi:10.1097/00004647-199801000-00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor blockers etanercept, infliximab and adalimumab. J Dermatolog Treat (2004) 15(5):280–94. doi:10.1080/09546630410017275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab (1996) 16:932–40. doi:10.1097/00004647-199609000-00017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Emsley HCA, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry (2005) 76:1366–72. doi:10.1136/jnnp.2004.054882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Plane JM, Shen Y, Pleasure DE. Prospects for minocycline neuroprotection. Arch Neurol (2010) 67(12):1442–8. doi:10.1001/archneurol.2010.191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against HI injury. Ann Neurol (2002) 52(1):54–61. doi:10.1002/ana.10242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Fox C, Dingman A, Derugin N, Wendland MF, Manabat C, Ji S, et al. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab (2005) 25(9):1138–49. doi:10.1038/sj.jcbfm.9600121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Diguet E, Gross CE, Tison F, Bezard E. Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Exp Neurol (2004) 189:1–4. doi:10.1016/j.expneurol.2004.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Tsuji M, Wilson MA, Lange MS, Johnston MV. Minocycline worsens HI brain injury in a neonatal mouse model. Exp Neurol (2004) 189:58–65. doi:10.1016/j.expneurol.2004.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Yang L, Sugama S, Chirichigno JW, Gregorio J, Lorenzl S, Shin DH, et al. Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res (2003) 74:278–85. doi:10.1002/jnr.10709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Sriram K, Miller DB, O’Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-α. J Neurochem (2006) 96:706–18. doi:10.1111/j.1471-4159.2005.03566.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience (2003) 121(3):619–28. doi:10.1016/S0306-4522(03)00558-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Kim H-S, Suh Y-H. Minocycline and neurodegenerative diseases. Behav Brain Res (2009) 196(2):168–79. doi:10.1016/j.bbr.2008.09.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Parashos SA, Swearingen CJ, Biglan KM, Bodis-Wollner I, Liang GS, Ross GW, et al. Determinants of the timing of symptomatic treatment in early Parkinson disease: the National Institutes of Health Exploratory Trials in Parkinson disease (NET-PD) experience. Arch Neurol (2009) 66(9):1099–104. doi:10.1001/archneurol.2009.159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Gordon PH, Moore DH, Gelinas DF, Qualls C, Meister ME, Werner J, et al. Placebo controlled phase I/II studies of minocycline in amyotrophic lateral sclerosis. Neurology (2004) 62(10):1845–7. doi:10.1212/01.WNL.0000125321.92112.7E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, et al. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology (2004) 46(3):404–11. doi:10.1016/j.neuropharm.2003.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Kishi Y, Ohta S, Kasuya N, Sakita S, Ashikaga T, Isobe M. Ibudilast: a non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall. Cardiovasc Drug Rev (2001) 19(3):215–25. doi:10.1111/j.1527-3466.2001.tb00066.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Kalueff AV, Eremin KO, Tuohimaa P. Mechanisms of neuroprotective action of vitamin D3. Biochemistry (2004) 69(7):738–41.
165. Meehan TF, DeLuca HF. The vitamin D receptor is necessary for 1alpha,25-dihydroxyvitamin D(3) to suppress experimental autoimmune encephalomyelitis in mice. Arch Biochem Biophys (2002) 408(2):200–4. doi:10.1016/S0003-9861(02)00580-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Garcion E, Nataf S, Berod A, Darcy F, Brachet P. 1,25-Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat CNS during experimental allergic encephalomyelitis. Brain Res Mol Brain Res (1997) 45(2):255–67. doi:10.1016/S0169-328X(96)00260-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Garcion E, Sindji L, Nataf S, Brachet P, Darcy F, Montero-Menei CN. Treatment of experimental autoimmune encephalomyelitis in rat by 1,25-dihydroxyvitamin D3 leads to early effects within the CNS. Acta Neuropathol (2003) 105(5):438–48.
168. Hayes CE, Cantorna MT, DeLuca HF. Vitamin D and multiple sclerosis. Proc Soc Exp Biol Med (1997) 216(1):21–7. doi:10.3181/00379727-216-44153A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. d’Hellencourt CL, Montero-Menei CN, Bernard R, Couez D. Vitamin D3 inhibits proinflammatory cytokines and nitric oxide production by the EOC13 microglial cell line. J Neurosci Res (2003) 71:575–82. doi:10.1002/jnr.10491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Asanuma M, Miyazaki I, Ogawa N. Neuroprotective effects of nonsteroidal anti-inflammatory drugs on neurodegenerative diseases. Curr Pharm Des (2004) 10(6):695–700. doi:10.2174/1381612043453072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Garcia-Segura LM, Balthazart J. Steroids and neuroprotection: new advances. Front Neuroendocrinol (2009) 30(2):5–9. doi:10.1016/j.yfrne.2009.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Tutak E, Satar M, Zorludemir S. Neuroprotective effects of indomethacin and aminoguanidine in the newborn rats with HI cerebral injury. Neurochem Res (2005) 30(8):937–42. doi:10.1007/s11064-005-5978-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Vaughan CJ, Delanty N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke (1999) 30:1969–73. doi:10.1161/01.STR.30.9.1969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Rudolphi KA, Schubert P. Modulation of neuronal and glial cell function by adenosine and neuroprotection in vascular dementia. Behav Brain Res (1997) 83:123–8. doi:10.1016/S0166-4328(97)86055-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Plaschke K, Grant M, Weigand MA, Züchner J, Martin E, Bardenheuer HJ. Neuromodulatory effect of propentofylline on rat brain under acute and long-term hypoperfusion. Br J Pharmacol (2001) 133:107–16. doi:10.1038/sj.bjp.0704061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Haskó G, Pacher P, Vizi ES, Illes P. Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci (2005) 26(10):511–6. doi:10.1016/j.tips.2005.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Haskó G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol (2004) 25(1):33–9. doi:10.1016/j.it.2003.11.003
178. Villapol S, Fau S, Renolleau S, Biran V, Charriaut-Marlangue C, Baud O. Melatonin promotes myelination by decreasing white matter inflammation after neonatal stroke. Pediatr Res (2011) 69:51–5. doi:10.1203/PDR.0b013e3181fcb40b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Robertson NJ, Faulkner S, Fleiss B, Bainbridge A, Andorka C, Price D, et al. Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain (2013) 136:90–105. doi:10.1093/brain/aws285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Sun Y, Calvert JW, Zhang JH. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke (2005) 36:1672–8. doi:10.1161/01.STR.0000173406.04891.8c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Juul SE, Beyer RP, Bammler TK, McPherson RJ, Wilkerson J, Farin FM. Microarray analysis of high-dose recombinant erythropoietin treatment of unilateral brain injury in neonatal mouse hippocampus. Pediatr Res (2009) 65:485–92. doi:10.1203/PDR.0b013e31819d90c8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Zacharias R, Schmidt M, Kny J, Sifringer M, Bercker S, Bittigau P, et al. Dose-dependent effects of erythropoietin in propofol anesthetized neonatal rats. Brain Res (2010) 1343:14–9. doi:10.1016/j.brainres.2010.04.081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Kumral A, Tugyan K, Gonenc S, Genc K, Genc S, Sonmez U, et al. Protective effects of erythropoietin against ethanol-induced apoptotic neurodegeneration and oxidative stress in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res (2005) 160:146–56. doi:10.1016/j.devbrainres.2005.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Chattopadhyay A, Choudhury TD, Bandyopadhyay D, Datta AG. Protective effect of erythropoietin on the oxidative damage of erythrocyte membrane by hydroxyl radical. Biochem Pharmacol (2000) 59:419–25. doi:10.1016/S0006-2952(99)00277-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Petzelt C, Blom P, Schmehl W, Müller J, Kox WJ. Prevention of neurotoxicity in hypoxic cortical neurons by the noble gas xenon. Life Sci (2003) 72:1909–18. doi:10.1016/S0024-3205(02)02439-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Ma D, Hossain M, Chow A, Arshad M, Battson RM, Sanders RD, et al. Xenon and hypothermia combine to provide neuroprotection from neonatal asphyxia. Ann Neurol (2005) 58:182–93. doi:10.1002/ana.20547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Hobbs C, Thoresen M, Tucker A, Aquilina K, Chakkarapani E, Dingley J. Xenon and hypothermia combine additively, offering long-term functional and histopathologic neuroprotection after neonatal hypoxia/ischemia. Stroke (2008) 39:1307–13. doi:10.1161/STROKEAHA.107.499822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Dingley J, Hobbs C, Ferguson J, Stone J, Thoresen M. Xenon/hypothermia neuroprotection regimens in spontaneously breathing neonatal rats after HI insult: the respiratory and sedative effects. Anesth Analg (2008) 106:916–23. doi:10.1213/ane.0b013e3181618669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Thoresen M, Hobbs CE, Wood T, Chakkarapani E, Dingley J. Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal HI. J Cereb Blood Flow Metab (2009) 29:707–14. doi:10.1038/jcbfm.2008.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Zhuang L, Yang T, Zhao H, Fidalgo AR, Vizcaychipi MP, Sanders RD, et al. The protective profile of argon, helium, and xenon in a model of neonatal asphyxia in rats. Crit Care Med (2012) 40:1724–30. doi:10.1097/CCM.0b013e3182452164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Chakkarapani E, Dingley J, Liu X, Hoque N, Aquilina K, Porter H, et al. Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann Neurol (2010) 68:330–41. doi:10.1002/ana.22016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Faulkner S, Bainbridge A, Kato T, Chandrasekaran M, Kapetanakis AB, Hristova M, et al. Xenon augmented hypothermia reduces early lactate/N-acetyl aspartate and cell death in perinatal asphyxia. Ann Neurol (2011) 70:133–50. doi:10.1002/ana.22387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Navath RS, Kurtoglu YE, Wang B, Kannan S, Romero R, Kannan RM. Dendrimer-drug conjugates for tailored intracellular drug release based on glutathione levels. Bioconjug Chem (2008) 19:2446–55. doi:10.1021/bc800342d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J, et al. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci Transl Med (2012) 4:130ra46. doi:10.1126/scitranslmed.3003162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Balakrishnan B, Nance E, Johnston MV, Kannan R, Kannan S. Nanomedicine in cerebral palsy. Int J Nanomedicine (2013) 8:4183–95. doi:10.2147/IJN.S35979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: hypoxia-ischemic encephalopathy, inflammation, developing brain, microglia, immune dysregulation
Citation: Bhalala US, Koehler RC and Kannan S (2015) Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain. Front. Pediatr. 2:144. doi: 10.3389/fped.2014.00144
Received: 03 November 2014; Accepted: 22 December 2014;
Published online: 14 January 2015.
Edited by:
Hitesh Singh Sandhu, The University of Tennessee Health Science Center, USAReviewed by:
Jhuma Sankar, All India Institute of Medical Sciences, IndiaMichael Shoykhet, Washington University in St. Louis School of Medicine, USA
Copyright: © 2015 Bhalala, Koehler and Kannan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Utpal S. Bhalala, Anesthesiology and Critical Care Medicine, Johns Hopkins Hospital, Bloomberg Children’s Center, 1800 Orleans Street, Suite 6349B, Baltimore, MD 21287, USA e-mail: ubhalal1@jhmi.edu