Missing cells: pathophysiology, diagnosis, and management of (pan)cytopenia in childhood
 Miriam Erlacher1,2
Miriam Erlacher1,2
 Brigitte Strahm1*
Brigitte Strahm1*
- 1Division of Pediatric Hematology and Oncology, Department of Pediatrics and Adolescent Medicine, University Medical Center of Freiburg, Freiburg, Germany
- 2Freiburg Institute for Advanced Studies, University of Freiburg, Freiburg, Germany
Peripheral blood cytopenia in children can be due to a variety of acquired or inherited diseases. Genetic disorders affecting a single hematopoietic lineage are frequently characterized by typical bone marrow findings, such as lack of progenitors or maturation arrest in congenital neutropenia or a lack of megakaryocytes in congenital amegakaryocytic thrombocytopenia, whereas antibody-mediated diseases such as autoimmune neutropenia are associated with a rather unremarkable bone marrow morphology. By contrast, pancytopenia is frequently associated with a hypocellular bone marrow, and the differential diagnosis includes acquired aplastic anemia, myelodysplastic syndrome, inherited bone marrow failure syndromes such as Fanconi anemia and dyskeratosis congenita, and a variety of immunological disorders including hemophagocytic lymphohistiocytosis. Thorough bone marrow analysis is of special importance for the diagnostic work-up of most patients. Cellularity, cellular composition, and dysplastic signs are the cornerstones of the differential diagnosis. Pancytopenia in the presence of a normo- or hypercellular marrow with dysplastic changes may indicate myelodysplastic syndrome. More challenging for the hematologist is the evaluation of the hypocellular bone marrow. Although aplastic anemia and hypocellular refractory cytopenia of childhood (RCC) can reliably be differentiated on a morphological level, the overlapping pathophysiology remains a significant challenge for the choice of the therapeutic strategy. Furthermore, inherited bone marrow failure syndromes are usually associated with the morphological picture of RCC, and the recognition of these entities is essential as they often present a multisystem disease requiring different diagnostic and therapeutic approaches. This paper gives an overview over the different disease entities presenting with (pan)cytopenia, their pathophysiology, characteristic bone marrow findings, and therapeutic approaches.
Introduction
(Pan)cytopenia in childhood can be caused by a variety of underlying diseases, including hematological and non-hematological entities. Overlapping phenotypes and pathophysiologies pose a major diagnostic challenge. However, an accurate and rapid diagnosis is essential for adequate therapy planning, surveillance, and genetic counseling. Bone marrow (BM) analysis is of special importance for the diagnostic work-up of cytopenias affecting one or more lineages. In the following, we will describe the individual disorders, their underlying pathophysiology and clinical characteristics, as well as typical BM findings.
The Hierarchy of Hematopoiesis
All peripheral blood cells originate from hematopoietic stem cells (HSCs) that reside in fetal liver and fetal or adult BM. HSCs are able to self-renew, which is essential for maintenance of lifelong hematopoiesis. They do not commit directly to single lineages, but instead differentiate into multipotent progenitors, common lymphoid and (erythro-) myeloid progenitors, which in turn give rise to more differentiated precursor cells. Lineage commitment is determined by a delicate network of transcription factors and epigenetic mechanisms that establish differentiation into the corresponding lineage while suppressing maturation toward other lineages (1). The hierarchical tree of hematopoiesis is depicted in Figure 1, but there is evidence that such a linear and rigid model is oversimplified (2). Also, a specific HSC subset has been reported to directly evolve into megakaryocytes, at least in the murine system (3). Depending on the differentiation stage affected, disturbances can occur in single or multiple lineages (Figure 1).
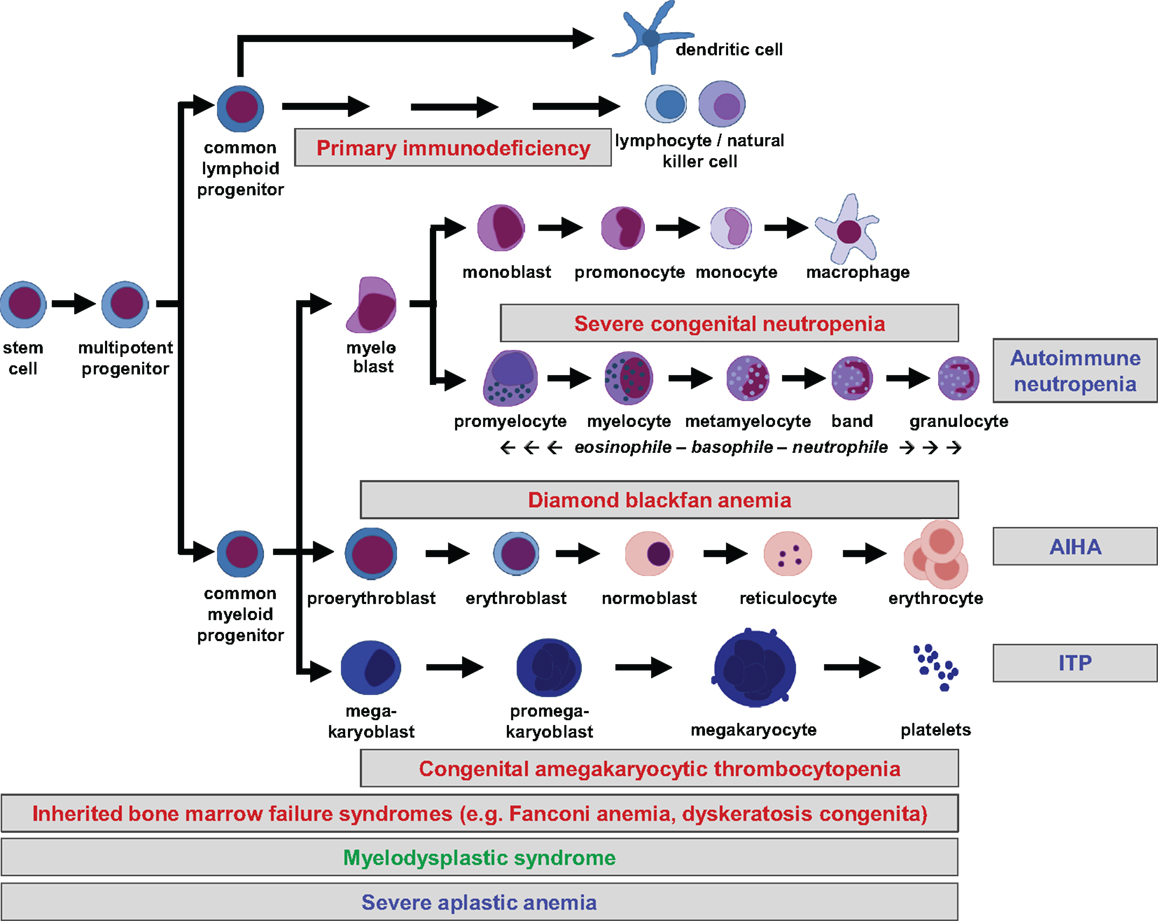
Figure 1. Hierarchical tree of human hematopoiesis. Disturbances leading to cytopenias can affect single or multiple lineages and be caused by cell-intrinsic or extrinsic mechanisms. Intrinsic defects are caused by inherited or acquired mutations, while extrinsic defects can be caused by autoreactive lymphocytes. A selection of frequent pediatric disorders is shown. Labeling: inherited defects: red; acquired mutations: green; autoimmune disorders: blue. AIHA, autoimmune hemolytic anemia; ITP, idiopathic thrombocytopenic purpura.
Signs and Symptoms of Cytopenia
The condition caused by low erythrocyte numbers and hemoglobin concentration is called anemia and is clinically characterized by paleness, weakness, and general malaise. Severe anemia due to rapid hemoglobin drop (i.e., blood loss, hemolysis) may lead to cardiovascular symptoms such as tachycardia and arterial hypotension. Anemia due to bone marrow failure usually has insidious manifestation. Reticulocytosis points toward active red cell production while lack of reticulocytes is indicative for insufficient erythropoiesis. Other informative parameters are mean corpuscular volume (MCV) and HbF levels as signs of stress hematopoiesis or low haptoglobin with increased unconjugated bilirubin in the presence or absence of a positive antiglobulin test indicating hemolysis (4).
Platelets are fragments of membrane and cytoplasm derived from megakaryocytes and important for hemostasis. Thrombocytopenia is characterized by cutaneous and mucosal bleeding signs, such as epistaxis, petechia, and purpura. Platelet size and morphology can help classifying the underlying disease. Platelets are abnormally small in patients with Wiskott–Aldrich syndrome (WAS) and large in patients with Bernard–Soulier syndrome or May–Hegglin anomaly (5). Platelet size diminishes over time, and therefore higher platelet volume may indicate the presence of less mature platelets in cases of increased degradation (e.g., idiopathic thrombocytopenic purpura, ITP) (6).
Lack of neutrophils, neutropenia, is characterized by an increased susceptibility to bacterial infections. Risk of infections correlates with granulocyte counts, with severe neutropenia (<500/μl) being associated with a high risk of life-threatening infections (7). Depending on the extent of neutropenia and the frequency of severe infections, prophylactic antibiotic treatment and/or G-CSF application might be considered. Response to G-CSF is indicative for the myelopoietic capacity of the BM.
In line with the many different types and developmental stages of lymphocytes, lymphopenia can affect an entire lineage (i.e., B, T, and/or NK cells) or individual mature cell types (e.g., regulatory T cells). Type of lacking cells as well as abundance of other lymphocyte types determine the phenotype of the individual disease. Phenotypes can be highly variable and range from (severe) combined immunodeficiency to mildly increased susceptibility to specific infections. Primary immune diseases can also be associated with immune dysregulation (8).
Neonatal cytopenias are particularly challenging due to the extremely diverse spectrum of differential diagnoses. Impairment of fetal erythropoiesis or thrombopoiesis due to inherited defects or prenatal infections can result in hydrops fetalis or perinatal hemorrhagic complications, respectively. Ineffective fetal hematopoiesis within the BM can lead to persistent extramedullary hematopoiesis in liver, spleen, and occasionally in the skin (9). Affected newborns are characterized by hepatosplenomegaly and may have skin infiltrations (so called “blueberry muffin” babies). The latter condition is indicative for hemolytic diseases, malignant BM infiltrations, infantile malignant osteopetrosis, or congenital infections (10). Neonatal erythroderma might indicate the presence of severe immune dysregulation as seen in Omenn’s syndrome, a hyperinflammatory variant of leaky SCID (11).
A detailed medical history is essential in all cases of cytopenias and should include details of concomitant health problems, susceptibility to bleeding or infections, previous exposure to drugs or toxins, and recent foreign travel. Family history should focus on hematological and immune disorders, cancer susceptibility, or unexplained infant deaths. Thorough physical examination should include the assessment of signs of lymphoproliferation (i.e., hepatosplenomegaly and lymphadenopathy), as well as dysmorphic features, and stigmata, indicating the presence of a syndromic disease.
Cytopenia Affecting a Single Lineage
In principle, single lineage-cytopenias may be caused by insufficient production or premature depletion of mature cells of the respective hematopoietic lineage. In the majority of cases, the latter is mediated by autoreactive antibodies, resulting in ITP, autoimmune neutropenia (AIN), or autoimmune hemolytic anemia (AIHA), respectively (12). Frequently, autoantibodies are formed as a reaction to infections or vaccinations due to the so-called “molecular mimicry” between pathogen components and blood cell antigens (13). Similarly, heparin-induced thrombocytopenia (HIT) is caused by antibodies specific for heparin-binding platelets (14). Generally, these disorders show normal BM morphology or hyperactive hematopoiesis in the corresponding lineage. Most tests for autoantibodies have a low sensitivity and specificity, and are therefore not required for the confirmation of the diagnosis, except for AIHA (15). Since the BM is normal in ITP or AIN and thus has the capacity of increased cell production if stressed, signs and symptoms are frequently milder compared to patients with hypoproliferative deficiencies of these cell lineages. Even in the complete absence of peripheral granulocytes, patients rarely suffer from life-threatening infections. In contrast to these clinically mostly benign autoimmune disorders, AIHA, especially in childhood, can be life-threatening because of rapid hemoglobin decline (16).
Furthermore, a premature destruction of red blood cells may be caused by defective hemoglobin, metabolic enzymes, or membrane components. These syndromes can be diagnosed by hemoglobin electrophoresis, determination of enzymatic activity or osmotic fragility tests, respectively, and will not be discussed here in more detail. Increased destruction of platelets and the subsequent risk of bleeding are also characteristic features of inherited disorders, such as Wiskott–Aldrich syndrome (WAS) and X-linked thrombocytopenia (XLT). While platelet generation has been shown to be unaffected in this syndrome, cytoskeletal changes occurring in circulating platelets lead to their premature degradation (17).
Similar to the single lineage-cytopenias caused by premature cell loss, disorders with inadequate cell generation can affect all individual lineages, i.e., megakaryopoiesis, erythropoiesis, and myelopoiesis. These disorders, their pathophysiologies, diagnostic characteristics, and symptoms are summarized in Table 1. Disorders with insufficient generation of lymphocytes result in (severe) combined or milder immunodeficiencies. Since they require distinct diagnostic approaches and therapies, these syndromes are beyond the scope of this review and were discussed elsewhere (18).
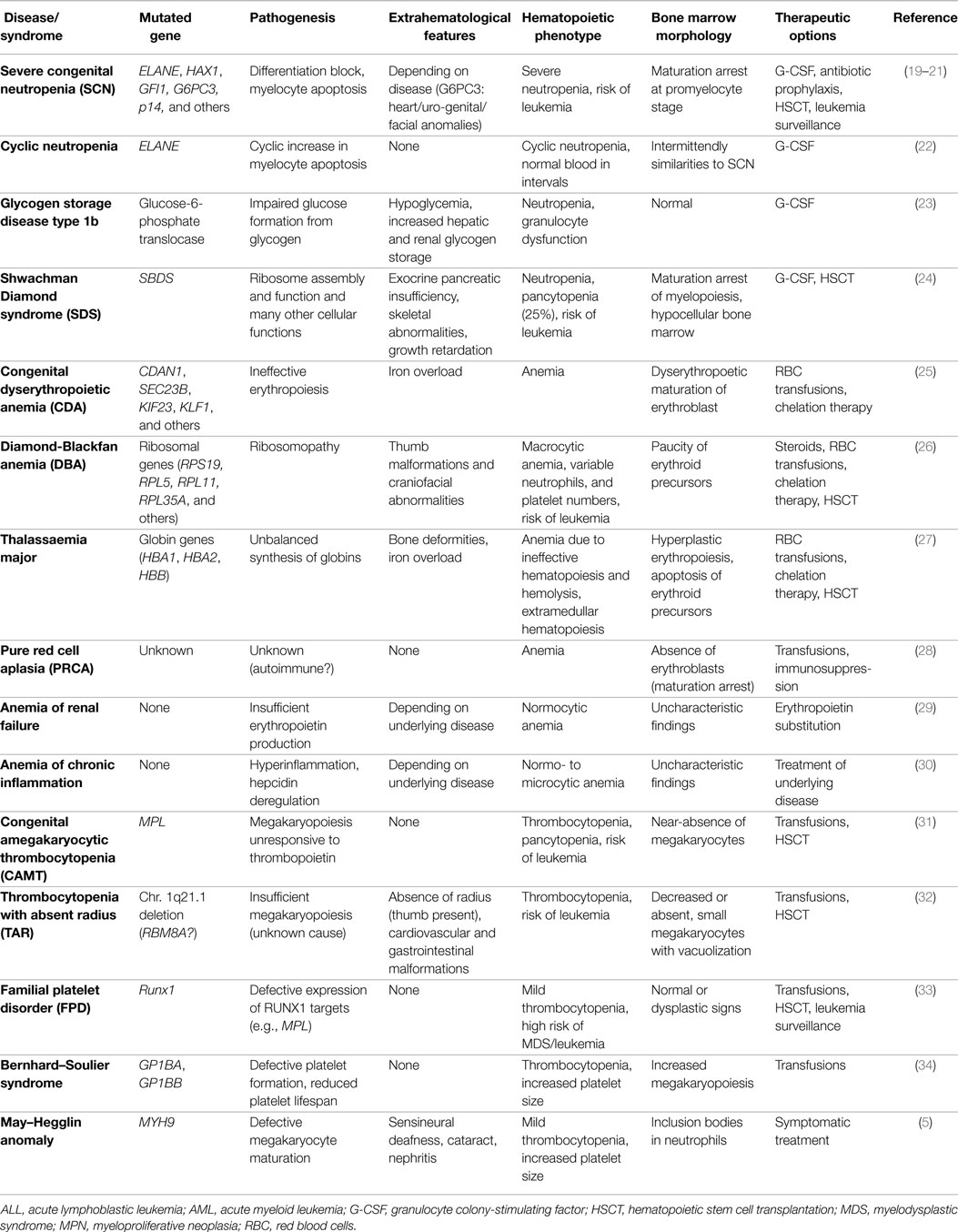
Table 1. Syndromes characterized by inadequate formation of mature blood cells resulting in single lineage cytopenia.
When More Lineages are Affected
Cytopenias affecting two or three blood lineages, the latter called pancytopenia (greek , pan = involving all), can also be caused by deregulation of either cell generation or degradation. As an additional layer of classification, the disorders can be divided into inherited or acquired BM failures. It is important to emphasize that inherited BM failure syndromes (IBMFS) as well as acquired BM failure can present in all age groups, and that in some cases an isolated cytopenia preceeds the development of pancytopenia. This requires particular attention and repeated reevaluation by the attending physician or hematologist. Isolated thrombocytopenia can initially be diagnosed as ITP, but additional anemia or neutropenia over time might indicate the presence of systemic autoimmune disorders such as systemic lupus erythematosus (SLE). Similarly, isolated neutropenia can be considered to be secondary to an infection, but the subsequent decrease of other blood cells should rapidly lead to BM analysis. In the following, we will describe the different forms of pancytopenia in more detail.
Pancytopenia as Consequence of Autoimmunity and Immune Dysregulation
While single-line immune-mediated degradation of platelets, erythrocytes, or neutrophils is frequent, combinations thereof are rare and often serious (35, 36). The combination of AIHA and thrombocytopenia, occurring either simultaneously or sequentially, is called Evans syndrome and is often a manifestation of the autoimmune lymphoproliferative syndrome (ALPS) (37). ALPS is caused by germline or somatic mutations in FAS, FASL, or CASP10, resulting in insufficient apoptosis of activated autoreactive lymphocytes via the extrinsic pathway (38, 39). Rarely, ALPS can be caused by RAS mutations resulting in defective intrinsic apoptosis of lymphocytes (40). Concomitant lymphoproliferation is characteristic for ALPS. Similarly, cytopenias can occur in acquired, multifactorial autoimmune syndromes such as SLE or the primary antiphospholipid syndrome (36). Germline syndromes characterized by autoimmunity are the IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked) syndrome caused by lack of regulatory T cells and the APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy) syndrome caused by insufficient induction of central (thymic) tolerance (41, 42). Other primary immunodeficiencies frequently leading to autoimmune cytopenia are leaky SCID, WAS, hyper-IgM syndrome, and common variable immunodeficiency (CVID) (36). Indicative of the presence of such syndromes are lymphoproliferation, autoantibodies, oligoclonal T cells, increased complement consumption, and signs of autoimmunity or autoinflammation affecting other organs (i.e., dermatitis, glomerulonephritis, and inflammatory bowel disease). Bone marrow findings are usually not characteristic and merely indicate increased cellular turnover.
Immune dysregulation is also the cause of pancytopenia in patients with hemophagocytic lymphohistiocytosis (HLH). This life-threatening syndrome is characterized by impaired pathogen elimination, hyperinflammation, and hystiocytic and lymphoid tissue infiltration. It can occur as familial disease (FHL with PRF1, UNC13D, STX11, or STXBP2 mutations), in syndromes characterized by additional albinism (i.e., Griscelli syndrome type II, Hermansky-Pudlak syndrome type II, and Chediak-Higashi syndrome) and secondary to infection, rheumatic, or neoplastic disorders. Secondary HLH is also known as macrophage activation syndrome (MAS). Deregulation of T and NK cell cytotoxicity and/or lysosomal trafficking are underlying mechanisms of HLH (43). Pancytopenia is not mediated by autoantibodies but instead by macrophage hyperactivation, resulting in hemophagocytosis and cytokine-mediated marrow suppression. Accordingly, hemophagocytosis in BM is one of the diagnostic criteria, next to hypertriglyceridemia, hypofibrinogenemia, and increased levels of ferritin and soluble IL2 receptor. Degranulation and cytotoxicity assays as well as genetic analysis confirm the diagnosis (44).
Extrinsic Conditions Associated with Impaired Hematopoiesis
Certain extrinsic, environmental conditions can interfere significantly with blood formation, either pre- or postnatally. The most frequent causes of impaired hematopoiesis are infections. Congenital TORCH infections (i.e., toxoplasmosis, rubella, cytomegalovirus, herpes simplex, and others) often result in decreased maturation of megakaryocytes and platelet formation, in combination with increased immune-mediated platelet destruction (45, 46). Parvovirus B19 infections lead to apoptosis and cell cycle arrest in infected fetal erythroblasts, thereby resulting in fetal anemia and hydrops (47). Also postnatally, parvovirus B19 can transiently affect erythroid progenitors. While healthy children are only mildly affected, children with hemolytic anemia and immunocompromised patients might develop aplastic crisis and persistent anemia, respectively (48). Many other viral infections are associated with transient hematopoietic depression of one or more lineages. Important infections to be considered in the differential diagnosis of peripheral cytopenias are hepatitis C, HIV (in children nowadays mostly due to vertical transmission), and Helicobacter pylori, all causing thrombocytopenia and some of them anemia. Visceral leishmaniasis, in contrast, results in an HLH-like disorder with massive phagocytosis of blood components (49, 50).
Other causes of suppressed hematopoiesis are nutritional deficiencies observed in anorexia nervosa or B12, folate, and iron deficiency. While mild deficiencies frequently cause anemia, severe lack of essential food components can result in complete BM failure and immunodeficiency (51–53).
Finally, hematopoiesis can be restricted by pathological microenvironmental conditions. In Gaucher disease, BM dysfunction is caused by accumulation of glucocerebrosides in macrophages and results in (pan)cytopenia (54). By contrast, osteopetrosis is caused by reduced bone resorption leading to bone sclerosis and, among other symptoms, restriction of the medullar cavity. This results in BM failure and compensatory extramedullary hematopoiesis. Different disease subtypes have been reported, with infantile malignant osteopetrosis being the most severe form (55).
Inherited Intrinsic Defects of Hematopoiesis
Various inborn genetic defects result in hematopoietic failure, and most of them are not restricted to hematopoiesis but also affect different organ systems. Traditionally, Fanconi anemia (FA), dyskeratosis congenita (DC), congenital amegakaryocytic thrombocytopenia (CAMT), thrombocytopenia with absent radii (TAR), Diamond Blackfan anemia (DBA), Shwachman Diamond syndrome (SDS), and severe congenital neutropenia (SCN) have been subsumed as inherited BM failure syndromes (IBMFS). BM failure can occur at different ages and has highly variable presentations. Disease kinetics and degree of severity depend on the underlying gene mutation and presumably on concomitant polymorphisms and environmental influences, such as life style, infections, and exposition to toxins (56). Underlying pathogenetic mechanisms are heterogeneous and comprise metabolic dysfunction, inhibition of differentiation, ribosomal dysfunction, DNA repair deficiency, and telomere maintenance.
In DBA, SDS, SCN, CAMT, and TAR, primarily one hematopoietic lineage is affected. These syndromes, their underlying pathogenesis, and phenotype, as well as characteristic BM findings are summarized in Table 1. However, it has to be kept in mind that patients with CAMT or SDS frequently show a progress from isolated thrombocytopenia or neutropenia, respectively, to complete BM failure (31). In DBA, erythropoiesis is primarily affected but full BM failure also occurs at low frequency (26, 56). Ribosomal dysfunction has been shown to induce excessive TP53 activation leading to apoptosis and being most harmful to highly proliferating erythroid progenitor cells (24, 26).
In the following, we will focus on syndromes affecting more than one hematopoietic lineage and frequently resulting in severe BM failure and pancytopenia. One of the most severe forms of hematopoietic failure is reticular dysgenesis, a rare, autosomal-recessive syndrome. Erythro- and megakaryopoiesis have been described to be unaffected in some patients, but newborns present with complete absence of neutrophils and early onset SCID. Due to the lack of both innate and adaptive immune functions, reticular dysgenesis is rapidly fatal unless hematopoietic stem cell transplantation (HSCT) is performed. Mutations in the AK2 gene have been identified to be causative for some cases, but most remain unsolved (57, 58).
The most frequent syndromes characterized by severe BM failure are FA and DC. In FA, mutations are found in 15 different genes (FANCA, FANCC, FANCG, FANCD2, and others), all involved in DNA damage repair, particularly in resolution of DNA interstrand cross-links during replication. Patients may present with congenital abnormalities, such as short stature, microphthalmia, thumb and radius deformities, café-au-lait spots and heart, renal and genitourinary malformations. Hematopoietic failure often emerges in childhood or adolescence, and most patients are only diagnosed with FA at the onset of pancytopenia (59). When treated with mitomycin C, FA cells accumulate chromosomal breaks and undergo G2 arrest, which is used as a diagnostic test (60). DC is a disease caused by mutations in genes affecting telomere elongation and maintenance (DKC1, TERC, TERT, TINF2, and others). Although the disease was first characterized by its mucocutaneous symptoms (i.e., skin pigmentation, nail dystrophy, and mucosal leukoplakia), it is now known that the development of BM failure may preceed the mucocutaneous manifestations. Age of onset and disease phenotype are highly variable and at least partially depend on characteristic mutations (61). By contrast, the diagnostic work-up invariably reveals very short telomeres at time of presentation. Unresolved DNA damage and critically short telomeres induce DNA damage checkpoint activation in FA and DC cells, respectively, with TP53 being a critical mediator (62). As a consequence, cells stop proliferation, and senescence or apoptosis are induced. Together, these pathways confer protection from malignant transformation but at the same time they contribute to BM failure and pancytopenia. As a consequence, FA and DC cells have a high selective pressure to inactivate the DNA damage checkpoint, as reflected by their high propensity to develop secondary MDS and AML (Tables 2 and 3) (63). The highest risk is observed in FA patients where accumulation of DNA damage and unresolved chromosomal aberrations rapidly lead to malignant transformation (64).
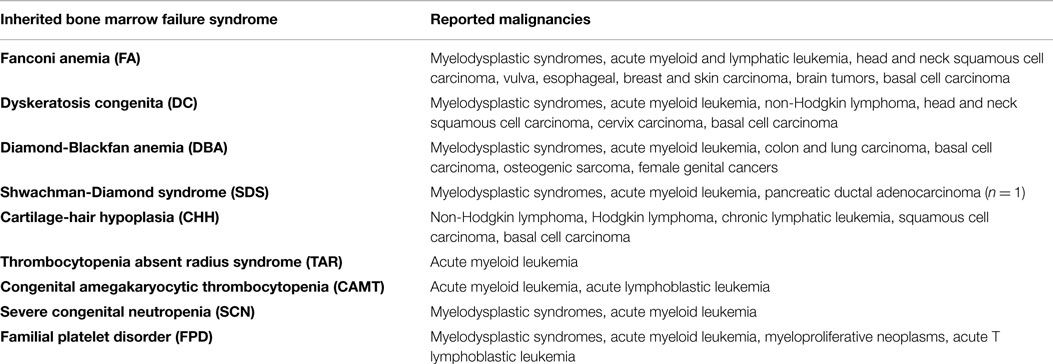
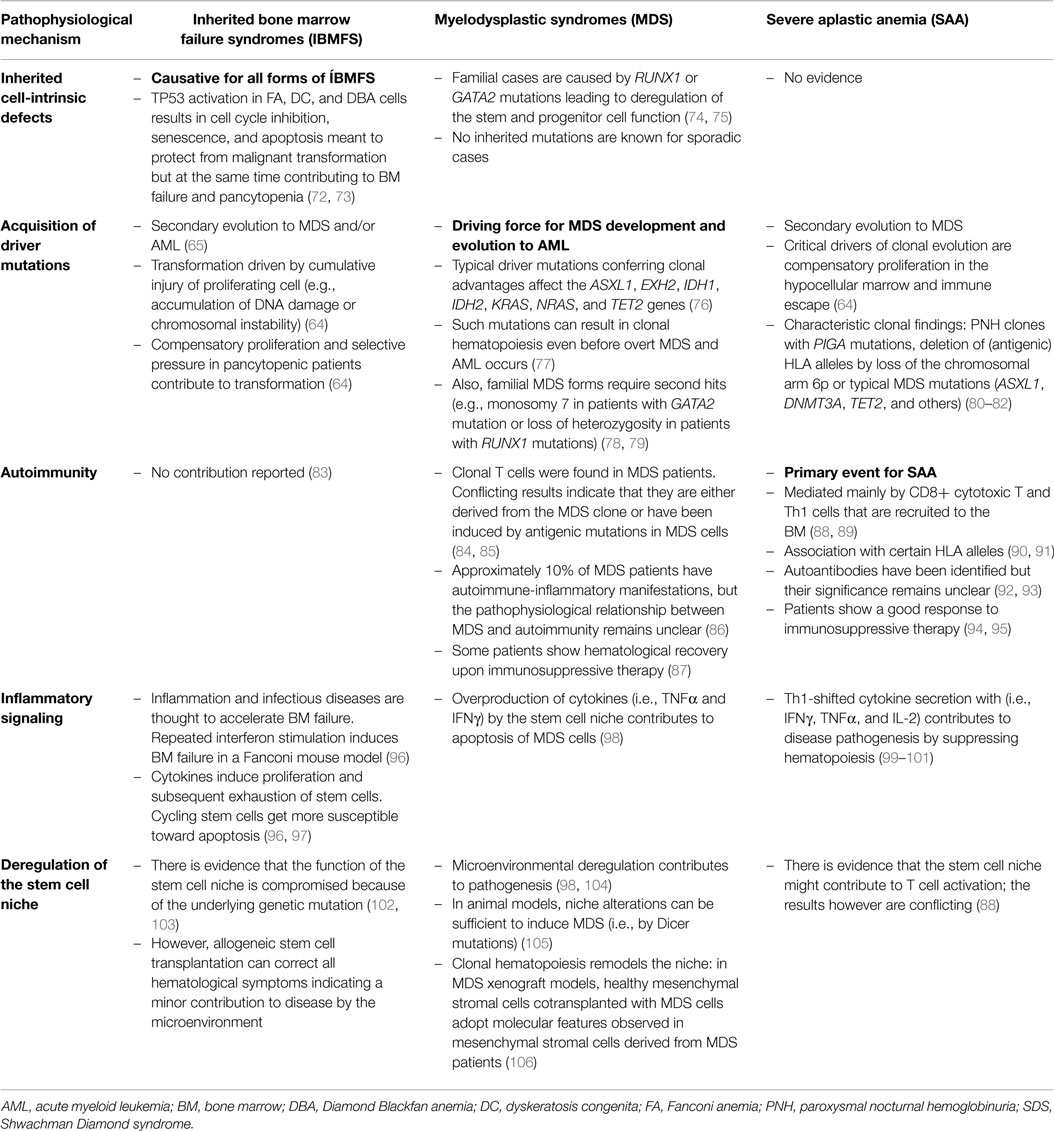
Table 3. The different pathophysiological mechanisms and their relative contributions to inherited bone marrow failure syndromes, severe aplastic anemia, and myelodysplastic syndromes.
Other genetic diseases associated with pancytopenia are Seckel and Pearson syndromes. Seckel syndrome represents a DNA damage repair deficiency characterized by BM failure, dwarfism, microcephaly, mental retardation, and skeletal malformations. Among others, ATR mutations have been identified to be responsible for this rare syndrome (107). Pearson syndrome is caused by loss of mitochondrial DNA and thus one of the so called mitochondriopathies. Next to failure to thrive, exocrine pancreatic dysfunction, and susceptibility to metabolic imbalance, patients frequently suffer from anemia, thrombocytopenia, and/or neutropenia. BM analysis typically shows sideroblastic anemia and vacuolization of hematopoietic precursor cells (108).
It is hardly surprising that DNA damage deficiencies, telomeropathies, ribosomopathies, or mitochondriopathies do not only disturb hematopoiesis but also affect developmental programs and adult tissues. Many of the syndromes described here are additionally characterized by an increased susceptibility to malignancies, including hematological malignancies and solid tumors (Table 2) (65–71). It is obvious that both, the multisystemic nature of disease and the tumor susceptibility, have to be taken into account during patient care and therapy planning. It is therefore of great importance to recognize the underlying cause of BM failure even in patients with mild or no extrahematological symptoms (56, 109). Pitfalls and diagnostic approaches especially in disorders with hypocellular BM are discussed below.
Acquired Disorders of Hematopoiesis
Myelodysplastic Syndrome
Myelodysplastic syndrome (MDS) is a clonal disorder originating from a hematopoietic stem/progenitor cell that supposedly has acquired driver mutations. Due to deregulation of differentiation and increased susceptibility to apoptosis, MDS is characterized by ineffective hematopoiesis in one or more lineages. BM analysis shows characteristic dysplastic cells, such as micromegakaryocytes, binucleated erythroid precursors, and hypo- or hypersegmented neutrophils. MDS has a high propensity to further progress to more advanced disease, including MDS-related acute myeloid leukemia (MDR-AML). This evolution is caused by additional subclonal driver mutations that have been acquired in a stepwise manner and result in further increase in proliferation and impairment of differentiation (110).
In general, MDS is a disease of old age, with an incidence of 50 cases per 100,000 persons aged over 70 years. As shown recently, even healthy, aged individuals frequently have clonal expansion of hematopoietic cells harboring driver mutations (i.e., DNMT3A, JAK2, TET2, and others), although they do not (yet) suffer from overt MDS or leukemia (77). Risk of MDS is further increased in people exposed to chemo- or radiotherapy earlier in life with a life-time risk of 2–10% after treatment with alkylating agents, topoisomerase II inhibitors, or ionizing irradiation (111).
By contrast, childhood MDS is rare, and differs from adult MDS in several aspects. For this reason, the WHO 2008 classification incorporated a classification specific for childhood MDS allowing unambiguous classification of most patients (112). Depending on the number of blasts in peripheral blood (PB) and BM, childhood MDS is classified as refractory cytopenia of childhood (RCC; PB blasts <2%, BM blasts <5%), refractory anemia with excess blasts (RAEB; PB blasts 2–10%, and/or BM blasts 5–19%), RAEB in transformation (RAEB-T; PB and/or BM blasts 20–29%), and MDR-AML (>30% BM blasts) (113). The most common subtype is RCC (50%). While adult MDS patients usually present with isolated anemia (“refractory anemia”), affected children frequently present with thrombocytopenia (<150,000/μl; 75%), neutropenia (<1,000/μl; 50%), and/or anemia (Hb <10 g/dl; 50%). HbF and MCV are frequently elevated (114). In >80% of all RCC cases, BM analysis reveals a marked decrease of cellularity. The remaining 20% of patients with RCC have a normo- or hypercellular BM (114).
Common cytogenetic abnormalities of childhood MDS are monosomy 7 (approximately 30%) and trisomy 8. Loss of chromosome 5q, frequently seen in adults, is only rarely found in children. A structurally complex karyotype (≥3 chromosomal aberrations including at least one structural aberration) is rare but invariably associated with poor prognosis (115–117). Chromosomal aberrations do not represent initiating events but indicate disease progression. Along this line, karyotypic evolution is usually accompanied by progression to more advanced MDS forms. The incidence of cytogenetic abnormalities is lowest in RCC with hypocellular BM. Patients with RCC and monosomy 7 are at high risk of progression with a cumulative incidence of 80% at 6 years from diagnosis and a median time to progression of 1.9 years (118). By contrast, RCC patients with a normal karyotype or trisomy 8 may have stable disease for many years. Patients with advanced MDS are at risk for further disease progression and have an indication for early HSCT (114). In infants, monosomy 7 or del(7q) has also been reported to disappear spontaneously, however, only in rare cases (119).
Next to sporadic cases, MDS can arise secondary to inherited or acquired BM disorders or in the context of chemo/radiotherapy. Importantly, all IBMFS described above have a risk to transform into secondary MDS, as described above in more detail for FA and DC (Table 2) (64). Lifetime risk of IBMFS patients is highly variable and ranges from very high in patients with FA (40–50% by the age of 40 years) to very low in DBA patients (65). Patients with other inherited syndromes might develop MDS directly without hypocellular prophase (120). GATA2 haploinsufficiency is the most frequent reason for such familiar MDS. Patients show variable disease complexes with extrahematological symptoms, such as lymphedema and deafness, and 40% develop MDS during adolescence and early adulthood. Preceding hematological and immunological symptoms are heterogeneous and include monocytopenia, mild neutropenia, and DC, B and/or NK cell deficiency. Depending on the predominant symptoms, the syndrome was named DCML (dendritic cell, monocyte, B and NK lymphoid) deficiency, Emberger syndrome, or MonoMAC (121). Similarly, patients with heterozygous germline RUNX1 mutation have a high propensity to develop MDS (20–50%). Since precedent hematological symptoms are limited to qualitative and quantitative platelet defects, the syndrome is called familial platelet disorder (FPD) (33). It is not yet fully understood, why patients with RUNX1 and GATA2 mutations are at such high risk for MDS (120).
Severe aplastic anemia
While MDS is an intrinsic disorder of hematopoiesis, severe aplastic anemia (SAA) is an autoimmune process. In contrast to autoimmune cytopenias affecting mature blood cells, immature and multipotent cells are targeted in SAA resulting in the severe phenotype characterized by pancytopenia and BM aplasia (88, 94). Evidence for the immune pathophysiology of the disease was first derived from clinical observations such as the response to immunosuppressive therapy (IST) or successful autologous recovery after failed allogeneic HSCT (122, 123). Since then, much effort has been invested in characterization of the immune phenotype, and certain HLA alleles have been associated with an increased risk (90, 91). Autoantibodies against different antigens (e.g., antimoesin or kinectin) have been detected in SAA patients but their significance remains unknown (92, 93). Detailed analysis of the T cell compartment revealed a recruitment of activated T cells to the BM (124) and oligoclonality in some cases (125–128). CD8+ cells isolated from SAA patients were able to suppress colony formation in vitro (129). Th1-shifted cytokine secretion with elevated IFNγ, TNFα, and IL-2 levels is thought to contribute to disease pathogenesis by suppressing hematopoiesis (89, 99–101).
Although SAA is primarily immune-mediated, acquisition of somatic mutations and clonal hematopoiesis seem to be relevant phenomena. Clonal evolution is thought to be driven mainly by two mechanisms: (i) proliferative pressure to compensate for hematopoietic failure, and (ii) immune escape.
Best described is the presence of a so-called paroxysmal nocturnal hemoglobinuria (PNH) clone in patients with SAA. In PNH, the clonal cell population harbors a mutation in the PIGA gene and thus lacks cell surface proteins linked to a glycosylphosphatidylinositol anchor. In clinically manifest PNH, this leads to complement-mediated hemolysis and the risk of thrombosis (80). It is possible that PIGA mutations occur to escape autoimmunity directed against glycosylphosphatidylinositol-bound antigens. Alternatively, the clone might exist a priori and be positively selected when other hematopoietic cells are hit by autoreactive T cells in SAA. Also, copy number-neutral loss of heterozygosity of chromosome 6p is observed disproportionately frequent in SAA patients (81). This is thought to be a mechanism of immune-escape by deletion of antigenic HLA alleles. Finally, SAA patients have been shown to carry somatic mutations similar to those mutated in MDS (i.e., ASXL1, DNMT3A, TET2, and others), indicating transformation into secondary MDS (82, 130).
B-cell precursor acute lymphoblastic leukemia
About 2% of patients with B-cell precursor acute lymphoblastic leukemia (ALL) present with pancytopenia and hypocellular BM. Usually, this prophase is transient and goes into spontaneous remission before ALL emerges with a latency of up to 9 months. Hematopoietic cells in the aplastic phase have been shown to carry the same gene mutations as the leukemic blasts (131, 132).
Inherited Bone Marrow Failure, MDS, and SAA: The Diagnostic Challenge of a Hypocellular Marrow
One of the major challenges in pediatric hematology is the correct diagnosis of patients with pancytopenia and hypocellular BM. Once infections and other more frequent conditions resulting in a transient suppression of hematopoieisis have been excluded, the main differential diagnosis includes SAA, hypoplastic MDS, and IBMFS, such as FA and DC. Careful diagnostic work-up and accurate distinction of these entities are mandatory because they require different strategies for therapy, surveillance, and counseling. In particular, IBMFS have to be excluded since they often present as multisystem and tumor-susceptibility syndromes with limited tolerance to various therapies (56, 64, 109). The diagnostic challenge is not only due to similar clinical presentation but also caused by a relevant overlap in the underlying pathophysiology. Table 3 shows different pathophysiological mechanisms contributing to IBMFS, MDS-RCC, and SAA to variable degrees. Further problems for the diagnostic work-up are scarce cell numbers isolated from hypocellular BM and lack of discriminating functional tests.
Knowing these hurdles, the following diagnostic criteria and workflow should be followed:
(i) BM analysis: next to aspiration cytology, histological analysis of a trephine biopsy should be performed. RCC and SAA can be reliably differentiated in trephine biopsies (114, 133) and the respective criteria, as defined in the WHO classification 2008, are depicted in Table 4 (82, 113). Additionally, cytogenetic analysis with metaphase cytogenetics and FISH should be performed. Chromosomal aberrations indicate the presence of MDS. However, >60% of all RCC cases have a normal karyotype (114). Next generation sequencing will be helpful to detect clonal driver mutations indicative for MDS-RCC, but this method is still expensive and reserved for research questions.
(ii) Exclusion of IBMFS: IBMFS and RCC frequently have overlapping morphological features. Thus, careful past medical and family history, as well as physical examination, is required to exclude presence of a multisystem disease. FA and DC should be excluded by mitomycin C stimulation and telomere measurements, respectively. Genetic testing can confirm the suspected diagnosis, but gene mutations cannot be detected in all cases. In case of the diagnosis of a defined IBMFS or a presentation that is highly suspicious for an inherited disorder, possible related stem cell donors should be carefully evaluated for signs and symptoms of the respective disease. Patients with strong evidence of multisystem disease but without genetic diagnosis might benefit from functional tests such as chromosomal breakage, cell cycle arrest, ribosomal profiling, or others. However, such tests might be technically challenging and difficult to interpret and thus are not part of standard diagnostic work-up.

Table 4. Morphological criteria for severe aplastic anemia and myelodysplastic syndromes, type refractory cytopenia of childhood (EWOG-MDS 2008) (13, 82, 113).
Since disorders of hypocellular BM are rare, diagnostic work-up should be performed by experienced hematologists and hematopathologists, and patients should be treated after consultation with reference centers. This ensures best care for affected patients and their families and prevents underdiagnosis of RCC and telomeropathies in SAA cohorts (109, 134–136).
It is also important to repeatedly reevaluate patients with IBMFS and SAA, since both disorders are at risk to progress into secondary MDS. Secondary MDS can be diagnosed by an increase in blast cells or BM cellularity, despite persistent pancytopenia, and by acquisition of chromosomal aberrations.
Therapeutic Approaches and Limitations
Treatment of (pan)cytopenia is based on a few symptomatic or curative therapeutic options. Symptomatic treatment includes the transfusion of blood products, such as red blood cells and platelets, and prevention or treatment of infections with antimicrobial drugs, and if indicated immunoglobulin substitutions. Granulocyte or T cell transfusions are technically feasible but restricted to very few life-threatening situations. Hematopoietic growth factors may be used to stimulate hematopoiesis and overcome reduced blood cell production. G-CSF is the therapy of choice for patients with SCN and has changed the fate of majority of children with this serious condition (19, 137). Today, allogeneic HSCT is limited to patients who do not respond to G-CSF or develop MDS (138). Although the situation is less clear, selected patients with SDS might benefit from G-CSF in case of severe neutropenia (66). The thrombopoietin analog eltrombopag has been successfully used in chronic ITP (139) and surprisingly induced multilineage responses in an early study in patients with refractory SAA (140). Other, less specific stimulants of hematopoiesis are the steroid prednisolone or the androgens, danazol and oxymetholone, used for DBA or DC, FA, and SAA patients, respectively (26, 141–144).
Some of the disorders described in this review require immune-modulatory or suppressive therapies. These include high dose-immunoglobulins, anti-thymocyte globulin (ATG), steroids, cyclosporine, mycophenolic acid, and others. Detailed treatment schedules for ITP, AIHA, ALPS, SLE, HLH, and others are beyond the scope of this review and described elsewhere.
Immunosuppressive therapy traditionally has been the therapy of choice for patients with SAA with no matched related donor (94, 145). The best established regimen consists of ATG in combination with cyclosporine A (95). In 2011, a randomized trial demonstrated that the source of ATG (horse vs rabbit) is relevant and the treatment with ATG derived from horses is more effective compared to products derived from rabbits (146). These results have subsequently been confirmed in multiple observational studies (147, 148). Using the combination of horse ATG and cyclosporine, the response rate is approximately 60–70% with a risk of relapse or clonal evolution of 10–30 or 10–20%, respectively (145). Attempts to improve efficacy were based on additional treatment with sirolimus, mycophenoalte mofetil, or danazol, and did not result in significant improvements. Also, the addition of G-CSF did not result in better response rates or improved survival, and therefore is not generally recommended. Based on the observation that autoimmunity contributes to the pathophysiology of MDS (Table 3) and that adult patients with hypoplastic MDS have successfully been treated with IST (149), selected pediatric patients with hypocellular RCC have successfully been treated with IST (87, 150, 151).
Hematopoietic stem cell transplantation is a curative treatment for patients with SAA and MDS, and the hematological manifestation of IBMFS. Patients with SAA traditionally were transplanted following a conditioning regimen consisting of cyclophosphamide/ATG in the presence of a matched sibling donor with excellent results (152). Based on less favorable results, HSCT from an unrelated donor was only performed in patients with non-response to IST, relapse, or clonal evolution. Lately, the results of HSCT from unrelated donors have improved considerably and upfront primary HSCT from a well-matched unrelated donor might be considered in selected cases if the donor is available in an acceptable timeframe (153–155).
In RCC, the indication for HSCT is based on the severity of pancytopenia (ANC <1000/μl and/or transfusion dependency) and the presence of cytogenetic aberrations (monosomy 7, 7q-, complex karyotype). Patients with hypocellular RCC with normal karyotype are successfully treated with a reduced intensity regimen, whereas patients with cytogenetic aberrations or a normo-/hypercellular marrow require a more intensive regimen. Patients with advanced MDS should be transplanted as soon as possible following an intensive conditioning regimen (118, 156, 157).
In IBMFS, allogeneic HSCT is able to treat the hematological, and immunological manifestations of the disease. However, extrahematological manifestations, such as pulmonary fibrosis in DC, are not only not treated by this therapeutic approach but may also be an insurmountable obstacle for HSCT. In addition, the majority IBMFS are multisystem disorders with a complex pathophysiology that result in an increased sensitivity to chemotherapeutic drugs requiring a specific adaption of conditioning regimens (158). The careful evaluation of possible related stem cell donors is of special importance in familial disease.
Finally, it has to be mentioned that HSCT is also the only curative treatment for all forms of primary immunodeficiencies and familial HLH. Experimental approaches such as targeted gene correction are under evaluation with regard to efficacy and safety, but not yet suitable for broad application.
In summary, it is still a great challenge to find the correct diagnosis for patients with (pan)cytopenia, especially in the presence of a hypocellular BM. Better definition and differentiation between the individual inherited and acquired disorders are required to ensure adequate medical care, surveillance, and genetic counseling for affected children and their families. To this end, the underlying pathophysiological mechanisms have to be investigated in more detail. Furthermore, functional tests and biomarkers with higher specificity for individual diseases have to be developed. Finally, we recommend that all affected children should be registered in registries and/or clinical trials and treated according to general guidelines.
Author Contributions
ME and BS wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell (2012) 10:120–36. doi: 10.1016/j.stem.2012.01.006
2. Laurenti E, Dick JE. Molecular and functional characterization of early human hematopoiesis. Ann N Y Acad Sci (2012) 1266:68–71. doi:10.1111/j.1749-6632.2012.06577.x
3. Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature (2013) 502:232–6. doi:10.1038/nature12495
5. Balduini CL, Savoia A. Genetics of familial forms of thrombocytopenia. Hum Genet (2012) 131:1821–32. doi:10.1007/s00439-012-1215-x
6. Blajchman MA, Senyi AF, Hirsh J, Genton E, George JN. Hemostatic function, survival, and membrane glycoprotein changes in young versus old rabbit platelets. J Clin Invest (1981) 68:1289–94. doi:10.1172/JCI110375
7. Skokowa J, Germeshausen M, Zeidler C, Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hematol (2007) 14:22–8. doi:10.1097/00062752-200701000-00006
8. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
9. Mostoufi-Zadeh M, Weiss LM, Driscoll SG. Nonimmune hydrops fetalis: a challenge in perinatal pathology. Hum Pathol (1985) 16:785–9. doi:10.1016/S0046-8177(85)80249-5
10. Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J (2008) 14:8.
11. Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol (2008) 122:1082–6. doi:10.1016/j.jaci.2008.09.037
12. Valent P. Low blood counts: immune mediated, idiopathic, or myelodysplasia. Hematology Am Soc Hematol Educ Program (2012) 2012:485–91. doi:10.1182/asheducation-2012.1.485
13. Rinaldi M, Perricone C, Ortega-Hernandez OD, Perricone R, Shoenfeld Y. Immune thrombocytopaenic purpura: an autoimmune cross-link between infections and vaccines. Lupus (2014) 23:554–67. doi:10.1177/0961203313499959
14. McKenzie SE, Sachais BS. Advances in the pathophysiology and treatment of heparin-induced thrombocytopenia. Curr Opin Hematol (2014) 21:380–7. doi:10.1097/MOH.0000000000000066
15. Youinou P, Jamin C, Le Pottier L, Renaudineau Y, Hillion S, Pers JO. Diagnostic criteria for autoimmune neutropenia. Autoimmun Rev (2014) 13:574–6. doi:10.1016/j.autrev.2014.01.001
16. Bass GF, Tuscano ET, Tuscano JM. Diagnosis and classification of autoimmune hemolytic anemia. Autoimmun Rev (2014) 13:560–4. doi:10.1016/j.autrev.2013.11.010
17. Haddad E, Cramer E, Rivière C, Rameau P, Louache F, Guichard J, et al. The thrombocytopenia of Wiskott Aldrich syndrome is not related to a defect in proplatelet formation. Blood (1999) 94:509–18.
18. Sponzilli I, Notarangelo LD. Severe combined immunodeficiency (SCID): from molecular basis to clinical management. Acta Biomed (2011) 82:5–13.
19. Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, et al. Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol (2003) 72:82–93. doi:10.1002/ajh.10255
20. Zeidler C, Germeshausen M, Klein C, Welte K. Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol (2009) 144:459–67. doi:10.1111/j.1365-2141.2008.07425.x
21. Klein C. Congenital neutropenia. Hematology Am Soc Hematol Educ Program (2009):344–50. doi:10.1182/asheducation-2009.1.344
22. Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Rodger E, Kelley ML, et al. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol (2015) 22:3–11. doi:10.1097/MOH.0000000000000105
23. Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med (2014) 16:e1. doi:10.1038/gim.2014.128
24. Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood (2011) 118:4305–12. doi:10.1182/blood-2011-06-353938
25. Shalev H, Kapelushnik J, Moser A, Dgany O, Krasnov T, Tamary H. A comprehensive study of the neonatal manifestations of congenital dyserythropoietic anemia type I. J Pediatr Hematol Oncol (2004) 26:746–8. doi:10.1097/00043426-200411000-00011
26. Ball S. Diamond Blackfan anemia. Hematology Am Soc Hematol Educ Program (2011) 2011:487–91. doi:10.1182/asheducation-2011.1.487
27. Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol (2009) 16:187–94. doi:10.1097/MOH.0b013e32832990a4
28. Sawada K, Hirokawa M, Fujishima N. Diagnosis and management of acquired pure red cell aplasia. Hematol Oncol Clin North Am (2009) 23:249–59. doi:10.1016/j.hoc.2009.01.009
29. Gangat N, Wolanskyj AP. Anemia of chronic disease. Semin Hematol (2013) 50:232–8. doi:10.1053/j.seminhematol.2013.06.006
30. Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med (2015) 21:221–30. doi:10.1038/nm.3814
31. Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer (2011) 57:199–203. doi:10.1002/pbc.22927
32. Albers CA, Newbury-Ecob R, Ouwehand WH, Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr Opin Genet Dev (2013) 23:316–23. doi:10.1016/j.gde.2013.02.015
33. Owen C. Insights into familial platelet disorder with propensity to myeloid malignancy (FPD/AML). Leuk Res (2010) 34:141–2. doi:10.1016/j.leukres.2009.07.037
34. Diz-Kucukkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome. Hematology Am Soc Hematol Educ Program (2013) 2013:268–75. doi:10.1182/asheducation-2013.1.268
35. Martino R, Muñiz-Díaz E, Arilla M, Ibáñez M, Altés A, Guanyabens C, et al. Combined autoimmune cytopenias. Haematologica (1995) 80:305–10.
36. Seidel MG. Autoimmune and other cytopenias in primary immunodeficiencies: pathomechanisms, novel differential diagnoses, and treatment. Blood (2014) 124:2337–44. doi:10.1182/blood-2014-06-583260
37. Seif AE, Manno CS, Sheen C, Grupp SA, Teachey DT. Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood (2010) 115:2142–5. doi:10.1182/blood-2009-08-239525
38. Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr (2012) 24:1–8. doi:10.1097/MOP.0b013e32834ea739
39. Fleisher TA, Oliveira JB. Monogenic defects in lymphocyte apoptosis. Curr Opin Allergy Clin Immunol (2012) 12:609–15. doi:10.1097/ACI.0b013e3283588da0
40. Oliveira JB. The expanding spectrum of the autoimmune lymphoproliferative syndromes. Curr Opin Pediatr (2013) 25:722–9. doi:10.1097/MOP.0000000000000032
41. Verbsky JW, Chatila TA. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr Opin Pediatr (2013) 25:708–14. doi:10.1097/MOP.0000000000000029
42. Moraes-Vasconcelos D, Costa-Carvalho BT, Torgerson TR, Ochs HD. Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J Clin Immunol (2008) 28(Suppl 1):S11–9. doi:10.1007/s10875-008-9176-5
43. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematology Am Soc Hematol Educ Program (2013) 2013:605–11. doi:10.1182/asheducation-2013.1.605
44. Lehmberg K, Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol (2013) 160:275–87. doi:10.1111/bjh.12138
45. Hohlfeld P, Forestier F, Kaplan C, Tissot JD, Daffos F. Fetal thrombocytopenia: a retrospective survey of 5,194 fetal blood samplings. Blood (1994) 84: 1851–6.
46. Ulusoy E, Tüfekçi O, Duman N, Kumral A, Irken G, Oren H. Thrombocytopenia in neonates: causes and outcomes. Ann Hematol (2013) 92:961–7. doi:10.1007/s00277-013-1726-0
47. de Jong EP, de Haan TR, Kroes AC, Beersma MF, Oepkes D, Walther FJ. Parvovirus B19 infection in pregnancy. J Clin Virol (2006) 36:1–7. doi:10.1016/j.jcv.2006.01.004
48. Chisaka H, Morita E, Yaegashi N, Sugamura K. Parvovirus B19 and the pathogenesis of anaemia. Rev Med Virol (2003) 13:347–59. doi:10.1002/rmv.395
49. Hussein H, Phillips N, Riste M. Visceral leishmaniasis with haemophagocytic lymphohistiocytosis: the importance of scrutinising your samples. Br J Haematol (2013) 162:146. doi:10.1111/bjh.12412
50. Bode SF, Bogdan C, Beutel K, Behnisch W, Greiner J, Henning S, et al. Hemophagocytic lymphohistiocytosis in imported pediatric visceral leishmaniasis in a nonendemic area. J Pediatr (2014) 165:147–53. doi:10.1016/j.jpeds.2014.03.047
51. Zeb JA, Zahid B, Ahmad S, Gul Z. Pancytopenia in children: a 6-year spectrum of patients admitted to Pediatric Department of Rehman Medical Institute, Peshawar. Pak J Med Sci. (2013) 29:1153–7. doi:10.12669/pjms.295.3865
52. Belen B, Hismi BO, Kocak U. Severe vitamin B12 deficiency with pancytopenia, hepatosplenomegaly and leukoerythroblastosis in two Syrian refugee infants: a challenge to differentiate from acute leukaemia. BMJ Case Rep (2014) 2014. doi:10.1136/bcr-2014-203742
53. Erlacher M, Grünert SC, Cseh A, Steinfeld R, Salzer U, Lausch E, et al. Reversible pancytopenia and immunodeficiency in a patient with hereditary folate malabsorption. Pediatr Blood Cancer (2015) 62(6):1091–4. doi:10.1002/pbc.25364
54. Ida H, Rennert OM, Ito T, Maekawa K, Eto Y. Type 1 Gaucher disease: phenotypic expression and natural history in Japanese patients. Blood Cells Mol Dis (1998) 24:73–81. doi:10.1006/bcmd.1998.0172
55. Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis (2009) 4:5. doi:10.1186/1750-1172-4-5
56. Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program (2007) 2007(1):29–39. doi:10.1182/asheducation-2007.1.29
57. Pannicke U, Hönig M, Hess I, Friesen C, Holzmann K, Rump EM, et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet (2009) 41:101–5. doi:10.1038/ng.265
58. Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, et al. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet (2009) 41:106–11. doi:10.1038/ng.278
59. Soulier J. Fanconi anemia. Hematology Am Soc Hematol Educ Program (2011) 2011:492–7. doi:10.1182/asheducation-2011.1.492
60. Cervenka J, Arthur D, Yasis C. Mitomycin C test for diagnostic differentiation of idiopathic aplastic anemia and Fanconi anemia. Pediatrics (1981) 67:119–27.
61. Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program (2011) 2011:480–6. doi:10.1182/asheducation-2011.1.480
62. Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood (2006) 107:2680–5. doi:10.1182/blood-2005-07-2622
63. Ceccaldi R, Briot D, Larghero J, Vasquez N, Dubois d’Enghien C, Chamousset D, et al. Spontaneous abrogation of the G(2)DNA damage checkpoint has clinical benefits but promotes leukemogenesis in Fanconi anemia patients. J Clin Invest (2011) 121:184–94. doi:10.1172/JCI43836
64. Bagby GC, Meyers G. Bone marrow failure as a risk factor for clonal evolution: prospects for leukemia prevention. Hematology Am Soc Hematol Educ Program (2007) 2007(1):40–6. doi:10.1182/asheducation-2007.1.40
65. Alter BP, Giri N, Savage SA, Peters JA, Loud JT, Leathwood L, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol (2010) 150:179–88. doi:10.1111/j.1365-2141.2010.08212.x
66. Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood (2006) 107:4628–35. doi:10.1182/blood-2005-11-4370
67. Dhanraj S, Manji A, Pinto D, Scherer SW, Favre H, Loh ML, et al. Molecular characteristics of a pancreatic adenocarcinoma associated with Shwachman-Diamond syndrome. Pediatr Blood Cancer (2013) 60:754–60. doi:10.1002/pbc.24453
68. Tamary H, Nishri D, Yacobovich J, Zilber R, Dgany O, Krasnov T, et al. Frequency and natural history of inherited bone marrow failure syndromes: the Israeli Inherited Bone Marrow Failure Registry. Haematologica (2010) 95:1300–7. doi:10.3324/haematol.2009.018119
69. Camitta BM, Rock A. Acute lymphoidic leukemia in a patient with thrombocytopenia/absent radii (Tar) syndrome. Am J Pediatr Hematol Oncol (1993) 15:335–7.
70. Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood (2012) 119:3815–9. doi:10.1182/blood-2011-08-375972
71. Taskinen M, Ranki A, Pukkala E, Jeskanen L, Kaitila I, Mäkitie O. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A (2008) 146A:2370–5. doi:10.1002/ajmg.a.32478
72. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature (2000) 408:433–9. doi:10.1038/35044005
73. Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell (2012) 11:36–49. doi:10.1016/j.stem.2012.05.013
74. de Pater E, Kaimakis P, Vink CS, Yokomizo T, Yamada-Inagawa T, van der Linden R, et al. Gata2 is required for HSC generation and survival. J Exp Med (2013) 210:2843–50. doi:10.1084/jem.20130751
75. Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature (2010) 464:112–5. doi:10.1038/nature08761
76. Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood (2013) 122:4021–34. doi:10.1182/blood-2013-09-381665
77. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med (2014) 20:1472–8. doi:10.1038/nm.3733
78. Hirabayashi S, Strahm B, Urbaniak S, Karow A, Cseh A, van den Heuvel-Eibrink MM, et al. High frequency of GATA2 mutations in children with non-familial MDS and monosomy 7. 54th ASH Annual Meeting. Atlanta (2012). Ref Type: Generic.
79. Michaud J, Wu F, Osato M, Cottles GM, Yanagida M, Asou N, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood (2002) 99:1364–72. doi:10.1182/blood.V99.4.1364
80. van den Heuvel-Eibrink MM. Paroxysmal nocturnal hemoglobinuria in children. Paediatr Drugs (2007) 9:11–6. doi:10.2165/00148581-200709010-00002
81. Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y, et al. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood (2011) 118:6601–9. doi:10.1182/blood-2011-07-365189
82. Kulasekararaj AG, Jiang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood (2014) 124:2698–704. doi:10.1182/blood-2014-05-574889
83. Korthof ET, Svahn J, Peffault de Latour R, Terranova P, Moins-Teisserenc H, Socié G, et al. Immunological profile of Fanconi anemia: a multicentric retrospective analysis of 61 patients. Am J Hematol (2013) 88:472–6. doi:10.1002/ajh.23435
84. Epling-Burnette PK, Painter JS, Rollison DE, Ku E, Vendron D, Widen R, et al. Prevalence and clinical association of clonal T-cell expansions in Myelodysplastic Syndrome. Leukemia (2007) 21:659–67. doi:10.1038/sj.leu.2404590
85. de Vries AC, Langerak AW, Verhaaf B, Niemeyer CM, Stary J, Schmiegelow K, et al. T-cell receptor Vbeta CDR3 oligoclonality frequently occurs in childhood refractory cytopenia (MDS-RC) and severe aplastic anemia. Leukemia (2008) 22:1170–4. doi:10.1038/leu.2008.23
86. Giannouli S, Voulgarelis M. A comprehensive review of myelodysplastic syndrome patients with autoimmune diseases. Expert Rev Clin Immunol. (2014) 10:1679–88. doi:10.1586/1744666X.2014.970181
87. Yoshimi A, Baumann I, Führer M, Bergsträsser E, Göbel U, Sykora KW, et al. Immunosuppressive therapy with anti-thymocyte globulin and cyclosporine A in selected children with hypoplastic refractory cytopenia. Haematologica (2007) 92:397–400. doi:10.3324/haematol.10683
88. Zeng Y, Katsanis E. The complex pathophysiology of acquired aplastic anemia. Clin Exp Immunol (2015) 180(3):361–70. doi:10.1111/cei.12605
89. Kordasti S, Marsh J, Al-Khan S, Jiang J, Smith A, Mohamedali A, et al. Functional characterization of CD4+ T cells in aplastic anemia. Blood (2012) 119:2033–43. doi:10.1182/blood-2011-08-368308
90. Chen C, Lu S, Luo M, Zhang B, Xiao L. Correlations between HLA-A, HLA-B and HLA-DRB1 allele polymorphisms and childhood susceptibility to acquired aplastic anemia. Acta Haematol (2012) 128:23–7. doi:10.1159/000337094
91. Führer M, Durner J, Brünnler G, Götte H, Deppner C, Bender-Götze C, et al. HLA association is different in children and adults with severe acquired aplastic anemia. Pediatr Blood Cancer (2007) 48:186–91. doi:10.1002/pbc.20785
92. Hirano N, Butler MO, Von Bergwelt-Baildon MS, Maecker B, Schultze JL, O’Connor KC, et al. Autoantibodies frequently detected in patients with aplastic anemia. Blood (2003) 102:4567–75. doi:10.1182/blood-2002-11-3409
93. Takamatsu H, Espinoza JL, Lu X, Qi Z, Okawa K, Nakao S. Anti-moesin antibodies in the serum of patients with aplastic anemia stimulate peripheral blood mononuclear cells to secrete TNF-alpha and IFN-gamma. J Immunol (2009) 182:703–10. doi:10.4049/jimmunol.182.1.703
94. Young NS. Current concepts in the pathophysiology and treatment of aplastic anemia. Hematology Am Soc Hematol Educ Program (2013) 2013:76–81. doi:10.1182/asheducation-2013.1.76
95. Führer M, Rampf U, Baumann I, Faldum A, Niemeyer C, Janka-Schaub G, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood (2005) 106:2102–4. doi:10.1182/blood-2005-03-0874
96. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature (2015) 520(7548):549–52. doi:10.1038/nature14131
97. Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211:245–62. doi:10.1084/jem.20131043
98. Bulycheva E, Rauner M, Medyouf H, Theurl I, Bornhäuser M, Hofbauer LC, et al. Myelodysplasia is in the niche: novel concepts and emerging therapies. Leukemia (2015) 29:259–68. doi:10.1038/leu.2014.325
99. Li J, Zhao Q, Xing W, Feng J, Wu H, Li H, et al. Interleukin-27 enhances the production of tumour necrosis factor-alpha and interferon-gamma by bone marrow T lymphocytes in aplastic anaemia. Br J Haematol (2011) 153:764–72. doi:10.1111/j.1365-2141.2010.08431.x
100. Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood (2002) 100:1185–91. doi:10.1182/blood-2002-01-0035
101. Liu CY, Fu R, Wang HQ, Li LJ, Liu H, Guan J, et al. Fas/FasL in the immune pathogenesis of severe aplastic anemia. Genet Mol Res (2014) 13:4083–8. doi:10.4238/2014.May.30.3
102. Balakumaran A, Mishra PJ, Pawelczyk E, Yoshizawa S, Sworder BJ, Cherman N, et al. Bone marrow skeletal stem/progenitor cell defects in dyskeratosis congenita and telomere biology disorders. Blood (2015) 125:793–802. doi:10.1182/blood-2014-06-566810
103. Lecourt S, Vanneaux V, Leblanc T, Leroux G, Ternaux B, Benbunan M, et al. Bone marrow microenvironment in fanconi anemia: a prospective functional study in a cohort of fanconi anemia patients. Stem Cells Dev (2010) 19:203–8. doi:10.1089/scd.2009.0062
104. Santamaría C, Muntión S, Rosón B, Blanco B, López-Villar O, Carrancio S, et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica (2012) 97:1218–24. doi:10.3324/haematol.2011.054437
105. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature (2010) 464:852–7. doi:10.1038/nature08851
106. Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell (2014) 14:824–37. doi:10.1016/j.stem.2014.02.014
107. O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet (2003) 33:497–501. doi:10.1038/ng1129
108. Seneca S, De Meirleir L, De Schepper J, Balduck N, Jochmans K, Liebaers I, et al. Pearson marrow pancreas syndrome: a molecular study and clinical management. Clin Genet (1997) 51:338–42. doi:10.1111/j.1399-0004.1997.tb02484.x
109. Shimamura A. Clinical approach to marrow failure. Hematology Am Soc Hematol Educ Program (2009) 2009(1):329–37. doi:10.1182/asheducation-2009.1.329
110. Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer (2007) 7:118–29. doi:10.1038/nrc2047
111. Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood (2000) 96:3932–8.
112. Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press (2008). Ref Type: Generic.
113. Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia (2003) 17:277–82. doi:10.1038/sj.leu.2402765
114. Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program (2011) 2011:84–9. doi:10.1182/asheducation-2011.1.84
115. Luna-Fineman S, Shannon KM, Atwater SK, Davis J, Masterson M, Ortega J, et al. Myelodysplastic and myeloproliferative disorders of childhood: a study of 167 patients. Blood (1999) 93:459–66.
116. Passmore SJ, Chessells JM, Kempski H, Hann IM, Brownbill PA, Stiller CA. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol (2003) 121:758–67. doi:10.1046/j.1365-2141.2003.04361.x
117. Göhring G, Michalova K, Beverloo HB, Betts D, Harbott J, Haas OA, et al. Complex karyotype newly defined: the strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood (2010) 116:3766–9. doi:10.1182/blood-2010-04-280313
118. Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KR, et al. Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7. Blood (2003) 102:1997–2003. doi:10.1182/blood-2002-11-3444
119. Mantadakis E, Shannon KM, Singer DA, Finklestein J, Chan KW, Hilden JM, et al. Transient monosomy 7: a case series in children and review of the literature. Cancer (1999) 85:2655–61. doi:10.1002/(SICI)1097-0142(19990615)85:12<2655::AID-CNCR23>3.0.CO;2-W
120. West AH, Godley LA, Churpek JE. Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci (2014) 1310:111–8. doi:10.1111/nyas.12346
121. Collin M, Dickinson R, Bigley V. Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol (2015) 169:173–87. doi:10.1111/bjh.13317
122. Gluckman E, Marmont A, Speck B, Gordon-Smith EC. Immunosuppressive treatment of aplastic anemia as an alternative treatment for bone marrow transplantation. Semin Hematol (1984) 21:11–9.
123. Speck B, Cornu P, Jeannet M, Nissen C, Burri HP, Groff P, et al. Autologous marrow recovery following allogeneic marrow transplantation in a patient with severe aplastic anemia. Exp Hematol (1976) 4:131–7.
124. Ren J, Hou XY, Ma SH, Zhang FK, Zhen JH, Sun L, et al. Elevated expression of CX3C chemokine receptor 1 mediates recruitment of T cells into bone marrow of patients with acquired aplastic anaemia. J Intern Med (2014) 276:512–24. doi:10.1111/joim.12218
125. Kochenderfer JN, Kobayashi S, Wieder ED, Su C, Molldrem JJ. Loss of T-lymphocyte clonal dominance in patients with myelodysplastic syndrome responsive to immunosuppression. Blood (2002) 100:3639–45. doi:10.1182/blood-2002-01-0155
126. Wlodarski MW, Gondek LP, Nearman ZP, Plasilova M, Kalaycio M, Hsi ED, et al. Molecular strategies for detection and quantitation of clonal cytotoxic T-cell responses in aplastic anemia and myelodysplastic syndrome. Blood (2006) 108:2632–41. doi:10.1182/blood-2005-09-3902
127. Zeng W, Maciejewski JP, Chen G, Young NS. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J Clin Invest (2001) 108:765–73. doi:10.1172/JCI200112687
128. Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet (2004) 364:355–64. doi:10.1016/S0140-6736(04)16724-X
129. Xing L, Liu C, Fu R, Wang H, Wang J, Liu X, et al. CD8+HLA-DR+ T cells are increased in patients with severe aplastic anemia. Mol Med Rep. (2014) 10:1252–8. doi:10.3892/mmr.2014.2344
130. Babushok DV, Perdigones N, Perin JC, Olson TS, Ye W, Roth JJ, et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet (2015) 208(4):115–28. doi:10.1016/j.cancergen.2015.01.007
131. Hasle H, Heim S, Schroeder H, Schmiegelow K, Ostergaard E, Kerndrup G. Transient pancytopenia preceding acute lymphoblastic leukemia (pre-ALL). Leukemia (1995) 9:605–8.
132. Villarreal-Martinez L, Jaime-Perez JC, Rodriguez-Martinez M, Gonzalez-Llano O, Gomez-Almaguer D. Acute lymphoblastic leukemia of childhood presenting as aplastic anemia: report of two cases. Rev Bras Hematol Hemoter. (2012) 34:165–7. doi:10.5581/1516-8484.20120037
133. Baumann I, Führer M, Behrendt S, Campr V, Csomor J, Furlan I, et al. Morphological differentiation of severe aplastic anaemia from hypocellular refractory cytopenia of childhood: reproducibility of histopathological diagnostic criteria. Histopathology (2012) 61:10–7. doi:10.1111/j.1365-2559.2011.04156.x
134. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med (2005) 352:1413–24. doi:10.1056/NEJMoa042980
135. Fibbe WE. Telomerase mutations in aplastic anemia. N Engl J Med (2005) 352:1481–3. doi:10.1056/NEJMe058015
136. Dumitriu B, Feng X, Townsley DM, Ueda Y, Yoshizato T, Calado RT, et al. Telomere attrition and candidate gene mutations preceding monosomy 7 in aplastic anemia. Blood (2015) 125:706–9. doi:10.1182/blood-2014-10-607572
137. Welte K, Zeidler C, Reiter A, Müller W, Odenwald E, Souza L, et al. Differential effects of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in children with severe congenital neutropenia. Blood (1990) 75:1056–63.
138. Connelly JA, Choi SW, Levine JE. Hematopoietic stem cell transplantation for severe congenital neutropenia. Curr Opin Hematol (2012) 19:44–51. doi:10.1097/MOH.0b013e32834da96e
139. Zimmer J, Hentges F, Andres E. Eltrombopag in thrombocytopenia. N Engl J Med (2008) 358:1072–3. doi:10.1056/NEJMc073571
140. Olnes MJ, Scheinberg P, Calvo KR, Desmond R, Tang Y, Dumitriu B, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med (2012) 367:11–9. doi:10.1056/NEJMoa1200931
141. Islam A, Rafiq S, Kirwan M, Walne A, Cavenagh J, Vulliamy T, et al. Haematological recovery in dyskeratosis congenita patients treated with danazol. Br J Haematol (2013) 162:854–6. doi:10.1111/bjh.12432
142. Scheckenbach K, Morgan M, Filger-Brillinger J, Sandmann M, Strimling B, Scheurlen W, et al. Treatment of the bone marrow failure in Fanconi anemia patients with danazol. Blood Cells Mol Dis (2012) 48:128–31. doi:10.1016/j.bcmd.2011.11.006
143. Jaime-Pérez JC, Colunga-Pedraza PR, Gómez-Ramírez CD, Gutiérrez-Aguirre CH, Cantú-Rodríguez OG, Tarín-Arzaga LC, et al. Danazol as first-line therapy for aplastic anemia. Ann Hematol (2011) 90:523–7. doi:10.1007/s00277-011-1163-x
144. Rose SR, Kim MO, Korbee L, Wilson KA, Ris MD, Eyal O, et al. Oxandrolone for the treatment of bone marrow failure in Fanconi anemia. Pediatr Blood Cancer (2014) 61:11–9. doi:10.1002/pbc.24617
145. Samarasinghe S, Marsh J, Dufour C. Immune suppression for childhood acquired aplastic anemia and myelodysplastic syndrome: where next? Haematologica (2014) 99:597–9. doi:10.3324/haematol.2014.105569
146. Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med (2011) 365:430–8. doi:10.1056/NEJMoa1103975
147. Marsh JC, Bacigalupo A, Schrezenmeier H, Tichelli A, Risitano AM, Passweg JR, et al. Prospective study of rabbit antithymocyte globulin and cyclosporine for aplastic anemia from the EBMT Severe Aplastic Anaemia Working Party. Blood (2012) 119:5391–6. doi:10.1182/blood-2012-02-407684
148. Yoshimi A, Niemeyer CM, Fuhrer MM, Strahm B. Comparison of the efficacy of rabbit and horse antithymocyte globulin for the treatment of severe aplastic anemia in children. Blood (2013) 121:860–1. doi:10.1182/blood-2012-10-461509
149. Sloand EM, Wu CO, Greenberg P, Young N, Barrett J. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol (2008) 26:2505–11. doi:10.1200/JCO.2007.11.9214
150. Yoshimi A, van den Heuvel-Eibrink MM, Baumann I, Schwarz S, Simonitsch-Klupp I, de Paepe P, et al. Comparison of horse and rabbit antithymocyte globulin in immunosuppressive therapy for refractory cytopenia of childhood. Haematologica (2014) 99:656–63. doi:10.3324/haematol.2013.095786
151. Hasegawa D, Manabe A, Yagasaki H, Ohtsuka Y, Inoue M, Kikuchi A, et al. Treatment of children with refractory anemia: the Japanese Childhood MDS Study Group trial (MDS99). Pediatr Blood Cancer (2009) 53:1011–5. doi:10.1002/pbc.22121
152. Locatelli F, Porta F, Zecca M, Pedrazzoli P, Maccario R, Giani S, et al. Successful bone marrow transplantation in children with severe aplastic anemia using HLA-partially matched family donors. Am J Hematol (1993) 42:328–33. doi:10.1002/ajh.2830420315
153. Samarasinghe S, Steward C, Hiwarkar P, Saif MA, Hough R, Webb D, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol (2012) 157:339–46. doi:10.1111/j.1365-2141.2012.09066.x
154. Maury S, Balère-Appert ML, Pollichieni S, Oneto R, Yakoub-Agha I, Locatelli F, et al. Outcome of patients activating an unrelated donor search for severe acquired aplastic anemia. Am J Hematol (2013) 88:868–73. doi:10.1002/ajh.23522
155. Fuhrer M. Risk-adapted procedures for HSCT from alternative donor in children with severe aplastic anaemia. Bone Marrow Transplant (2008) 42(Suppl 2):S97–100. doi:10.1038/bmt.2008.293
156. Strahm B, Locatelli F, Bader P, Ehlert K, Kremens B, Zintl F, et al. Reduced intensity conditioning in unrelated donor transplantation for refractory cytopenia in childhood. Bone Marrow Transplant (2007) 40:329–33. doi:10.1038/sj.bmt.1705730
157. Strahm B, Nöllke P, Zecca M, Korthof ET, Bierings M, Furlan I, et al. Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia (2011) 25:455–62. doi:10.1038/leu.2010.297
Keywords: cytopenia, childhood, bone marrow failure, hypocellular bone marrow, myelodysplastic syndrome, refractory cytopenia of childhood, severe aplastic anemia
Citation: Erlacher M and Strahm B (2015) Missing cells: pathophysiology, diagnosis, and management of (pan)cytopenia in childhood. Front. Pediatr. 3:64. doi: 10.3389/fped.2015.00064
Received: 06 May 2015; Accepted: 25 June 2015;
Published: 13 July 2015
Edited by:
Markus G. Seidel, Medical University Graz, AustriaReviewed by:
Jan-Henning Klusmann, Hannover Medical School, GermanyMilen Minkov, Rudolfstiftung Hospital, Austria
Copyright: © 2015 Erlacher and Strahm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brigitte Strahm, Department of Pediatrics and Adolescent Medicine, Division of Pediatric Hematology and Oncology, University Medical Center of Freiburg, Mathildenstr. 1, Freiburg 79106, Germany, brigitte.strahm@uniklinik-freiburg.de