Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience
 C. Alexander Valencia1*
C. Alexander Valencia1*
 Ammar Husami1
Ammar Husami1
 Jennifer Holle1
Judith A. Johnson1
Jennifer Holle1
Judith A. Johnson1
 Yaping Qian2
Yaping Qian2
 Abhinav Mathur1
Chao Wei1
Abhinav Mathur1
Chao Wei1
 Subba Rao Indugula1
Subba Rao Indugula1
 Fanggeng Zou1
Haiying Meng1
Fanggeng Zou1
Haiying Meng1
 Lijun Wang1
Lijun Wang1
 Xia Li1
Xia Li1
 Rachel Fisher1
Tony Tan1
Amber Hogart Begtrup1
Rachel Fisher1
Tony Tan1
Amber Hogart Begtrup1
 Kathleen Collins1
Kathleen Collins1
 Katie A. Wusik1
Katie A. Wusik1
 Derek Neilson1
Derek Neilson1
 Thomas Burrow1
Elizabeth Schorry1
Thomas Burrow1
Elizabeth Schorry1
 Robert Hopkin1
Robert Hopkin1
 Mehdi Keddache1
Mehdi Keddache1
 John Barker Harley3,4
Kenneth M. Kaufman3,4
John Barker Harley3,4
Kenneth M. Kaufman3,4  Kejian Zhang1
Kejian Zhang1
- 1Division of Human Genetics, Cincinnati Children’s Hospital Medical Center and Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA
- 2Myriad Genetics Laboratories, Inc., Salt Lake City, UT, USA
- 3Center for Autoimmune Genomics and Etiology (CAGE), Cincinnati Children’s Hospital Medical Center and Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, USA
- 4US Department of Veterans Affairs Medical Center, Cincinnati, OH, USA
Background: There are limited reports of the use of whole exome sequencing (WES) as a clinical diagnostic tool. Moreover, there are no reports addressing the cost burden associated with genetic tests performed prior to WES.
Objective: We demonstrate the performance characteristics of WES in a pediatric setting by describing our patient cohort, calculating the diagnostic yield, and detailing the patients for whom clinical management was altered. Moreover, we examined the potential cost-effectiveness of WES by examining the cost burden of diagnostic workups.
Methods: To determine the clinical utility of our hospital’s clinical WES, we performed a retrospective review of the first 40 cases. We utilized dual bioinformatics analyses pipelines based on commercially available software and in-house tools.
Results: Of the first 40 clinical cases, we identified genetic defects in 12 (30%) patients, of which 47% of the mutations were previously unreported in the literature. Among the 12 patients with positive findings, seven have autosomal dominant disease and five have autosomal recessive disease. Ninety percent of the cohort opted to receive secondary findings and of those, secondary medical actionable results were returned in three cases. Among these positive cases, there are a number of novel mutations that are being reported here. The diagnostic workup included a significant number of genetic tests with microarray and single-gene sequencing being the most popular tests. Significantly, genetic diagnosis from WES led to altered patient medical management in positive cases.
Conclusion: We demonstrate the clinical utility of WES by establishing the clinical diagnostic rate and its impact on medical management in a large pediatric center. The cost-effectiveness of WES was demonstrated by ending the diagnostic odyssey in positive cases. Also, in some cases it may be most cost-effective to directly perform WES. WES provides a unique glimpse into the complexity of genetic disorders.
Introduction
Mendelian diseases account for a significant number of pediatric disorders. Recently, a systematic review of the records of 5,747 consecutive admissions in 1996 to Rainbow Babies and Children’s Hospital (Cleveland, OH, USA) found an underlying disorder with a significant genetic component in 71% of admitted children (1). Diagnosing patients with complex phenotypes generally involves physical examination, detailed family history, complementary tests such as radiography and metabolite analysis, and genetic testing. A significant proportion of patients undergo extensive genetic testing including karyotyping, array-based comparative genomic hybridization, Sanger sequencing, and multigene next-generation sequencing panels but still remain undiagnosed (2). Accurate diagnosis potentially benefits patients and their families by altering clinical management, predicting recurrence risks, providing prognosis, and ending the diagnostic odyssey that is invasive, time consuming and costly. Clinical diagnosis is therapeutic in its own right for the patient/family, as a result of ending the “diagnostic odyssey” – quite apart from the tangible clinical benefits.
The advent of next-generation sequencing technologies has provided an opportunity to affordably screen a patient’s entire exome to establish genetic basis of disease (3–9). The “exome” is the component of the genome that predominantly encodes protein; these segments are referred to as “exons” and can include non-coding exons. The exome comprises about 1% of the genome and is, so far, the component most likely to include interpretable mutations that result in clinical phenotypes. Whole exome sequencing (WES) involves determination of the DNA sequence of most of these protein-encoding exons and may include some DNA regions that encode RNA molecules that are not involved in protein synthesis. The utility of WES to identify variants causative of Mendelian disorders has been clearly demonstrated in identifying novel candidate genes for Miller syndrome, Fowler syndrome, Perrault Syndrome, and many other disorders (10–17). However, the clinical utility of exome sequencing in pediatric patients needs further examination (18–22). Herein, we report a series of our first 40 consecutive pediatric cases that were referred for WES in a clinical laboratory. We demonstrate the clinical utility of WES in a pediatric setting by describing our patient cohort, calculating the diagnostic yield, detailing the cases in which clinical management was altered, and potential cost-effectiveness of WES as a single test by examining the number and types of genetic tests that were performed prior to WES that add to the cost of diagnostic workups.
Patients, Materials, and Methods
Clinical Samples
Forty pediatric patients referred by medical specialists (Medical geneticists 77%, Immunologists 15%, Cardiologists 3%, and others 3%) for exome sequencing have had the analysis and results disclosure completed. The patients in this cohort had diverse clinical features and these are summarized in Table 1. Before referral, all patients had undergone extensive diagnostic evaluations (e.g., aCGH microarray, targeted gene tests/panels, metabolic screening, clinical genetic evaluations, and other laboratory workup) that did not lead to a unifying diagnosis. Consent for clinical WES was obtained from the patients and/or their family. Internal review board (IRB) approval was obtained at Cincinnati Children’s Hospital Medical Center (CCHMC) for this retrospective study.
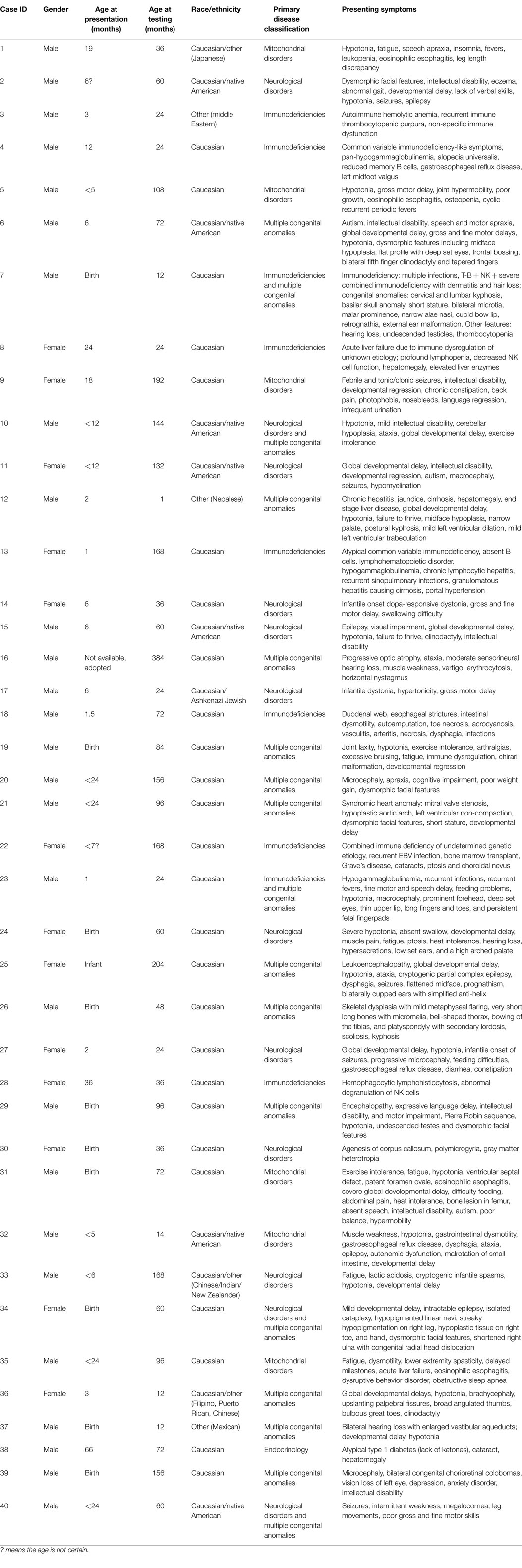
Table 1. Patient demographic information and detailed description of presenting symptoms.
Whole Exome Sequencing and Sanger Confirmation
WES and analysis protocols were developed and validated by the CCHMC molecular genetics laboratory of the Division of Human Genetics. Briefly, genomic DNA samples from patients were fragmented by sonication, ligated to Illumina multiplexing paired-end adapters, amplified by means of a polymerase-chain reaction and hybridized to biotin-labeled NimbleGen V3 exome capture reagent (Roche NimbleGen). Hybridization was achieved at 47°C for 64–72 h. After washing and reamplification, paired-end sequencing (2 × 100 bp) was performed on the Illumina HiSeq 2500 platform to provide average sequence coverage of more than 100×, with more than 97% of the target bases having at least 10× coverage. Clinically relevant variants, from proband and parental samples (whenever available), were confirmed by Sanger sequencing.
Data Analysis and Annotation
To aid in the clinical interpretation of variants, data were annotated and analyzed using two pipelines: (1) next gene pipeline (NGP) and the (2) genome analysis toolkit (GATK)/golden helix pipeline (GGHP). For NPG, output data from the Illumina HiSeq 2500 were converted from bcl files to FastQ files using the Illumina consensus assessment of sequence and variation (CASAVA) software, version 1.8, and mapped to the reference haploid human-genome sequence (hg19) with NextGene 2.3.4 using default settings. Passing quality control/quality assurance (QC/QA) parameters included >75 million reads, >100× average coverage, >95% at 10× coverage of the target exome, and >200 million unique molecules for each case. Variant calls, which differed from the reference sequence was obtained with NextGene 2.3.4. NextGENe genotyping settings were as follows: non-synonymous variants are generated for CDS ± 20 bp and heterozygous, homozygous and wildtype calls had allele percentages of >20 and <80%, >80 and <20%, respectively. Alamut HT 1.1.8 was used for variant annotation. In-house developed scripts were applied for variant prioritization based on phenotype-genotype correlation (Phenomizer and Pheno2Gene, a CCHMC-developed tool), presence in the literature, inheritance modeling, frequency in exome sequencing project (ESP) and exome aggregation consortium (ExAC) population databases (<1%, unless literature reported with a higher frequency or other lines of evidence supported pathogenicity of variants), mutation type, in silico predictions (POLYPHEN, SIFT, and Grantham Scale), and presence of the variant in functional domains. Variants were excluded from the analysis if they had a coverage of <10×.
For GGHP, reads generated by the sequencer were aligned to the human genome with BWA 0.5.9. Then, the aligned reads were checked for quality (QC/QA parameters). Upon passing, the aligned data were preprocessed to reduce sequencing biases using Picard 1.5.3 and GATK Appistry 2013.2. The data were compared to the reference sequence, along with standard control samples and other samples in the batch in order to generate a final list of variants for the exome test using with GATK 7.7.4. The output variant calls were in the standard VCF format. The variants were called with GATK using the following commands: -T Unified Genotyper-dcov 1000-stand_call_conf 30.0-stand_emit_conf 30.0 – min_base_quality_score 20 -A Depth Of Coverage -A Indel Type -A QualByDepth -A ReadPosRankSumTest -A FisherStrand -A MappingQualityRankSumTest -l INFO -glm. The VCF file was analyzed using Golden Helix Software (ver. 7.7.4) as previously reported (23). Briefly, quality filtering to exclude variants was based on alternative allele ratio, genotype quality score value (GQ <20 excluded), and read depth (depth <15× excluded). Genetic filters included keeping variants that had a <1% allele frequency and fit Mendelian inheritance models.
Data Interpretation and Reporting
Common and unique variants from both analysis pipelines that remained after filtering were further classified as deleterious mutations, variants of unknown clinical significance (VUCS), or benign variants in the context of their relatedness to the patient’s phenotype, and pathogenic mutations on the American College of Medical Genetics (ACMG) gene list were classified as medically actionable secondary findings. Stringent conditions were employed to identify causative mutations. Specifically, we followed the ACMG guidelines to denote deleterious mutations and binned them into the ACMG category 1 (previously reported to be deleterious) or category 2 (predicted to be deleterious) (24). Previously reported or predicted to be deleterious mutations in genes associated with the patient’s phenotypes were prioritized. Each causative mutation was scrutinized by thorough literature review and database searches. Moreover, the pathogenicity of novel and rare variants was assessed by in silico prediction programs. Additionally, patterns of familial segregation were examined for correlation. Moreover, pathogenic variants were identified based on the ACMG secondary finding recommendations (if the family chose to receive this information) (25). At the final stage, clinical exome sequencing data interpretation was performed by a team represented by clinical molecular and medical geneticists, pediatric subspecialists, and genetic counselors. The criteria for a full or partial molecular diagnosis were defined as follows: (1) Full molecular diagnosis – Gene variant(s) that is classified as likely pathogenic or pathogenic explains most or all of the clinical features of the patient. (2) Partial molecular diagnosis – Gene variant(s) that is classified as likely pathogenic or pathogenic explains one or several of the clinical features of the patient.
Results
Cohort Description
Of the 40 patients, 30, 17, 22, and 25% were patients with primary phenotypes related to multiple congenital anomalies, immunodeficiency, neurological, and mitochondrial disorders, respectively (Table 1; Figure 1, Primary indication). Thirteen percent had clinical features of more than two of the broad aforementioned categories. All patients were under 17 years of age at the time of exome analysis (average age 83.2 months) and much younger at the time of clinical presentation (average age 5.3 months) (Table 1).
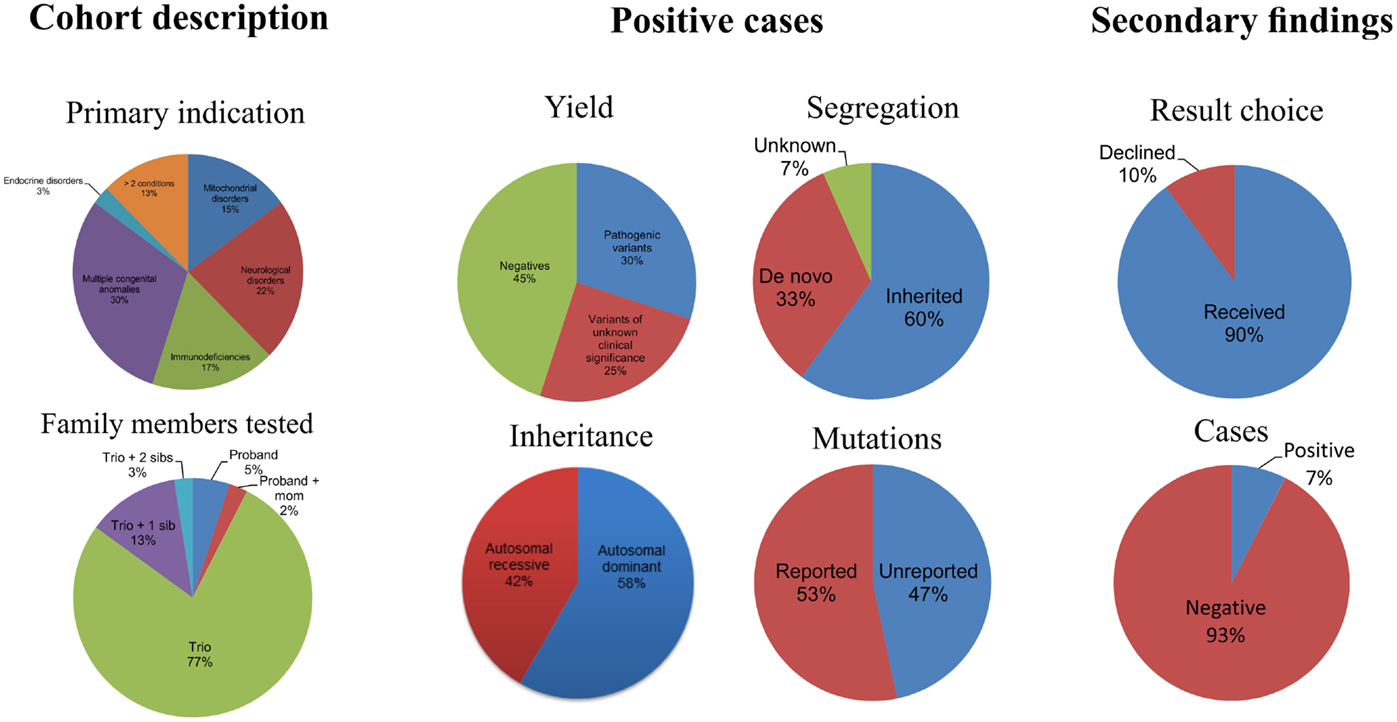
Figure 1. Descriptive statistics of the patient cohort and the positive exome cases.
Exome Sequencing
As quality control/quality assurance parameters we measured the average coverage, percentage at 10× and 20× per family (Table S1 in Supplementary Material). The typically QC/QA acceptable average coverage was >100× and >95% at 10×.The mean average coverage for the cohort was 125.76×. In addition, the mean percentage coverage at 10× and 20× was 96.85 and 95.40%, respectively. There were several exceptions to the 100× average coverage, namely cases 82, 80, 36, and 38, and after several attempts the average coverage was lower, but other QC/QA parameters including the 95% at 10× were all met. Thus, in rare occasions, exceptions may be made if the sample is limiting, DNA quality is less than optimal, several attempts are made to meet all parameters, or if most of the other QC/QA parameters were met.
On average 32,171 single-nucleotide variants (pre-filtering) and small insertion and deletion, changes were identified in each patient’s exome by comparison with the current reference h19 haploid human genome sequence (Table S1 in Supplementary Material). On average 2031 (post-filtering) of potential clinically useful variants were kept after filters were applied (Table S1 in Supplementary Material). This number has continued to decrease by the utilization of internal normal controls and for current cases it is ~500 variants. Typically, three to eight variants were submitted for Sanger sequencing for each proband and family member(s). More than 84% of the variants selected for potential reporting were confirmed by means of Sanger sequencing. The remaining 16% were proven to be false positives due to unequal allele fractions, poor mapping scores, sequence homology, and insertion and deletion erroneous calls. Within the false positive variants, significant proportions were deletions (16%) and insertions (12%) and the remaining were single nucleotide variants (72%).
Diagnoses Using Exome Data
Of the 40 probands, 12 carried 18 mutant alleles at 15 different chromosomal loci that satisfied criteria for a full or partial molecular diagnosis (Table 2). The overall rate of a positive molecular diagnosis was 30% (Figure 1, Yield). This group included seven patients with autosomal dominant disease and five with autosomal recessive disease (Figure 1, Inheritance). A diverse group of disorders were represented in the positive patients (Table 2), including primary immunodeficiency, Ehlers–Danlos syndrome and multiple congenital anomalies, such as Otofaciocervical syndrome, Acromesomelic dysplasia Maroteaux type, and Bainbridge-Ropers syndrome.
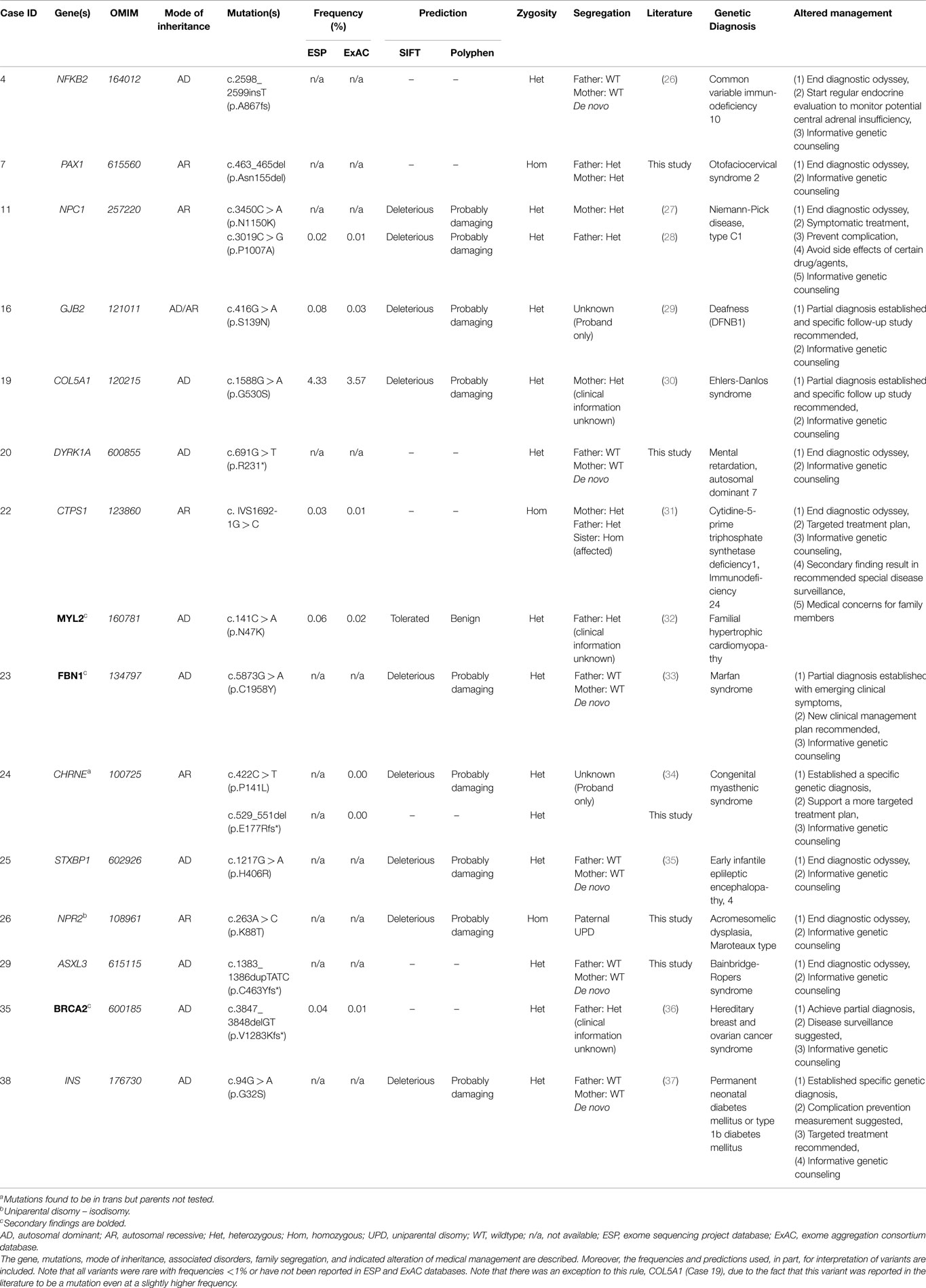
Table 2. Positive exome cases with pathogenic variants and secondary findings.
A full range of mutation types were observed in this cohort: three frameshift, two in-frame, two nonsense, two splicing, and eight missense mutations (Table 2). Missense (50%) and frameshift (19%) mutations made up the highest percentages of changes (Figure 1, Mutation type). The majority of these mutations were inherited (58%); however, a significant percentage of de novo mutations, defined as mutations present in the proband and not in the parents, were observed (29%) (Figure 1, Segregation). Moreover, 47% of the mutations were previously unreported in the peer-reviewed literature and variant databases (Figure 1, Literature). A total of seven patients had autosomal dominant disorders; of which, three were novel variants that had not been described in the peer-reviewed literature. For the five patients with autosomal recessive disease, parental studies indicated that four had inherited mutant alleles from each carrier parent. The remaining patient, for whom parental samples were not available, was found to have mutations in trans by allele-specific PCR.
Variants of unknown clinical significance were found in a number of patients (Table 3). These variants were placed on this table because of pathogenicity prediction inconsistency, higher minor allele frequency, and/or minimal published literature information; however, these genes/variants may explain part of the phenotype. For example, case 6 had two POLR3B variants classified as VUCS. c.1958A > T is a rare variant with frequencies less than 1% in ESP and ExAC. On the other hand, c.2218A > G is a variant with a higher frequency, but the predictions are inconsistent with one being probably damaging. Interestingly, both variants supported a compound heterozygous model and the gene is associated with the phenotype described for this patient. Similarly, cases 21, 34, 37, and 39 had higher variant frequencies, but with other lines of evidence that supported their pathogenicity such as a second allele in a compound heterozygous model, predictions that were deleterious and phenotype consistency. Importantly, these cases illustrate the complexity of variant interpretation and that frequency is only one of the variables used to bin them into pathogenicity categories. For cases 11 and 16, in addition to pathogenic variants, compound heterozygous variants classified as VUCS were found in genes which may contribute to the complex phenotypes in these patients. Only VUCSs were observed in cases 1, 2, 3, 6, 21, 23, and 24. These variants may be reclassified in the future once these genes/variants are better understood.
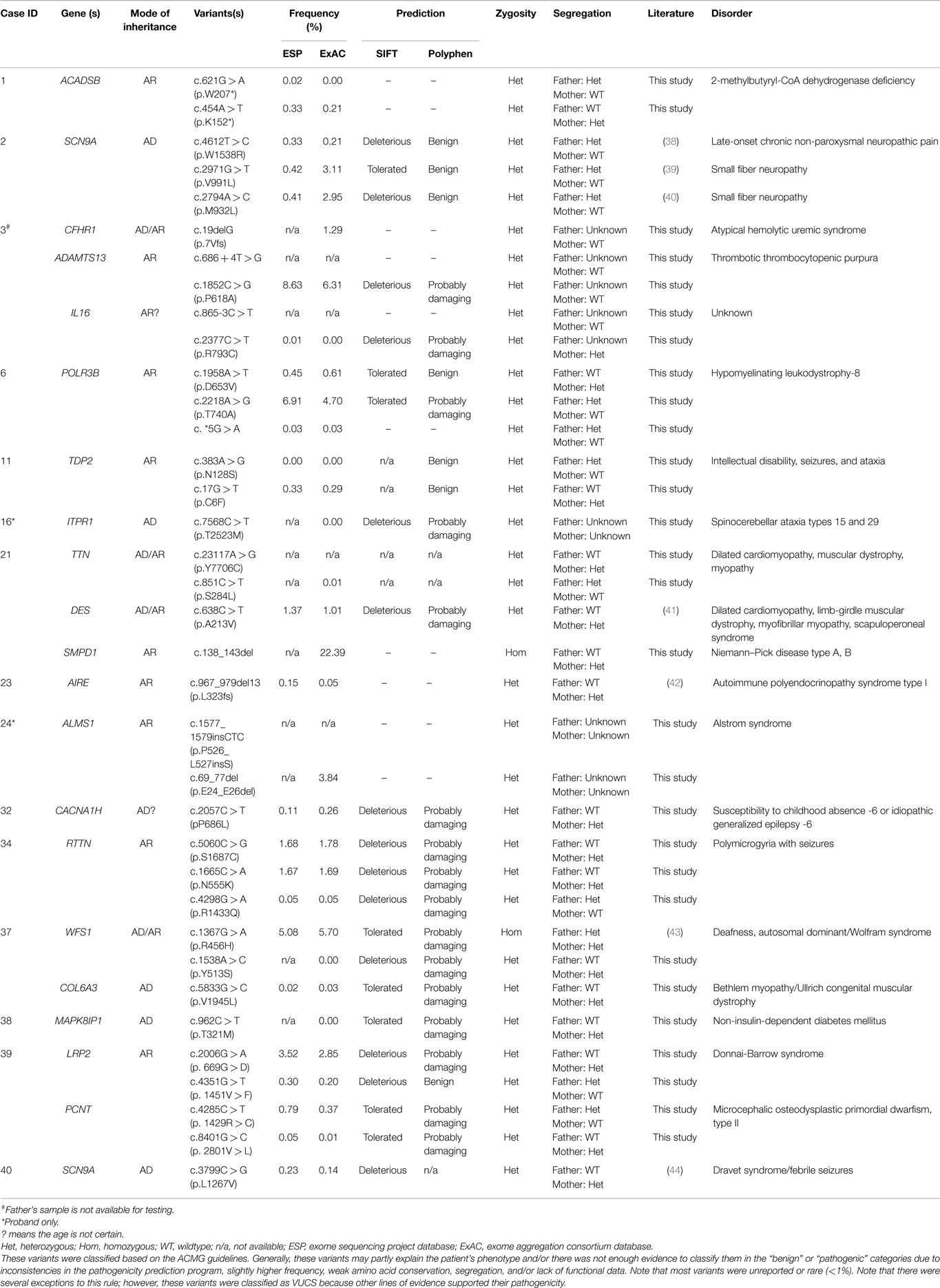
Table 3. Variants of unknown clinical significance.
Altered Management of Patients
In positive cases, WES results led to a change in the medical management of the patients (Table 2). Willig et al. defined medical management in a broader sense, namely, usefulness of genomic sequencing, return of results before discharge or death, genetic or reproductive counseling change, subspecialty consult initiation, medication change, procedure change, diet change, palliative care initiation, imaging change, and patient transfer to different facility, in a study aiming to identify Mendelian disorders in critically ill infants (45). For example, patient 38 was a six-year-old male with insulin-dependent diabetes, cataracts, and hyperglycemia, who did not fit the classic patterns of type 1 diabetes mellitus, maturity onset diabetes of the young (MODY), non-ketotic hyperosmolar syndrome, or neonatal diabetes. Upon exome sequencing, he was found to be heterozygous for a de novo missense mutation in the insulin gene (INS) which is associated with permanent neonatal diabetes mellitus or type 1b diabetes mellitus. Identifying the exact genetic cause of a patient’s diabetes is important as it directs future monitoring and treatment. Testing for monogenic diabetes is important and should be considered in any pediatric patient with antibody negative diabetes. In monogenic diabetes (GCK mutations excluded), monitoring for long-term complications such as retinopathy, neuropathy, and proteinuria should be considered at diagnosis given the long prodrome of unrecognized hyperglycemia. As science advances, patient 38 may be an ideal candidate for beta cell transplantation as the risk for islet cell antibody-mediated attack would be negligible. In addition to changing imaging and/or medication and initiation of subspecialty consults, a broader meaning of altered management was used in other exome positive cases which included receiving genetic counseling and ending the diagnostic odyssey (Table 2).
Secondary Findings
In addition to the diagnostic findings, 36 of 40 patients chose to receive secondary finding results. Three of the 36 patients had reported medically actionable secondary findings in a total of three genes (Table 2; Figure 1). Genes MYL2, FBN1, and BRCA2 were among the medically actionable genes recently recommended for reporting by the ACMG (25). For example, Patient 23 was 2 years old at the time of testing. He presented with hypogammaglobulinemia, recurrent infections, fine motor and speech delay, and feeding problems. Dysmorphic features included macrocephaly, prominent forehead, deep set eyes, thin upper lip, long digits, and persistent fetal finger pads. Although WES did not identify the cause of the primary clinical features, it importantly identified a de novo FBN1 mutation which gave the patient a secondary genetic diagnosis of Marfan syndrome. Upon the time of results return, he had developed hypermobility, gait abnormality, and abnormal posture. He was referred for cardiologic and ophthalmologic evaluations and for subsequent management for Marfan syndrome.
Cost Analysis
To address the cost of genetic testing prior to WES and consequently the potential cost-effectiveness of WES as a single test, we examined the number and type of genetic tests performed in our cohort prior to WES (Figure 2). Strikingly, there were 19 (48%) patients who had at least four genetic tests prior to WES (Figure 2A). From these 19 patients, three patients had a large number of genetic tests (>10 tests). The mean genetic test number for the cohort was ~4. Interestingly, microarray (63%) and single gene sequencing (63%) tests were the most frequently performed in this cohort, followed by karyotype and multi-gene panel sequencing. Therefore, the cost of genetic testing before WES is significant.
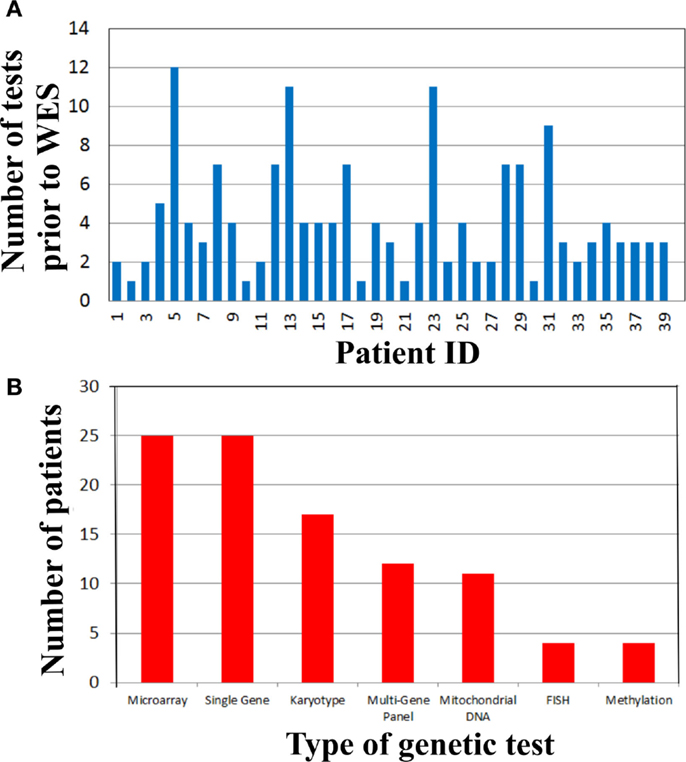
Figure 2. Genetic testing cost analysis of cohort prior to whole exome sequencing. (A) Number of genetic tests performed before whole exome sequencing per patient. (B) Types of genetic tests completed in the cohort prior to whole exome sequencing.
Discussion
On applying WES to the diagnoses of 40 unselected consecutive patients, we observed an overall molecular diagnostic yield of 30%, which is slightly higher than the positive rates of reported clinical exome tests of ~25% (Figure 1) (18, 19). This may be the result of different categories of presentation, sample bias, sample size, employment of a dual analysis pipeline, and phenotyping. The inclusion of dual analysis pipelines (NGP and GGHP) allowed the test to be more sensitive and in some cases showed that variants not captured by one pipeline were indeed detected by the other. Alternatively, because patients with multiple congenital anomalies had the highest diagnostic yield of any group (50%) (Figure 1; Table 2), referring physicians may have selected patients most likely to have a genetic etiology based on striking phenotypic attributes. Moreover, WES provided a diagnosis to 17% of patients with both neurological disorders and immunodeficiencies. About 8% of patients had both multiple congenital anomalies and either immunodeficiency or neurological disorders. Our results suggest that these three groups of patients are good candidates for testing with WES.
The utility of WES is higher than karyotype analysis (5–15%), chromosomal microarray analysis (15–20%), and may be comparable to disease-specific gene panels (46–48). The diagnostic yield of single gene tests vary from 0 to 64% depending on phenotype specificity and availability of complementary diagnostic tests (49). Similarly, previous studies that have employed next-generation sequencing approaches have shown varying positive rates. NGS panel positive rates for muscular dystrophy, sporadic intellectual disability, severe intellectual disability, immunodeficiencies, and retinitis pigmentosa have been reported to be 41, 31, 13, 15, and 82%, respectively (19, 49, 50). Thus, the WES positive rate is comparable to many other genetic tests that are considered part of the routine diagnostic workup.
Time-consuming, extensive and costly clinical diagnostic workups were performed before ordering the clinical WES test. This workup included a significant number of genetic tests (Figure 2). In fact, there were 19 (48%) patients who had at least four genetic tests prior to WES (Figure 2A) and microarray and single gene sequencing were the most frequently performed genetic tests (Figure 2B). These data demonstrated that costly genetic testing was performed prior to WES and in that in some cases (>10 genetic tests) the combination of genetic tests was even more expensive than WES itself. One explanation for this is that older technologies were employed in the diagnostic workup of some of these patients because those were the only tests available at that time. The cost of reaching a diagnosis became astronomical when the clinical work of non-genetic testing was added to the entire diagnostic testing equation. For example, patient 29 had chromosome and microarray analyses, mitochondrial studies, very long chain fatty acid study, brain MRI, spine MRI, muscle biopsy, and an EEG. He had consultations with a myriad of specialists including geneticists, ENT, orthopedics, and neurologists. This patient has a mutation in ASXL3, which is associated with the newly recognized Bainbridge-Ropers syndrome that would not have been identified by conventional genetic testing because it was not available at the time of diagnosis. Currently, one clinical laboratory started offering this gene as a clinical test in the United States in March of 2015. In some instances, WES may be the most cost-effective way to reach a diagnosis and guide appropriate management by significantly reducing the time to diagnosis and cost of testing, if it is implemented at an earlier time in the diagnostic workup of complex genetic cases. To improve clinical and cost outcomes more broadly, diagnostic algorithms that include WES testing need to be created and implemented in the near future.
WES testing is more comprehensive than other single gene and panel tests, but it is laborious and therefore uses expensive human resources. However, it is difficult to quantify these costs because analysis pipelines are constantly evolving and making the process faster by utilizing a growing powerful internal and public control datasets as an example. Caution should be taken when interpreting cost analysis numbers because exome sequencing and its complementary analysis is likely to continue to decrease in the near future, perhaps with the introduction of new sequencing technologies and clever algorithms that will find candidate variants quickly.
Even though microarrays, karyotyping, FISH, and methylation are non-sequencing assays, they were included in the cost analysis because there are known examples of overlapping clinical features caused by sequence mutations found by WES or other mutation types detected by the aforementioned techniques. The typical indications for microarray deletion/duplication analysis (SNP microarray) include developmental delay, intellectual disability, congenital anomalies, and dysmorphic features. These clinical characteristics were observed in several of our WES cases and microarray testing was pertinent and ruled out large deletion/duplication events and thus WES was the next step of the diagnostic workup because there are single gene mutations that are responsible for similar clinical characteristics (Table 1). In addition, another potential use of SNP microarray analysis is to identify loss of heterozygosity (LOH) regions in consanguineous cases. This information in conjunction with WES testing may aid in identifying the regions to be examined for homozygous variants. However, this information is not crucial because WES analysis includes a homozygous genotyping model and would identify candidate homozygous variants. Choosing to do other genetic testing prior to WES analysis and any other genetic testing permutations depend on specific clinical scenarios, thus, inclusion or exclusion of non-sequencing tests in cost analyses must be taken with caution.
Interestingly, 33% of our 12 patients with positive results were based on disease–gene discoveries made within the past 2 years (Table 2). Specifically, four patients, including those with mutations in NFKB2, PAX1, CTPS1, and ASXL3, would not have received a specific clinical diagnose if their exome data had been analyzed earlier than 2 years ago, prior to published works describing the gene–disease associations (31, 51–54). During this explosion of genomic data, it is likely that we will see the literature and databases populated with more cases that will aid in the interpretation of clinical exome data. There is also the possibility of an evolving phenotype that might at some point alter or add to the diagnosis in some patients (19). This new information may aid in the interpretation of VUCS variant calls (Table 3). However, in other cases extensive future research, which includes RNA-expression studies, needs to be performed to clarify the significance of VUCS variants.
Variants in multiple genes with described clinical phenotypes were identified in several patients. In a patient with common variable immunodeficiency (CVID)-like symptoms, Lindsley et al. addressed the functional consequences of mutations in NFKB2 [c.2598_2599insT (p.A867fs)] and TNFRSF13B [c.706G > T (p.E236*)] (Tables 2 and 3) (26). They reported the comprehensive immune evaluation of patient 4 and provide evidence that aberrant NFKB2 signaling not only causes humoral immune deficiency, but also interferes with the TCR-mediated proliferation of T cells. These observations expand the known phenotype associated with NFKB2 mutations. Unexpectedly, Western blot analysis of lysate from fresh, unstimulated PBMCs from the patient and father revealed a normal TNFRSF13B full-length protein. Yang et al. described multiple “hits” in four patients in their initial report and in a recent larger study, consisting of a cohort of 2000 consecutive exome cases, the same group reported a 4.6% positive rate for patients with multiple affected genes (19, 20). For atypical phenotypes, WES can expand the differential to include conditions that would otherwise not be considered. Additionally, WES is bound to expand the phenotype spectrum by the characterization of more complex and atypical cases.
The medical management of patients was altered after obtaining a positive exome test. Willig et al. have defined medical management in a broader sense in a study aiming to identify Mendelian disorders in critically ill infants (45). The definition of medical management includes usefulness of genomic sequencing, return of results before discharge or death, genetic or reproductive counseling change, subspecialty consult initiation, medication change, procedure change, diet change, palliative care initiation, imaging change, and patient transfer to different facility. From the traditional definition of medical management perspective, identifying the exact genetic cause of a patient 38’s antibody negative diabetes was important to direct future monitoring and treatment. Monitoring retinopathy, neuropathy, and proteinuria were considered at diagnosis (Table 2). Similarly, WES allowed change of management for patient 23 by being referred for cardiologic and ophthalmologic evaluations after the secondary finding of Marfan syndrome. In the broader sense of medical management, positive WES results altered the management of corresponding patients because they received genetic counseling, ended the diagnostic odyssey, changed imaging and/or medication, and initiated subspecialty consults to mention a few points.
Negative results for cases in which a diagnosis was not reached may be due to the lack of understanding of the exome, technical limitations, and patient selection bias. There are a large number of coding genes that have not been associated with human diseases and much needed genetic and functional studies should be done to elucidate their function. It is possible that some of these genes of unknown clinical significance (GUCS) may help explain the clinical features in several of our negative cases in the future. Thus, it is important to perform re-analysis on exome data annually to interrogate for newly discovered genes with human disease associations. Moreover, mutations may be located in non-coding regions, such as regulatory or deep intronic regions, that cannot be detected by WES. Moreover, mutations may be located in low coverage coding regions. Due to WES being a hybridization-based assay, the presence of multiple pseudogenes, homologous regions, or repetitive regions poses a technical challenge that obscures the presence of variants (55). In addition, large deletion/duplication mutations, complex rearrangements, trinucleotide repeats, and imprinting changes may be missed by WES and may represent a significant portion of mutations in the negative cases. It is possible that the clinical presentations of some of the patients could be explained by polygenic effects (complex disease) or a non-genetic etiology.
In conclusion, the use of WES to analyze 40 consecutive clinical cases yielded a diagnosis in 30% of these cases, which demonstrates the utility of this technology as a diagnostic test for pediatric patients with a wide variety of disease presentations. Positive WES results allowed clinicians to complete the genetic workup, end the diagnostic odyssey and provide appropriate medical management and more informative genetic counseling to families. Importantly, a number of novel mutations are being reported here. The cost-effectiveness of WES testing is evident by the reduction of time to diagnosis and cost of other testing and in some cases WES may be warranted as a first-tier test. Although there are technical challenges with NGS, WES provides a unique glimpse into the complexity of genetic disorders as well as the challenges in diagnosing them. However, healthcare system integration and routine adoption of WES need more careful consideration and future research.
Author Contributions
Drs. CV and KZ conceptualized and initiated the study, collected, analyzed, and interpreted the data, wrote and reviewed the manuscript; Mrs. AH, AM, TT, Mses. JJ and JH, and Drs. YQ, MK, KK, and JH developed the WES test and reviewed the manuscript; Drs. CW, SI, FZ, HM, XL, LW, and AB performed the data analyses and reviewed the manuscript; Mses. KC and KW, and Drs. TB, ES, DN, and RH provided patient’s clinical information and reviewed the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank Drs. Taosheng Huang, Nancy Leslie, Howard Saal, and many other physicians that referred patients to our institution for WES analyses. We want to thank the molecular genetics laboratory staff that performed the clinical WES tests. Funding sources: JH and KK are partially supported by the National Human Genome Research Institute (U01 HG006828).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/article/10.3389/fped.2015.00067
Abbreviations
ACMG, American College of Medical Genetics; CAP, College of American Pathologists; CASAVA, consensus assessment of sequence and variation; IRB, Institutional Review Board; MODY, maturity onset diabetes of the young; VUCS, variant of unknown clinical significance; WES, whole exome sequencing.
References
1. McCandless SE, Brunger JW, Cassidy SB. The burden of genetic disease on inpatient care in a children’s hospital. Am J Hum Genet (2004) 74:121–7. doi:10.1086/381053
2. Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, et al. The national institutes of health undiagnosed diseases program: insights into rare diseases. Genet Med (2012) 14:51–9. doi:10.1038/gim.0b013e318232a005
3. Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature (2009) 461:272–6. doi:10.1038/nature08250
4. Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DCY, Nazareth L, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med (2010) 362:1181–91. doi:10.1056/NEJMoa0908094
5. Vissers LELM, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, et al. A de novo paradigm for mental retardation. Nat Genet (2010) 42:1109–12. doi:10.1038/ng.712
6. Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med (2011) 3:87re3. doi:10.1126/scitranslmed.3002243
7. Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med (2012) 63:35–61. doi:10.1146/annurev-med-051010-162644
8. De Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med (2012) 367:1921–9. doi:10.1056/NEJMoa1206524
9. Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet (2012) 380:1674–82. doi:10.1016/S0140-6736(12)61480-9
10. Hoischen A, van Bon BWM, Gilissen C, Arts P, van Lier B, Steehouwer M, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet (2010) 42:483–5. doi:10.1038/ng.581
11. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron (2010) 68:857–64. doi:10.1016/j.neuron.2010.11.036
12. Krawitz PM, Schweiger MR, Rödelsperger C, Marcelis C, Kölsch U, Meisel C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet (2010) 42:827–9. doi:10.1038/ng.653
13. Lalonde E, Albrecht S, Ha KCH, Jacob K, Bolduc N, Polychronakos C, et al. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum Mutat (2010) 31:918–23. doi:10.1002/humu.21293
14. Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med (2010) 363:2220–7. doi:10.1056/NEJMoa1002926
15. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet (2010) 42:790–3. doi:10.1038/ng.646
16. Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet (2010) 42:30–5. doi:10.1038/ng.499
17. Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet (2010) 87:282–8. doi:10.1016/j.ajhg.2010.07.007
18. Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med (2014) 16(12):922–31. doi:10.1038/gim.2014.58
19. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med (2013) 369:1502–11. doi:10.1056/NEJMoa1306555
20. Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA (2014) 312(18):1870–9. doi:10.1001/jama.2014.14601
21. Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA (2014) 312:1880–7. doi:10.1001/jama.2014.14604
22. Ellard S, Kivuva E, Turnpenny P, Stals K, Johnson M, Xie W, et al. An exome sequencing strategy to diagnose lethal autosomal recessive disorders. Eur J Hum Genet (2015) 23:401–4. doi:10.1038/ejhg.2014.120
23. Patel ZH, Kottyan LC, Lazaro S, Williams MS, Ledbetter DH, Tromp H, et al. The struggle to find reliable results in exome sequencing data: filtering out mendelian errors. Front Genet (2014) 5:16. doi:10.3389/fgene.2014.00016
24. Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. Molecular subcommittee of the ACMG laboratory quality assurance committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med (2008) 10:294–300. doi:10.1097/GIM.0b013e31816b5cae
25. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med (2013) 15:565–74. doi:10.1038/gim.2013.73
26. Lindsley AW, Qian Y, Valencia CA, Shah K, Zhang K, Assa’ad A. Combined immune deficiency in a patient with a novel NFKB2 mutation. J Clin Immunol (2014) 34:910–5. doi:10.1007/s10875-014-0095-3
27. Sun X, Marks DL, Park WD, Wheatley CL, Puri V, O’Brien JF, et al. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am J Hum Genet (2001) 68:1361–72. doi:10.1086/320599
28. Greer WL, Dobson MJ, Girouard GS, Byers DM, Riddell DC, Neumann PE. Mutations in NPC1 highlight a conserved NPC1-specific cysteine-rich domain. Am J Hum Genet (1999) 65:1252–60. doi:10.1086/302620
29. Marlin S, Garabédian EN, Roger G, Moatti L, Matha N, Lewin P, et al. Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch Otolaryngol Head Neck Surg (2001) 127:927–33. doi:10.1001/archotol.127.8.927
30. Giunta C, Nuytinck L, Raghunath M, Hausser I, De Paepe A, Steinmann B. Homozygous Gly530Ser substitution in COL5A1 causes mild classical Ehlers-Danlos syndrome. Am J Med Genet (2002) 109:284–90. doi:10.1002/ajmg.10373
31. Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature (2014) 510:288–92. doi:10.1038/nature13386
32. Andersen PS, Havndrup O, Bundgaard H, Moolman-Smook JC, Larsen LA, Mogensen J, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J Med Genet (2001) 38:E43. doi:10.1136/jmg.38.12.e43
33. Ogawa N, Imai Y, Takahashi Y, Nawata K, Hara K, Nishimura H, et al. Evaluating Japanese patients with the Marfan syndrome using high-throughput microarray-based mutational analysis of fibrillin-1 gene. Am J Cardiol (2011) 108:1801–7. doi:10.1016/j.amjcard.2011.07.053
34. Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, et al. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit. Neuron (1996) 17:157–70. doi:10.1016/S0896-6273(00)80289-5
35. Saitsu H, Kato M, Okada I, Orii KE, Higuchi T, Hoshino H, et al. STXBP1 mutations in early infantile epileptic encephalopathy with suppression-burst pattern. Epilepsia (2010) 51:2397–405. doi:10.1111/j.1528-1167.2010.02728.x
36. Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet (1996) 12:333–7. doi:10.1038/ng0396-333
37. Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A (2007) 104:15040–4. doi:10.1073/pnas.0707291104
38. Dabby R, Sadeh M, Gilad R, Lampl Y, Cohen S, Inbar S, et al. Chronic non-paroxysmal neuropathic pain – novel phenotype of mutation in the sodium channel SCN9A gene. J Neurol Sci (2011) 301:90–2. doi:10.1016/j.jns.2010.10.006
39. Faber CG, Hoeijmakers JGJ, Ahn H-S, Cheng X, Han C, Choi J-S, et al. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol (2012) 71:26–39. doi:10.1002/ana.22485
40. Yuan R, Zhang X, Deng Q, Si D, Wu Y, Gao F, et al. Two novel SCN9A gene heterozygous mutations may cause partial deletion of pain perception. Pain Med (2011) 12:1510–4. doi:10.1111/j.1526-4637.2011.01237.x
41. Kostareva A, Sjoberg G, Gudkova A, Smolina N, Semernin E, Shlyakhto E, et al. Desmin A213V substitution represents a rare polymorphism but not a mutation and is more prevalent in patients with heart dilation of various origins. Acta Myol (2011) 30(1):42–5.
42. Millar S, Carson D. Clinical phenotypes of autoimmune polyendocrinopathycandidiasis-ectodermal dystrophy seen in the Northern Ireland paediatric population over the last 30 years. Ulster Med J (2012) 81(3):118–22.
43. Kytövuori L, Seppänen A, Martikainen MH, Moilanen JS, Kamppari S, Särkioja T, et al. WFS1 variants in finnish patients with diabetes mellitus, sensorineural hearing impairment or optic atrophy, and in suicide victims. J Hum Genet (2013) 58:495–500. doi:10.1038/jhg.2013.29
44. Singh NA, Pappas C, Dahle EJ, Claes LRF, Pruess TH, De Jonghe P, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet (2009) 5:e1000649. doi:10.1371/journal.pgen.1000649
45. Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med (2015) 3(5):377–87. doi:10.1016/S2213-2600(15)00139-3
46. Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the quality standards subcommittee of the American academy of neurology and the practice committee of the child neurology society. Neurology (2003) 60:367–80. doi:10.1212/01.WNL.0000031431.81555.16
47. Shaffer LG, American College of Medical Genetics Professional Practice and Guidelines Committee. American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation. Genet Med (2005) 7(9):650–4. doi:10.1097/01.gim.0000186545.83160.1e
48. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet (2010) 86:749–64. doi:10.1016/j.ajhg.2010.04.006
49. Valencia CA, Ankala A, Rhodenizer D, Bhide S, Littlejohn MR, Keong LM, et al. Comprehensive mutation analysis for congenital muscular dystrophy: a clinical PCR-based enrichment and next-generation sequencing panel. PLoS One (2013) 8:e53083. doi:10.1371/journal.pone.0053083
50. Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol (2014) 133:529–34. doi:10.1016/j.jaci.2013.08.032
51. Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet (2013) 93:812–24. doi:10.1016/j.ajhg.2013.09.009
52. Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MAA. Novel NFKB2 mutation in early-onset CVID. J Clin Immunol (2014) 34:686–90. doi:10.1007/s10875-014-0064-x
53. Pohl E, Aykut A, Beleggia F, Karaca E, Durmaz B, Keupp K, et al. A hypofunctional PAX1 mutation causes autosomal recessively inherited otofaciocervical syndrome. Hum Genet (2013) 132:1311–20. doi:10.1007/s00439-013-1337-9
54. Bainbridge MN, Hu H, Muzny DM, Musante L, Lupski JR, Graham BH, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med (2013) 5:11. doi:10.1186/gm415
Keywords: whole exome sequencing, next generation sequencing, diagnosis, children, clinical utility, pediatrics
Citation: Valencia CA, Husami A, Holle J, Johnson JA, Qian Y, Mathur A, Wei C, Indugula SR, Zou F, Meng H, Wang L, Li X, Fisher R, Tan T, Hogart Begtrup A, Collins K, Wusik KA, Neilson D, Burrow T, Schorry E, Hopkin R, Keddache M, Harley JB, Kaufman KM and Zhang K (2015) Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience. Front. Pediatr. 3:67. doi: 10.3389/fped.2015.00067
Received: 13 May 2015; Accepted: 13 July 2015;
Published: 03 August 2015
Edited by:
M. Z. A. Bhuiyan, University Hospital Lausanne, SwitzerlandReviewed by:
Peter Turnpenny, Royal Devon and Exeter NHS Foundation Trust, UKAlex Vincent Postma, Academic Medical Center, Netherlands
Xusheng Wang, St. Jude Children’s Research Hospital, USA
Copyright: © 2015 Valencia, Husami, Holle, Johnson, Qian, Mathur, Wei, Indugula, Zou, Meng, Wang, Li, Fisher, Tan, Hogart Begtrup, Collins, Wusik, Neilson, Burrow, Schorry, Hopkin, Keddache, Harley, Kaufman and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Alexander Valencia, Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, MLC7016, Cincinnati, OH 45229, USA, alexander.valencia@cchmc.org