The Centenary of Immune Thrombocytopenia – Part 1: Revising Nomenclature and Pathogenesis
 Rita Consolini
Rita Consolini Annalisa Legitimo
Annalisa Legitimo
- Laboratory of Immunology, Department of Clinical and Experimental Medicine, Division of Pediatrics, University of Pisa, Pisa, Italy
The natural history of the immune thrombocytopenia (ITP) is interesting and intriguing because it traces different steps underlying autoimmune diseases. The review points out the main steps that have accompanied the stages of its history and the consequential changes related to its terminology. ITP is an autoimmune disease resulting from platelet antibody-mediated destruction and impaired megakaryocyte and platelet production. However, research advances highlight that a complex dysregulation of the immune system is involved in the pathogenesis of this condition. The review examines the role of the multiple immune components involved in the autoimmunity process, focusing on the more recent mechanisms, which could be new promising therapeutic targets for ITP patients.
Introduction
Immune thrombocytopenia (ITP) is a blood disorder characterized by a low platelet count and mucocutaneous bleeding. The severity of bleeding generally correlates with the severity of thrombocytopenia. Its estimated incidence is 100 cases out of a million people per year; about half of these cases occurs in previously healthy children, where it represents the most frequent blood disorder. The etiology of ITP is still unknown, and the diagnosis continued to be one of exclusion: the appropriate methodological approach will be described in the second part of the review. Since the beginning of the century, its long history brought new insights about the pathogenesis of the disorder, leading to better refining of both terminology and clinical states. Although the platelet destruction by autoantibodies is considered the main pathogenetic mechanism, a complex dysregulation of the immune system is observed in ITP patients. Autoantibodies also may fix complement to platelets, T-cell and cytokine profiles are shifted to type 1 immune response, and the monocytic phagocytic system appears to be excessively activated, resulting in a pathogenic loop (1). In addition, several negative immune regulators such as T and B regulatory cells (Tregs, Bregs) and tolerogenic dendritic cells (DCs) are dysfunctional, enable to efficiently suppress the pathogenic process (1). The recently discovered T follicular helper cells (TFHs) are reported to be involved in B cell recruitment and differentiation in the spleen of ITP patients (2). Therefore, the autoimmune attack on platelets (and megakaryocytes) is amplified by multiple mechanisms. Together with the antibody suppression of megakaryopoiesis and thrombopoiesis, also other mechanisms such as abnormal apoptosis and thrombopoietin (TPO) dysregulation are described (3–7). Further, genetic factors are thought to play a role in susceptibility to developing ITP (8, 9). The knowledge of the multiple immune defects has opened new therapeutic opportunities that we will discuss in the next second part of the review.
The Intriguing History of ITP
The history of ITP is interesting and intriguing because it traces the different steps underlying autoimmune diseases. It starts with the identification of platelets into the blood, goes round the definition of the immuno-pathogenetic mechanisms, and ends by opening the window for new therapeutic options in the field of autoimmune disorders (Table 1). Many evolving concepts have subtended the pathophysiology of ITP. The historical perspective of ITP has been detailed in the outstanding review of Stasi and Newland (10), where the readers can found exciting suggestions for the comprehension of developing steps of ITP pathophysiology, as if it were a model of autoimmune disorder. Our review summarizes the essential points that mark the history of this disease. In 1915, Frank proposed that ITP resulted from the suppression of platelet production by megakaryocytes (MKs), due to a toxic factor produced by the spleen. The following year, based on the evidence that splenectomy normalized the platelet count in many patients with ITP, a medical student called P. Kaznelson, radically changed Frank’s hypothesis. He suggested that thrombocytopenia was due to an increased platelet destruction in the spleen, focusing on the critical role of the spleen in the pathogenesis of ITP. Furthermore, the mechanism of the spleen platelet destruction remained to be clarified, if by the direct destruction or by the release of an inhibiting factor. In 1938, Troland and Lee experienced that the injection of a substance (thrombocytopen) into the rabbits, extracted from spleen of patients with ITP, consistently produced a rapid, although transient, fall of the platelet count. Despite of this, other Authors affirmed that the disease was due to a fundamental abnormality of the spleen, which “exerted an unusual effect upon the production of platelets from the megakaryocytes in the marrow” (11). In 1951, Harrington–Hollingsworth experiments unequivocally settled the debate about the mechanism of the ITP thrombocytopenia, i.e. peripheral platelet destruction versus impaired platelet production. They demonstrated that healthy volunteers (Harrington included!), who received a blood transfusion from patient with ITP, became thrombocytopenic, concluding that ITP was characterized by reduced platelet survival due to a plasmatic factor. In the same year, Evans et al. suggested that the thrombopoietic factor was an antiplatelet antibody, identified as immunoglobulin (Ig) of G isotype (12). Its increase was subsequently demonstrated in 90% of chronic ITP patients (13). In 1982, new investigations about the antibody components of ITP, performed by Van Leeuwen et al., demonstrated that ITP patients produced autoantibodies against two platelet glycoproteins (GP), GPIIb and GPIIIa. Since that time, several laboratories have provided direct evidence for the presence of autoantibodies against GPIIb/IIIa and other platelet antigens in PTI (14–17). In 1994, the primary regulator of platelet production, called thrombopoietin, was purified and cloned (18–20); its role in the pathophysiology of ITP, underlying inappropriate thrombopoiesis in most patients, has been suggested (21, 22). In 1996, Wright et al. elucidated the cross-reactive nature of these autoantibodies, introducing in the ITP pathogenesis, the hypothesis of “molecular mimicry,” resulting in loss of tolerance and the activation of the autoimmunity mechanisms. Since the early 1980s, several studies have supported the hypothesis that thrombocytopenia might result not only from antibody platelet destruction but also from antibody against megakaryocytes (23–25). The role of autoantibodies in patients with ITP has been further defined by in vitro experiments, supporting the view that autoantibodies in ITP either suppressed MKs production or maturation and platelet release (26, 27). Other investigators confirmed the involvement of megakaryocytes, by showing in ITP patients, extensive megakaryocytic abnormalities suggestive of a mechanism of “non-classical apoptosis” (28). Already since 1991, Semple et al. suggested that not only the antibody response but also T cell compartment was involved with multiple abnormalities in the pathogenesis of ITP. In particular, they showed that the disorder might be the result of an abnormal T helper (Th) cell defect, that could direct autoreactive B cells to produce autoantibodies. Therefore, ITP was considered a disease due to an immune system multi-dysfunction, in which not only the T helper cells were involved, but also T regulatory cells that might play a role in the persistence of disease through the loss of tolerance (29, 30). Olsson et al., in 2003, brilliantly discovered that cytotoxic T (Tc) cells in ITP patients, deprived of platelet antibodies, could cause thrombocytopenia (31). Subsequent studies in 2007, suggested that CD8+ T cells lead to impaired platelet production, by suppressing autologous MK apoptosis (32). From the year 1951, that marked the beginning of the era of the immunologic character of ITP, focusing as central theme “the autoantibody-platelet destruction,” ITP pathogenesis appears to be multifactorial, resulting as a complex interplay involving multiple components of the immune system. The considerable advances in immunology over the past two decades expanded our knowledge about the immune defects underlying the ITP pathogenesis and allowed the development of “targeted therapies.”
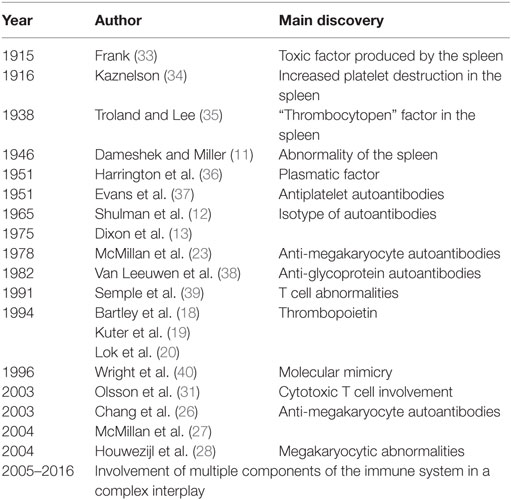
Table 1. The historical view of ITP.
New Terminology and Changes of Definition of the Different Phases of ITP
Based on the above described story, in 2009, the International Working Group (IWG), following the Consensus Conference in Vicenza, Italy (the Vicenza Consensus Conference, October 2007), proposed a new terminology, with the aim to underline the autoimmune pathogenesis of ITP [(41); Figure 1]. IWG decided to maintain the acronym ITP, recognizing its historical origin, but changing the meaning. The acronym ITP means “IMMUNE THROMBOCYTOPENIA”: the letter “P” today means “Primary” and replaces the outdated “Purpura,” which is considered an inappropriate term to describe the disorder, being the symptoms (like purpura) absent in most cases. Instead, the term “primary” refers to the absence of a recognized cause of the disease. Additionally, the IWG clearly distinguished between primary and secondary forms, in which thrombocytopenia is due to an underlying disease present at diagnosis (42). Unless otherwise specified, ITP refers to the primary form of immune thrombocytopenia. Along with the change of terminology concerning the disease definition, also the definition of its different phases has been changed. Traditionally, the definitions “acute ITP” and “chronic ITP” have been used to identify respectively a self-limited form of the disease (e.g., secondary to viral infection in children) and a lasting for more than 6 months form. Currently, as reliable predictive clinical and laboratory signs of disease duration have not yet been identified, the term “newly diagnosed ITP” is used to define all cases at diagnosis (41). In most cases (85%), the disease is self-limited, reaching a complete remission in 3–6 months from diagnosis. It is typically observed in children, both male and female with the same incidence, showing (in 50–60% of the cases) a recent history of viral disease. Twenty percent of cases do not achieve spontaneous remission or do not reach or maintain remission after treatment in the period lasting between 3 and 12 months from diagnosis; the new term “persistent ITP” is introduced to define these ITP patients. Furthermore, the term “chronic ITP” is used for patients with ITP lasting for more than 12 months. The only term that is historically maintained is “refractory ITP,” referred to the chronic form that is not responsive to the treatment.
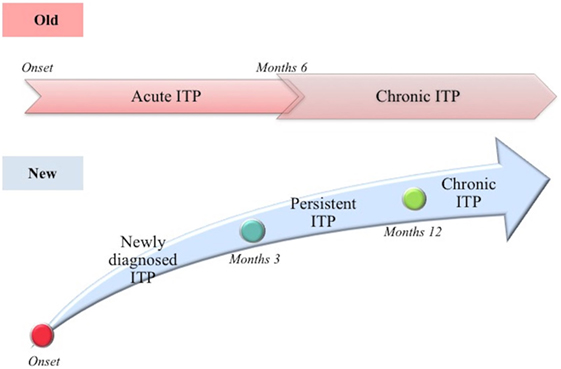
Figure 1. Old and new definition of the different stages of disease (immune thrombocytopenia).
Pathogenesis
Immune thrombocytopenia is historically considered an autoimmune disease attributed to platelet destruction due to antibodies secreted by autoreactive B cells. Moreover, continued research provided evidence that an impaired platelet production is also important in the pathogenesis of ITP, leading new insights into the comprehension of the complex immunopathogenic mechanisms underlying this disease (43, 44). The triggering event of ITP is unknown, and it is still unclear whether the immune defects associated with ITP play causative roles in the disease or are secondary epiphenomena (evoked by the structural platelet glycoprotein modifications) brought on by the inflammation associated with the disorder (44). Furthermore, a complex array of the immune mechanisms involved in the ITP autoimmunity has been delineated in the recent years.
Effects of Autoantibodies on Platelets and Megakaryocytes
Classically, the primary event of ITP is the arising of autoantibodies opsonizing platelets that leads to a markedly enhanced Fc-receptor (Fc-R)-mediated clearance of platelets by the monocytic phagocytic system (macrophages and dendritic cells) residing in the spleen (and liver) (43). Both platelet destruction and impaired platelet production are primarily mediated by IgG autoantibodies, although IgM and IgA may be involved (45, 46), mainly directed against platelet membrane glycoprotein complexes, such as GPIIb/IIIa and GPIb/IX (2, 47); other GP (Ia–IIa, IV, and V) have also been identified (46). Furthermore, it has been demonstrated that the two most frequent platelet membrane glycoprotein targets, GPIIb heterodimer and GPIb/IX complex, were also expressed on megakaryocytes during the early stages of differentiation (48). Moreover, autoantibodies were also found to bind MKs (23). In the late 1960s, animal models provided the first demonstration that autoantibodies might not only cause peripheral destruction of mature platelets but also directly affect MKs. Subsequently, in a study of pediatric patients suffering from ITP, a significant suppression of in vitro MK production was observed, when the plasma samples contained anti-GPIb/IX antibodies alone or in combination with anti-GPIIb/IIIa antibodies (26). By using MK cultures derived from human CD34+ cell, it has been recently demonstrated that antiplatelet antibodies inhibited proplatelet formation by MK and, therefore, their ability to release platelets (49). Additionally the discover of an autoantibody-antigen target, the Mpl TPO receptor (the cellular homolog of the myeloproliferative leukemia virus oncogene), allowed to demonstrate, by using plasma derived from thrombocytopenic ITP patients in MK colony-forming assays, the suppression of MK proliferation by anti-Mpl autoantibodies (50).
The Role of Complement
There are clear indications that complement activation is involved in the disappearance of platelets from the circulation of patients with ITP (51), although its role and that one of complement receptors in the pathogenesis of ITP are poorly understood (52). The autoantibodies, specifically bound to platelet antigens, can fix complement (C3) on platelet and megakaryocyte (MK) membranes, triggering cell destruction through complement system, either by enhancing clearance or direct cell destruction. Subsequent studies have described the intrinsic ability of platelets to activate both classical (53, 54) and alternative (52, 55) pathways of complement.
Other Mechanisms of Impaired Thrombopoiesis
Platelet kinetic studies suggested that both impaired platelet production/destruction might be determined not only by autoantibody suppression of both megakaryopoiesis and thrombopoiesis, but also by other mechanisms such as abnormal apoptosis and TPO dysregulation (3, 4). Although an increased number of mature megakaryocytes compared to normal controls are observed in ITP patients, the maturation program of these cells appears to be impaired. The histopathology and ultrastructural abnormalities of bone marrow of the majority of patients are consistent with an apoptosis-like programed cell death (28). Elevated apoptosis markers and an increased proportion of megakaryocytes with activated caspase-3 have been observed in bone marrow biopsies of ITP patients (28). TPO is the primary regulator of megakaryocytopoiesis and thrombopoiesis, supporting cell survival, cell cycling, and modulating apopotosis and cell cycle regulators (46). Patients with ITP, considering their often extremely low platelet count, characteristically exhibited lower than expected levels of endogenous TPO (eTPO) (56–58). It has also been observed that, although in the presence of similar platelet counts, eTPO levels in ITP patients were markedly lower than those observed in patients with aplastic anemia (5, 48, 59). The reason of this unexpected observation could be explained because of binding to Mpl on the increased MK mass with subsequent internalization and degradation or secondary to TPO bounds to platelets targeted for destruction (46). Furthermore, the observation of reduced TPO levels in the presence of low platelet count suggested impaired platelet production and failure to compensate for the thrombocytopenia (48). However, other studies comparing eTPO and platelets counts in ITP patients reported conflicting results (5, 6). Although the prevailing model of steady-state TPO regulation is based on the circulating TPO concentration inversely proportional “to Mpl mass,” a novel physiological feedback mechanism has been documented. Grozovsky et al., in 2015, demonstrated that the circulatory lifespan of platelets was determined by sialic acid loss that triggers platelet removal by the hepatic Ashwell–Morell receptor (AMR). The AMR is a transmembrane heteroligomeric glycoprotein complex composed of ASGPR1 (CLEC4H1, HL-1) and ASGPR2 (CLEC4H2, HL-2) subunits, which are highly conserved among mammalian species (7, 60). The AMR-mediated removal of desialylated aged platelets regulates TPO synthesis in the liver by recruiting janus kinase2 (JAK2) and transcrption3 (STAT3) phosphorylation (7). These data contribute to explain “the plasma TPO-level discrepancies” observed in human pathologic conditions, characterized by thrombocytopenia and liver disease, abovementioned. In fact, the antibody-mediated clearance of sialylated platelets would avoid the AMR platelet removal and, therefore, reduce TPO expression (7). These studies opened an interesting window of opportunity, indicating TPO receptor agonists (TPO-RA) as an alternative treatment option for children with chronic ITP. Clinical trials in children are ongoing and data are emerging on safety and efficacy of these agents (61). Although the autoantibodies-mediating accelerated platelet clearance from the circulation continues to be the central theme in the current understanding of ITP, its pathogenesis appears to be multifactorial, resulting as a complex interplay involving multiple components of the immune system (Figures 2 and 3).
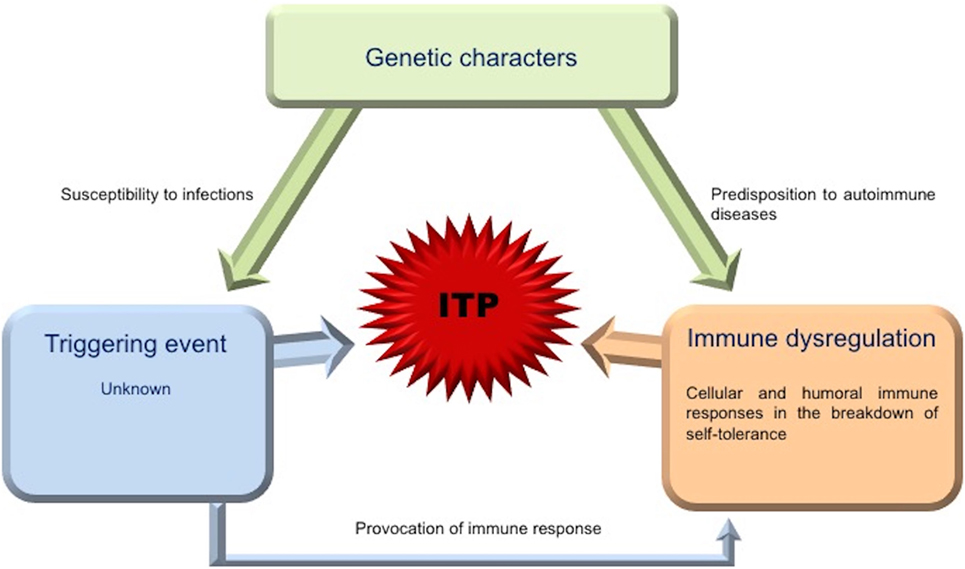
Figure 2. Picture of interplay of contributing factors in ITP. The triggering event for ITP is substantially unknown. Genetic factors may play an important role in the development of the disease, being associated with increased susceptibility to infections or predisposition to autoimmune diseases. The generalized immune dysregulation, with an imbalance between the immunoregulatory elements (cells, receptors, cytokines, and other signaling molecules), regulatory and effector T cells, drives pathogenic T and B cell effector responses against platelets and megakaryocytes.
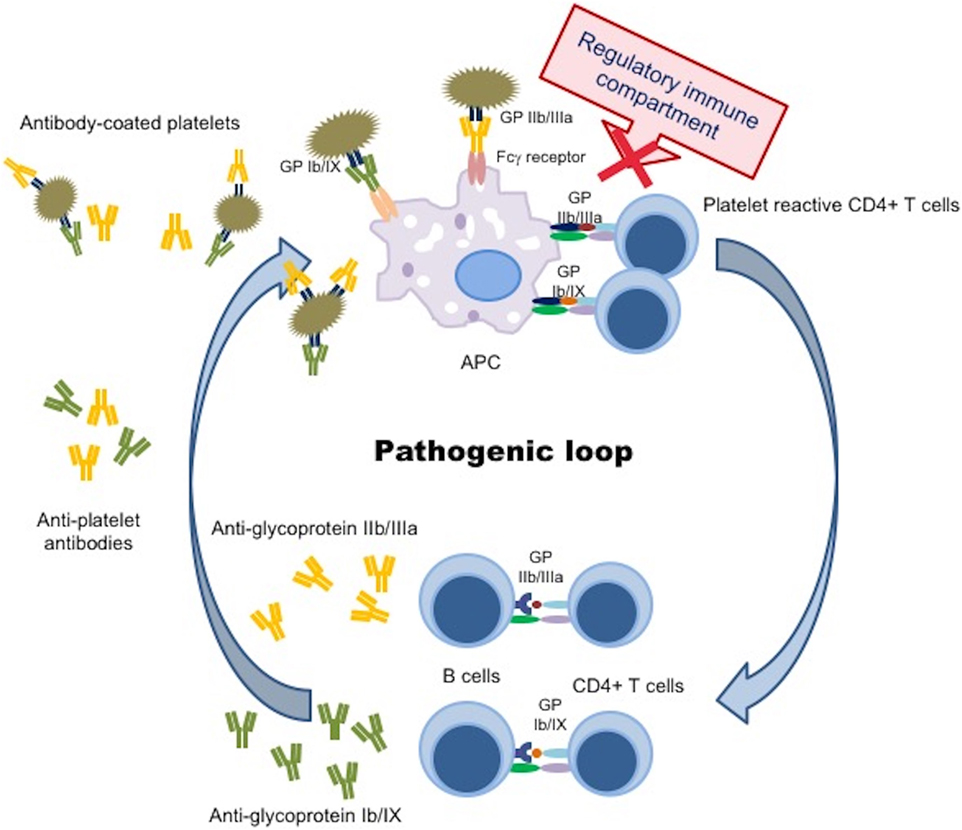
Figure 3. Pathogenic loop in immune thrombocytopenia. Schematic picture of a continuous pathogenic loop carried out by APC, autoreactive CD4+ T cells, and autoantibody-producing B cells that maintains antiplatelet antibody production in ITP patients. A defect in the immune regulatory compartment (DCs, Tregs, Bregs) did not suppress the interaction between APC and autoreactive CD4+ T cells, resulting in prevention from a trigger of the pathogenic loop. APC, antigen presenting cell; GP, glycoprotein.
The Role of Antigen-Presenting Cells
Following the exposition to the cell surface of “cryptic” antigens, the appearance of a wide spectrum of new peptides can therefore induce an “acquired cell recognition of new self-determinants” by the antigen-presenting cells (APC), playing a crucial role not only in the arising but also mainly in the progression of the ITP (62). These cells in normal conditions play the primary function of recognizing, binding to and internalizing protein antigens for endosomal proteolytic degradation. After these peptides complexed with MHC molecules are presented to T cells in conjunction with signaling costimulatory events (i.e., CD28/B7 and CD80/86 interactions between APC and T cell), and in the presence of appropriate cytokines, the T cell become properly activated (44). The local microenvironment and extrinsic stimuli can influence APC phenotype and function; in certain conditions, such as inflammation, APC abnormalities, in number and function, lead to abnormal processing or presentation of self antigens or inappropriate T cell activation. Dendritic cells, even referred as professional APC, are the most specialized among all leukocytes in the presentation of antigen. Peripheral blood DCs are classified in two broad groups according to their lineage, myeloid DCs (mDCs), often referred as conventional DCs, and plasmacytoid DCs (pDCs) (63). The mDC subset plays specific functions in the initiation of adaptive immune response, being critical for the activation of effector T cells and differentiation of naive CD4+ T cells into Th1 cells. The pDCs produce high levels of type I interferon (IFN-α/β/ω), reflecting their important function in antiviral immune responses (1, 63–65). The type I IFN is a pleiotropic cytokine not only with antiviral properties but also with capability to regulate pDC survival, mDC differentiation, mDC-mediated cytotoxic T cell responses, cross presentation, upregulation of costimulatory MHC molecules, and activation of natural killer (NK) and B cells (63, 66–69). Plasmacytoid DCs also display pro-inflammatory and immunosuppressive tolerogenic properties (70). Recently, low number of circulating pDCs has been shown in untreated patients with ITP (65). These authors explained this result as an abnormality of their distribution and suggested that pDCs could be mobilized into peripheral inflamed tissues, particularly to the spleen. Together with the pDC number alteration, also the dysfunction of DCs has been demonstrated to contribute to the development of ITP (71). In experiments performed by pulsing DCs from ITP patients and healthy donors, with autologous/allogeneic fresh and aged platelets, Catani et al. (71) showed that the DCs from patients stimulated T-cell proliferation in a greater degree than those one from healthy donors. They concluded that this might be due to an increased expression of the costimulatory antigen CD86 on the surface of DC patients (71). The importance of the ITP dysregulation of the APC-T cell cross talk has been reported by Zhong et al. (72).
The Role of Cytotoxic Cells
Cytotoxic T lymphocytes (CTLs) are cells able to kill neoplastic or infected cells with bacteria or viruses by programing these cells to undergo apoptosis (73). In ITP patients, with no identifiable anti-platelets autoantibodies, CTLs have been shown to increase both platelet and MK lysis, suggesting the role of these cells in mediating thrombocytopenia (31, 32, 74–76). Apoptosis and perforin/granzyme-mediated cytotoxicity constitute the main pathway used by CTLs to destruct autologous platelets (75). A variety of T cell and cytokine abnormalities have been observed in patients with ITP, suggesting that T cell-mediated peripheral platelet destruction and MK destruction/inhibition could be involved in the pathogenetic mechanism of thrombocytopenia (26, 27, 31, 44, 77–79). Interleukin-27 (IL-27), a member of IL-12 family, is a cytokine with multiple immunomodulatory functions, showing both pro-inflammatory and anti-inflammatory effects (80). Liu et al. demonstrated low plasma and mRNA expression levels of IL27 in active ITP patients, suggesting that this cytokine might be involved in the pathogenesis of ITP (81). Recent data suggested that IL-27 could inhibit platelet destruction by negatively regulating CTL cytotoxicity toward autologous platelets in ITP (80); this negative regulation was obtained by decreasing granzyme B expression whereas granzyme A and perforin were not affected (80). Based on these experiments, it has been suggested that IL-27 might have a therapeutic role in ITP patients (80). Thus, in addition to platelet-specific autoantibodies, also CTLs and aberrant cytokine profiles can be involved in platelet destruction, playing important role in the pathogenesis of ITP (73, 80).
T Follicular Helper Cells
T follicular helper cells are the specialized providers of B cell help; they are the key cell type required for the formation of germinal centers (GCs) and the generation of long-lived serological memory. TFHs have emerged to play a key role in regulating the humoral immune response that occurs with autoimmune diseases, infectious diseases, and tumors (82). TFHs are characterized by low expression levels of cytokines, IFN-γ, IL-4, and IL-17, characteristic of Th1, Th2, and Th17 cells respectively and by the expression of effector molecules that are critical for their development and function, including chemokine receptor 5 (CXCR5), inducible costimulator (ICOS), programed death-1 (PD-1), surface receptors of IL-21, IL-6, CD40, and transcription factors Bcl-6 and c-Maf (2, 83). It has been demonstrated that human splenic TFHs are the main producers of interleukin IL-21 (2), a cytokine playing a critical role in T cell and B cell homeostasis. In B cells, IL-21 signaling is involved in germinal center formation, Ig-secreting B cells, and antibody response (83, 84), while in CD4 T cells, it promotes the development of Th17 and TFHs themselves (83–85).
In ITP patients, splenic TFH frequency is higher than healthy donors and correlates with germinal center and plasma cell percentages that are also increased (2). Although the role of the inflammatory environment in the increase in TFH frequency could not be completely excluded, Audia et al. (2) strongly suggested that the expansion of TFHs could be involved in B-cell recruitment and differentiation in the spleen of ITP patients, through IL-21 secretion and by the interaction of CD154 with CD40, contributing to the development of plasma cells producing antiplatelet autoantibodies. Therefore, TFHs and associated molecules, such as IL-21 and CD40, could be new promising therapeutic targets for ITP patients.
The Loss of Tolerance
The Th1/Th2 Balance
Immune homeostasis is maintained via a balance of two types of responses, defined by cytokine secretion profiles. The responses of type 1, characterized primarily by IL2, IFNγ, TNFα, and TNFβ1 production, are involved in response to intracellular pathogens and generally promote pro-inflammatory, cell-mediated, complement fixing phenotypes. On the other hand, type 2 reactions, characterized by IL4, IL5, IL6, IL10, and IL13 cytokine production, function in the fight against extracellular pathogens, and typically elicit an immediate-type hypersensitivity response (43). The polarization of the immune system toward either type 1 or type 2 immunity is dependent on the level of the cytokines. In ITP, type 1/type 2 ratio is unbalanced, shifted toward a type 1 phenotype, favoring autoreactive B cell development (a Th2 phenotype being observed in patients during remission or during intravenous immunoglobulin treatment) (1, 43, 86, 87). This has been attributed to the reduction of peripheral Th2 cells, of Tc2 cells and of CD25brightFoxp3+ Tregs either in number or in function (30, 43, 87–90). Zhong et al. (72) showed that the CD16+ monocyte subset from the peripheral blood of chronic ITP promoted the expansion of the Th1 subset via IL12 secretion, while inhibited the proliferation of Th17 cells and the induction of Tregs. The Th1/Th2 balance in ITP is showed in Figure 4.
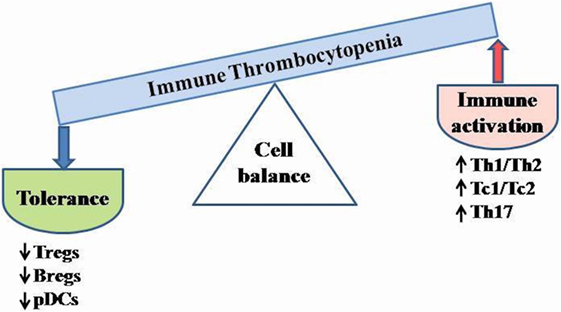
Figure 4. Schematic cell imbalance in ITP. Dysregulation of T cell activity and cytokine abnormalities are critical for ITP development. The unbalanced type 1/type 2 ratio leads to autoreactive B cell differentiation. Reduced number and/or impaired function of Tregs, Bregs, and tolerogenic DCs may contribute to enhance the immune activation. Bregs, B regulatory cells; pDCs, plasmacytoid dendritic cells; Tregs, T regulatory cells; Th, T helper cells; Tc, T cytotoxic cells.
Treg and Th17 Compartments
Tregs are a subpopulation of T cells specialized in immune suppression and contribute to the maintenance of peripheral immune tolerance; their defects are thought to play a role in the pathogenesis of various autoimmune diseases (91). In the last decade, data from numerous studies have demonstrated a dysregulation of Treg cell population in ITP (29, 30, 91–102). Treg deficiency was reported in both adults and children with ITP (91): the lowest Treg frequency was detected in patients with acute phase of the disease and/or with low platelet count, while Tregs increase was observed in remission phase. Interestingly, a failure of the cross communication between DCs and Tregs in patients with ITP has been reported (103). These authors showed that the decreased expression and/or activity of the immunomodulatory enzyme indoelamine 2,3-dioxygenase 1 (IDO1) by DCs, contributed to the reduced conversion from T cells into Tregs, suggesting the importance of the cross talk between DCs and the adaptative immune system in the pathophysiology of immune thrombocytopenia (103). T cell-related pro-inflammatory cytokines may play a pivotal role in immune dysregulation during active ITP. In the past years, increased levels of macrophage colony-stimulating factor (M-CSF) (causing a stimulus for to the platelet antigen presentation by macrophages to T cells), reduced levels of transforming growth factor (TGF)-β production have been described in children with chronic ITP (104). IL10 is a cytokine with anti-inflammatory properties; initially described as a product of Th2 cells able to inhibit cytokine synthesis of Th1 cells (105), IL-10 is now known to be produced by many different types of cells, including Tregs (106). Based on data showing that IL10 producing Tregs contributed to the effective control of several autoimmune diseases (107), a recent study investigated the role of this cytokine in newly diagnosed ITP patients (106). The Authors identified a numerical and functional defect of Tregs associated with an excessive activation T effector cells, suggesting that the assessed insufficient secretion of IL-10 could compromise the inhibitory capability of Tregs against T effectors cells and play a major role in the exuberant CD4+ T cell immune response of ITP (106). More attention has been deserved to IL17, a cytokine produced by Th17 cells, a newly defined CD4+ Th subset; Th17 cells modulate the pro-inflammatory response by producing, as well as IL17, also IL6, IL21, tumor necrosis factor (TNF), and other mediators (108–110). Thus, while Treg cells play a fundamental role in the maintenance of immune tolerance to prevent autoimmune disease, Th17 cells play the opposite role. Indeed, it has been demonstrated that Th17 cells are more potent than Th1 cells in inducing autoimmune disorders (111–113). Several reports support a role for IL17 in the pathogenesis of ITP. Rocha et al. (110) suggested that increased levels of IL17 and of Th17-related cytokines found in adult patients with chronic ITP might contribute to the disease pathogenesis. The Treg/Th17 imbalance has been found associated with disease activity among adult ITP patients, implying an association between Th17 cell elevation and the development of ITP and suggesting a prognostic role of this cytokine (114). Wang et al. (115) found that IL17 levels were elevated in pediatric patients with chronic ITP and positively correlated with INFγ expression, suggesting that both Th17 and Th1 might be simultaneously involved in the dysregulation of cellular immunity observed in pediatric patients. However, other groups did not report significant differences either in the serum levels or in the expression of IL17 on peripheral blood mononuclear cells between ITP patients and normal controls (116, 117), arguing that IL17 might not play an important role in the pathogenesis of adult patients with chronic ITP. These conflicting results concerning the IL17 role underline the variability in the clinical phenotype and evolution of the disease duration among the ITP population. The IL23/IL17 axis is critical for the development of inflammatory diseases. IL23, which is mainly secreted by APCs, is the pivotal mediator responsible for the differentiation of naive CD4+ T cells into Th17 cells; the importance of the IL23 cytokine and of its receptor in both Th17 maturation and function has been demonstrated (113, 118, 119). Several studies reported that the IL23/Th17 pathway was strongly involved in the pathogenesis of many autoimmune diseases, such as rheumatoid arthritis (120), multiple sclerosis (121), primary biliary cirrhosis (122), and inflammatory bowel disease (123). However, less is known with regard to the levels of expression and synthesis of these two cytokines in patients with ITP. The role of IL23/Th17 pathway in adult ITP patients has been recently investigated (113). These Authors, found higher levels of IL17 and IL23 in patients with ITP than in controls and a decrease of both cytokines after effective treatment, suggesting that IL23 was engaged in the development of ITP through enhancement of the Th17 response.
The Role of Bregs
Interestingly, Bregs are a distinct B cell population with potent immunoregulatory properties; among them, the most well subset characterized is the IL10-producing subset (B10 cells) (1, 124, 125), which plays an important role in autoimmune diseases (1, 87, 126, 127). Recent findings showed that IL10 produced by B10 cells is required in combination with different costimulatory molecules for the differentiation and maintenance of Tregs and for inhibition of Th17 cells (87, 128, 129). Recently, it has been demonstrated that a dysregulation of Breg compartment was an additional defect in the immune network in ITP patients (1, 87). Hua et al. (87) demonstrated that the number of B10 cells positively correlated with both Treg count and Treg/Th17 ratio, suggesting that the ability of these cells to regulate functional T cell subsets might be impaired in ITP patients.
Genetic Susceptibility
Only a small number of individuals develop ITP after exposure to infectious agents. Such variability could be explained by differences in genetic factors associated with the host defenses to pathogens, such as genes encoding inflammatory cytokines (9). In the recent past, genomic studies have allowed the knowledge of genetic factors influencing the development of ITP chronic pattern. Abnormal polymorphisms have been identified within the genes for several inflammatory cytokines. Satoh et al. (8) identified polymorphisms at TNFβ (+252 GG) gene. As TNFβ (+252) influences the capacity of T and B cells to produce TNFβ, by checking the autoreactive T- and B-cell responses to platelet membrane antigens, the Authors suggested that these polymorphisms might play a role in an individual’s susceptibility to ITP, by promoting the specific autoantibody response. Interestingly, a recent study demonstrated that the heterozygous variant (AG) genotype of TNFβ was associated with persistent ITP (130). Reports about TNF-α-308 gene polymorphisms are conflicting in both adult and pediatric ITP (130–132). IL4 (VNTR intron 3) and IL10 (−627) polymorphisms have also been associated with the pathogenesis of ITP and believed to contribute to the susceptibility of developing ITP (133–135). CNR2 is a gene encoding CB2, a cannabinoid receptor expressed on various immune cells and involved in immune regulation by modulating Th1 and Th2 balance and cell migration and by inhibiting pro-inflammatory cytokine production (9, 136, 137). CNR2 functional variant (Q63R) has been described to influence childhood ITP toward the chronic course of the disease (9, 138). Recent works demonstrated that stress-related pathways might contribute to the pathogenesis of ITP. In particular, over expression of vanin-1 (VNN-1), an oxidative stress sensor, has been demonstrated to be strongly associated with progression to chronic disease in both pediatric and adult patients (9, 139–141).
Conclusion
Since the beginning of the century, new insights have been provided about the pathogenesis of ITP. Consequently, the nomenclature concerning both its definition and evolutionary steps are modified. Although the accelerated platelet destruction by platelet autoantibodies is considered the hallmark of ITP pathogenesis, research advances highlight the complex immune mechanism underlying the disease. The continuous fine understanding of the contribution of each immune defect (e.g., the checkpoint tolerance defect variously involved in ITP patients) could explain the clinical diversity and the different tendency to perpetuate the disease observed in the ITP population and similarly open new windows of opportunities for targeted therapy and “tailored treatment.”
Author Contributions
All Authors contributed to the work presented in this paper. RC wrote the paper. All Authors reviewed the paper, and provided approval of the final version.
Conflict of Interest Statement
The authors declare that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kuwana M. Dysregulated negative immune regulators in immune thrombocytopenia. VOXS (2014) 9:217–22. doi:10.1111/voxs.12065
2. Audia S, Rossato M, Santegoets K, Spijkers S, Wichers C, Bekker C, et al. Splenic TFH expansion participates in B-cell differentiation and antiplatelet-antibody production during immune thrombocytopenia. Blood (2014) 124(18):2858–66. doi:10.1182/blood-2014-03-563445
3. Isaka Y, Kambayashi J, Kimura K, Matsumoto M, Uehara A, Hashikawa K, et al. Platelet production, clearance and distribution in patients with idiopathic thrombocytopenic purpura. Thrombosis Res (1990) 60:121–31. doi:10.1111/j.1365-2141.2007.06737.x
4. Louwes H, Zeinali Lathori OA, Vellenga E, de Wolf JT. Platelet kinetic studies in patients with idiopathic thrombocytopenic purpura. Am J Med (1999) 106:430–4. doi:10.1016/S0002-9343(99)00054-6
5. Gu J, Lu L, Xu R, Chen X. Plasma thrombopoietin levels in patients with aplastic anemia and idiopathic thrombocytopenic purpura. Chin Med J (2002) 115:983–6.
6. Kuwana M, Okazaki Y, Satoh T, Asahi A, Kajihara M, Ikeda Y. Initial laboratory findings useful for predicting the diagnosis of idiopathic thrombocytopenic purpura. Am J Med (2005) 118:1026–33. doi:10.1016/j.amjmed.2004.12.027
7. Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, et al. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat Med (2015) 21(1):47–54. doi:10.1038/nm.3770
8. Satoh T, Pandey JP, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y, et al. Single nucleotide polymorphisms of the inflammatory cytokine genes in adults with chronic immunethrombocytopenic purpura. Br J Haematol (2004) 124:796–801. doi:10.1111/j.1365-2141.2004.04843.x
9. Yacobovich J, Revel-Vilk S, Tamary H. Childhood immune thrombocytopenia-who will spontaneously recover? Semin Hematol (2013) 50(Suppl 1):S71–4. doi:10.1053/j.seminhematol.2013.03.013
10. Stasi R, Newland AC. ITP: a historical perspective. Br J Haematol (2011) 153(4):437–50. doi:10.1111/j.1365-2141.2010.08562.x
11. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood (1946) 1(1):27–50. doi:10.1182/blood-2015-11-682948
12. Shulman NR, Marder VJ, Weinrach RS. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann N Y Acad Sci (1965) 124:499–542. doi:10.1111/j.1749-6632.1965.tb18984.x
13. Dixon R, Rosse W, Ebbert L. Quantitative determination of antibody in idiopathic thrombocytopenic purpura. Correlation of serum and platelet-bound antibody with clinical response. N Engl J Med (1975) 292:230–6. doi:10.1056/NEJM197501302920503
14. Woods VL Jr, Kurata Y, Montgomery RR, Tani P, Mason D, Oh EH, et al. Autoantibodies against platelet glycoprotein Ib in patients with chronic immune thrombocytopenic purpura. Blood (1984) 64:156–60.
15. Woods VL Jr, Oh EH, Mason D, McMillan R. Autoantibodies against the platelet glycoprotein IIb/IIIa complex in patients with chronic ITP. Blood (1984) 63:368–75.
16. Kiefel V, Santoso S, Weisheit M, Mueller-Eckhardt C. Monoclonal antibody specific immobilization of platelet antigens (MAIPA): a new tool for the identification of platelet-reactive antibodies. Blood (1987) 70:1722–6.
17. McMillan R, Tani P, Millard F, Berchtold P, Renshaw L, Woods VL Jr. Platelet associated and plasma anti-glycoprotein autoantibodies in chronic ITP. Blood (1987) 70:1040–5.
18. Bartley TD, Bogenberger J, Hunt P, Li YS, Lu HS, Martin F, et al. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell (1994) 77:1117–24. doi:10.1016/0092-8674(94)90450-2
19. Kuter DJ, Beeler DL, Rosenberg RD. The purification of megapoietin: a physiological regulator of megakaryocyte growth and platelet production. Proc Nat Acad Sci U S A (1994) 91:11104–8. doi:10.1073/pnas.91.23.11104
20. Lok S, Kaushansky K, Holly RD, Kuijper JL, Lofton-Day CE, Oort PJ, et al. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature (1994) 369:565–8. doi:10.1038/369565a0
21. Emmons RV, Reid DM, Cohen RL, Meng G, Young NS, Dunbar CE, et al. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood (1996) 87:4068–71.
22. Kosugi S, Kurata Y, Tomiyama Y, Tahara T, Kato T, Tadokoro S, et al. Circulating thrombopoietin level in chronic immune thrombocytopenic purpura. Br J Haematol (1996) 93:704–6. doi:10.1046/j.1365-2141.1996.d01-1702.x
23. McMillan R, Luiken GA, Levy R, Yelenosky R, Longmire RL. Antibody against megakaryocytes in idiopathic thrombocytopenic purpura. JAMA (1978) 239(23):2460–2. doi:10.1001/jama.1978.03280500056020
24. Rabellino EM, Levene RB, Leung LL, Nachman RL. Human megakaryocytes II. Expression of platelet proteins in early marrow megakaryocytes. J Exp Med (1981) 154:88–100. doi:10.1084/jem.154.1.88
25. Vinci G, Tabilio A, Deschamps JF, Van Haeke D, Henri A, Guichard J, et al. Immunological study of in vitro maturation of human megakaryocytes. Br J Haematol (1984) 56:589–605. doi:10.1111/j.1365-2141.1984.tb02184.x
26. Chang M, Nakagawa PA, Williams SA, Schwartz MR, Imfeld KL, Buzby JS, et al. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood (2003) 102:887–95. doi:10.1182/blood-2002-05-1475
27. McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood (2004) 103:1364–9. doi:10.1182/blood-2003-08-2672
28. Houwezijl EJ, Blom NR, van der Want JJ, Esselink MT, Koornstra JJ, Smit JW, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood (2004) 103:500–6. doi:10.1182/blood-2003-01-0275
29. Stasi R, Cooper N, Del Poeta G, Stipa E, Laura Evangelista M, Abruzzese E, et al. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell depleting therapy with rituximab. Blood (2008) 112:1147–50. doi:10.1182/blood-2007-12-129262
30. Yu J, Heck S, Patel V, Levan J, Yu Y, Bussel JB, et al. Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura. Blood (2008) 112:1325–8. doi:10.1182/blood-2008-01-135335
31. Olsson B, Andersson PO, Jernås M, Jacobsson S, Carlsson B, Carlsson LM, et al. T cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med (2003) 9:1123–4. doi:10.1038/nm921
32. Li S, Wang L, Zao C, Li L, Peng J, Hou M. CD8+ T cells suppress autologous megakaryocyte apoptosis in idiopathic thrombocytopenic purpura. Br J Haematol (2007) 139(4):605–11. doi:10.1111/j.1365-2141.2007.06737.x
33. Frank E. Die essentielle thrombopenie (Konstitutionelle Purpura-Pseudohemophilie), I: Klinische Bild. Berl Klin Wchnschr. (1915) 52:454–8.
34. Kaznelson P. Verschwinden der hämorrhagischen Diathese bei einem Falle von ‘essentieller Thrombopenie’ (Frank) nach Milzexstirpation: Splenogene thrombolytische Purpura. Wien Klin Wochenschr (1916) 29:1451–4.
35. Troland CE, Lee FC. Thrombocytopen, a substance in the extract from the spleen of patients with idiopathic thrombocytopenic purpura that reduces the number of blood platelets. JAMA (1938) 111:221–6. doi:10.1001/jama.1938.02790290007003
36. Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med (1951) 38:1–10.
37. Evans RS, Takahashi K, Duane RT, Payne R, Liu C. Primary thrombocytopenic purpura and acquired hemolytic anemia; evidence for a common etiology. AMA Arch Inter Med (1951) 87:48–65. doi:10.1001/archinte.1951.03810010058005
38. Van Leeuwen EF, van der Ven JT, Engelfriet CP, von dem Borne AE. Specificity of autoantibodies in autoimmune thrombocytopenia. Blood (1982) 59:23–6.
39. Semple JW, Bruce S, Freedman J. Suppressed natural killer cell activity in patients with chronic autoimmune thrombocytopenic purpura. Am J Hematol (1991) 37:258–62. doi:10.1002/ajh.2830370409
40. Wright JF, Blanchette VS, Wang H, Arya N, Petric M, Semple JW, et al. Characterization of platelet-reactive antibodies in children with varicella-associated acute immune thrombocytopenic purpura (ITP). Br J Haematol (1996) 95:145–52. doi:10.1046/j.1365-2141.1996.d01-1872.x
41. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood (2009) 113(11):2386–93. doi:10.1182/blood-2008-07-162503
42. Liebman HA, Stasi R. Secondary immune Thrombocytopenic purpura. Curr Opin Haematol (2007) 14:557–73. doi:10.1097/MOH.0b013e3282ab9904
43. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematol Am Soc Hematol Educ Program (2012) 2012:306–12. doi:10.1182/asheducation-2012.1.306
44. McKenzie CGJ, Guo L, Freedman J, Semple JW. Cellular immune dysfunction in immune thrombocytopenia (ITP) Br. J Haematol (2013) 163(1):10–23. doi:10.1111/bjh.12480
45. He R, Reid DM, Jones CE, Shulman NR. Spectrum of Ig classes, specificities, and titers of serum antiglycoproteins in chronic idiopathic thrombocytopenic purpura. Blood (1994) 83:1024–32.
46. Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist (2009) 14:12–21. doi:10.1634/theoncologist.2008-0132
47. Tomiyama Y, Kosugi S. Autoantigenic epitopes on platelet glycoproteins. Int J Hematol (2005) 81(2):100–5. doi:10.1532/IJH97.04193
48. Nugent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol (2009) 146:585–96. doi:10.1111/j.1365-2141.2009.07717.x
49. Iraqi M, Perdomo J, Yan F, Choi PY, Chong BH. Immune thrombocytopenia: antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica (2015) 100(5):623–32. doi:10.1080/19420862.2015.1075681
50. Kuwana M, Okazaki Y, Kajihara M, Kaburaki J, Miyazaki H, Kawakami Y, et al. Autoantibody to c-Mpl (thrombopoietin receptor) in systemic lupus erythematosus: relationship to thrombocytopenia with megakaryocytic hypoplasia. Arthritis Rheum (2002) 46:2148–59. doi:10.1002/art.10420
51. Verschoor A, Langer HF. Crosstalk between platelets and the complement system in immune protection and disease. Thromb Haemost (2013) 110(5):910–9. doi:10.1160/TH13-02-0102
52. Peerschke EI, Andemariam B, Yin W, Bussel JB. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with immune thrombocytopenic purpura. Br J Haematol (2009) 148:638–45. doi:10.1111/j.1365-2141.2009.07995.x
53. Peerschke EI, Yin W, Grigg SE, Ghebrehiwet BJ. Blood platelets activate the classical pathway of human complement. J Thromb Haemost (2006) 4(9):2035–42. doi:10.1111/j.1538-7836.2006.02065.x
54. Peerschke EI, Panicker S, Bussel J. Classical complement pathway activation in immune thrombocytopenia purpura: inhibition by a novel C1s inhibitor. Br J Haematol (2016) 173(6):942–5. doi:10.1111/bjh.13648
55. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med (2005) 201:871–9. doi:10.1084/jem.20041497
56. Nichol JL. Endogenous TPO (eTPO) levels in health and disease: possible clues for therapeutic intervention. Stem Cells (1998) 16(Suppl 2):165–75. doi:10.1002/stem.5530160719
57. Aledort LM, Hayward CP, Chen MG, Nichol JL, Bussel J, ITP Study Group. Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am J Hematol (2004) 76(3):205–13. doi:10.1002/ajh.20104
58. Rank A, Weigert O, Ostermann H. Management of chronic immune thrombocytopenic purpura: targeting insufficient megakaryopoiesis as a novel therapeutic principle. Biologics (2010) 4:139–45. doi:10.2147/BTT.S3436
59. Kosar A, Haznedaroglu IC, Buyukasik Y, Ozcebe O, Kirazli S, Dundar S. Circulating thrombopoietin and interleukin-6 in newly diagnosed autoimmune versus aplastic thrombocytopenia. Haematologica (1998) 83:1055–6.
60. Grewal PK. The Ashwell-Morell receptor. Methods Enzymol (2010) 479:223–41. doi:10.1016/S0076-6879(10)79013-3
61. Garzon AM, Mitchell WB. Use of thrombopoietin receptor agonists in childhood immune thrombocytopenia. Front Pediatr (2015) 3:70. doi:10.3389/fped.2015.00070
62. Catani L. Recent advances in the pathology of idiopathic thrombocytopenic purpura. Hematol Meeting Rep (2009) 3(3):55–63. doi:10.4081/hmr.v3i3.572
63. Legitimo A, Consolini R, Failli A, Orsini G, Spisni R. Dendritic cell defects in the colorectal cancer. Hum Vaccin Immunother (2014) 10(11):3224–35. doi:10.4161/hv.29857
64. Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol (2008) 8:594–606. doi:10.1038/nri2358
65. Saito A, Yokohama A, Osaki Y, Ogawa Y, Nakahashi H, Toyama K, et al. Circulating plasmacytoid dendritic cells in patients with primary and Helicobacter pylori-associated immune thrombocytopenia. Eur J Haematol (2012) 88(4):340–9. doi:10.1111/j.1600-0609.2011.01745.x
66. LeBon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14:461–70. doi:10.1016/S1074-7613(01)00126-1
67. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood (2002) 99:3263–71. doi:10.1182/blood
68. LeBon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8 C T cells stimulated by virus-induced type I interferon. Nat Immunol (2003) 4:1009–15. doi:10.1038/ni978
69. Mathan TS, Figdor CG, Buschow SI. Human plasmacytoid dendritic cells: from molecules to intercellular communication network. Front Immunol (2013) 4:372. doi:10.3389/fimmu.2013.00372
70. Schettini J, Mukherjee P. Physiological role of plasmacytoid dendritic cells and their potential use in cancer immunity. Clin Dev Immunol (2008) 2008:106321. doi:10.1155/2008/106321
71. Catani L, Fagioli ME, Tazzari PL, Ricci F, Curti A, Rovito M, et al. Dendritic cells of immune thrombocytopenic purpura (ITP) show increased capacity to present apoptotic platelets to T lymphocytes. Exp Hematol (2006) 34(7):879–87. doi:10.1016/j.exphem.2006.03.009
72. Zhong H, Bao W, Li X, Miller A, Seery C, Haq N, et al. CD16+ monocytes control T-cell subset development in immune thrombocytopenia. Blood (2012) 120(16):3326–35. doi:10.1182/blood-2012-06-434605
73. Ji X, Zhang L, Peng J, Hou M. T cell immune abnormalities in immune thrombocytopenia. J Hematol Oncol (2014) 7:72. doi:10.1186/s13045-014-0072-6
74. Cooper N, Bussel J. The pathogenesis of immune thrombocytopaenic purpura. Br J Haematol (2006) 133:364–74. doi:10.1111/j.1365-2141.2006.06024.x
75. Zhang F, Chu X, Wang L, Zhu Y, Li L, Ma D, et al. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur J Haematol (2006) 76(5):427–31. doi:10.1111/j.1600-0609.2005.00622.x
76. Zhao C, Li X, Zhang F, Wang L, Peng J, Hou M. Increased cytotoxic T-lymphocyte-mediated cytotoxicity predominant in patients with idiopathic thrombocytopenic purpura without platelet autoantibodies. Haematologica (2008) 93(9):1428–30. doi:10.3324/haematol.12889
77. Cines DB, Bussel JB, Liebman HA, LuningPrak ET. The ITP syndrome: pathogenic and clinical diversity. Blood (2009) 26:6511–21. doi:10.1182/blood-2009-01-129155
78. Chow L, Aslam R, Speck ER, Kim M, Cridland N, Webster ML, et al. A murine model of severe immune thrombocytopenia is induced by antibody-and CD8+ T cell-mediated responses that are differentially sensitive to therapy. Blood (2010) 6:1247–53. doi:10.1182/blood-2009-09-244772
79. Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood (2010) 2:168–86. doi:10.1182/blood-2009-06-225565
80. Zhou H, Qiu JH, Wang T, Yu YY, Liu XN, Li X, et al. Interleukin 27 inhibits cytotoxic T-lymphocyte-mediated platelet destruction in primary immune thrombocytopenia. Blood (2014) 124(22):3316–9. doi:10.1182/blood-2014-06-580084
81. Liu XG, Ren J, Yu Y, Sun L, Shi Y, Qin P, et al. Decreased expression of interleukin-27 in immune thrombocytopenia. Br J Haematol (2011) 153(2):259–67. doi:10.1111/j.1365-2141.2011.08614.x
82. Yang X, Yang J, Chu Y, Xue Y, Xuan D, Zheng S, et al. T follicular helper cells and regulatory B cells dynamics in systemic lupus erythematosus. PLoS One (2014) 9:e88441. doi:10.1371/journal.pone.0088441
83. Xie J, Cui D, Liu Y, Jin J, Tong H, Wang L, et al. Changes in follicular helper T cells in idiopathic thrombocytopenic purpura patients. Int J Biol Sci (2015) 11(2):220–9. doi:10.7150/ijbs.10178
84. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29(1):127–37. doi:10.1016/j.immuni.2008.06.001
85. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29:138–49. doi:10.1016/j.immuni.2008.05.009
86. Neunert CE. Current management of immune thrombocytopenia. Hematology Am Soc Hematol Educ Program (2013) 2013:276–82. doi:10.1182/asheducation-2013.1.276
87. Hua F, Ji L, Zhan Y, Li F, Zou S, Chen L, et al. Aberrant frequency of IL-10-producing B cells and its association with Treg/Th17 in adult primary immune thrombocytopenia patients. Biomed Res Int (2014) 2014:571302. doi:10.1155/2014/571302
88. Panitsas FP, Theodoropoulou M, Kouraklis A, Karakantza M, Theodorou GL, Zoumbos NC, et al. Adult chronic idiopathic thrombocytopenic purpura (ITP) is the manifestation of a type-1 polarized immune response. Blood (2004) 103(7):2645–7. doi:10.1182/blood-2009-01-129155
89. Wang T, Zhao H, Ren H, Guo J, Xu M, Yang R, et al. Type 1 and type 2 T-cell profiles in idiopathic thrombocytopenic purpura. Haematologica (2005) 90(7):914–23.
90. Teke HU, Gunduz E, Akay OM, Gülbas Z. Abnormality of regulatory T-cells in remission and non-remission idiopathic thrombocytopaenic purpura patients. Platelets (2013) 24(8):625–31. doi:10.3109/09537104.2012.748188
91. Nishimoto T, Kuwana M. CD4+CD25+Foxp3+ regulatory T cells in the pathophysiology of immune thrombocytopenia. Semin Hematol (2013) 50(Suppl 1):S43–9. doi:10.1053/j.seminhematol.2013.03.018
92. Sakakura M, Wada H, Tawara I, Nobori T, Sugiyama T, Sagawa N, et al. Reduced CD4+CD25+ T cells in patients with idiopathic thrombocytopenic purpura. Thromb Res (2007) 120:187–93. doi:10.1016/j.thromres.2006.09.008
93. Liu B, Zhao H, Poon MC, Han Z, Gu D, Xu M, et al. Abnormality of CD4(+)CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. Eur J Haematol (2007) 78:139–43. doi:10.1111/j.1600-0609.2006.00780.x
94. Ling Y, Cao X, Yu Z, Ruan C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. Eur J Haematol (2007) 79:310–6. doi:10.1111/j.1365-2249.2009.03913.x
95. Olsson B, Ridell B, Carlsson L, Jacobsson S, Wadenvik H. Recruitment of T cells into bone marrow of ITP patients possibly due to elevated expression of VLA-4 and CX3CR1. Blood (2008) 112(4):1078–84. doi:10.1182/blood-2008-02-139402
96. Bao W, Bussel JB, Heck S, He W, Karpoff M, Boulad N, et al. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood (2010) 116:4639–45. doi:10.1182/blood-2010-04-281717
97. Li Z, Mou W, Lu G, Cao J, He X, Pan X, et al. Low-dose rituximab combined with short-term glucocorticoids upregulates Treg cell levels in patients with immune thrombocytopenia. Int J Hematol (2011) 93:91–8. doi:10.1007/s12185-010-0753-z
98. Aboul-Fotoh L-M, Abdel Raheem MM, El-Deen MA, Osman AM. Role of CD4+CD25+ T cells in children with idiopathic thrombocytopenic purpura. J Pediatr Hematol Oncol (2011) 33:81–5. doi:10.1097/MPH.0b013e3181f46b82
99. Audia S, Samson M, Guy J, Janikashvili N, Fraszczak J, Trad M, et al. Immunologic effects of rituximab on the human spleen in immune thrombocytopenia. Blood (2011) 118:4394–400. doi:10.1182/blood-2011-03-344051
100. Zahran AM, Elsayh KI. CD4+CD25+High FoxP3+ regulatory T cells, B lymphocytes, and T lymphocytes in patients with acute ITP in Assiut Children Hospital. Clin Appl Thromb Hemost (2012) 20(1):61–7. doi:10.1177/1076029612454937
101. Daridon C, Loddenkemper C, Spieckermann S, Kühl AA, Salama A, Burmester GR, et al. Splenic proliferative lymphoid nodules distinct from germinal centers are sites of autoantigen stimulation in immune thrombocytopenia. Blood (2012) 120:5021–31. doi:10.1182/blood-2012-04-424648
102. Arandi N, Mirshafiey A, Jeddi-Tehrani M, Shaghaghi M, Sadeghi B, Abolhassani H, et al. Alteration in frequency and function of CD4+CD25+FOXP3+ regulatory T cells in patients with immune thrombocytopenic purpura. Iran J Allergy Asthma Immunol (2014) 13(2):85–92.
103. Catani L, Sollazzo D, Trabanelli S, Curti A, Evangelisti C, Polverelli N, et al. Decreased expression of indoleamine 2,3-dioxygenase 1 in dendritic cells contributes to impaired regulatory T cell development in immune thrombocytopenia. Ann Hematol (2013) 92(1):67–78. doi:10.1007/s00277-012-1556-5
104. Zhou B, Zhao H, Yang RC, Han ZC. Multi-dysfunctional pathophysiology in ITP. Crit Rev Oncol Hematol (2005) 54:107–16. doi:10.1016/j.critrevonc.2004.12.004
105. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med (1989) 170:2081–95. doi:10.1084/jem.170.6.2081
106. Li F, Ji L, Wang W, Hua F, Zhan Y, Zou S, et al. Insufficient secretion of IL-10 by Tregs compromised its control on over-activated CD4+ T effector cells in newly diagnosed adult immune thrombocytopenia patients. Immunol Res (2015) 61(3):269–80. doi:10.1007/s12026-014-8620-2
107. Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS. Regulatory T-cells in autoimmune diseases: challenges, controversies and yet unanswered questions. Autoimmun Rev (2015) 14(2):105–16. doi:10.1016/j.autrev.2014.10.012
109. Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology (2011) 134:8–16. doi:10.1111/j.1365-2567.2011.03465.x
110. Rocha AM, Souza C, Rocha GA, de Melo FF, Clementino NC, Marino MC, et al. The levels of IL-17A and of the cytokines involved in Th17 cell commitment are increased in patients with chronic immune thrombocytopenia. Haematologica (2011) 96:1560–4. doi:10.3324/haematol.2011.046417
111. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13:668–77. doi:10.1016/j.autrev.2013.12.004
112. Wang X, Zheng XY, Ma C, Wang XK, Wu J, Adem A, et al. Mitigated Tregs and augmented Th17 cells and cytokines are associated with severity of experimental autoimmune neuritis. Scand J Immunol (2014) 80(3):180–90. doi:10.1111/sji.12260
113. Ye X, Zhang L, Wang H, Chen Y, Zhang W, Zhu R, et al. The role of IL-23/Th17 pathway in patients with primary immune thrombocytopenia. PLoS One (2015) 10(1):e0117704. doi:10.1371/journal.pone.0117704
114. Ji L, Zhan Y, Hua F, Li F, Zou S, Wang W, et al. The ratio of Treg/Th17 cells correlates with the disease activity of primary immune thrombocytopenia. PLoS One (2012) 7(12):e50909. doi:10.1371/journal.pone.0050909
115. Wang JD, Chang TK, Lin HK, Huang FL, Wang CJ, Lee HJ. Reduced expression of transforming growth factor-beta1 and correlated elevation of interleukin-17 and interferon-gamma in pediatric patients with chronic primary immune thrombocytopenia (ITP). Pediatr Blood Cancer (2011) 57:636–40. doi:10.1002/pbc.22984
116. Guo ZX, Chen ZP, Zheng CL, Jia HR, Ge J, Gu DS, et al. The role of Th17 cells in adult patients with chronic idiopathic thrombocytopenic purpura. Eur J Haematol (2009) 82:488–9. doi:10.1111/j.1600-0609.2009.01229.x
117. Sollazzo D, Trabanelli S, Curti A, Vianelli N, Lemoli RM, Catani L. Circulating CD4+CD161+CD196+ Th17 cells are not increased in immune thrombocytopenia. Haematologica (2011) 96:632–4. doi:10.3324/haematol.2010.038638
118. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi:10.1084/jem.20041257
119. Toussirot E. The IL23/Th17 pathway as a therapeutic target in chronic inflammatory diseases. Inflamm Allergy Drug Targets (2012) 11:159–68. doi:10.2174/187152812800392805
120. Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine (2008) 41(2):84–91. doi:10.1016/j.cyto.2007.09.014
121. Romme Christensen J, Bornsen L, Hesse D, Krakauer M, Sorensen PS, Søndergaard HB, et al. Cellular sources of dysregulated cytokines in relapsing-remitting multiple sclerosis. J Neuroinflammation (2012) 9:215. doi:10.1186/1742-2094-9-215
122. Qian C, Jiang T, Zhang W, Ren C, Wang Q, Qin Q, et al. Increased IL-23 and IL-17 expression by peripheral blood cells of patients with primary biliary cirrhosis. Cytokine (2013) 64:172–80. doi:10.1016/j.cyto.2013.07.005
123. Fransen K, van Sommeren S, Westra HJ, Veenstra M, Lamberts LE, Modderman R, et al. Correlation of genetic risk and messenger RNA expression in a Th17/IL23 pathway analysis in inflammatory bowel disease. Inflamm Bowel Dis (2014) 20:777–82. doi:10.1097/MIB.0000000000000013
124. Dilillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann N Y Acad Sci (2010) 1183:38–57. doi:10.1111/j.1749-6632.2009.05137.x
125. Kalampokis I, Yoshizaki A, Tedder TF. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res Ther (2013) 15(Suppl 1):S1. doi:10.1186/ar3907
126. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4 + T cell immunity. Nat Rev Immunol (2010) 10(4):236–47. doi:10.1038/nri2729
127. Salinas GF, Braza F, Brouard S, Tak P, Baeten D. The role of B lymphocytes in the progression from autoimmunity to autoimmune disease. Clin Immunol (2013) 146(1):34–45. doi:10.1016/j.clim.2012.10.005
128. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Muñoz-Suano A, Kamanaka M, et al. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol (2011) 186:5569–79. doi:10.4049/jimmunol.1100284
129. Carter NA, Rosser EC, Mauri C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res Ther (2012) 14(1):R32. doi:10.1186/ar3736
130. Yadav DK, Tripathi AK, Kumar A, Agarwal J, Prasad KN, Gupta D, et al. Association of TNF-α-308G>A and TNF-β +252A>G genes polymorphisms with primary immune thrombocytopenia: a North Indian study. Blood Coagul Fibrinolysis (2016) 27(7):791–6. doi:10.1097/MBC.0000000000000492
131. Sarpatwari A, Bussel JB, Ahmed M, Erqou S, Semple JW, Newland AC, et al. Single nucleotide polymorphism (SNP) analysis demonstrates a significant association of tumour necrosis factor-alpha (TNFA) with primary immune thrombocytopenia among Caucasian adults. Hematology (2011) 16:243–8. doi:10.1179/102453311X13025568941808
132. El Sissy MH, El Sissy AH, Elanwary S. Tumor necrosis factor-α-308G/A gene polymorphism in Egyptian children with immune thrombocytopenic purpura. Blood Coagul Fibrinolysis (2014) 25(5):458–63. doi:10.1097/MBC.0000000000000089
133. Wu KH, Peng CT, Li TC, Wan L, Tsai CH, Lan SJ, et al. Interleukin 4, interleukin 6 and interleukin 10 polymorphisms in children with acute and chronic immune thrombocytopenic purpura. Br J Haematol (2005) 128(6):849–52. doi:10.1111/j.1365-2141.2005.05385.x
134. Saitoh T, Kasamatsu T, Inoue M, Mitsui T, Koiso H, Yokohama A, et al. Interleukin-10 gene polymorphism reflects the severity of chronic immune thrombocytopenia in Japanese patients. Int J Lab Hematol (2011) 33:526–32. doi:10.1111/j.1751-553X.2011.01320.x
135. Makhlouf MM, Abd Elhamid SM. Expression of IL4 (VNTR intron 3) and IL10 (-627) genes polymorphisms in childhood immune thrombocytopenic purpura. Lab Med (2014) 45(3):211–9. doi:10.1309/LMB0QC5T1RXTTRZQ
136. Galiegue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem FEBS (1995) 232:54–61. doi:10.1111/j.1432-1033.1995.tb20780.x
137. Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med (2009) 11:e3. doi:10.1017/S1462399409000957
138. Rossi F, Mancusi S, Bellini G, Roberti D, Punzo F, Vetrella S, et al. CNR2 functional variant (Q63R) influences childhood immune thrombocytopenic purpura. Haematologica (2011) 96:1883–5. doi:10.3324/haematol.2011.045732
139. Hang B, Lo C, Shen L, Jones C, Cusmano-Ozog K, Park-Snyder S, et al. The role of vanin-1 and oxidative stress-related pathways in distinguishing acute and chronic pediatric ITP. Blood (2011) 117:4569–79. doi:10.1182/blood-2010-09-304931
140. Zhang B, Lo C, Shen L, Sood R, Jones C, Cusmano-Ozog K, et al. The role of vanin-1 and oxidative stress-related pathways in distinguishing acute and chronic pediatric ITP. Blood (2011) 117:4569–79. doi:10.1182/blood-2010-09-304931
Keywords: immune thrombocytopenia, pathogenesis, autoimmune disease, megakaryocytopoiesis, platelets
Citation: Consolini R, Legitimo A and Caparello MC (2016) The Centenary of Immune Thrombocytopenia – Part 1: Revising Nomenclature and Pathogenesis. Front. Pediatr. 4:102. doi: 10.3389/fped.2016.00102
Received: 07 June 2016; Accepted: 07 September 2016;
Published: 19 October 2016
Edited by:
Claudio Pignata, University of Naples Federico II, ItalyReviewed by:
Pietro Merli, IRCCS Bambino Gesù Ospedale Pediatrico, ItalyMarkus Bender, University Hospital Würzburg, Germany
Copyright: © 2016 Consolini, Legitimo and Caparello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rita Consolini, rita.consolini@med.unipi.it