
- 1 Rheumatology Unit, Department of Medicine, Karolinska Institutet, Stockholm, Sweden
- 2 Actar AB, Solna, Sweden
Microsomal prostaglandin E synthase-1 (mPGES-1) is a well-recognized target for the development of novel anti-inflammatory drugs that can reduce symptoms of inflammation in rheumatic diseases and other inflammatory conditions. In this review, we focus on mPGES-1 in rheumatic diseases with the aim to cover the most recent advances in the understanding of mPGES-1 in rheumatoid arthritis, osteoarthritis, and inflammatory myopathies. Novel findings regarding regulation of mPGES-1 cell expression as well as enzyme inhibitors are also summarized.
Introduction
Microsomal prostaglandin E synthase-1 (mPGES-1) catalyzes the glutathione dependent oxidoreduction of prostaglandin H2 into PGE2 (Jakobsson et al., 1999; Pettersson et al., 2005; Figure 1).
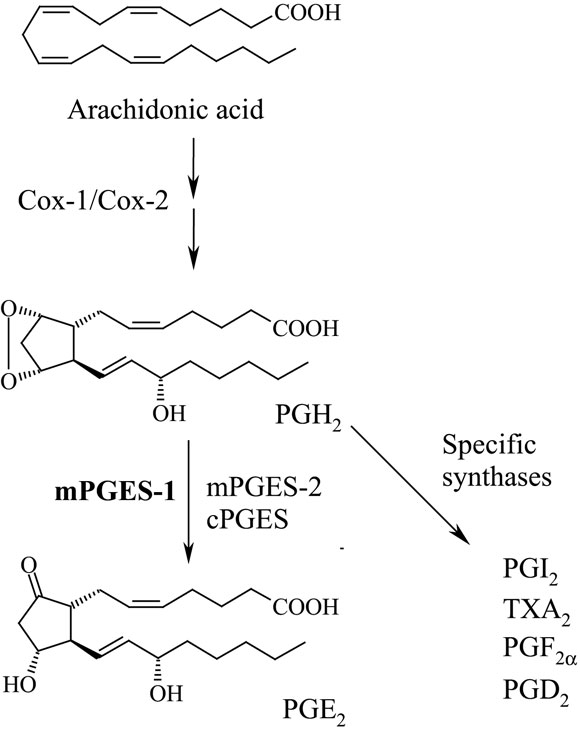
Figure 1. Biosynthesis of prostaglandin E2. Cyclooxygenase (Cox)-1 or 2 produce PGH2 that can be converted into PGE2 by mPGES-1. Alternatively, mPGES-2 or cytosolic PGES catalyze the same reaction and likely such PGE2 is more involved in physiological reactions. Different cells produce distinct prostaglandin profiles which reflect their expression profiles of specific prostaglandin synthases. For instance, endothelial cells produce both PGE2 and PGI2 whereas platelets predominantly produce TXA2.
The substrate for mPGES-1, prostaglandin endoperoxide H2 (PGH2) is generated by cyclooxygenase (Cox)-1 or 2 by oxidation of arachidonic acid in turn released from phospholipids by various phospholipases. PGH2 is also converted to PGE2 by two other enzymes, cytosolic PGES (cPGES) and mPGES-2, structurally and biologically distinct from mPGES-1 (Murakami et al., 2002; Tanioka et al., 2003). During inflammation the prostaglandin cascade is activated by calcium signaling and cytokine dependent enzyme inductions. In particular, Cox-2 and mPGES-1 are rapidly induced within hours by pro-inflammatory cytokines in macrophages and fibroblasts (Murakami et al., 2000; Stichtenoth et al., 2001). In many situations this cellular activation results in significant increase of PGE2 biosynthesis. PGE2 mediates its effects via G-protein coupled receptors EP1 to EP4. If produced via mPGES-1, PGE2 usually causes inflammation including swelling, fever and inflammatory pain (Engblom et al., 2003; Trebino et al., 2003; Kamei et al., 2004), while cPGES or mPGES-2 seems to constitutively produce PGE2 possibly important for physiological reactions (Murakami et al., 2003; Tanioka et al., 2003). Therefore, mPGES-1 is today a well- recognized target for the development of novel anti-inflammatory drugs that can reduce symptoms of inflammation in rheumatic diseases and other inflammatory conditions, hopefully without causing stomach ulcerations or severe cardiovascular side effects as traditional non-steroidal anti-inflammatory drugs (NSAIDs) or selective Cox-2 inhibitors may do (Samuelsson et al., 2007). In this review, we focus on mPGES-1 in rheumatic diseases with the aim to cover the most recent advances in the understanding of mPGES-1 in rheumatoid arthritis (RA), osteoarthritis (OA), and inflammatory myopathies.
Rheumatoid Arthritis
In patients with RA, the PGE2 levels in the synovial fluid are markedly elevated (Punzi et al., 1989), and both activated synovial tissue cells and recruited synovial fluid cells might contribute to the release of PGE2 into the synovial fluid. Indeed, mPGES-1 is markedly expressed in RA synovial tissue (Westman et al., 2004) and even stronger up-regulated in patients with active than with inactive RA (Murakami et al., 2003). Within the synovial tissue, mPGES-1 was mainly localized to the synovial lining cells (Figure 2A). High expression was also found in sublining layer in scattered mononuclear fibroblast-like cells, mononuclear infiltrates and endothelial cells of some blood vessels. Phenotypic analysis using immunofluorescence also demonstrated that mPGES-1 was expressed in synovial fibroblasts and macrophages, the abundant cell populations present in hyperplastic RA synovial tissue. No expression of mPGES-1 was observed in T or B lymphocytes (Westman et al., 2004).
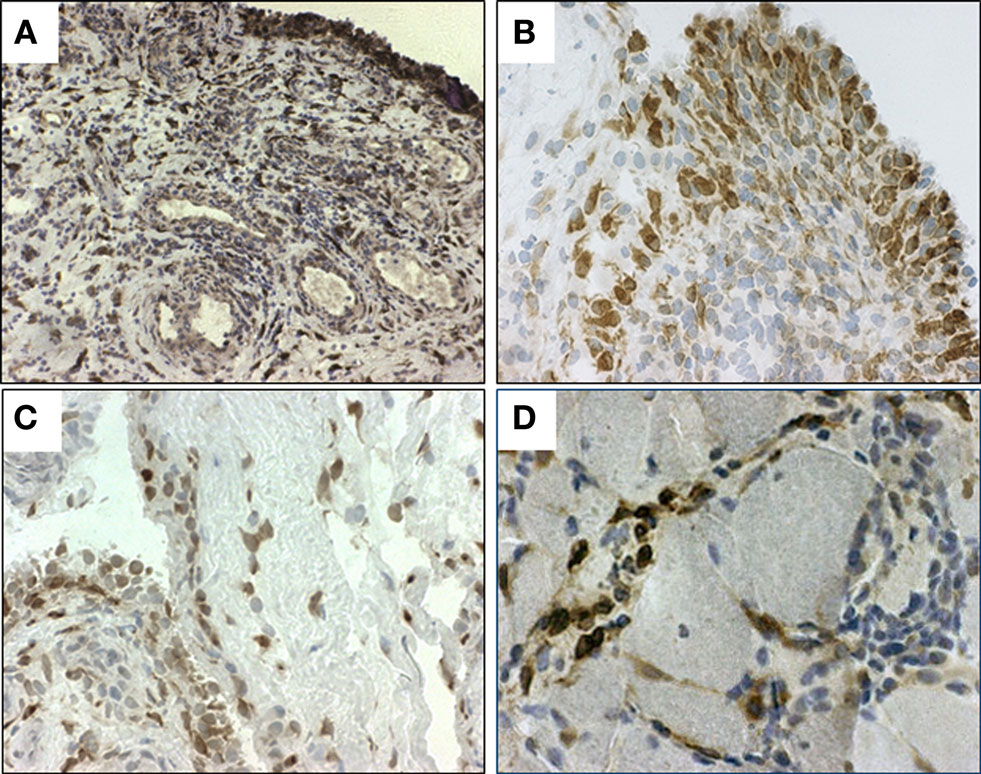
Figure 2. Immunohistochemical staining (brown) for microsomal PGES (mPGES)-1 in representative (A,B) synovial tissue from RA patients, (C) synovial tissue from OA patient and (D) in muscle tissue from a patient with polymyositis. Original magnification ×250 (A) and ×500 (B–D).
The distribution pattern of mPGES-1 positive cells largely reflects the localization of Cox-2 positive cells in RA synovial tissue. As demonstrated with double immunofluorescence, mPGES-1 and Cox-2 were co-localized in cells of the synovial lining layer and sublining cells and in synovial fluid mononuclear cells (Korotkova et al., 2005). The colocalization of these enzymes has been reported in the same subcellular compartments, the perinuclear membrane, and endoplasmic reticulum (Murakami et al., 2000; Lazarus et al., 2002), which provides basis for the preferred functional coupling between mPGES-1 and Cox-2 (Murakami et al., 2000; Thoren and Jakobsson, 2000) although Cox-1 may also serve as provider of PGH2 in some situations (Dieter et al., 2000; Murakami et al., 2000). This focused up-regulation of mPGES-1 and Cox-2 in RA synovial cells is probably the cause of the abundant PGE2 biosynthesis in the joints in turn contributing to inflammatory and destructive processes. Interestingly, studies in vitro and in experimental arthritis model have reported that cytosolic phospholipase A2 (cPLA2), that supplies arachidonic acid to downstream Cox, is also activated in arthritic tissue and the activity showed a correlation with severity of the arthritis (Hulkower et al., 1994; Tai et al., 2010).
The expression of cPGES was also observed in synovial lining cells, sublining mononuclear and fibroblast-like cells, mononuclear infiltrates, and some blood vessels (Westman et al., 2004). The staining for mPGES-2 revealed its expression in synovial lining and sublining cells, and the staining pattern was similar in RA patients despite different pathological states (Murakami et al., 2003). The presence of the housekeeping cPGES and mPGES-2 in RA synovial tissue implies that they might produce PGE2 required for the maintenance of homeostasis.
In order to better understand what mechanisms regulate the overexpression of Cox and mPGES-1 in RA we have studied the effects of anti-rheumatic drugs. Intra-articular treatment of patients with glucocorticoids significantly reduced mPGES-1 as well as both Cox-1 and Cox-2 expression in arthritic synovial tissue ex vivo. In contrast, treatment of RA patients with TNF blockers did not suppress either mPGES-1 or Cox expression in synovial tissues, implying that other mechanisms function in sustaining the inflammation independently of TNF (Korotkova et al., 2005).
In vitro Regulation of mPGES-1 Expression in Cells from RA Joint
The results of in vitro experiments have provided convincing evidence that the expression of mPGES-1 in RA joints might be up-regulated by a wide range of stimuli. Initially, the induction of mPGES-1 was demonstrated in response to the pro-inflammatory cytokines IL-1β, TNF, or LPS. In synovial fluid mononuclear cells isolated from RA patients, the expression of mPGES-1 and Cox-2 was substantially up-regulated in response to LPS and accompanied by enhanced PGE2 release (Korotkova et al., 2005). Treatment of RA synovial fibroblasts with IL-1β and TNF in vitro caused a coordinated up-regulation of mPGES-1 and Cox-2 with concomitant abundant PGE2 production, but there was no effect on cPGES expression (Stichtenoth et al., 2001; Kojima et al., 2002). Moreover, the early release of PGE2 from these cells may further increase the expression of mPGES-1 via an autocrine positive feed-back loop (Kojima et al., 2003). In addition, in rodent primary osteoblasts mPGES-1 and Cox-2 were strongly induced by bone resorptive cytokines IL-1β and TNF, as well as by fibroblast growth factor 2 (FGF-2) and LPS, resulting in an enhanced biosynthesis of PGE2. This suggests that cytokine-induced mPGES-1 may be a potent regulator of bone resorption in RA via PGE2 production (Murakami et al., 2000; Saegusa et al., 2003; Inada et al., 2006).
Recent studies have elucidated additional mechanisms involved in regulation of mPGES-1 expression in the RA joint. Epidermal growth factor (EGF) is constitutively produced by RA synovial fibroblasts and found in the synovial fluid of RA patients at high levels (Bucala et al., 1991; Kusada et al., 1993). EGF stimulates the release of inflammatory mediators and the growth of synovial cells suggesting its involvement in the pathogenesis of this disease (Kusada et al., 1993; Satoh et al., 2001). EGF increases both Cox-2 and mPGES-1 mRNA expression in synovial fibroblasts from RA patients and induces PGE2 production via the ERK1/MAPK and NFkB pathways (Nah et al., 2010). Another molecule that contributes to mPGES-1 up-regulation is adiponectin, one of the adipokines produced by fat cells. Adiponectin is strongly up-regulated in synovial fluid and synovial tissue from RA patients and exerts significant pro-inflammatory effects (Ehling et al., 2006). Interestingly, RA synovial fibroblasts exposed to adiponectin released high amounts of PGE2 by induction of the enzymes mPGES-1 and Cox-2 (Kusunoki et al., 2010).
Recently, a direct role in inflammation and joint destruction in RA was suggested for microparticles, abundantly present in the synovial fluid of inflamed joints (Distler et al., 2005). Microparticles are small membrane-coated vesicles released from activated or dying cells by exocytic budding and display surface proteins from their parental cells. Microparticles derived from leukocytes strongly induced mPGES-1 and Cox-2 expression in RA synovial fibroblasts thereby stimulating the production of PGE2 (Jungel et al., 2007).
The induction of mPGES-1 is markedly suppressed by anti-inflammatory glucocorticoids. In studies examining RA synovial fibroblasts, synovial fluid monocytes, and OA chondrocytes, treatment with dexamethasone (Dex) decreased mPGES-1 mRNA, protein expression, and enzyme activity induced by pro-inflammatory stimuli in dose-dependent manner (Stichtenoth et al., 2001; Kojima et al., 2002; Korotkova et al., 2005; Shimpo et al., 2009). However, the inhibition of mPGES-1 by Dex was weaker compared with that of Cox-2 in IL-1ß stimulated RA synovial fibroblasts (Kojima et al., 2002). Interestingly, in OA chondrocytes, PGE2 recovered mPGES-1 expression from suppression by Dex, whereas it did not restore the expression of Cox-2 in the presence of Dex (Shimpo et al., 2009). These results suggest that different mechanisms might be involved in the inhibition of mPGES-1 and Cox-2 by glucocorticoids. Glucocorticoids suppress Cox-2 expression both by transcriptional (via inhibition of transcription factors such as AP-1 and NF-κB) and post-transcriptional mechanisms involving mRNA destabilization (Newton et al., 1998). The inhibitory effect of Dex on mPGES-1 expression may be at least partly explained by suppression of the NFkB and AP-1, playing a role in induction of mPGES-1 (Jungel et al., 2007). In addition, the putative promoter of the mouse mPGES-1 gene contains three glucocorticoid response elements that may regulate mPGES-1 expression (Naraba et al., 2002). However, further studies are required to better define the molecular mechanisms of mPGES-1 suppression by glucocorticoids.
Osteoarthritis
In patients with OA, cartilage spontaneously releases more PGE2 than normal cartilage (Amin et al., 1997). In OA cartilage mPGES-1 mRNA and protein levels are markedly elevated compared to the normal cartilage, and mPGES-1 immunostaining demonstrates that mPGES-1 is located in the chondrocytes of OA cartilage and the synovial lining cells of OA synovial tissue (Kojima et al., 2004, 2005; Li et al., 2005). We have also detected mPGES-1 expression in OA synovial lining layer and sublining cells (Gheorghe, unpublished observation, Figure 2). Thus, overexpression of mPGES-1 in OA synovial tissue and cartilage might contribute to chronic inflammation, pain, and catabolic processes related to OA.
Long-term treatment with traditional NSAIDs or selective Cox inhibitors decreases PGE2 production in OA cartilage via direct inhibition of Cox activity and suppression of Cox-2 and mPGES-1 expression. In cultured chondrocytes, NSAIDs inhibit IL-1β-induced mPGES-1, Cox-2 expression, and PGE2 synthesis (Alvarez-Soria et al., 2008). The mechanisms behind the inhibition of mPGES-1 and Cox-2 expression by NSAID are not entirely elucidated. There is evidence that PGE2 promotes its own synthesis in the synoviocytes by stabilization of Cox-2 mRNA (Faour et al., 2001) and by increased mPGES-1 expression (Kojima et al., 2003). Hence, suppression of induced PGE2 biosynthesis by NSAID potentially might prevent further up-regulation of mPGES-1 and Cox-2 expression via inhibition of this autocrine positive feed-back loop.
Treatment of chondrocytes or synovial fibroblasts from OA patients with the pro-inflammatory cytokine IL-1β results in a strong induction of mPGES-1 and increased release of PGE2 (Cheng et al., 2004; Masuko-Hongo et al., 2004; Li et al., 2005). TNF and IL-17 also up-regulate mPGES-1 expression in chondrocytes, though less potently, whereas other cytokines, such as IL-4, IL-6, IL-8, IL-10, and IFNg have no effect (Kojima et al., 2004; Li et al., 2005). Interestingly, at low levels (0.1 pg/ml–0.5 ng/ml) the cytokines IL-1β, TNF, and IL-17 alone show little effect on mPGES-1 expression and PGE2 production. However, the combination of these cytokines at low concentrations displayed synergistic effects on mPGES-1 expression in OA chondrocytes. In contrast, the levels of cPGES and mPGES-2 were not changed by these treatments (Li et al., 2005).
Similarly to RA, several other factors besides the pro-inflammatory cytokines are implicated in the induction of mPGES-1 in the OA joint. The increase of mPGES-1 expression in OA cartilage is at least partly related to hypoxia and activity of hypoxia activated factor 1α (HIF1α; Grimmer et al., 2007). Advanced glycation end products (AGEs) extensively accumulate in cartilage collagen with age and predispose to the development of OA (DeGroot et al., 2004). Nah et al. (2008) have shown that treatment of primary human OA chondrocytes with AGE induces mPGES-1 and Cox-2 expression and elevates PGE2 production.
Recently a possible link between OA and obesity has been shown in several studies, highlighting the critical role of mechanical loading and the potential role of adipokines in cartilage degradation (Pottie et al., 2006; Gosset et al., 2008). Interestingly, both mechanical loading and some adipokines have been demonstrated to induce mPGES-1 in cartilage. The mechanical compression of mouse cartilage explants significantly elevated PGE2 release, which was associated with induced Cox-2 and mPGES-1 expression. Thus, both Cox-2 and mPGES-1 are encoded by mechanosensitive genes and implicated in compression-induced PGE2 production (Gosset et al., 2006). Adipose tissue cells secrete a number of adipokines, proteins with the structural and functional properties of cytokines, which possess potent pro-inflammatory and pro-degradative effects and might contribute to OA progression (Pottie et al., 2006). A newly identified adipokine visfatin potently induces mPGES-1 and suppresses 15-PGDH expression, leading to enhanced PGE2 production by human and mouse articular chondrocytes (Gosset et al., 2008).
Inflammatory Myositis
The expression and localization of mPGES-1 and other enzymes related to PGE2 biosynthetic pathway were also examined in muscle tissue from patients with polymyositis or dermatomyositis. A significantly enhanced expression of mPGES-1 (Figure 2D), Cox-2, and Cox-1 was detected in myositis muscle tissue compared to healthy muscle tissue, suggesting their role in the pathogenesis of inflammatory myositis. Double immunofluorescence demonstrated a predominant expression of mPGES-1 in macrophages (Korotkova et al., 2008). Conventional immunosuppressive treatment of myositis patients with oral glucocorticoids and DMARDs led to a significant down-regulation of Cox-2 while mPGES-1 expression in inflamed muscle was not affected (Korotkova et al., 2008). The divergent effects of the treatment on Cox-2 and mPGES-1 expression may be explained by the differences in the mechanisms of glucocorticoid actions on the enzymes. In addition, the suppression of mPGES-1 may require higher glucocorticoid concentration, as previously observed after intra-articular treatment with high doses of glucocorticoids (Korotkova et al., 2005).
Taken together, these studies demonstrate that mPGES-1 is strongly up-regulated in rheumatic diseases by a variety of stimuli. Thus, the inhibition of PGE2 biosynthesis, preferably by targeting mPGES-1, might act synergistically with different anti-rheumatic therapies for optimal anti-inflammatory control.
Lessons from Experimental Models in Rodents
The important role of mPGES-1 as a novel therapeutic target for treatment of RA was further strengthened in experimental models of arthritis. A strong up-regulation of mPGES-1 mRNA and protein levels was demonstrated in the inflamed paws of rats with adjuvant-induced arthritis during the full course of the disease (Mancini et al., 2001; Claveau et al., 2003). Moreover in arthritic rats mPGES-1 was also induced in endothelial cells along the blood–brain barrier and the paraventricular nucleus of the hypothalamus suggesting the involvement of mPGES-1 in the activation of the central nervous symptoms associated with chronic autoimmune disease (Engblom et al., 2002a). Centrally produced PGE2 binds EP receptors present on relevant deep neural structures and elicits the central nervous illness response such as fever, fatigue, anorexia–cachexia, hyperalgesia, and dysregulation of the hypothalamus–pituitary adrenal axis (Engblom et al., 2002b).
Generation of mice with genetic deletion of mPGES-1 (mPGES-1 KO) has provided valuable insights into the role of mPGES-1 in inflammatory arthritis. In mPGES-1 KO mice the incidence of collagen-induced arthritis (CIA), as well as specific parameters of arthritis severity including synovial hyperplasia, infiltration of inflammatory cells, cartilage destruction, and bone erosion were significantly reduced compared with those in wild type (WT) mice (Trebino et al., 2003; Kojima et al., 2008). Moreover, during the development of CIA, pain perception after mechanical stimulation was significantly reduced in mPGES-1 KO mice compared to that in WT mice (Kojima et al., 2008). The reduction in observed inflammatory features in mPGES-1 KO mice was also associated with significantly lower serum levels of anti-collagen II antibody and reduced levels of total IgG and IgM (Kojima et al., 2008). The decreased antibody response seen in mPGES-1 KO mice was not due to impairment of T cell and B cell numbers or functions. It has been suggested that mPGES-1 deficiency and reduced PGE2 may result in an altered cytokine profile and tissue environment in which antigen-presenting cells encounter antigen and interact with T cells and B cells (Kojima et al., 2008).
The severity of collagen antibody-induced arthritis was also lower in mPGES-1 KO mice relatively to WT mice and associated with milder cartilage degeneration and lower bone resorption likely due to the reduced number of osteoclasts (Kamei et al., 2004). The critical role of mPGES-1 for bone losses related to inflammation was confirmed in mice. In WT mice, LPS administration resulted in reduced bone density and thickness and increased bone resorption associated with enhanced number of osteoclasts, while mPGES-1 KO mice were resistant to such LPS-induced bone loss (Inada et al., 2006).
Thus, targeting mPGES-1 in arthritic mice reduces inflammation, humoral immune response, and protects them from pain and joint destruction, replicating many of the favorable anti-inflammatory effects by traditional NSAIDs and Cox-2 inhibitors. Furthermore, Cox-2 has an important role in the resolution of inflammation via generation of anti-inflammatory eicosanoids like 15-deoxy-Δ12,14-PGJ2, resolvins, and protectins, and its inhibition within this period might propagate inflammation. Moreover, although PGE2 is a classic promoter of inflammation, some anti-inflammatory and pro-resolving effects of PGE2 have also been reported (Gomez et al., 2005; Chan and Moore, 2010). Therefore, it is important to examine whether mPGES-1 is the source of PGE2 during the resolution phase of inflammation.
Interestingly, a different expression profile of Cox-2 and mPGES-1 was detected during the resolution phase of normal wound repair (Kapoor et al., 2007). Following the up-regulation during the inflammation phase, the expression of mPGES-1 dropped rapidly to the basal levels during the resolution phase, while Cox-2 expression was maintained throughout the initiation and repair processes. In mice with CIA, blocking Cox-2 during the resolution phase perpetuated inflammation resulting in increased destruction of the joints (Chan and Moore, 2010). In contrast, mPGES-1 was restricted to the inflammation phase, suggesting that mPGES-1 inhibition may possibly be able to limit the severity of inflammation without interfering with resolution of arthritis (Chan and Moore, 2010). However, further investigation is needed to establish that mPGES-1 does not affect the resolution process.
It was suggested that selective blockage of mPGES-1 would also avoid the typical for NSAIDs adverse side effects such as the quite common GI side effects or the severe but less common cardiovascular side effects. Indeed, genetic deletion of mPGES-1, in contrast to deletion, disruption, or pharmacological inhibition of Cox-2, did not result in accelerated thrombogenesis or elevated blood pressure in normolipidemic mice (Cheng et al., 2006). Furthermore, lack of mPGES-1 delayed atherosclerosis development and attenuated aortic aneurysm formation in hyperlipidemic mice with genetic deletion of the receptor for low density lipoprotein (LDLR −/−; Wang et al., 2006, 2008).
Another concern is the effect of NSAIDs and mPGES1 inhibition on the blood pressure. Several studies have shown that blood pressure was similar in mPGES1-deficient and WT mice on control or high- or low-salt diets (Cheng et al., 2006; Francois et al., 2007) and in response to infusion of angiotensin II (Wang et al., 2008). In contrast, other studies have demonstrated that blockage of mPGES-1 augmented hypertensive response to high-salt feeding or infusion of angiotensin II in mice (Jia et al., 2006, 2008). This discrepancy probably reflects the differences in genetic background of mice which significantly influence the generation of other vasoactive prostanoids and blood pressure response to mPGES-1 deficiency (Facemire et al., 2010).
Deficiency in mPGES-1 and reduced PGE2 formation may also have unfavorable effects on the cardiac response to myocardial infarction (MI). Lack of mPGES-1 resulted in eccentric cardiomyocyte hypertrophy and compromised the left ventricular remodeling after acute MI in mice resulting in a mild heart failure. In contrast, cardiac mass, infarct size, and mortality after MI were similar in mPGES-1 KO and WT mice (Degousee et al., 2008). However, no head to head comparisons have been performed comparing Cox-2 KO with mPGES-1 KO mice in this model. In line, compared to genetic deletion of mPGES-1 the treatment of WT mice with Cox-2 inhibitor celecoxib exerted an increased cardiac risk under acute ischemia condition augmenting myocardial injury and reducing post-MI survival (Wu et al., 2009). The results suggested that selective inhibition of mPGES-1 derived PGE2 but not other PGs suppressible by Cox-2 inhibitors might have reduced cardiac risk compared to selective inhibition of Cox-2 (Wu et al., 2009).
However, it remains to be investigated whether selective pharmacological inhibition of mPGES-1 will result in the same effects as conferred by mPGES-1 gene deletion studies. Direct comparisons with Cox-2 inhibitors will also provide important insights about the relative and putative side effects elicited by mPGES1 inhibition.
mPGES-1 Inhibitors
Several selective inhibitors of mPGES-1 activity or expression have been identified and some of them have been evaluated in different inflammatory models (Xu et al., 2008; Guerrero et al., 2009; Bruno et al., 2010; Mbalaviele et al., 2010). The inhibitor of mPGES-1 expression, BTH (4-benzo[b]thiophen-2-yl-3-bromo-5-hydroxy-5H-furan-2-one) synthesized at Departamento de Farmacologia, Universitat de Valencia, Spain, was studied in acute and chronic inflammatory models (mouse air pouch and CIA, respectively; Guerrero et al., 2009). In mouse air pouch exudates, BTH suppressed PGE2 production via down-regulation of mPGES-1 expression without any effect on Cox-1 and Cox-2 expression or activity. Treatment of mice with BTH strongly reduced CIA incidence and alleviated severe arthritis, diminishing the inflammatory cell infiltration, cartilage erosion, and proteoglycan loss. These clinical effects were associated with reduced mPGES-1 expression and suppressed PGE2 production in paw homogenates. The inhibitory effect of BTH on mPGES-1 expression can be partly explained by its inhibitory action on the activation of NFkB (Guerrero et al., 2007), which might contribute to suppression of other pro-inflammatory mediators and needs to be elucidated, thus the selectivity of this compound must be questioned.
Among selective inhibitors of mPGES-1 enzyme activity, two inhibitors have been evaluated in cell-based systems relevant to RA, i.e., human monocytes and synovial fibroblasts (Bruno et al., 2010; Mbalaviele et al., 2010). AF3442, a lead compound from a series of carbazole benzamides synthesized in Angelini Research Center, Rome, Italy, is a potent inhibitor of human mPGES-1 activity (IC50 of 0.06 μM). In LPS-induced human monocytes AF3442 significantly reduced PGE2 formation without redirection of PGH2 metabolism to other prostanoids (Bruno et al., 2010). PF-9184, synthesized at Pfizer, Inc. (St. Louis, MO, USA), is another potent and selective inhibitor of human mPGES-1 from an oxicam series (IC50 of 16.5 nM) and shows low potency against rat mPGES-1. PF-9184 decreased PGE2 biosynthesis by RA synovial fibroblasts induced with IL-1β for 24 h. In addition, unexpected inhibition of the prostacyclin metabolite 6-keto PGF1a was detected, presumably due to interference with feed-back mechanisms on Cox-2 expression (Mbalaviele et al., 2010). At experimental conditions analyzing a direct effect of this inhibitor on mPGES-1 activity, PF-9184 significantly suppressed PGE2 generation, while shunting PGH2 into PGI2 and PGF2a pathways respectively (Mbalaviele et al., 2010).
MF63, a lead compound from a series of phenanthrene imidazole inhibitors made by Merck Frost, Quebec, Canada, is a selective inhibitor of mPGES-1 with high degree of selectivity over mPGES-2, PGI2, PGD2, and thromboxane (TX) synthases, Cox-1, Cox-2, 5-lipoxygenase, and various prostanoid and leukotriene receptors. It is potent inhibitor of human (IC50 of 1.3 nM) and guinea pig (IC50 of 0.9 nM) mPGES-1, but displays little activity toward the rat or mouse enzyme (Xu et al., 2008). Therefore in vivo efficacy of MF63 was evaluated in a knock-in (KI) mouse, expressing human mPGES-1 as well as in guinea pig. In the KI mouse, MF63 selectively suppressed PGE2, but not other prostanoids in LPS-induced air pouch model and inhibited LPS-induced hyperalgesia by 50% at 10 mg/kg and 80% at 100 mg/kg. In the guinea pig, MF63 strongly inhibited LPS-induced pyresis and hyperalgesia and relieved chronic osteoarthritic pain at 30–100 mg/kg. Under these concentrations MF63 did not cause gastrointestinal toxic effects in the KI mouse and non-human primates (Xu et al., 2008).
The inter-species differences of inhibitor potencies make it more difficult to evaluate if a human enzyme inhibitor have target or off-target effects in relevant animal models. The 3D structure of mPGES-1 revealed that the active surface falls in between transmembrane helices one and four of neighboring monomers (Jegerschold et al., 2008). Recently we demonstrated the combined role of three individual amino acids, different among human and mice/rat mPGES-1 located in transmembrane helix 4, that together accounted for most of the observed species difference for the utilized mPGES1 inhibitor (Pawelzik et al., 2010). Thus, this could pose a problem in the development of mPGES-1 inhibitors. However, as also demonstrated inhibitors with potencies to both rodent and human species exist (Pawelzik et al., 2010).
Conclusion
mPGES-1 constitutes a novel target for anti-inflammatory drugs. The enzyme is overexpressed in RA, OA, and myositis. Anti-rheumatic drugs including TNF blockers or oral glucocorticoids are ineffective in down-regulating this enzyme suggesting that local inflammation persists while the overall symptoms are under control. The latter finding also suggests that combination therapies of biologicals and mPGES1 inhibitors may be more effective than single therapies. Inhibitors against mPGES-1 are being developed by several laboratories but so far no clinical trial data have been reported. The suitability of mPGES-1 as a drug target and the pros and cons comparing with Cox inhibition is a rather complex matter and not discussed in details in this review. In short, one may state that mice devoid of mPGES-1 display fewer or milder phenotypes that relate to putative side effects of an enzyme inhibitor as compared with Cox-2 deleted mice. Clearly, any future mPGES-1 inhibitor will have to be monitored for effects on the GI tract and cardiovascular system, the latter being of particular interests since mPGES-1 inhibition may be used in order to prevent ischemic events while disadvantageous post-MI.
Conflict of Interest Statement
Per-Johan Jakobsson is a member of the board of directors for NovaSAID AB.
Acknowledgments
This study is supported by The Swedish Research Council, The Swedish Rheumatism Association, King Gustaf V 80 years Foundation, The Swedish Society of Medicine, Karolinska Institutet Foundation, The Swedish County Council and Marianne and Marcus Wallenberg foundation.
References
Alvarez-Soria, M. A., Herrero-Beaumont, G., Moreno-Rubio, J., Calvo, E., Santillana, J., Egido, J., and Largo, R. (2008). Long-term NSAID treatment directly decreases COX-2 and mPGES-1 production in the articular cartilage of patients with osteoarthritis. Osteoarthr. Cartil. 16, 1484–1493.
Amin, A. R., Attur, M., Patel, R. N., Thakker, G. D., Marshall, P. J., Rediske, J., Stuchin, S. A., Patel, I. R., and Abramson, S. B. (1997). Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J. Clin. Invest. 99, 1231–1237.
Bruno, A., Di Francesco, L., Coletta, I., Mangano, G., Alisi, M. A., Polenzani, L., Milanese, C., Anzellotti, P., Ricciotti, E., Dovizio, M., Di Francesco, A., Tacconelli, S., Capone, M. L., and Patrignani, P. (2010). Effects of AF3442 [N-(9-ethyl-9H-carbazol-3-yl)-2-(trifluoromethyl)benzamide], a novel inhibitor of human microsomal prostaglandin E synthase-1, on prostanoid biosynthesis in human monocytes in vitro. Biochem. Pharmacol. 79, 974–981.
Bucala, R., Ritchlin, C., Winchester, R., and Cerami, A. (1991). Constitutive production of inflammatory and mitogenic cytokines by rheumatoid synovial fibroblasts. J. Exp. Med. 173, 569–574.
Chan, M. M., and Moore, A. R. (2010). Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J. Immunol. 184, 6418–6426.
Cheng, S., Afif, H., Martel-Pelletier, J., Pelletier, J. P., Li, X., Farrajota, K., Lavigne, M., and Fahmi, H. (2004). Activation of peroxisome proliferator-activated receptor gamma inhibits interleukin-1beta-induced membrane-associated prostaglandin E2 synthase-1 expression in human synovial fibroblasts by interfering with Egr-1. J. Biol. Chem. 279, 22057–22065.
Cheng, Y., Wang, M., Yu, Y., Lawson, J., Funk, C. D., and Fitzgerald, G. A. (2006). Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J. Clin. Invest. 116, 1391–1399.
Claveau, D., Sirinyan, M., Guay, J., Gordon, R., Chan, C. C., Bureau, Y., Riendeau, D., and Mancini, J. A. (2003). Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J. Immunol. 170, 4738–4744.
Degousee, N., Fazel, S., Angoulvant, D., Stefanski, E., Pawelzik, S. C., Korotkova, M., Arab, S., Liu, P., Lindsay, T. F., Zhuo, S., Butany, J., Li, R. K., Audoly, L., Schmidt, R., Angioni, C., Geisslinger, G., Jakobsson, P. J., and Rubin, B. B. (2008). Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation 117, 1701–1710.
DeGroot, J., Verzijl, N., Wenting-van Wijk, M. J., Jacobs, K. M., Van El, B., Van Roermund, P. M., Bank, R. A., Bijlsma, J. W., TeKoppele, J. M., and Lafeber, F. P. (2004). Accumulation of advanced glycation end products as a molecular mechanism for aging as a risk factor in osteoarthritis. Arthritis Rheum. 50, 1207–1215.
Dieter, P., Scheibe, R., Jakobsson, P. J., Watanabe, K., Kolada, A., and Kamionka, S. (2000). Functional coupling of cyclooxygenase 1 and 2 to discrete prostanoid synthases in liver macrophages. Biochem. Biophys. Res. Commun. 276, 488–492.
Distler, J. H., Pisetsky, D. S., Huber, L. C., Kalden, J. R., Gay, S., and Distler, O. (2005). Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum. 52, 3337–3348.
Ehling, A., Schaffler, A., Herfarth, H., Tarner, I. H., Anders, S., Distler, O., Paul, G., Distler, J., Gay, S., Scholmerich, J., Neumann, E., and Muller-Ladner, U. (2006). The potential of adiponectin in driving arthritis. J. Immunol. 176, 4468–4478.
Engblom, D., Ek, M., Andersson, I. M., Saha, S., Dahlstrom, M., Jakobsson, P. J., Ericsson-Dahlstrand, A., and Blomqvist, A. (2002a). Induction of microsomal prostaglandin E synthase in the rat brain endothelium and parenchyma in adjuvant-induced arthritis. J. Comp. Neurol. 452, 205–214.
Engblom, D., Ek, M., Saha, S., Ericsson-Dahlstrand, A., Jakobsson, P. J., and Blomqvist, A. (2002b). Prostaglandins as inflammatory messengers across the blood–brain barrier. J. Mol. Med. 80, 5–15.
Engblom, D., Saha, S., Engstrom, L., Westman, M., Audoly, L. P., Jakobsson, P. J., and Blomqvist, A. (2003). Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat. Neurosci. 6, 1137–1138.
Facemire, C. S., Griffiths, R., Audoly, L. P., Koller, B. H., and Coffman, T. M. (2010). The impact of microsomal prostaglandin e synthase 1 on blood pressure is determined by genetic background. Hypertension 55, 531–538.
Faour, W. H., He, Y., He, Q. W., de Ladurantaye, M., Quintero, M., Mancini, A., and Di Battista, J. A. (2001). Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J. Biol. Chem. 276, 31720–31731.
Francois, H., Facemire, C., Kumar, A., Audoly, L., Koller, B., and Coffman, T. (2007). Role of microsomal prostaglandin E synthase 1 in the kidney. J. Am. Soc. Nephrol. 18, 1466–1475.
Gomez, P. F., Pillinger, M. H., Attur, M., Marjanovic, N., Dave, M., Park, J., Bingham, C. O. III, Al-Mussawir, H., and Abramson, S. B. (2005). Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-kappaB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J. Immunol. 175, 6924–6930.
Gosset, M., Berenbaum, F., Levy, A., Pigenet, A., Thirion, S., Saffar, J. L., and Jacques, C. (2006). Prostaglandin E2 synthesis in cartilage explants under compression: mPGES-1 is a mechanosensitive gene. Arthritis Res. Ther. 8, R135.
Gosset, M., Berenbaum, F., Salvat, C., Sautet, A., Pigenet, A., Tahiri, K., and Jacques, C. (2008). Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum. 58, 1399–1409.
Grimmer, C., Pfander, D., Swoboda, B., Aigner, T., Mueller, L., Hennig, F. F., and Gelse, K. (2007). Hypoxia-inducible factor 1alpha is involved in the prostaglandin metabolism of osteoarthritic cartilage through up-regulation of microsomal prostaglandin E synthase 1 in articular chondrocytes. Arthritis Rheum. 56, 4084–4094.
Guerrero, M. D., Aquino, M., Bruno, I., Riccio, R., Terencio, M. C., and Paya, M. (2009). Anti-inflammatory and analgesic activity of a novel inhibitor of microsomal prostaglandin E synthase-1 expression. Eur. J. Pharmacol. 620, 112–119.
Guerrero, M. D., Aquino, M., Bruno, I., Terencio, M. C., Paya, M., Riccio, R., and Gomez-Paloma, L. (2007). Synthesis and pharmacological evaluation of a selected library of new potential anti-inflammatory agents bearing the gamma-hydroxybutenolide scaffold: a new class of inhibitors of prostanoid production through the selective modulation of microsomal prostaglandin E synthase-1 expression. J. Med. Chem. 50, 2176–2184.
Hulkower, K. I., Wertheimer, S. J., Levin, W., Coffey, J. W., Anderson, C. M., Chen, T., DeWitt, D. L., Crowl, R. M., Hope, W. C., and Morgan, D. W. (1994). Interleukin-1 beta induces cytosolic phospholipase A2 and prostaglandin H synthase in rheumatoid synovial fibroblasts. Evidence for their roles in the production of prostaglandin E2. Arthritis Rheum. 37, 653–661.
Inada, M., Matsumoto, C., Uematsu, S., Akira, S., and Miyaura, C. (2006). Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. J. Immunol. 177, 1879–1885.
Jakobsson, P. J., Thoren, S., Morgenstern, R., and Samuelsson, B. (1999). Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U.S.A. 96, 7220–7225.
Jegerschold, C., Pawelzik, S. C., Purhonen, P., Bhakat, P., Gheorghe, K. R., Gyobu, N., Mitsuoka, K., Morgenstern, R., Jakobsson, P. J., and Hebert, H. (2008). Structural basis for induced formation of the inflammatory mediator prostaglandin E2. Proc. Natl. Acad. Sci. U.S.A. 105, 11110–11115.
Jia, Z., Guo, X., Zhang, H., Wang, M. H., Dong, Z., and Yang, T. (2008). Microsomal prostaglandin synthase-1-derived prostaglandin E2 protects against angiotensin II-induced hypertension via inhibition of oxidative stress. Hypertension 52, 952–959.
Jia, Z., Zhang, A., Zhang, H., Dong, Z., and Yang, T. (2006). Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ. Res. 99, 1243–1251.
Jungel, A., Distler, O., Schulze-Horsel, U., Huber, L. C., Ha, H. R., Simmen, B., Kalden, J. R., Pisetsky, D. S., Gay, S., and Distler, J. H. (2007). Microparticles stimulate the synthesis of prostaglandin E(2) via induction of cyclooxygenase 2 and microsomal prostaglandin E synthase 1. Arthritis Rheum. 56, 3564–3574.
Kamei, D., Yamakawa, K., Takegoshi, Y., Mikami-Nakanishi, M., Nakatani, Y., Oh-Ishi, S., Yasui, H., Azuma, Y., Hirasawa, N., Ohuchi, K., Kawaguchi, H., Ishikawa, Y., Ishii, T., Uematsu, S., Akira, S., Murakami, M., and Kudo, I. (2004). Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J. Biol. Chem. 279, 33684–33695.
Kapoor, M., Kojima, F., Yang, L., and Crofford, L. J. (2007). Sequential induction of pro- and anti-inflammatory prostaglandins and peroxisome proliferators-activated receptor-gamma during normal wound healing: a time course study. Prostaglandins Leukot. Essent. Fatty Acids 76, 103–112.
Kojima, F., Kapoor, M., Yang, L., Fleishaker, E. L., Ward, M. R., Monrad, S. U., Kottangada, P. C., Pace, C. Q., Clark, J. A., Woodward, J. G., and Crofford, L. J. (2008). Defective generation of a humoral immune response is associated with a reduced incidence and severity of collagen-induced arthritis in microsomal prostaglandin E synthase-1 null mice. J. Immunol. 180, 8361–8368.
Kojima, F., Kato, S., and Kawai, S. (2005). Prostaglandin E synthase in the pathophysiology of arthritis. Fundam. Clin. Pharmacol. 19, 255–261.
Kojima, F., Naraba, H., Miyamoto, S., Beppu, M., Aoki, H., and Kawai, S. (2004). Membrane-associated prostaglandin E synthase-1 is upregulated by proinflammatory cytokines in chondrocytes from patients with osteoarthritis. Arthritis Res. Ther. 6, R355–R365.
Kojima, F., Naraba, H., Sasaki, Y., Beppu, M., Aoki, H., and Kawai, S. (2003). Prostaglandin E2 is an enhancer of interleukin-1beta-induced expression of membrane-associated prostaglandin E synthase in rheumatoid synovial fibroblasts. Arthritis Rheum. 48, 2819–2828.
Kojima, F., Naraba, H., Sasaki, Y., Okamoto, R., Koshino, T., and Kawai, S. (2002). Coexpression of microsomal prostaglandin E synthase with cyclooxygenase-2 in human rheumatoid synovial cells. J. Rheumatol. 29, 1836–1842.
Korotkova, M., Helmers, S. B., Loell, I., Alexanderson, H., Grundtman, C., Dorph, C., Lundberg, I. E., and Jakobsson, P. J. (2008). Effects of immunosuppressive treatment on microsomal prostaglandin E synthase 1 and cyclooxygenases expression in muscle tissue of patients with polymyositis or dermatomyositis. Ann. Rheum. Dis. 67, 1596–1602.
Korotkova, M., Westman, M., Gheorghe, K. R., af Klint, E., Trollmo, C., Ulfgren, A. K., Klareskog, L., and Jakobsson, P. J. (2005). Effects of antirheumatic treatments on the prostaglandin E2 biosynthetic pathway. Arthritis Rheum. 52, 3439–3447.
Kusada, J., Otsuka, T., Matsui, N., Hirano, T., Asai, K., and Kato, T. (1993). Immuno-reactive human epidermal growth factor (h-EGF) in rheumatoid synovial fluids. Nippon Seikeigeka Gakkai Zasshi 67, 859–865.
Kusunoki, N., Kitahara, K., Kojima, F., Tanaka, N., Kaneko, K., Endo, H., Suguro, T., and Kawai, S. (2010). Adiponectin stimulates prostaglandin E(2) production in rheumatoid synovial fibroblasts. Arthritis Rheum. 62, 1641–1649.
Lazarus, M., Kubata, B. K., Eguchi, N., Fujitani, Y., Urade, Y., and Hayaishi, O. (2002). Biochemical characterization of mouse microsomal prostaglandin E synthase-1 and its colocalization with cyclooxygenase-2 in peritoneal macrophages. Arch. Biochem. Biophys. 397, 336–341.
Li, X., Afif, H., Cheng, S., Martel-Pelletier, J., Pelletier, J. P., Ranger, P., and Fahmi, H. (2005). Expression and regulation of microsomal prostaglandin E synthase-1 in human osteoarthritic cartilage and chondrocytes. J. Rheumatol. 32, 887–895.
Mancini, J. A., Blood, K., Guay, J., Gordon, R., Claveau, D., Chan, C. C., and Riendeau, D. (2001). Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J. Biol. Chem. 276, 4469–4475.
Masuko-Hongo, K., Berenbaum, F., Humbert, L., Salvat, C., Goldring, M. B., and Thirion, S. (2004). Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 50, 2829–2838.
Mbalaviele, G., Pauley, A. M., Shaffer, A. F., Zweifel, B. S., Mathialagan, S., Mnich, S. J., Nemirovskiy, O. V., Carter, J., Gierse, J. K., Wang, J. L., Vazquez, M. L., Moore, W. M., and Masferrer, J. L. (2010). Distinction of microsomal prostaglandin E synthase-1 (mPGES-1) inhibition from cyclooxygenase-2 inhibition in cells using a novel, selective mPGES-1 inhibitor. Biochem. Pharmacol. 79, 1445–1454.
Murakami, M., Nakashima, K., Kamei, D., Masuda, S., Ishikawa, Y., Ishii, T., Ohmiya, Y., Watanabe, K., and Kudo, I. (2003). Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 278, 37937–37947.
Murakami, M., Nakatani, Y., Tanioka, T., and Kudo, I. (2002). Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 68–69, 383–399.
Murakami, M., Naraba, H., Tanioka, T., Semmyo, N., Nakatani, Y., Kojima, F., Ikeda, T., Fueki, M., Ueno, A., Oh, S., and Kudo, I. (2000). Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275, 32783–32792.
Nah, S. S., Choi, I. Y., Lee, C. K., Oh, J. S., Kim, Y. G., Moon, H. B., and Yoo, B. (2008). Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology (Oxford) 47, 425–431.
Nah, S. S., Won, H. J., Ha, E., Kang, I., Cho, H. Y., Hur, S. J., Lee, S. H., and Baik, H. H. (2010). Epidermal growth factor increases prostaglandin E2 production via ERK1/2 MAPK and NF-kappaB pathway in fibroblast like synoviocytes from patients with rheumatoid arthritis. Rheumatol. Int. 30, 443–449.
Naraba, H., Yokoyama, C., Tago, N., Murakami, M., Kudo, I., Fueki, M., Oh-Ishi, S., and Tanabe, T. (2002). Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J. Biol. Chem. 277, 28601–28608.
Newton, R., Seybold, J., Kuitert, L. M., Bergmann, M., and Barnes, P. J. (1998). Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J. Biol. Chem. 273, 32312–32321.
Pawelzik, S. C., Uda, N. R., Spahiu, L., Jegerschold, C., Stenberg, P., Hebert, H., Morgenstern, R., and Jakobsson, P. J. (2010). Identification of key residues determining species differences in inhibitor binding of microsomal prostaglandin E synthase-1. J. Biol. Chem. 285, 29254–29261.
Pettersson, P. L., Thoren, S., and Jakobsson, P. J. (2005). Human microsomal prostaglandin E synthase 1: a member of the MAPEG protein superfamily. Meth. Enzymol. 401, 147–161.
Pottie, P., Presle, N., Terlain, B., Netter, P., Mainard, D., and Berenbaum, F. (2006). Obesity and osteoarthritis: more complex than predicted! Ann. Rheum. Dis. 65, 1403–1405.
Punzi, L., Schiavon, F., Cavasin, F., Ramonda, R., Gambari, P. F., and Todesco, S. (1989). The influence of intra-articular hyaluronic acid on PGE2 and cAMP of synovial fluid. Clin. Exp. Rheumatol. 7, 247–250.
Saegusa, M., Murakami, M., Nakatani, Y., Yamakawa, K., Katagiri, M., Matsuda, K., Nakamura, K., Kudo, I., and Kawaguchi, H. (2003). Contribution of membrane-associated prostaglandin E2 synthase to bone resorption. J. Cell. Physiol. 197, 348–356.
Samuelsson, B., Morgenstern, R., and Jakobsson, P. J. (2007). Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol. Rev. 59, 207–224.
Satoh, K., Kikuchi, S., Sekimata, M., Kabuyama, Y., Homma, M. K., and Homma, Y. (2001). Involvement of ErbB-2 in rheumatoid synovial cell growth. Arthritis Rheum. 44, 260–265.
Shimpo, H., Sakai, T., Kondo, S., Mishima, S., Yoda, M., Hiraiwa, H., and Ishiguro, N. (2009). Regulation of prostaglandin E(2) synthesis in cells derived from chondrocytes of patients with osteoarthritis. J. Orthop. Sci. 14, 611–617.
Stichtenoth, D. O., Thoren, S., Bian, H., Peters-Golden, M., Jakobsson, P. J., and Crofford, L. J. (2001). Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J. Immunol. 167, 469–474.
Tai, N., Kuwabara, K., Kobayashi, M., Yamada, K., Ono, T., Seno, K., Gahara, Y., Ishizaki, J., and Hori, Y. (2010). Cytosolic phospholipase A2 alpha inhibitor, pyrroxyphene, displays anti-arthritic and anti-bone destructive action in a murine arthritis model. Inflamm. Res. 59, 53–62.
Tanioka, T., Nakatani, Y., Kobayashi, T., Tsujimoto, M., Oh-ishi, S., Murakami, M., and Kudo, I. (2003). Regulation of cytosolic prostaglandin E2 synthase by 90-kDa heat shock protein. Biochem. Biophys. Res. Commun. 303, 1018–1023.
Thoren, S., and Jakobsson, P. J. (2000). Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur. J. Biochem. 267, 6428–6434.
Trebino, C. E., Stock, J. L., Gibbons, C. P., Naiman, B. M., Wachtmann, T. S., Umland, J. P., Pandher, K., Lapointe, J. M., Saha, S., Roach, M. L., Carter, D., Thomas, N. A., Durtschi, B. A., McNeish, J. D., Hambor, J. E., Jakobsson, P. J., Carty, T. J., Perez, J. R., and Audoly, L. P. (2003). Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. U.S.A. 100, 9044–9049.
Wang, M., Song, W. L., Cheng, Y., and Fitzgerald, G. A. (2008). Microsomal prostaglandin E synthase-1 inhibition in cardiovascular inflammatory disease. J. Intern. Med. 263, 500–505.
Wang, M., Zukas, A. M., Hui, Y., Ricciotti, E., Pure, E., and FitzGerald, G. A. (2006). Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 14507–14512.
Westman, M., Korotkova, M., af Klint, E., Stark, A., Audoly, L. P., Klareskog, L., Ulfgren, A. K., and Jakobsson, P. J. (2004). Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum. 50, 1774–1780.
Wu, D., Mennerich, D., Arndt, K., Sugiyama, K., Ozaki, N., Schwarz, K., Wei, J., Wu, H., Bishopric, N. H., and Doods, H. (2009). Comparison of microsomal prostaglandin E synthase-1 deletion and COX-2 inhibition in acute cardiac ischemia in mice. Prostaglandins Other Lipid Mediat. 90, 21–25.
Xu, D., Rowland, S. E., Clark, P., Giroux, A., Cote, B., Guiral, S., Salem, M., Ducharme, Y., Friesen, R. W., Methot, N., Mancini, J., Audoly, L., and Riendeau, D. (2008). MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J. Pharmacol. Exp. Ther. 326, 754–763.
Keywords: prostaglandin E synthase, rheumatoid arthritis, osteoarthritis, inflammatory myositis, inhibitors
Citation: Korotkova M and Jakobsson P-J (2011) Microsomal prostaglandin E synthase-1 in rheumatic diseases. Front. Pharmacol. 1:146. doi: 10.3389/fphar.2010.00146
Received: 04 September 2010;
Paper pending published: 24 September 2010;
Accepted: 22 December 2010;
Published online: 20 January 2011.
Edited by:
Dieter Steinhilber, Goethe Universitat, GermanyReviewed by:
Daniel Poeckel, Cellzome Aktiengesellschaft, GermanyOliver Werz, Friedrich Schiller University Jena, Germany
Sabine Grösch, Johan Wolfgang Goethe University, Germany
Copyright: © 2011 Korotkova and Jakobsson. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Per-Johan Jakobsson, Rheumatology Unit, Department of Medicine, Karolinska Institutet, Karolinska University Hospital, CMM L8:04, 171 76 Stockholm, Sweden. e-mail: per-johan.jakobsson@ki.se