 Luc Dannhoffer* Arnaud Billet Mathilde Jollivet Patricia Melin-Heschel Christelle Faveau Frédéric Becq
Luc Dannhoffer* Arnaud Billet Mathilde Jollivet Patricia Melin-Heschel Christelle Faveau Frédéric Becq
- Institut de Physiologie et Biologie Cellulaires, UMR 6187, Université de Poitiers, CNRS, Poitiers, France
Cystic fibrosis (CF) is a major inherited disorder involving abnormalities of fluid and electrolyte transport in a number of different organs due to abnormal function of cystic fibrosis transmembrane conductance regulator (CFTR) protein. We recently identified a family of CFTR activators, which contains the hit: RP107 [7-n-butyl-6-(4-hydroxyphenyl)[5H]-pyrrolo[2,3-b]pyrazine]. Here, we further evaluated the effect of the chemical modifications of the RP107-OH radical on CFTR activation. The replacement of the OH radical by a fluorine atom at position 2 (RP193) or 4 (RP185) significantly decreased the toxicity of the compounds without altering the ability to activate CFTR, especially for RP193. The non-toxic compound RP193 has no effect on cAMP production but stimulates the channel activity of wild-type CFTR in stably transfected CHO cells, in human bronchial epithelial NuLi-1 cells, and in primary culture of human bronchial epithelial cells (HBEC). Whole-cell and single patch-clamp recordings showed that RP193 induced a linear, time- and voltage-independent current, which was fully inhibited by two different and selective CFTR inhibitors (CFTRinh-172 and GPinh5a). Moreover, RP193 stimulates CFTR in temperature-rescued CuFi-1 (F508del/F508del) HBEC and in CHO cells stably expressing G551D-CFTR. This study shows that it is feasible to reduce cytotoxicity of chemical compounds without affecting their potency to activate CFTR and to rescue the class 2 F508del-CFTR and class 3 G551D-CFTR CF mutant activities.
Introduction
Cystic fibrosis (CF; MIM#219700) disease results from mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein. CFTR, which is especially expressed at the apical membrane of the airway epithelial cells (Kreda et al., 2005; Regnier et al., 2008), acts as cAMP-activated chloride channel as well as a regulator of other ion channels like the canonical Transient Receptor Potential 6, TRPC6 (Antigny et al., 2011). In the lung, CFTR dysfunctions carry abnormal ion transports leading to airway surface liquid volume depletion in proximal and distal airways allowing chronic bacterial infections and finally severe lung damages (Boucher, 1999; Tarran, 2004; Blouquit et al., 2006).
Over 1600 mutations of the CFTR gene have been identified and several classes of mutations according to the fate of the final product have been proposed (Welsh and Smith, 1993). Based on this classification, it is in theory possible to predict a strategy for developing a personalized CF therapy corresponding to the class of the CFTR mutation (Becq, 2010). The majority of severe CFTR mutations belongs to class 1–3 leading to reduced chloride permeability in affected epithelia in response to cAMP agonists. Class 1 includes non-sense, frame shift, and splice site mutations (e.g., stop mutations such as G542X) with unstable transcripts and failure of CFTR translation. Class 2 includes the most common CFTR mutation F508del. The biogenesis of F508del mutants is extremely inefficient with its retention in the endoplasmic reticulum (ER) but it is feasible to rescue the abnormal trafficking by in vitro cooling CF cells or pharmacological correctors (reviewed in Becq, 2010). Importantly, sufficient amount of rescued F508del-CFTR at the plasma membrane conduct chloride ions despite a gating defect (Dalemans et al., 1991; Li et al., 1993; Dormer et al., 2005). Patients with a class 3 mutation of CFTR (e.g., G551D, G1349D) have also a severe form of CF, because they do not have or reduced activation of the CFTR chloride function at the plasma membrane. Class 4 mutants present defective permeation, class 5 mutants a reduced synthesis, and class 6 is characterized by altered apical membrane residence time of CFTR channels at the membrane (Kreindler, 2010).
Such classification could be very helpful for mutation-oriented search of personalized CFTR modulators. For example, the identification of pharmacological correctors of the processing defect of CFTR could be developed only for patients diagnosed with class 2 CFTR mutations such as carrying at least one F508del mutation. Agents directly stimulating the ion channel activity of CFTR might be adapted to class 3 or 4 mutations and to class 2 in combination with a trafficking corrector.
Our group is interested in developing novel modulators for class 2 and 3 CFTR defect activity. One family of CFTR modulators, the pyrrolo[2,3-b]pyrazines, which belong to a type of derivatives initially described as cyclin-dependent kinase (CDK)/glycogen synthase kinase-3 (GSK-3) inhibitors, which act by competing with ATP for binding to the kinase active site, has been identified (Mettey et al., 2003; Noel et al., 2006). One compound of this family, RP107 [7-n-butyl-6-(4-hydroxyphenyl)[5H]-pyrrolo[2,3-b]pyrazine], has been more particularly investigated (Noel et al., 2006) because we observed that the length of the alkyl chain is essential for CFTR activation with the following potency: (CH2)3-CH3 ≫ H ≅ CH3 ≅ (CH2)2-CH3 ≅ (CH2)4-CH3. Based on these observations, we decided to conserve the butyl chain in R1 position and to modify only determinants on the phenyl ring. Four different compounds (RP146, 173, RP185, and RP193) have been synthesized and their effect on CFTR activity measured.
Materials and Methods
Chemistry
1H NMR spectra were recorded on a Bruker Advance DPX 300 spectrometer at 300 mHz. Chemical shifts (ppm) were reported relative to tetramethylsilane (TMS). Coupling constants (J) were reported in Hertz (Hz), and s, d, t, q, m, and bs referred to singlet, doublet, triplet, quartet, multiplet, and broad singlet, respectively. Infrared spectra (IR) were recorded on an ATI Mattson genesis series FTIR. Elemental analyses were indicated by the symbol of the elements, and the results were within ±0.4% (for C, H, N) of the theoretical values unless otherwise noted; they were performed on a Perkin Elmer Elemental Analyzer (2400). Melting points were measured in open capillary tubes on an Electrothermal 9200 apparatus and are uncorrected.
All experiments involving butyllithium (BuLi) were carried out in dried apparatus under an atmosphere of dry oxygen-free nitrogen. Tetrahydrofuran (THF) was distilled from benzophenone–sodium. Diisopropylamine and heterocycles were distilled and stored over barium oxide. Butyllithium (2.5 M solution in hexane) was supplied by Acros. Alkylpyrazines and alkylpyridines were prepared according to usual procedures. Sodium hydride was an 95% dispersion in mineral oil. Matrex silica gel 60 Å, 20–45 μm, Merck alumina gel 90 Å, 63–200 μm were employed for column chromatographies.
General Procedure for the Preparation of 4,7-Diazaindoles
These compounds were prepared as previously described (Mettey et al., 2003). Physical and spectral data for compound RP108 has been reported (Mettey et al., 2003; Noel et al., 2006).
7-n-butyl-6-(4-fluorophenyl)-5H-pyrrolo[2,3-b]pyrazine (RP185)
Yield 18%; white cotton; mp 161.9°C; IR (KBr, cm-1) 3164, 3053, 2958, 2851; 1H NMR (CDCl3) δ (ppm) 11.63 (bs, 1H), 8.46 (s, 1H), 8.05 (s, 1H), 7.70–7.74 (m, 2H), 7.29 (t, J = 8.1 Hz, 2H), 2.99 (t, J = 7.7 Hz, 2H), 1.71–1.79 (m, 2H), 1.39–1.46 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H). Anal. (C16H16N3F) C, H, N.
7-n-butyl-6-(2-fluorophenyl)-5H-pyrrolo[2,3-b]pyrazine (RP193)
Yield 15%; white powder; mp 136.3°C; IR (KBr, cm-1) 3131, 3058, 2959, 2853, 1469, 1224, 755; 1H NMR (CDCl3) δ (ppm) 10.81 (bs, 1H), 8.45 (s, 1H), 8.11 (s, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.46–7.53 (m, 1H), 7.26–7.37 (m, 2H), 2.93 (t, J = 7.8 Hz, 2H), 1.7–1.8 (m, 2H), 1.32–1.44 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). Anal. (C16H16N3F) C, H, N.
General Procedure for the Preparation of Compounds, from Their Methoxy Precursors
Demethylation was performed with HBr, according to our previously described method (Mettey et al., 2003). Physical and spectral data for compound RP107 has been reported (Mettey et al., 2003; Noel et al., 2006).
7-n-Butyl-6-(3-hydroxyphenyl)-5H-pyrrolo[2,3-b]pyrazine hydrobromide (RP146)
Yield 71%; mp 196.0°C (dec); IR (KBr, cm-1) 3249, 3092, 2954, 2924, 2859, 2729; 1H NMR (DMSO-d6) δ (ppm) 12.97 (s, 1H), 8.95 (bs, 1H), 8.63 (d, J = 3 Hz, 1H), 8.60 (d, J = 3 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 7.27 (d, J = 7.6 Hz, 1H), 7.26 (s, 1H), 7.05 (dd, J = 7.6 and 1.6 Hz, 1H), 3.01 (t, J = 7.7 Hz, 2H), 1.66–1.95 (m, 2H), 1.38–1.51 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H). Anal. (C16H17N3O, HBr) C, H, N.
7-n-Butyl-6-(2-hydroxyphenyl)-5H-pyrrolo[2,3-b]pyrazine hydrobromide (RP173)
Yield 80%; yellow powder; mp 243.0°C (dec); IR (KBr, cm-1) 3072, 2951, 2639; 1H NMR (DMSO-d6) δ (ppm) 12.58 (bs, 1H), 9.95 (bs, 1H), 8.49 (d, J = 3 Hz, 1H), 8.45 (d, J = 3 Hz, 1H), 7.34–7.38 (m, 2H), 7.06 (d, J = 8.5 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 5.15 (bs, 1H), 2.76 (t, J = 7.6 Hz, 2H), 1.51–1.61 (m, 2H), 1.17–1.29 (m, 2H), 0.78 (t, J = 7.3 Hz, 3H). Anal. (C16H17N3O, HBr) C, H, N.
Elemental analyses
(RP173) Calcd for C16H17N3O, HBr: C, 55.18; H, 5.21; N, 12.07
Found: C, 55.02; H, 5.21; N, 12.02
(RP185) Calcd for C16H16N3F: C, 71.35; H, 5.99; N, 15.60
Found: C, 71.29; H, 5.99; N, 15.50
(RP193) Calcd for C16H16N3F: C, 71.35; H, 5.99; N, 15.60
Found: C, 71.13; H, 5.94; N, 15.58
Cell Line Culture
All cell cultures were grown at 37°C in 5% CO2 in standard culture conditions as follows. CHO cells stably transfected with pNUT vector alone or containing wild-type CFTR (wt-CFTR-CHO) and the mutant G551D-CFTR (Tabcharani et al., 1991; Becq et al., 1994, 1999) were cultured in α-minimal essential medium-GlutaMAX containing 7% of fetal bovine serum, 50 IU/ml penicillin and 50 μg/ml streptomycin, and methotrexate for cell selection (wt-CFTR-CHO: 100 μM; G551D-CHO: 20 μM; pNUT-CHO: 20 μM). CuFi-1 and NuLi-1 cells derived from human bronchial epithelium from a CF patient (CuFi-1, F508del/F508del-CFTR mutant genotype) and a non-CF subject (NuLi-1, wild-type CFTR). These cell lines were grown on human placental collagen type VI (Sigma, St. Louis, MO, USA) coated flasks in BEGM medium (Cambrex Bio Science Walkersville, MD, USA), as previously described (Zabner et al., 2003). Cells were seeded in 24-well plates for iodide efflux and in 35-mm plastic dishes for whole-cell and cell-attached patch-clamp recordings. Culture media were renewed every 2 days.
Primary Cultures of Human Bronchial Epithelial Cells
Fragments of non-CF lungs were obtained from three patients who underwent lung resection for cancer (three males, mean age: 61.3 ± 5.5 years). Tissues were received and processed within 2 h after surgery. All procedures were performed in compliance with the current French legislation. Human bronchi were dissected free of lung parenchyma. Bronchial epithelial cells were harvested using enzymatic isolation procedures to establish primary cultures, as previously described (Blouquit et al., 2002; Dannhoffer et al., 2009). Briefly, fragment of bronchi were incubated 24 h at 4°C with 0.1% protease and 0.01% deoxyribonuclease in DMEM/Ham’s F-12 medium. Enzymatic digestion was neutralized by 10% fetal bovine serum; cells were then centrifuged at 600 rpm for 6 min at room temperature (RT). Epithelial isolated cells were seeded at high density on home-made permeable collagen supports affixed to an orifice drilled in polycarbonate cups (Blouquit et al., 2002; Dannhoffer et al., 2009). They reached confluence between 8 and 12 days after seeding. The culture medium consisted of a 1:1 mix of Dulbecco’s modified Eagle’s medium-Ham’s F-12 medium supplemented with: 5 μg/ml insulin, 7.5 μg/ml transferrin, 10−6 M hydrocortisone, 2 μg/ml endothelial cell growth supplement, 25 ng/ml epithelial growth factor, 3.10−8 M triiodothyronine, 2.5 mM L-glutamine, and 100 IU/ml penicillin and 100 μg/ml streptomycin.
Iodide Efflux
Screening of molecules and concentration response curves were determined by measuring the rate of 125I efflux with a high-capacity robotic system (MultiProbe II EXT; PerkinElmer Life Sciences) adapted to the determination of iodide efflux as described previously (Marivingt-Mounir et al., 2004). Time-dependent rates (k = peak rate, min−1) of 125I efflux were calculated from the following equation: k = ln (125It1/125It2)/(t1–t2), where 125It is the intracellular 125I at time t and t1 and t2 are successive time points. Relative rates were calculated: kpeak (peak rate of efflux) - kbasal (basal rate of efflux; min−1), i.e., the maximal value for the time-dependent rate (kpeak, min−1) excluding the third point used to establish the baseline (kbasal, min−1). Concentration-dependent activation curves were constructed as percentage maximal activation (set at 100%) transformed from the calculated relative rates. In some experiments, chloride transport inhibitors were present in the loading solution and in the efflux buffer. The activity of CFTR-dependent iodide efflux was stimulated by 1 μM forskolin or by a cocktail containing 1 μM forskolin +100 μM RP derivatives (146, 173, 185, or 193). The effect of RP193 100 μM without forskolin on CFTR activity was also investigated.
Cytotoxicity Assay
To determine effects of compounds on wt-CFTR-CHO cells viability, colorimetric 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich) assay was performed as previously described (Routaboul et al., 2007). Cell survival was evaluated by measuring the mitochondrial-dependent reduction of MTT. Cells seeded in 96-well plates were incubated during 24 h in the presence of 100 μM of the test compound (RP107, RP108, RP146, RP173, RP185, or RP193) at 37°C, and the MTT test was performed the day after. At each point of treatment, cells were washed with PBS (in mM: 130 NaCl, 2 KCl, 1.5 Na2HPO4, and 8 KH2PO4, pH 7.4). The medium was removed and 100 μl of MTT solution (0.5 mg/ml in PBS) were added to each well. After 4 h of incubation at 37°C, the supernatant was removed and the purple formazan crystals were dissolved by addition of 100 μl dimethylsulfoxide (DMSO, Sigma-Aldrich). The plates were agitated, and the optical density was read at the wavelength of 570 nm and a reference wavelength at 630 nm in a microplate reader (SpectraCount microplate photometer, Packard). Report values were the mean of three replicates and are expressed as percentage of the control values.
Determination of cAMP
cAMP levels were measured using the cAMP-Screen ELISA System (Applied Biosystems, Bedford, MA, USA). Wt-CFTR-CHO cells were incubated with drugs for 10 min at 37°C. Reactions were terminated, processed, and quantitated according to manufacturer’s instructions.
Whole-Cell Patch-Clamp Recordings
Whole-cell patch-clamp experiments were performed on wt-CFTR-CHO and K1-CHO, which did not expressed CFTR, cells at RT. Currents were recorded with a RK-400 patch-clamp amplifier (Biologic, Claix, France). Current–voltage (I/V) relationships were constructed by clamping the membrane potential to −40 mV and by pulses from −100 to +100 mV with 20 mV increments. Pipettes were pulled from borosilicate glass capillary tubing (GC150-TF10; Clark Electromedical Inc., Reading, UK) using a two-step vertical puller from Narishige (Tokyo, Japan) and had a resistance from 3 to 5 MΩ. They were filled with the following solution (in mM: 1 NaCl, 113 L-aspartic acid, 113 CsOH, 1 MgCl2, 27 CsCl, 1 EGTA, 10 TES, and 3 MgATP, ex temporane, pH 7.2; 285 mOsm). They were connected to the head of the patch-clamp amplifier through an Ag–AgCl pellet. The bath solution contained in mM: 145 NaCl, 4 CsCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 TES (pH 7.4; 340 mOsm). The liquid potential was corrected before seal establishment. Seal resistances ranging from 5 to 10 GΩ were obtained. Pipette capacitances were electronically compensated in cell-attached mode. Membrane capacitances were measured in the whole-cell mode by fitting capacitance currents obtained in response to a hyperpolarization of 10 mV, with a first-order exponential, and by integrating the surface of the capacitance current. Sample frequency was 3 kHz. Results were analyzed with the pClamp9 package software (Molecular Devices, Sunnyvale, CA, USA). Mean values of membrane capacitance were 39.1 ± 6.0 pF (n = 14) for wt-CFTR-CHO cells. For graphic representations, I/V relationship was normalized to 1 pF to remove variability due to differences in cell sizes. For time course experiments, current amplitude measured at +40 mV was plotted each 10 s.
Single-Channel Patch-Clamp Recordings
Single-channel currents were recorded from cell-attached patches. Pipettes were pulled from borosilicate glass capillary tubing (GC150-F10; Clark Electromedical Inc., Reading, UK) using a two-step vertical puller from Narishige (Tokyo, Japan) had a resistance of 15–30 MΩ. Seal resistances ranging from 10 to 20 GΩ were obtained. Experiments were performed at RT. Cells were stimulated with forskolin (1 μM) and RP193 (10 μM). Results were displayed conventionally with inward currents (outward flow of anions) indicated by downward deflections. In all the figures, dashed lines give the zero current baseline when the channels are in the closed state. Potentials were expressed as the bath potential minus the patch electrode potential. Single-channel recordings were sampled at 3 kHz and filtered at 100 Hz with an eight-pole Bessel filter. The pipette solution contained in mM: 150 NaCl, 2 MgCl2, and 10 TES (pH = 7.4); the bath contained: 145 NaCl, 4 KCl, 2 MgCl2, and 10 TES (pH = 7.4).
Transepithelial ion transports
Confluent human bronchial epithelial cell cultures were mounted in Ussing chambers, bathed on both sides with Krebs bicarbonate Ringer (KBR) solution warmed at 37°C, and gassed with 95% O2-5% CO2. KBR composition was in mM: 119.7 NaCl, 15 NaHCO3, 10 glucose, 4.5 KCl, 2 CaCl2, 1.5 NaH2PO4, 0.7 Na2HPO4, 0.23 MgCl2. The short-circuit current (Isc) was monitored continuously using a VCC MC2 voltage clamp (Physiologic Instruments Inc., San Diego, USA). One voltage-sensing electrode and one current-passing electrode were placed on each side of the culture. The transepithelial resistance (Rt) was determined by clamping the potential difference (PD) to +2 mV at 90 s intervals, recording the deflection in short-circuit current (Isc), and applying Ohm’s law. Cell cultures were allowed to equilibrate until stabilization of bioelectric variables, which required approximately 20–30 min. Basal bioelectric activity was monitored for an additional 10 min period prior to the addition of drugs. The pharmacological agent amiloride (an inhibitor of the epithelial sodium channel, ENaC) and forskolin (an activator of adenylate cyclase) were then added sequentially, and the bioelectric activity was allowed to stabilize 10–15 min before addition of the subsequent agent. Amiloride (10 μM) was added to the apical bath, forskolin (1 μM) was added to the apical and the basolateral baths and RP193 (100 and 200 μM) was added to the basolateral bath. Changes in Isc were calculated as the difference between the values measured immediately before the addition of reagents, and the values corresponding to the plateau phase after the addition of amiloride, forskolin, and RP193.
Chemicals
All culture media, hormones, growth factors, or antibiotics were from Invitrogen (Cergy-Pontoise, France) or Sigma (Saint Quentin Fallavier, France), and FBS was from PerbioScience (Brebières, France). Unless otherwise stated, chemicals were purchased from Sigma.
Results
Effect of the Newly Synthesized Compounds on CFTR
We first examined the effects of the modified pyrrolo[2,3-b]pyrazine derivatives (Figure 1A) on CHO cells stably expressing wt-CFTR with a cell-based primary screening assay using iodide efflux measurement (Marivingt-Mounir et al., 2004). All compounds stimulated CFTR with various half-maximal effective concentrations. For the most potent compound, RP193 we determined an EC50 of 270 nM. Then, for RP173 we obtained an EC50 of 0.2 μM. Finally for the two remaining compounds we found much higher values with or EC50 = 78 μM for RP185 and EC50 = 97 μM for RP146 (Figure 1B). Before extensive analysis of the activation of CFTR by these compounds, their cytotoxicity has been evaluated by measuring cellular dehydrogenase activity using the water-soluble tetrazolium salt MTT. Importantly, we observed that the two compounds with a fluorine atome on the phenyl ring, at the position 2 (RP193) or 4 (RP185) are not cytotoxic (Figure 1C). These experiments also revealed that the others compounds (RP107, RP108, RP146, and RP173) have a significantly higher cytotoxicity compared to the control conditions. Thus, among the pyrrolo[2,3-b]pyrazine derivatives tested here and previously (Noel et al., 2006) the compound 7-n-butyl-6-(2-fluorophenyl)-5H-pyrrolo[2,3-b]pyrazine named RP193 is the less toxic derivative and the most potent activator of wt-CFTR. Iodide efflux experiments were then performed with RP193 without forskolin 1 μM on wt-CFTR-CHO cells and revealed no stimulation of CFTR activity (Figure 1D). Further experiments revealed that potentiation of CFTR activity by RP193 (Figure 1D) was independent of intracellular cAMP levels (Figure 1E).
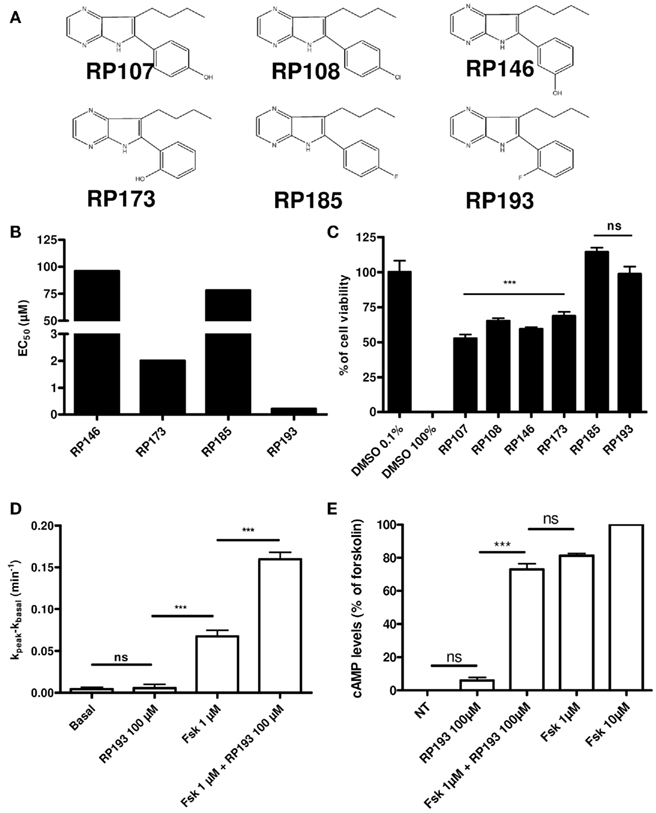
Figure 1. (A) Chemical structure of the 2 pyrrolo[2,3-b]pyrazines previously tested compounds (RP107 and RP108) and newly synthesized compounds (RP146, 173, 185, and 193). (B) EC50 of different compounds tested determinate by iodide efflux in wt-CFTR-CHO. (C) Evaluation of the cytotoxicity of the six different derivatives. The toxicity of the compounds was evaluated as described in the experimental section. Results are presented as percentage of cell viability for wt-CFTR-CHO cells treated 24 h with 100 μM of each compound; n = 20 for each compound, except for RP146, n = 3 and for DMSO 100% and DMSO 0.1%, n = 10. (D) Bar chart showing RP193 (100 μM), forskolin (1 μM), and forskolin (1 μM) + RP193 (100 μM) dependent iodide efflux in wt-CFTR-CHO cells. n = 4 for each. (E) Bar chart showing cAMP levels in wt-CFTR-CHO cells stimulated by RP193 (100 μM), forskolin (1 μM) + RP193 (100 μM), or forskolin (1 μM). Data are expressed as the mean ± SEM of 8–16 measurements normalized to cAMP levels in the presence of 10 μM forskolin (100%) and are representative of two independent experiments. Ns: no significant differences with respect to experimental condition in which DMSO 0.1% was added, ***p < 0.001 using the t-test.
Activation of a Linear CFTR Cl− Current by RP193
We performed whole-cell patch-clamp experiments to record CFTR chloride currents after stimulation of wt-CFTR-CHO cells by RP193. Characteristic whole-cell currents and the corresponding current/voltage plots in the absence and presence of RP193 are presented Figure 2. In the absence of any activator, no or very small current was recorded (Figures 2A,B,D, noted basal). Addition of low concentration of forskolin (1 μM) into the bath stimulated a time- voltage-independent and linear conductance with small amplitude, suggesting the presence of a functional CFTR (Figures 2A,B,D, noted fsk). Addition of 10 μM RP193 (Figures 2A,B,D, RP193) in the presence of 1 μM fsk induced a large and significant increase of the CFTR current amplitude compared to basal current and to forskolin-induced current. When measured at +40 mV the amplitude values are: basal, 1.8 ± 0.4 pA/pF; fsk, 18.4 ± 6.2 pA/pF; fsk + RP193, 113.8 ± 15.1 pA/pF. This current was fully and rapidly inhibited by two selective CFTR inhibitors; CFTR-inh172 (10 μM, current amplitude at +40 mV: 8.2 ± 5.2 pA/pF) and GPinh5a (100 pM, current amplitude at +40 mV: 11.6 ± 7.7 pA/pF; Figures 2A,B,D). As expected, we observed no effect of either forskolin (1 μM) or RP193 (10 μM) in CHO-K1 (Figure 2C). The time course of the activation and inhibition of CFTR currents from two different whole-cell recordings is shown in Figure 3. Addition of RP193, in presence of forskolin, induced a significant increase of the current within 5–7 min, which was fully and rapidly inhibited by the addition of either CFTR-inh172 (Figure 3A) or GPinh5a (Figure 3B).
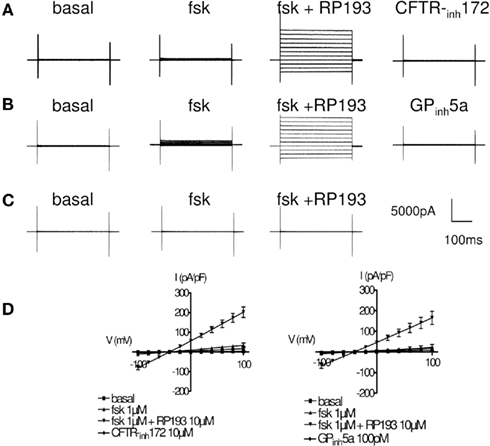
Figure 2. Activation of CFTR current in CHO cells. Representative current traces for CFTR are shown in the basal state and after successive addition of forskolin (1 μM), RP193 (10 μM), and specific CFTR inhibitor CFTR-inh172 (10 μM) (A) or GPinh5a (100 pM) (B). (C) Representative current traces recorded after the successive addition of forskolin (1 μM) and RP193 (10 μM) in CHO-K1. Step protocol consisted of 300 ms voltage from −100 to +100 mV from a holding potential of −40 mV. (D) I/V curves (mean ± SE) for CFTR current in the four experimental conditions for the three different protocols used.
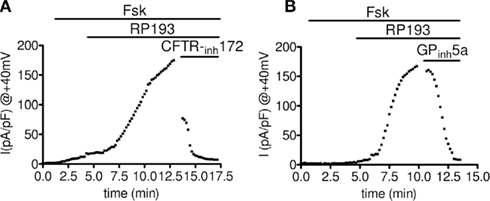
Figure 3. Representative time course of whole-cell activation and inhibition of CFTR current at +40 mV. (A) Sequential addition of Fsk (1 μM), RP193 (10 μM), and CFTR-inh172 (10 μM) and (B) sequential addition of Fsk (1 μM), RP193 (10 μM), and GPinh5a (100 pM). Horizontal bars represent time of perfusion of different drugs.
Activation of CFTR Chloride Channels by RP193
We performed single-channel patch-clamp experiments to record CFTR activity in the presence of RP193. In basal condition no spontaneous channel activity was detected in cell-attached configuration (Figure 4A, left part of the exemplar tracing). In the presence in the bath of a low concentration of forskolin (1 μM) we recorded little CFTR activity (Figure 4A, middle part of the exemplar tracing). Following addition to the bath of RP193 (10 μM) progressive and sustained CFTR activity was recorded (Figures 4A,B). After excision of the membrane in the presence of 1 μM forskolin and 10 μM RP193 in the bath, we observed the typical run down (Becq, 1996) of CFTR channels (Figure 4B) leading to complete closure of CFTR channels. The average unitary conductance of 8.1 ± 0.0 pS calculated in these experiments (n = 4) is consistent with previous data obtained using cAMP agonists (Becq et al., 1999; Kammouni et al., 1999; Figures 4C,D).
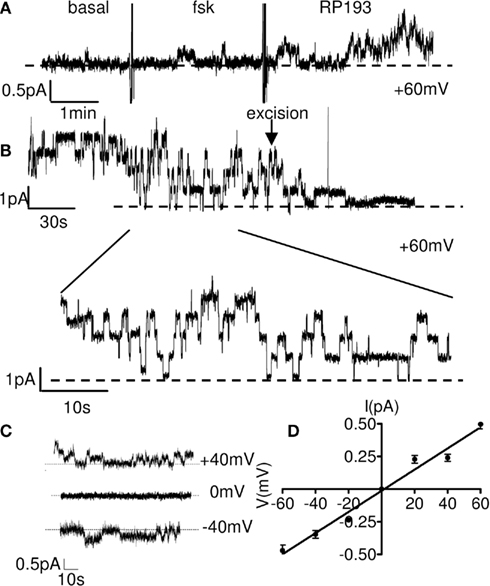
Figure 4. Single CFTR chloride channel activation by RP193 in wt-CFTR-CHO cells. (A) Continuous cell-attached recording obtained on a wt-CFTR-CHO showing the activation of CFTR chloride channel by 10 μM of RP193 in the bath in presence of forskolin (1 μM) at the potential of +60 mV. (B) Representative recording of rundown of CFTR after channel excision in presence of RP193 (10 μM). Insert depicts 1 min of channel opening recording in cell-attached configuration. (C) Representative recordings at various patch potentials as indicated in the presence of RP193 at (10 μM) in the bath and the associated I/V curve (D).
RP 193 Activates Two CFTR Mutants G551D- and F508del-CFTR Causing Severe CF Disease
To determine whether RP193 could activate CFTR mutants, we performed iodide efflux experiments with cells expressing two of the most frequent CF mutations, the class 3 CF mutation G551D-CFTR, studied in CHO cells stably expressing G551D-CFTR, and the class 2 CF mutation F508del-CFTR, studied in CuFi-1 cells derived from a human CF bronchial epithelium (F508del/F508del genotype). As CuFi-1 cells are CF bronchial epithelial cells, we also compared the effect of RP193 on NuLi-1 cells derived from a non-CF human bronchial epithelium. RP193 (100 μM) stimulated CFTR in NuLi-1 cells in presence of forskolin (10 μM; Figures 5A,B). This effect was abolished by the selective CFTR inhibitor CFTR-inh172 (10 μM, Figure 5B). RP193 did not stimulate iodide efflux in CuFi-1 cells with the F508del-CFTR mutation cultivated at 37°C (Figure 5C). Then, CuFi-1 cells were incubated for 24 h at low temperature (27°C) to rescue F508del-CFTR (Denning et al., 1992). In these conditions of culture and in presence of forskolin 10 μM, RP193 induced an iodide efflux significantly higher than with forskolin alone (Figures 5D,E). In further experiments, CuFi-1 cells were incubated 24 h at 37°C with the pharmacological corrector miglustat in order to rescue F508del-CFTR (Norez et al., 2006). Following this treatment of CuFi-1 cells, RP193 significantly induced a higher iodide efflux than in basal conditions or with forskolin alone (Figure 5F), confirming the functional rescue of F508del-CFTR proteins. This efflux was fully inhibited in presence of the CFTRinh-172 (10 μM, data not shown), and no effect of RP193 was observed in CuFi-1 cells non-treated with miglustat (Figure 5F). Thus RP193 is able to stimulate F508del-CFTR activity after maneuvers leading to the correction of its abnormal trafficking.
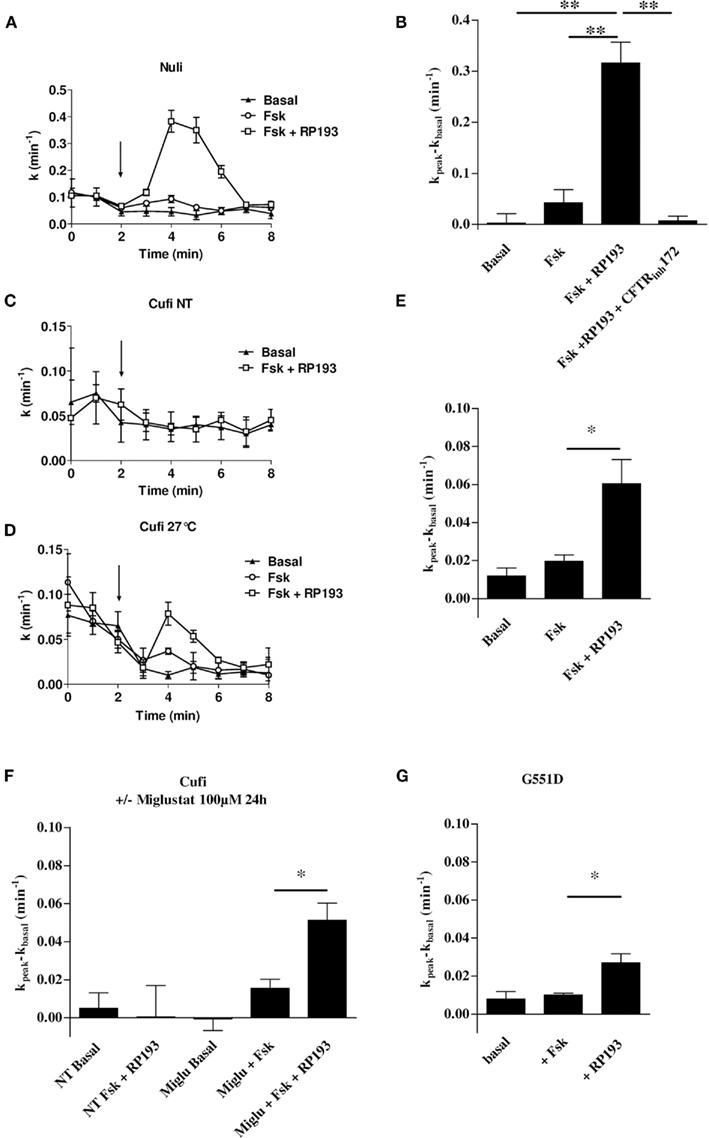
Figure 5. Examples of iodide efflux curves as function of time on NuLi-1 cells (A), CuFi-1 (C) cultured at 37°C and CuFi-1 treated by low temperature (24 h, 27°C) (D) in basal conditions, stimulated by forskolin (10 μM), or by forskolin (10 μM) and RP193 (100 μM). Bar chart showing iodide efflux experiments on NuLi-1 (B), CuFi-1 treated by low temperature (24 h, 27°C) (E), CuFi-1 treated with miglustat (24 h, 100 μM) (F) and G551D-CHO cells (G). Cells were successively treated with forskolin (10 μM) and RP193 (100 μM) and NuLi-1 cells were equally treated by CFTR-inh172 (10 μM), n = 4 for each. *p < 0.05 and **p < 0.01 using the t-test.
We also performed iodide efflux experiments on G551D-CFTR-CHO cells. No significant stimulation was observed compared to basal state with 10 μM forskolin alone (Figure 5G). However, RP193 10 μM stimulated an efflux in G551D-CFTR-CHO cells in presence of forskolin 10 μM (n = 4, Figure 5G) while RP193 alone did not have any effect (not shown). Taken together these experiments revealed that RP193 is able not only to activate wt-CFTR but also rescued F508del-CFTR and G551D-CFTR.
Activation of CFTR Chloride Channels by RP193 in Primary Cultures of Human Bronchial Epithelial Cells
Ten primary cultures of human bronchial epithelial cells (HBEC) were studied in Ussing chambers. The mean PD, Isc, and Rt of these cultures were respectively: −4.7 ± 0.6 mV, 53.4 ± 7.2 μA/cm2, and 98.8 ± 14.8 Ω cm2. Addition of amiloride (10 μM) to the luminal bath significantly reduced Isc in the preparations (−29.5 ± 4.5 μA/cm2). In the presence of amiloride, the addition of forskolin (1 μM) induced a significant increase in Isc in HBEC cultures (0.4 ± 0.1 μA/cm2). Successive addition of RP193 (100 and 200 μM) increased significantly Isc from respectively 0.5 ± 0.2 and 1.1 ± 0.3 μA/cm2 (Figure 6) indicating that RP193 is able to increased Isc in primary cultures of HBEC.
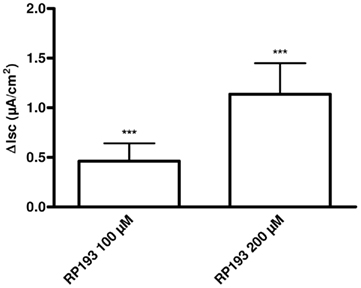
Figure 6. Bar chart representing Isc variations in primary culture of human bronchial epithelial cells in response to RP193 (100 and 200 μM) in presence of amiloride 10 μM and forskolin 1 μM, n = 10 for each. ***p < 0.001 using the t-test.
Discussion
Based on previously published data from our laboratory, one of the main objective of this ligand-based study was to evaluate the effects of the modification of determinants on the phenyl ring of the pyrrolo[2,3-b]pyrazines family on their potency to activate CFTR. Firstly, we translocated the 4-OH substituent of the phenyl ring of RP107, in position 2 (compound RP173) and in the position 3 (compound RP146). Secondly, based on results obtained with a 4-Cl substituent (RP108; Noel et al., 2006), we tested another halogen substituent at the same position on the phenyl ring: 4-F (RP185) and in position 2-F (RP193). Ability of these newly synthesized compounds to activate wt-CFTR, was assessed by iodide efflux, which allowed us to identify RP193 as a new cAMP-independent hit of the pyrrolo[2,3-b]pyrazines family (EC50 = 270 nM). Most importantly, RP193 appeared to be significantly less toxic than compounds developed previously as revealed by cytotoxicity assays. To complete our study of RP193 compound, we performed patch-clamp experiment in the whole-cell and the cell-attached configurations, which confirmed its ability to activate wt-CFTR. As lung pathology represents the most drastic syndrome in CF (Boucher, 1999), another important objective of this work was to evaluate the effect of RP193 on CFTR activity in cultures of human bronchial non-CF and CF epithelial cells. We then performed iodide efflux experiment on cells, which expressed two of the most common mutation encountered in CF patients. Our data demonstrated that RP193 was able to stimulate rescued F508del-CFTR and G551D-CFTR.
A recent study carried out in our laboratory demonstrated the ability of RP compounds previously described as CDK/GSK-3 inhibitors, to activate CFTR with a strong affinity (Noel et al., 2006). It was shown that RP107 was able to stimulate wt-CFTR with a submicromolar affinity by a cAMP-independent mechanism. The importance of the length of the alkyl chain was observed with the best activation of CFTR obtained with compounds having a butyl chain. This property was conserved in the newly synthesized compounds, and only the nature and/or the position of the substituent on the phenyl ring were changed. Firstly, translocation of the OH substituent at position 2 (RP173) or at position 3 (RP146) reduced the potency of the compound suggesting that the position of this substituent on the phenyl ring is an important determinant for the ability to activate CFTR.
Experiments performed with RP108 demonstrated efficiency to activate wt-CFTR of compound with a halogen substituent (Noel et al., 2006). Other studies demonstrated efficiency of compounds, like the benzimidazolone NS004 or 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid diamides, to activate CFTR with different halogen substituent in their structure (Gribkoff et al., 1994; Devor et al., 2000; Hirth et al., 2005; Murthy et al., 2005) suggesting that these derivatives could play an important role in the activation of CFTR. Substitution of the chlorine atom by another halogen, a fluorine atom, at position 4 (RP185) or position 2 (RP193) of the phenyl ring was then performed. It was shown that fluoride, the reduced form of fluorine, stimulated phosphorylated CFTR most probably by direct interaction with NBD2 domain and by slowing CFTR channel closure (Berger et al., 1998). Caci et al. (2003) also demonstrated activation of CFTR by a compound containing F residue. In their study, these authors demonstrated that UCCF-853 is an activator of CFTR-dependent Cl− secretion in intact human airway epithelium. Iodide efflux experiments revealed that our compounds potentiated CFTR activity with a higher potency for RP193. Marivingt-Mounir et al. (2004) already tried to introduce a fluorine atom in another class of CFTR activators, the benzo[c]quinoliziniums. However, no activation of CFTR was found with these compounds. Nevertheless, contrary to MPB104, the most active compound in their study, MPB with a fluorine substituent did not have a butyl chain, which appeared to be a key determinant for CFTR activation by RP agents.
Interestingly, RP107 and RP193 presented similar EC50 values for CFTR potentiation in iodide efflux (EC50 are respectively 150 and 270 nM). Nevertheless, cytotoxicity assays revealed that compounds with chlorine atom or hydroxyl radical are more toxic than compounds with fluorine atom whatever the position of the substituent is (2 or 4 on the phenyl ring). To our knowledge, there are no comparative studies of cell toxicity of compounds with or without a fluorine atom. The addition of the fluorine atom could then reduce cell toxicity of compounds that are still able to activate phosphorylated CFTR. These observations could provide some clue to generate new potent CFTR activators (importance of the OH radical) with less cytotoxicity (addition of a fluorine atom).
We demonstrated that RP193 is able to potentiate wt-CFTR in CHO cell line. It appeared then important to test this compound on CFTR mutant. First, we performed experiments on human bronchial epithelial cell expressing endogenous WT-CFTR or F508del-CFTR, the most common mutation in CF patients. RP193 potentiated WT-CFTR activity, as observed in other cell type, but it was also able to potentiate F508del-CFTR corrected either by low temperature incubation or by a 24-h-miglustat-treatment. Secondly, we performed experiments on G551D-CFTR, a class 3 mutation retrieved in patients with severe disease and which present no or reduced activation of the CFTR chloride function. Our studies revealed that in presence of a low forskolin concentration, RP193 potentiates the activity of G551D-CFTR. It should be noted that forskolin alone did not have any effect as previously demonstrated (Becq et al., 1994; Illek et al., 1999). Thus, the ability of RP193 compound to activate two of the most common CFTR mutants is very interesting in the perspective of developing CF pharmacotherapy. A recent study demonstrated that the use of pharmacological activator of CFTR could, in particular, improve lung function in patients with G551D-CFTR showing that pharmacological treatment in CF is a realistic objective (Accurso et al., 2010). Our approach developed here is clearly in this line.
In conclusion, our study emphasizes the importance of the presence of an OH radical in compounds in order to activate CFTR. We equally revealed that presence of a fluorine atom could reduce cytotoxicity without altering efficiency of the compound to stimulate phosphorylated CFTR. Finally, we identified RP193 as a new compound able to stimulate, through a cAMP-independent pathway, wt-CFTR, rescued F508del-CFTR, and G551D-CFTR activity in HBEC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank S. Mirval for technical assistance. This work was funded by the association Vaincre la mucoviscidose.
References
Accurso, F. J., Rowe, S. M., Clancy, J. P., Boyle, M. P., Dunitz, J. M., Durie, P. R., Sagel, S. D., Hornick, D. B., Konstan, M. W., Donaldson, S. H., Moss, R. B., Pilewski, J. M., Rubenstein, R. C., Uluer, A. Z., Aitken, M. L., Freedman, S. D., Rose, L. M., Mayer-Hamblett, N., Dong, Q., Zha, J., Stone, A. J., Olson, E. R., Ordonez, C. L., Campbell, P. W., Ashlock, M. A., and Ramsey, B. W. (2010). Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 363, 1991–2003.
Antigny, F., Norez, C., Dannhoffer, L., Bertrand, J., Raveau, D., Corbi, P., Jayle, C., Becq, F., and Vandebrouck, C. (2011). TRPC6 links Ca2+ mishandling to CFTR channel dysfunction in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 44, 83–90.
Becq, F. (1996). Ionic channel rundown in excised membrane patches. Biochim. Biophys. Acta 1286, 53–63.
Becq, F. (2010). Cystic fibrosis transmembrane conductance regulator modulators for personalized drug treatment of cystic fibrosis: progress to date. Drugs 70, 241–259.
Becq, F., Jensen, T. J., Chang, X. B., Savoia, A., Rommens, J. M., Tsui, L. C., Buchwald, M., Riordan, J. R., and Hanrahan, J. W. (1994). Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc. Natl. Acad. Sci. U.S.A. 91, 9160–9164.
Becq, F., Mettey, Y., Gray, M. A., Galietta, L. J., Dormer, R. L., Merten, M., Metaye, T., Chappe, V., Marvingt-Mounir, C., Zegarra-Moran, O., Tarran, R., Bulteau, L., Derand, R., Pereira, M. M., McPherson, M. A., Rogier, C., Joffre, M., Argent, B. E., Sarrouilhe, D., Kammouni, W., Figarella, C., Verrier, B., Gola, M., and Vierfond, J. M. (1999). Development of substituted benzo[c]quinolizinium compounds as novel activators of the cystic fibrosis chloride channel. J. Biol. Chem. 274, 27415–27425.
Berger, H. A., Travis, S. M., and Welsh, M. J. (1998). Fluoride stimulates cystic fibrosis transmembrane conductance regulator Cl- channel activity. Am. J. Physiol. 274, L305–L312.
Blouquit, S., Morel, H., Hinnrasky, J., Naline, E., Puchelle, E., and Chinet, T. (2002). Characterization of ion and fluid transport in human bronchioles. Am. J. Respir. Cell Mol. Biol. 27, 503–510.
Blouquit, S., Regnier, A., Dannhoffer, L., Fermanian, C., Naline, E., Boucher, R., and Chinet, T. (2006). Ion and fluid transport properties of small airways in cystic fibrosis. Am. J. Respir. Crit. Care Med. 174, 299–305.
Boucher, R. C. (1999). Molecular insights into the physiology of the “thin film” of airway surface liquid. J. Physiol. 516(Pt 3), 631–638.
Caci, E., Folli, C., Zegarra-Moran, O., Ma, T., Springsteel, M. F., Sammelson, R. E., Nantz, M. H., Kurth, M. J., Verkman, A. S., and Galietta, L. J. (2003). CFTR activation in human bronchial epithelial cells by novel benzoflavone and benzimidazolone compounds. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L180–L188.
Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Crystal, R. G., Pavirani, A., Lecocq, J. P., and Lazdunski, M. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528.
Dannhoffer, L., Blouquit-Laye, S., Regnier, A., and Chinet, T. (2009). Functional properties of mixed cystic fibrosis and normal bronchial epithelial cell cultures. Am. J. Respir. Cell Mol. Biol. 40, 717–723.
Denning, G. M., Anderson, M. P., Amara, J. F., Marshall, J., Smith, A. E., and Welsh, M. J. (1992). Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764.
Devor, D. C., Bridges, R. J., and Pilewski, J. M. (2000). Pharmacological modulation of ion transport across wild-type and deltaF508 CFTR-expressing human bronchial epithelia. Am. J. Physiol. Cell Physiol. 279, C461–C479.
Dormer, R. L., Harris, C. M., Clark, Z., Pereira, M. M., Doull, I. J., Norez, C., Becq, F., and McPherson, M. A. (2005). Sildenafil (Viagra) corrects deltaF508-CFTR location in nasal epithelial cells from patients with cystic fibrosis. Thorax 60, 55–59.
Gribkoff, V. K., Champigny, G., Barbry, P., Dworetzky, S. I., Meanwell, N. A., and Lazdunski, M. (1994). The substituted benzimidazolone NS004 is an opener of the cystic fibrosis chloride channel. J. Biol. Chem. 269, 10983–10986.
Hirth, B. H., Qiao, S., Cuff, L. M., Cochran, B. M., Pregel, M. J., Gregory, J. S., Sneddon, S. F., and Kane, J. L. Jr. (2005). Discovery of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid diamides that increase CFTR mediated chloride transport. Bioorg. Med. Chem. Lett. 15, 2087–2091.
Illek, B., Zhang, L., Lewis, N. C., Moss, R. B., Dong, J. Y., and Fischer, H. (1999). Defective function of the cystic fibrosis-causing missense mutation G551D is recovered by genistein. Am. J. Physiol. 277, C833–C839.
Kammouni, W., Moreau, B., Becq, F., Saleh, A., Pavirani, A., Figarella, C., and Merten, M. D. (1999). A cystic fibrosis tracheal gland cell line, CF-KM4. Correction by adenovirus-mediated CFTR gene transfer. Am. J. Respir. Cell Mol. Biol. 20, 684–691.
Kreda, S. M., Mall, M., Mengos, A., Rochelle, L., Yankaskas, J., Riordan, J. R., and Boucher, R. C. (2005). Characterization of wild-type and {delta}f508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell 16, 2154–2167.
Kreindler, J. L. (2010). Cystic fibrosis: exploiting its genetic basis in the hunt for new therapies. Pharmacol. Ther. 125, 219–229.
Li, C., Ramjeesingh, M., Reyes, E., Jensen, T., Chang, X., Rommens, J. M., and Bear, C. E. (1993). The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat. Genet. 3, 311–316.
Marivingt-Mounir, C., Norez, C., Derand, R., Bulteau-Pignoux, L., Nguyen-Huy, D., Viossat, B., Morgant, G., Becq, F., Vierfond, J. M., and Mettey, Y. (2004). Synthesis, SAR, crystal structure, and biological evaluation of benzoquinoliziniums as activators of wild-type and mutant cystic fibrosis transmembrane conductance regulator channels. J. Med. Chem. 47, 962–972.
Mettey, Y., Gompel, M., Thomas, V., Garnier, M., Leost, M., Ceballos-Picot, I., Noble, M., Endicott, J., Vierfond, J. M., and Meijer, L. (2003). Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J. Med. Chem. 46, 222–236.
Murthy, M., Pedemonte, N., MacVinish, L., Galietta, L., and Cuthbert, A. (2005). 4-Chlorobenzo[F]isoquinoline (CBIQ), a novel activator of CFTR and deltaF508 CFTR. Eur. J. Pharmacol. 516, 118–124.
Noel, S., Faveau, C., Norez, C., Rogier, C., Mettey, Y., and Becq, F. (2006). Discovery of pyrrolo[2,3-b]pyrazines derivatives as submicromolar affinity activators of wild type, G551D, and F508del cystic fibrosis transmembrane conductance regulator chloride channels. J. Pharmacol. Exp. Ther. 319, 349–359.
Norez, C., Noel, S., Wilke, M., Bijvelds, M., Jorna, H., Melin, P., DeJonge, H., and Becq, F. (2006). Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 580, 2081–2086.
Regnier, A., Dannhoffer, L., Blouquit-Laye, S., Bakari, M., Naline, E., and Chinet, T. (2008). Expression of cystic fibrosis transmembrane conductance regulator in the human distal lung. Hum. Pathol. 39, 368–376.
Routaboul, C., Norez, C., Melin, P., Molina, M. C., Boucherle, B., Bossard, F., Noel, S., Robert, R., Gauthier, C., Becq, F., and Decout, J. L. (2007). Discovery of alpha-aminoazaheterocycle-methylglyoxal adducts as a new class of high-affinity inhibitors of cystic fibrosis transmembrane conductance regulator chloride channels. J. Pharmacol. Exp. Ther. 322, 1023–1035.
Tabcharani, J. A., Chang, X. B., Riordan, J. R., and Hanrahan, J. W. (1991). Phosphorylation-regulated Cl- channel in CHO cells stably expressing the cystic fibrosis gene. Nature 352, 628–631.
Tarran, R. (2004). Regulation of airway surface liquid volume and mucus transport by active ion transport. Proc. Am. Thorac. Soc. 1, 42–46.
Welsh, M. J., and Smith, A. E. (1993). Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73, 1251–1254.
Keywords: cystic fibrosis, CFTR, activator and inhibitor, ion transport, epithelial cells
Citation: Dannhoffer L, Billet A, Jollivet M, Melin-Heschel P, Faveau C and Becq F (2011) Stimulation of wild-type, F508del- and G551D-CFTR chloride channels by non-toxic modified pyrrolo[2,3-b]pyrazine derivatives. Front. Pharmacol. 2:48. doi: 10.3389/fphar.2011.00048
Received: 08 June 2011; Accepted: 03 August 2011;
Published online: 23 August 2011.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Giovanni Zifaelli, Institute of Biophysics, National Research Council, ItalyMarc Chanson, University of Geneva, Switzerland
Copyright: © 2011 Dannhoffer, Billet, Jollivet, Melin-Heschel, Faveau and Becq. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Luc Dannhoffer, Institut de Physiologie et Biologie Cellulaires, UMR 6187, Université de Poitiers, CNRS, Pôle Biologie Santé Bât B36, BP 633, 1 rue Georges Bonnet, 86022 Poitiers, France. e-mail: luc.dannhoffer@ext.univ-poitiers.fr