 Dina Simkin1,2 Saïd Bendahhou1*
Dina Simkin1,2 Saïd Bendahhou1*- 1 UMR 6097, CNRS, TIANP, University of Nice Sophia-Antipolis, Nice, France
- 2 Interdepartmental Neuroscience, Rosalind Franklin University of Medicine and Science, North Chicago, IL, USA
Five inherited human disorders affecting skeletal muscle contraction have been traced to mutations in the gene encoding the voltage-gated sodium channel Nav1.4. The main symptoms of these disorders are myotonia or periodic paralysis caused by changes in skeletal muscle fiber excitability. Symptoms of these disorders vary from mild or latent disease to incapacitating or even death in severe cases. As new human sodium channel mutations corresponding to disease states become discovered, the importance of understanding the role of the sodium channel in skeletal muscle function and disease state grows.
Introduction
Voltage-gated sodium channels are essential in the generation and propagation of action potentials (APs) in excitable tissues such as muscle, heart, and nerve. Proper activity of these channels is crucial to the initiation of APs which ultimately lead to muscle contraction or neuronal firing. The necessity of Na+ channels is better emphasized by the existence of human inherited skeletal muscle disorders caused by mutations in the Na+ channel Nav1.4 which is specifically expressed in skeletal muscle. Mutations in the SCN4A gene encoding the human skeletal muscle Nav1.4 channel cause five different skeletal muscle disorders: potassium-aggravated myotonia (PAM), paramyotonia congenita (PMC), hyperkalemic periodic paralysis (HyperPP), hypokalemic periodic paralysis (HypoPP), and a form of congenital myasthenic syndrome (CMS). This review focuses on the role of Nav1.4 channel in skeletal muscle and the complex clinical symptoms of these disorders. Also the most recent findings of new Nav1.4 mutations causing a lethal form of myotonia will be discussed as well as treatment options for such disorders. An overview of skeletal muscle physiology is provided in order to illustrate the significance of ion channels within the skeletal muscle and their critical roles in muscle function.
Skeletal Muscle Physiology
Skeletal muscles have complex structures working in concert to provide the appropriate response to nerve impulse and metabolic processes. Specialized compartments within skeletal muscle fibers such as neuromuscular junctions, sarcolemma membrane, traverse tubules, and the sarcoplasmic reticulum (SR) provide the mechanical architecture required for the excitation–contraction coupling mechanism to take place.
At the neuromuscular junction, motoneuron activity is transferred to skeletal muscle generating an acetylcholine (ACh) dependent endplate potential. ACh is released from the nerve terminal and binds to nicotinic acetylcholine receptors (AChR). A large enough endplate potential can induce a sarcolemmal AP that propagates from the endplate to the tendon and through the transverse tubular (T-tubules) system which is mediated by the opening of the voltage-gated Nav1.4 Na+ channels. Na+ channels quickly inactivate and the depolarized potential enables the opening of delayed rectifier K+ channels which mediate outward K+ current during the repolarization stage of the muscle AP (Jurkat-Rott and Lehmann-Horn, 2005). High chloride channel (Cl−) conductance then takes over to enforce the final repolarization or to reduce the afterdepolarization of the skeletal muscle fiber. This afterdepolarization is skeletal muscle AP specific and consists of an early and late phase mediated by different ionic currents (Jurkat-Rott et al., 2006). The early phase is caused by the spread of the depolarization spike in the T-tubules while the late phase is considered to be caused by accumulation of K+ ions in the T-tubules which increases with frequency and duration of repetitive APs (Almers, 1980). Inward chloride conductance in the T-tubular system alleviates some of the depolarization caused by the extracellular K+ accumulation by producing a more negative membrane potential than K+ equilibrium, which stimulates inward potassium flux (Jurkat-Rott et al., 2006).
The contraction of the muscle occurs as a result of Ca2+ release from the SR which binds to troponin (a calcium binding protein which is part of the thin filaments necessary to produce muscle contraction) enabling filament sliding and contraction. The process, which allows Ca2+ release, is initiated by voltage changes of the AP. These changes will target in part the voltage sensor of the voltage-gated Cav1.1 Ca2+ channel (Dihydropyridine receptor or DHPR) leading to channel conformation rearrangements. The DHPR is believed to physically interact with a calcium release channel of the SR the ryanodine receptor (RYR) which releases calcium stores from the SR allowing calcium to bind to troponin (Rios et al., 1991). When the AP is over, the RYR close and Ca2+ is transported back to the SR via Ca2+ATPases (SERCA).
Skeletal Muscle Na+ Channel Structure and Gating
Voltage-gated sodium channels are large integral membrane proteins expressed densely at the neuromuscular junctions where they selectively conduct sodium ions into the muscle fibers in physiological conditions. The Nav1.4 channel is composed of a 260-kDa α-subunit which consists of four homologous domains (I–IV), and each domain has six transmembrane segments (S1–S6; Figure 1; Noda et al., 1984; George et al., 1992a,b). The Nav1.4 channels complex structure formed at the membrane incorporates several important gating domains facilitating the channel’ three different gating states: resting (closed), activated (open), and inactivated (closed). When a voltage change occurs at cell surface, voltage sensing domains at the S4 segments sense this change and shift their conformation within the membrane relaying this change to the channels internal activation gate and opening it in a very fast manner. Within milliseconds of this fast activation, a ball and chain gate located at the intracellular loop between domains III and IV blocks the intracellular pore of the channel allowing the channel to quickly inactivate (Armstrong and Bezanilla, 1977; West et al., 1992). This fast inactivation process is voltage dependent and occurs at a greater extent and for a longer duration when membrane potentials are more depolarized. Before the channel can be opened again, the internal gate must close and deactivate and the ball and chain gate must be released, this process is called recovery from fast inactivation. Recovery from fast inactivation requires a hyperpolarized membrane potential to last several milliseconds. In addition to fast inactivation, slow inactivation occurs when the channel has been activated repeatedly during exercise, this prevents the channel from being available for further activation for hundreds of milliseconds to seconds. The slow (on the order of seconds to minutes) inactivation process enables the muscle to recover more quickly from exercise.
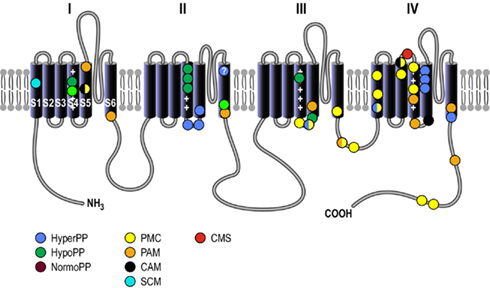
Figure 1. Basic structure of the voltage-gated sodium channel. Structural arrangement of the Nav1.4 channel α-subunit which is organized into four homologous domains (I to IV) and possesses six transmembrane segments (S1–S6), a pore-forming loop located between S5 and S6 segments, and cytosolic NH3 and COOH termini. The schematic diagram of the Nav1.4 channel shows the location of the mutations associated with hyperkalemic periodic paralysis (HyperPP), hypokalemic periodic paralysis (HypoPP), normokalemic periodic paralysis (NormoPP), paramyotonia congenita (PMC), potassium-aggravated myotonia (PAM), cold-aggravated myotonia (CAM), and congenital myasthenic syndrome (CMS).
Skeletal Muscle Pathophysiology
Over 40 Nav1.4 channel mutations leading to disease states have been found throughout each domain and segment of this channel (Figure 1 and Table 1). These Nav1.4 mutations produce changes that interfere with channel kinetics or function, producing changes in the micro or macroscopic electrophysiological properties of the skeletal muscle fibers thus underlying muscle hyperexcitability or inexcitability. Skeletal muscle Na+ channel disorders are non-dystrophic and consist of myotonia which presents as an increased muscular activity causing muscle stiffness, and periodic paralysis which presents as a decrease in muscle activity causing transient weakness or paralysis of the muscle.
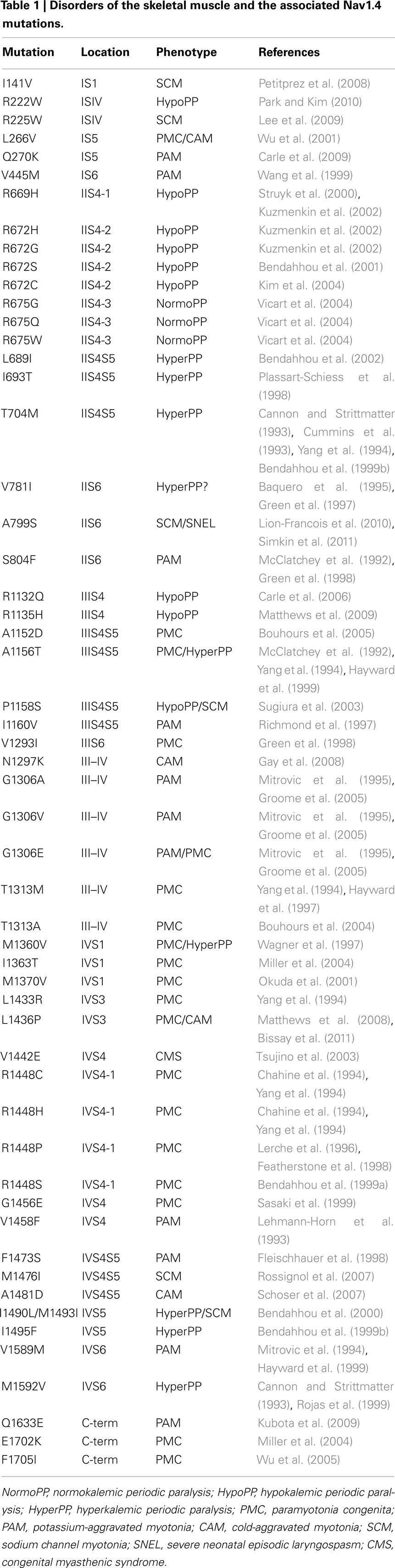
Table 1. Disorders of the skeletal muscle and the associated Nav1.4 mutations.
Myotonia
A common feature of myotonia is delayed relaxation of the muscle after voluntary contraction or mechanical stimulation, electrophysiologically characterized by highly organized repetitive electrical activity of the muscle fibers. Non-dystrophic myotonias can be caused by mutations in the Nav1.4 sodium channel which increases its function or the ClC-1 chloride channel decreasing channel function (Trip et al., 2008). Mutations causing up-regulation of the Nav1.4 sodium channel can cause PMC or PAM depending on the type of mutation and its functional effect on this channel. Both PMC and PAM cause repetitive APs and increase activity of the muscle leading to myotonia or muscle rigidity.
Potassium-aggravated Myotonia
Potassium-aggravated myotonia includes atypical myotonia congenita, moderate myotonia, myotonia fluctuans, myotonia permanens, acetazolamide-responsive myotonia, and painful myotonia which have overlapping clinical features. The prevalence of PAM is estimated at ∼1:400,000 (Lehmann-Horn et al., 2004). Myotonia fluctuans and moderate PAM patients exhibit stiffness during the first contractions after rest and thus complain that they are particularly stiff after rest. Upon further contractions, their muscles begin to warm up improving the initial stiffness, this occurrence is termed the warm-up phenomenon. However, other PAM patients become stiff within 10–30 min of strenuous work which may last for several hours (Lehmann-Horn et al., 2004).
Potassium-aggravated myotonia is exacerbated by potassium ingestion (but not cold temperatures) because increased K+ ingestion causes cellular depolarization (George, 2005). Also unlike other myotonias PAM does not present with any major weakness.
Eight Nav1.4 mutations causing PAM have been found in humans: V445M, S804F, I1160V, G1306A/V/E, V1458F, F1473S, and V1589M (McClatchey et al., 1992; Lehmann-Horn et al., 1993; Lerche et al., 1993; Mitrovic et al., 1994, 1995; Ptácek et al., 1994b; Richmond et al., 1997; Fleischhauer et al., 1998; Green et al., 1998; Hayward et al., 1999; Takahashi and Cannon, 1999; Wang et al., 1999; Groome et al., 2005). The clinical phenotypes of I1160V and V1589M mutations present as atypical myotonia congenita, G1306A presents myotonia fluctuans, and G1306E presents myotonia permanens. Most of these PAM mutations are situated in the interface between the cytosol and the membrane which is the location of the inactivation gate. An example of this is with the G1306 residue that is thought to act as the hinge for the inactivation gate. The more different the substituting mutation is from glycine the more severe are the clinical symptoms ranging from myotonia fluctuans (a benign form of myotonia) to myotonia permanens a severe myotonia (Lerche et al., 1993; Mitrovic et al., 1994).
Nav1.4 channel kinetics are altered by these mutations in such a way that the channel open probability is increased thus channel activity is up-regulated. With some of these PAM mutations, the rate of fast inactivation is slowed which allows the channels to stay open for a prolonged amount of time. However, the rate of recovery from inactivation is not found to be increased but deactivation is found to be slow for most of these PAM mutations. Most of these mutations have been reported to increase the size of the persistent Na+ current two to fourfold. The increase in this inward Na+ current generates after-depolarizations across the T-tubules and decreases the threshold required for AP generation consequently triggering repetitive AP and muscle contraction (Adrian and Marshall, 1976).
Paramyotonia Congenita
Paramyotonia congenita is different from PAM in that muscle stiffness is usually followed by flaccid weakness or paralysis (Lehmann-Horn et al., 2004). This form of myotonia is exacerbated by cold temperatures and muscle stiffness is increased with continued activity which is considered a paradoxical effect (Jackson et al., 1994; Vicart et al., 2005). Cold induced myotonia or weakness can last for several hours even if muscles are immediately re-warmed after short exposure to cold. The effect of temperature on cell excitability in PMC has been suggested to be a result of normal slowing of channel kinetics with cooling, and was not attributed to a consequence of altered Nav1.4 temperature sensitivity (Lerche et al., 1996; Fleischhauer et al., 1998; Dice et al., 2004; Webb and Cannon, 2008). In some cases of PMC, patients are sensitive to serum potassium levels, however, unlike with PAM these patients exhibit weakness provoked by a hypokalemic challenge (Ptácek et al., 1993b; Lehmann-Horn et al., 2004). The main characteristic of PMC is both cold exacerbated myotonia and muscle weakness. This suggests that the PMC mutations under different physiological conditions lead to an increased Na+ channel activity causing myotonia or inactivation of the channel with paralysis. The prevalence of PMC is ∼1:200,000 (Lehmann-Horn et al., 2004).
Sixteen mutations of the Nav1.4 channel have been found to cause PMC in humans: L266V, A1152D, A1156T, V1293I, G1306E, T1313M/A, M1360V, M1370V, L1433R, R1448C/H/P/S, G1456E, and F1705I (McClatchey et al., 1992; Ptácek et al., 1992; Lerche et al., 1993, 1996; Chahine et al., 1994; Yang et al., 1994; Mitrovic et al., 1995; Wagner et al., 1997; Featherstone et al., 1998; Green et al., 1998; Bendahhou et al., 1999a; Hayward et al., 1999; Sasaki et al., 1999; Okuda et al., 2001; Wu et al., 2001, 2005; Bouhours et al., 2004, 2005; Groome et al., 2005; Gay et al., 2008). However, Nav1.4 mutations A1156T and M1360V have been classified as both PMC and HyperPP. There are many similarities in electrophysiological properties and clinical observations between PMC and HyperPP. This led to a hypothesis that they are allelic disorders, supported by genetic linkage of this disease locus to the Nav1.4 channel gene on chromosome 17 (Fontaine et al., 1990; Ptácek et al., 1991). Most of the PMC mutations are located either on the inactivation particle (IFM; isoleucine, phenylalanine, methionine) between domains III and IV, or on domain IV itself at the S4 voltage sensor (Yang et al., 1994; Fleischhauer et al., 1998; Bouhours et al., 2004).
Functional expression revealed that in most cases, PMC mutations cause a decreased rate of channel inactivation and increased rate of recovery from inactivation and other mutations cause channel deactivation to be slowed (Chahine et al., 1994; Goldman, 1999; Jurkat-Rott et al., 2010). The functional effects of PMC seem to be more severe than those of PAM in that the time constant of fast inactivation is greater due to increased channel activity. This may lead to the clinical state of weakness of paralysis more efficiently than with PAM by depolarizing the resting potential and inactivating Na+ channels.
Severe Neonatal Episodic Laryngospasm
Recently a novel Nav1.4 myotonia mutation was identified causing severe neonatal episodic laryngospasm (SNEL) found to be lethal in newborns (Lion-Francois et al., 2010). These newborns presented with episodic apneas and apparent life threatening events that included myotonia of the laryngeal muscles preventing proper ventilation. One infant who was not treated in time with either carbamazepine or mexiletine died due to respiratory arrest during an apneic episode. A de novo A799S missense mutation of the Nav1.4 channel was found in this patient (Lion-Francois et al., 2010; Simkin et al., 2011). This mutation is located on the S6 transmembrane segment of domain II and results in such a severe phenotype by shifting the steady state of activation in a hyperpolarizing direction, slowing the kinetics of fast inactivation and deactivation, and dramatically increasing channel open probability (Simkin et al., 2011).
Na+ channel mutations have already been described in neonates (Tsujino et al., 2003; Gay et al., 2008). These patients had hypotonia and were harboring mutations I693I and N1297K. In addition to the myotonic events, these patients experienced muscle weakness as well. Although muscle weakness can be triggered in patients with Na+ channel myotonia, this clinical feature has not been observed in patients carrying the A799S mutation, making SNEL a distinct form of Na+ channel myotonia in newborn babies.
Periodic Paralysis
Periodic paralysis can be caused by mutations of the Nav1.4 sodium channel which decrease its function, the L-type calcium channel (DHPR), and the inwardly rectifying Kir2.1 channel by inhibiting its function in Andersen’s syndrome. In some cases of periodic paralysis, serum potassium levels are affected causing HyperPP, HypoPP, or normokalemic (where K+ levels remain normal) thus periodic paralysis is defined in terms of serum K+ levels.
Hyperkalemic Periodic Paralysis
Hyperkalemic periodic paralysis has been found in patients with sodium channel mutations that cause attacks of flaccid limb paralysis or weakness of the eye or throat muscles. HyperPP patients present with increased serum K+ levels during the episodes of weakness. The triggers of HyperPP include K+ ingestion, rest after exercise as well as cold temperatures, emotional stress, and fasting. HyperPP and PMC have overlapping symptoms because between episodes of periodic paralysis, HyperPP patients can experience a mild form of myotonia, which may be more pronounced before a paralytic attack. Also patients (which do not experience mild myotonia episodes between paralytic attacks) may be more prone to develop chronic progressive myopathy during midlife when paralytic attacks become more rare. Patients are usually diagnosed in the first decade of life and paralysis attacks increase in frequency and severity during puberty but begin to decrease after approximately 40 years of age. However, older individuals may develop permanent weakness related to the frequency and severity of prior attacks (Bradley et al., 1990; Ptácek et al., 1993a; Jurkat-Rott and Lehmann-Horn, 2007b). During a paralytic attack, patients may also experience respiratory insufficiency which could lead to death if not treated immediately. Attacks can last up to an hour and disappear as serum K+ levels decrease due to elimination by the kidney and reuptake by skeletal muscle cells. However, after an attack, as serum K+ levels drop, patients can become hypokalemic leading to misdiagnoses (Plassart et al., 1994; Jurkat-Rott and Lehmann-Horn, 2007b). Therefore, diagnoses are mostly based on the patient response to K+ rather than the level of K+ during an attack.
Several Nav1.4 mutations lead to HyperPP including L689I, I693T, T704M, A1156T, M1360V, I1490L, M1493I, I1495F, and M1592V (McClatchey et al., 1992; Cannon and Strittmatter, 1993; Cummins et al., 1993; Yang et al., 1994; Wagner et al., 1997; Plassart-Schiess et al., 1998; Bendahhou et al., 1999b, 2000, 2002; Hayward et al., 1999; Rojas et al., 1999). Mutations T704M and M1592V account for the majority of HyperPP cases (Venance et al., 2006). Most HyperPP mutations are located at the intracellular loop between transmembrane segments S4 and S5 of domains II and III or in the transmembrane segment S5 of domain IV. These areas (at least in domains III and IV) are thought to form a three-dimensional docking site for the fast inactivation particle. Structural malformations in those areas may reduce the affinity of the IFM particle to this docking site reducing fast inactivation of the Nav1.4 channel. Sodium channels typically open for several milliseconds, but HyperPP mutations cause the Nav1.4 channel to allow sodium current to flow even after tens of milliseconds (Cannon and Strittmatter, 1993). In the case of HyperPP, there is a persistent inward sodium current and sustained depolarization which leads to electrical inexcitability and muscle weakness (Lehmann-Horn et al., 1987). Also in excised fibers from patients there is an accumulation of intracellular Na+ consistent with increased inward sodium current (Weber et al., 2006). It is suggested that persistent Na+ conductance leads to membrane depolarization which drives K+ out of the cell into the extracellular space causing severe hyperkalemia and further depolarization of the muscle fibers and Na+ channel slow inactivation (Cannon et al., 1993; Cummins and Bendahhou, 2009). Indeed, it has been found that with HyperPP mutations L689I, I693T, T704M, and M1592V, the slow inactivation process (a process that modulates the availability of the channels) is impaired preventing the muscle from recovering after muscle contraction and subsequently causing paralysis, while in PMC the muscle is able to recover quicker (Cummins and Sigworth, 1996; Plassart-Schiess et al., 1998; Bendahhou et al., 1999b, 2002; Hayward et al., 1999; Cummins and Bendahhou, 2009; Platt and Griggs, 2009). Although, persistent current may underlay periodic paralysis and myotonia, other mechanisms have also been proposed. Some HyperPP mutations also shift the voltage dependence of activation in the negative direction which shifts the activation threshold and allows channels to open sooner (Cummins et al., 1993; Yang et al., 1994). This negative shift in activation leads to the rise of persistent currents that occur at voltage ranges between activation and fast inactivation or window currents (Cummins et al., 1993; Yang et al., 1994; Bendahhou et al., 1999b; Rojas et al., 1999). There are two mutations A1156T and M1360V that are associated with both HyperPP and PMC, and present with mixed phenotypes (Yang et al., 1994; Wagner et al., 1997). Neither slow inactivation nor activation is affected with these mutations, perhaps owing to the mixed clinical phenotype.
Hypokalemic Periodic Paralysis
Hypokalemic periodic paralysis can be caused by mutations of the Nav1.4 channel or the skeletal muscle calcium channel Cav1.1, or the inward rectifier potassium channel Kir2.1. HypoPP caused by Ca2+ channel mutations is referred to as HypoPP1 while HypoPP caused by Na+ channel mutations is referred to as HypoPP2 (Jurkat-Rott et al., 1994, 2000; Ptácek et al., 1994a; Sternberg et al., 2001; Fontaine et al., 2007). The major differences between HypoPP1 and HypoPP2 are that HypoPP1 has an earlier onset, HypoPP2 presents with myalgias and can be aggravated by acetazolamide (a carbonic anhydrase inhibitor typically used to treat periodic paralysis), and in muscle biopsies there is a predominance of tubular aggregates in HypoPP2 and vacuoles in HypoPP1 (Fontaine et al., 2007; Platt and Griggs, 2009). HypoPP is characterized by reversible attacks of muscle weakness with decreased serum K+ levels. During HypoPP attacks, patients can lose their muscle strength and reflexes and have subjective sensory symptoms. These attacks of weakness or paralysis are triggered by rest after strenuous exercise, ingestion of carbohydrates exposure to cold, or corticosteroid intake (Bendahhou et al., 2007). There is no evidence in electromyographic studies that HypoPP patients have any form of myotonia with the exception of eyelid myotonia which can sometimes even be present between attacks (Ptácek and Griggs, 1996). Attacks can last several hours or even days, unlike in HyperPP, and are more severe in nature. Patients are usually at their weakest during the later hours of the night and in the morning and progressively get stronger as the day goes by. Once an attack has resolved, patients appear completely normal. As patients get older, attack frequency decreases but some develop progressive persistent weakness in the form of proximal myopathy and can progress to debilitating weakness (to the point of wheelchair confinement) especially those who were untreated throughout previous attacks. Muscle biopsies from HypoPP patients reveal abnormal vacuoles resulting from SR dilatation (possibly as a result of osmotic fluxes) or atrophic changes even during attack free intervals. However, blood studies are unremarkable in these intervals.
A number of Nav1.4 mutations resulting in HypoPP2 have been found including in humans: R222W, R669H, R672H/G/S, R1129Q, R1132Q, R1135H, and P1158S (Struyk et al., 2000; Bendahhou et al., 2001; Kuzmenkin et al., 2002; Sugiura et al., 2003; Carle et al., 2006; Matthews et al., 2009; Hong et al., 2010). All Na+ or Ca2+ channel HypoPP mutations, are located on the voltage sensors of these two channels. Sodium channel HypoPP mutations are located primarily on the S4 voltage sensors of domains II and III where they neutralize positively charged residues hindering proper voltage sensor function (Kontis et al., 1997; Kuhn and Greeff, 1999; Matthews et al., 2009; Ruff, 2010). HypoPP Nav1.4 mutations (unlike other Nav1.4 mutations which cause a gain-of-function by enhancing activation or impairing inactivation) cause a channel loss of function by enhancing channel inactivation which, can be achieved by enhancing fast inactivation, slow inactivation or both reducing the availability of these sodium channels (Jurkat-Rott et al., 2000; Ruff and Cannon, 2000; Struyk et al., 2000; Bendahhou et al., 2001; Kuzmenkin et al., 2002). At normal muscle resting membrane potentials (RMP), about 70% of the Nav1.4 channels are available for activation which is enough to generate an AP. However, in HypoPP there is a significantly depolarized RMP which causes more Nav1.4 channels to enter the inactive and unavailable state for AP generation, while muscle is contracting, producing weakness. Other HypoPP Nav1.4 mutations allow fewer channels to enter the slow inactivation state when they need to rest while muscle is active which prevents the turnover from inactive to active channels to happen as needed for the muscle and thus more Na+ channels enter the inactive state subsequently causing paralysis. Nevertheless, this hypothesis of channel down regulation does not seem to explain the paradoxical sarcolemmal depolarization with hypokalemia or paralysis induced by insulin and glucose during HypoPP attacks (Tricarico et al., 1998; Ruff, 1999).
Recent studies propose that the depolarization seen with some HypoPP mutations can be due to a cationic leak through the voltage sensor created by the mutations called the gating pore current (channel up-regulation) which may cause a cellular pH imbalance due to proton movement into the fibers (Kuzmenkin et al., 2002; Carle et al., 2006; Sokolov et al., 2007; Struyk and Cannon, 2007; Struyk et al., 2008). This gating pore is distinct from the main sodium ion pore between segments S5 and S6 and is suggested to be formed by segments S1–S4 in each domain (making up four possible gating pores) perhaps by the mutation of the S4 segment (Yang et al., 1997; Sato et al., 2001; Long et al., 2005; Sokolov et al., 2005; Tombola et al., 2005). This gating current is a hyperpolarization activated current of monovalent cations which counteracts the K+ current and depolarizes the RMP thus it would explain why muscle fibers are depolarized during hypokalemia (Jurkat-Rott and Lehmann-Horn, 2007a; Jurkat-Rott et al., 2009). It has been suggested that the gating pore may lower the intracellular pH leading to (i) intracellular Na+ ion accumulation through the activation of transporters such as the Na+/H+ exchanger, and/or (ii) the inhibition of the inwardly rectifying K+ channels (see Matthews and Hanna, 2010). However, this has not been sufficiently elucidated because studies reporting on the gating currents corresponding to several HypoPP mutations were conducted in Xenopus oocytes only and have not been reproduced in mammalian cells (Sokolov et al., 2007; Struyk and Cannon, 2007).
Congenital Myasthenic Syndrome
Myasthenic syndrome (CMS) is a disorder with defective transmission of neuromuscular excitation resulting in muscle fatigue from defects in presynaptic, synaptic, or postsynaptic proteins (Engel et al., 2003). Patients experience fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis beginning at birth. CMS can be caused by several types of defects. In one case, nerve stimulation at physiological rates rapidly decreased the compound muscle APs but no abnormalities in RMP evoked quantal release synaptic potentials, AChR channel kinetics, or endplate ultrastructure was found. However, endplate potentials depolarizing the resting potential to −40 mV failed to excite APs (Tsujino et al., 2003).
In this case, a Nav1.4 channel mutation (V1442E) was found located at the extracellular linker between segments S3 and S4 of domain IV (Tsujino et al., 2003). Respiratory paralysis is not a common symptom of Nav1.4 mutation defects despite the fact that Nav1.4 is the predominant sodium channel in respiratory muscles. However, the CMS patient with the V1442E mutation did exhibit respiratory paralysis as well as paralysis of other muscles such as the bulbar muscles, limiting speech, and swallowing. This patient had also been on an apnea monitor since infancy and received ventilatory support during apneic attacks. In her early 1920s, the patient had limited ocular ductions, and weakness throughout her body worsened by activity. The V1442E mutation revealed an enhancement of fast inactivation even at hyperpolarized potentials and the availability of sodium channels was significantly reduced, even compared to HypoPP mutations, causing muscle AP failure even though the RMP was normal.
Treatments for Skeletal Muscle Sodium Channel Myotonias and Periodic Paralyzes
Myotonia
Treatment for myotonia is focused on reducing the involuntary AP bursts without blocking the voluntary high-frequency muscle stimulation. Although, it is important that PAM and PMC patients modify their lifestyle to avoid the triggers of their diseases such as potassium ingestion or cold temperatures, drug therapies are commonly used to relieve and prevent muscle stiffness (Table 2).
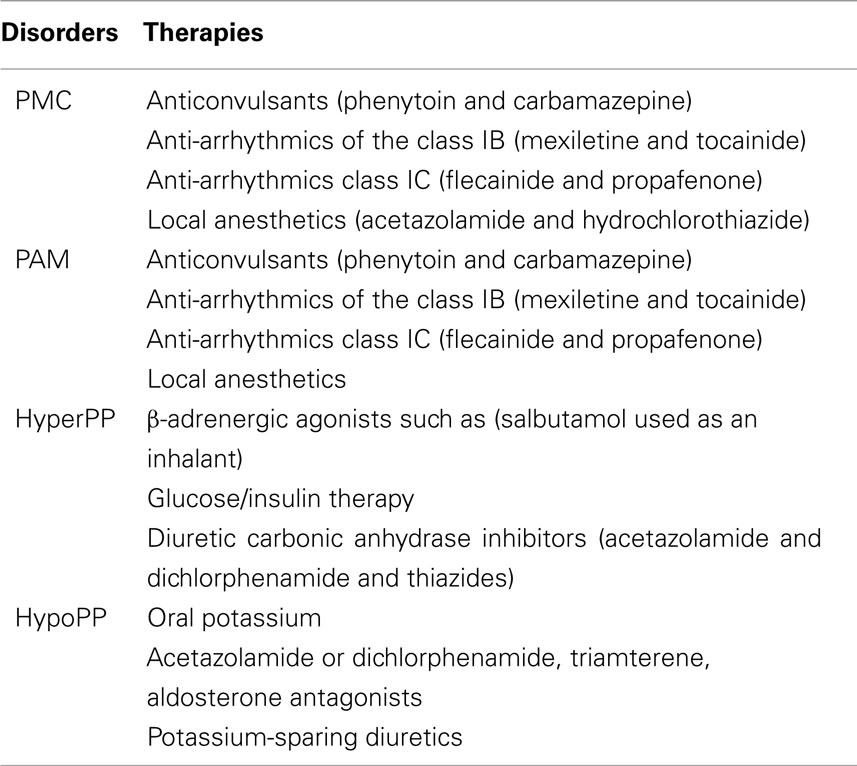
Table 2. Medication in the different forms of Na channel myotonia and paralysis.
For PMC and PAM patients, anticonvulsants (phenytoin and carbamazepine), anti-arrhythmics of the class IB (mexiletine and tocainide), class IC (flecainide and propafenone), and local anesthetics have been shown to have some efficacy relieving stiffness in PAM and preventing stiffness and weakness from occurring in PMC (Trip et al., 2006; Alfonsi et al., 2007). Agents such as mexiletine and flecainide (orally absorbed methylated lidocaine derivatives) prevent repetitive AP firing and myotonia by reversibly blocking sodium channels (Wang et al., 2004; De Bellis et al., 2006). This therapeutic effect enhances sodium channel inactivation and shifts the voltage dependence of steady state inactivation in a hyperpolarizing direction and slows the recovery from inactivation. In most cases, Nav1.4 mutations increased mexiletine sensitivity however, this was found to be dependent on the Nav1.4 mutation as in some cases mexiletine sensitivity was reduced contributing to the heterogeneity of clinical symptoms observed with mexiletine treatment (Fan et al., 1996; Featherstone et al., 1998; Takahashi and Cannon, 2001; Desaphy et al., 2003).
Since PMC patients present with severe myotonia that overshadows their attacks of weakness most of these patients rarely require medications for weakness. However, sometimes diuretic carbonic anhydrase inhibitor medications such as acetazolamide and hydrochlorothiazide can be given to reduce serum K+ levels or lower the pH and decrease the frequency and severity of paralytic events (Rudel et al., 1980; Ricker et al., 1983; Ptácek et al., 1993b; Ptácek and Griggs, 1996).
Periodic Paralysis
Patients with HyperPP often find that reducing their carbohydrate intake and avoiding strenuous exercise and cold improves their condition. Other treatments include β-adrenergic agonists such as salbutamol used as an inhalant on patients without cardiac arrhythmia or glucose/insulin therapy (Hanna et al., 1998). Diuretic carbonic anhydrase inhibitors (acetazolamide and dichlorphenamide) and thiazides seem to be the most effective in HyperPP (Sansone et al., 2008). Treatments that enhance slow inactivation of sodium channels or shift the voltage dependence of activation may be more effective in treating some HyperPP patients.
Treatment of HypoPP is better achieved by administrating oral potassium and by avoidance of carbohydrates and sodium in the diet. Increasing K+ levels usually helps to reduce the paradoxical membrane depolarization and shifts the resting potential to more normal hyperpolarized voltages. Administration of acetazolamide or dichlorphenamide has also proven to be useful, however in some cases these agents can exacerbate symptoms and triamterene (kidney sodium channel blocker used as a potassium-sparing diuretic) is used instead (Torres et al., 1981; Tawil et al., 2000; Sternberg et al., 2001; Venance et al., 2004). These carbonic anhydrase inhibitors also lower the intracellular sodium levels in addition to repolarizing the RMP (perhaps by opening calcium activated K+ channels; Tricarico et al., 1999). Other aldosterone antagonists and potassium-sparing diuretics are also used.
Conclusion
Skeletal muscle sodium channel disorders show significant intra and interfamilial phenotypical variability as well as variability in the functional electrophysiological properties of the Nav1.4 channel. It remains difficult to properly identify, classify, and treat myotonia and periodic paralysis patients due to an insufficient understanding of the mechanism by which these mutations bring about such variable phenotypes. Nav1.4 channel gating defects to produce gain-of-function which slows the rate of inactivation and or deactivation typically cause myotonia while Nav1.4 mutations that negatively shift the voltage dependence of activation typically cause HyperPP. Mutations that down-regulate Nav1.4 channel function by enhancing slow inactivation or the newly suggested mechanism of increased gating pore currents which depolarize muscle fibers in the presence of low extracellular K+ concentrations result in HypoPP. If gating pore currents do indeed underlie the abnormal depolarization associated with HypoPP, the “gating pore” may represent a novel therapeutic target for treating these patients. Nevertheless, functional studies may not lead to a full understanding of these diseases and careful analysis of clinical phenotypes, clinical trials, and genetic screening are clearly still lacking and needed. Understanding these different factors which underlie skeletal muscle sodium channel disorders may help to improve and develop new strategies of therapeutic treatment for these patients as well as treatments of related disorders of excitability.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the CentreNational de la RechercheScientifique (CNRS), Association Française contre les Myopathies (AFM) grant (Saïd Bendahhou), AFM and Châteaubriand fellowships to Dina Simkin.
References
Adrian, R. H., and Marshall, M. W. (1976). Action potentials reconstructed in normal and myotonic muscle fibres. J. Physiol. (Lond.) 258, 125–143.
Alfonsi, E., Merlo, I. M., Tonini, M., Ravaglia, S., Brugnoni, R., Gozzini, A., and Moglia, A. (2007). Efficacy of propafenone in paramyotonia congenita. Neurology 68, 1080–1081.
Almers, W. (1980). Potassium concentration changes in the transverse tubules of vertebrate skeletal muscle. Fed. Proc. 39, 1527–1532.
Armstrong, C. M., and Bezanilla, F. (1977). Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70, 567–590.
Baquero, J. L., Ayala, R. A., Wang, J., Curless, R. G., Feero, W. G., Hoffman, E. P., and Ebeid, M. R. (1995). Hyperkalemic periodic paralysis with cardiac dysrhythmia: a novel sodium channel mutation? Ann. Neurol. 37, 408–411.
Bendahhou, S., Cummins, T. R., Griggs, R. C., Fu, Y. H., and Ptácek, L. J. (2001). Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann. Neurol. 50, 417–420.
Bendahhou, S., Cummins, T. R., Hahn, A. F., Langlois, S., Waxman, S. G., and Ptácek, L. J. (2000). A double mutation in families with periodic paralysis defines new aspects of sodium channel slow inactivation. J. Clin. Invest. 106, 431–438.
Bendahhou, S., Cummins, T. R., Kula, R. W., Fu, Y. H., and Ptácek, L. J. (2002). Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology 58, 1266–1272.
Bendahhou, S., Cummins, T. R., Kwiecinski, H., Waxman, S. G., and Ptácek, L. J. (1999a). Characterization of a new sodium channel mutation at arginine 1448 associated with moderate Paramyotonia congenita in humans. J. Physiol. (Lond.) 518, 337–344.
Bendahhou, S., Cummins, T. R., Tawil, R., Waxman, S. G., and Ptácek, L. J. (1999b). Activation and inactivation of the voltage-gated sodium channel: role of segment S5 revealed by a novel hyperkalaemic periodic paralysis mutation. J. Neurosci. 19, 4762–4771.
Bendahhou, S., Fournier, E., Gallet, S., Menard, D., Larroque, M. M., and Barhanin, J. (2007). Corticosteroid-exacerbated symptoms in an Andersen’s syndrome kindred. Hum. Mol. Genet. 16, 900–906.
Bissay, V., Keymolen, K., Lissens, W., Laureys, G., Schmedding, E., and Keyser, J. D. (2011). Late onset painful cold-aggravated myotonia: three families with SCN4A L1436P mutation. Neuromuscul. Disord. 21, 590–593.
Bouhours, M., Luce, S., Sternberg, D., Willer, J. C., Fontaine, B., and Tabti, N. (2005). A1152D mutation of the Na+ channel causes paramyotonia congenita and emphasizes the role of DIII/S4-S5 linker in fast inactivation. J. Physiol. (Lond.) 565, 415–427.
Bouhours, M., Sternberg, D., Davoine, C. S., Ferrer, X., Willer, J. C., Fontaine, B., and Tabti, N. (2004). Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans. J. Physiol. (Lond.) 554, 635–647.
Bradley, W. G., Taylor, R., Rice, D. R., Hausmanowa-Petruzewicz, I., Adelman, L. S., Jenkison, M., Jedrzejowska, H., Drac, H., and Pendlebury, W. W. (1990). Progressive myopathy in hyperkalemic periodic paralysis. Arch. Neurol. 47, 1013–1017.
Cannon, S. C., Brown, R. H. Jr., and Corey, D. P. (1993). Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys. J. 65, 270–288.
Cannon, S. C., and Strittmatter, S. M. (1993). Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron 10, 317–326.
Carle, T., Fournier, E., Sternberg, D., Fontaine, B., and Tabti, N. (2009). Cold-induced disruption of Na+ channel slow inactivation underlies paralysis in highly thermosensitive paramyotonia. J. Physiol. (Lond.) 587, 1705–1714.
Carle, T., Lhuillier, L., Luce, S., Sternberg, D., Devuyst, O., Fontaine, B., and Tabti, N. (2006). Gating defects of a novel Na+ channel mutant causing hypokalemic periodic paralysis. Biochem. Biophys. Res. Commun. 348, 653–661.
Chahine, M., George, A. L. Jr., Zhou, M., Ji, S., Sun, W., Barchi, R. L., and Horn, R. (1994). Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron 12, 281–294.
Cummins, T. R., and Bendahhou, S. (2009). “Inherited disorders of skeletal muscle caused by voltage-gated sodium channel mutations,” in Biophysics of Ion Channels and Disease, ed. H. Duclohier (Kerala: Transworld Research Network), 1–27.
Cummins, T. R., and Sigworth, F. J. (1996). Impaired slow inactivation in mutant sodium channels. Biophys. J. 71, 227–236.
Cummins, T. R., Zhou, J., Sigworth, F. J., Ukomadu, C., Stephan, M., Ptácek, L. J., and Agnew, W. S. (1993). Functional consequences of a Na+ channel mutation causing hyperkalemic periodic paralysis. Neuron 10, 667–678.
De Bellis, M., De Luca, A., Rana, F., Cavalluzzi, M. M., Catalano, A., Lentini, G., Franchini, C., Tortorella, V., and Conte Camerino, D. (2006). Evaluation of the pharmacological activity of the major mexiletine metabolites on skeletal muscle sodium currents. Br. J. Pharmacol. 149, 300–310.
Desaphy, J. F., Pierno, S., De Luca, A., Didonna, P., and Camerino, D. C. (2003). Different ability of clenbuterol and salbutamol to block sodium channels predicts their therapeutic use in muscle excitability disorders. Mol. Pharmacol. 63, 659–670.
Dice, M. S., Abbruzzese, J. L., Wheeler, J. T., Groome, J. R., Fujimoto, E., and Ruben, P. C. (2004). Temperature-sensitive defects in paramyotonia congenita mutants R1448C and T1313M. Muscle Nerve 30, 277–288.
Engel, A. G., Ohno, K., Shen, X. M., and Sine, S. M. (2003). Congenital myasthenic syndromes: multiple molecular targets at the neuromuscular junction. Ann. N. Y. Acad. Sci. 998, 138–160.
Fan, Z., George, A. L. Jr., Kyle, J. W., and Makielski, J. C. (1996). Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. (Lond.) 496, 275–286.
Featherstone, D. E., Fujimoto, E., and Ruben, P. C. (1998). A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J. Physiol. (Lond.) 506, 627–638.
Fleischhauer, R., Mitrovic, N., Deymeer, F., Lehmann-Horn, F., and Lerche, H. (1998). Effects of temperature and mexiletine on the F1473S Na+ channel mutation causing paramyotonia congenita. Pflugers Arch. 436, 757–765.
Fontaine, B., Fournier, E., Sternberg, D., Vicart, S., and Tabti, N. (2007). Hypokalemic periodic paralysis: a model for a clinical and research approach to a rare disorder. Neurotherapeutics 4, 225–232.
Fontaine, B., Khurana, T. S., Hoffman, E. P., Bruns, G. A., Haines, J. L., Trofatter, J. A., Hanson, M. P., Rich, J., Mcfarlane, H., Yasek, D. M., Romano, D., Gusella, J., and Brown, R. (1990). Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science 250, 1000–1002.
Gay, S., Dupuis, D., Faivre, L., Masurel-Paulet, A., Labenne, M., Colombani, M., Soichot, P., Huet, F., Hainque, B., Sternberg, D., Fontaine, B., Gouyon, J. B., and Thauvin-Robinet, C. (2008). Severe neonatal non-dystrophic myotonia secondary to a novel mutation of the voltage-gated sodium channel (SCN4A) gene. Am. J. Med. Genet. A 146, 380–383.
George, A. L. Jr. (2005). Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115, 1990–1999.
George, A. L. Jr., Knittle, T. J., and Tamkun, M. M. (1992a). Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc. Natl. Acad. Sci. U.S.A. 89, 4893–4897.
George, A. L. Jr., Komisarof, J., Kallen, R. G., and Barchi, R. L. (1992b). Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Ann. Neurol. 31, 131–137.
Goldman, L. (1999). On mutations that uncouple sodium channel activation from inactivation. Biophys. J. 76, 2553–2559.
Green, D. S., George, A. L. Jr., and Cannon, S. C. (1998). Human sodium channel gating defects caused by missense mutations in S6 segments associated with myotonia: S804F and V1293I. J. Physiol. (Lond.) 510, 685–694.
Green, D. S., Hayward, L. J., George, A. L. Jr., and Cannon, S. C. (1997). A proposed mutation, Val781Ile, associated with hyperkalemic periodic paralysis and cardiac dysrhythmia is a benign polymorphism. Ann. Neurol. 42, 253–256.
Groome, J. R., Fujimoto, E., and Ruben, P. C. (2005). K-aggravated myotonia mutations at residue G1306 differentially alter deactivation gating of human skeletal muscle sodium channels. Cell. Mol. Neurobiol. 25, 1075–1092.
Hanna, M. G., Stewart, J., Schapira, A. H., Wood, N. W., Morgan-Hughes, J. A., and Murray, N. M. (1998). Salbutamol treatment in a patient with hyperkalaemic periodic paralysis due to a mutation in the skeletal muscle sodium channel gene (SCN4A). J. Neurol. Neurosurg. Psychiatr. 65, 248–250.
Hayward, L. J., Brown, R. H. Jr., and Cannon, S. C. (1997). Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys. J. 72, 1204–1219.
Hayward, L. J., Sandoval, G. M., and Cannon, S. C. (1999). Defective slow inactivation of sodium channels contributes to familial periodic paralysis. Neurology 52, 1447–1453.
Hong, D., Luan, X., Chen, B., Zheng, R., Zhang, W., Wang, Z., and Yuan, Y. (2010). Both hypokalaemic and normokalaemic periodic paralysis in different members of a single family with novel R1129Q mutation in SCN4A gene. J. Neurol. Neurosurg. Psychiatr. 81, 703–704.
Jackson, C. E., Barohn, R. J., and Ptácek, L. J. (1994). Paramyotonia congenita: abnormal short exercise test, and improvement after mexiletine therapy. Muscle Nerve 17, 763–768.
Jurkat-Rott, K., Fauler, M., and Lehmann-Horn, F. (2006). Ion channels and ion transporters of the transverse tubular system of skeletal muscle. J. Muscle Res. Cell. Motil. 27, 275–290.
Jurkat-Rott, K., Holzherr, B., Fauler, M., and Lehmann-Horn, F. (2010). Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch. 460, 239–248.
Jurkat-Rott, K., and Lehmann-Horn, F. (2005). Muscle channelopathies and critical points in functional and genetic studies. J. Clin. Invest. 115, 2000–2009.
Jurkat-Rott, K., and Lehmann-Horn, F. (2007a). Do hyperpolarization-induced proton currents contribute to the pathogenesis of hypokalemic periodic paralysis, a voltage sensor channelopathy? J. Gen. Physiol. 130, 1–5.
Jurkat-Rott, K., and Lehmann-Horn, F. (2007b). Genotype-phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics 4, 216–224.
Jurkat-Rott, K., Lehmann-Horn, F., Elbaz, A., Heine, R., Gregg, R. G., Hogan, K., Powers, P. A., Lapie, P., Vale-Santos, J. E., Weissenbach, J., and Fontaine, B. (1994). A calcium channel mutation causing hypokalemic periodic paralysis. Hum. Mol. Genet. 3, 1415–1419.
Jurkat-Rott, K., Mitrovic, N., Hang, C., Kouzmekine, A., Iaizzo, P., Herzog, J., Lerche, H., Nicole, S., Vale-Santos, J., Chauveau, D., Fontaine, B., and Lehmann-Horn, F. (2000). Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc. Natl. Acad. Sci. U.S.A. 97, 9549–9554.
Jurkat-Rott, K., Weber, M. A., Fauler, M., Guo, X. H., Holzherr, B. D., Paczulla, A., Nordsborg, N., Joechle, W., and Lehmann-Horn, F. (2009). K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc. Natl. Acad. Sci. U.S.A. 106, 4036–4041.
Kim, M. K., Lee, S. H., Park, M. S., Kim, B. C., Cho, K. H., Lee, M. C., Kim, J. H., and Kim, S. M. (2004). Mutation screening in Koreanhypokalemicperiodicparalysis patients: a novel SCN4A Arg672Cys mutation. Neuromuscul. Disord. 14, 727–731.
Kontis, K. J., Rounaghi, A., and Goldin, A. L. (1997). Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physiol. 110, 391–401.
Kubota, T., Kinoshita, M., Sasaki, R., Aoike, F., Takahashi, M. P., Sakoda, S., and Hirose, K. (2009). New mutation of the Na channel in the severe form of potassium-aggravated myotonia. Muscle Nerve 39, 666–673.
Kuhn, F. J., and Greeff, N. G. (1999). Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J. Gen. Physiol. 114, 167–183.
Kuzmenkin, A., Muncan, V., Jurkat-Rott, K., Hang, C., Lerche, H., Lehmann-Horn, F., and Mitrovic, N. (2002). Enhanced inactivation and pH sensitivity of Na(+) channel mutations causing hypokalaemic periodic paralysis type II. Brain 125, 835–843.
Lee, S. C., Kim, H. S., Park, Y. E., Choi, Y. C., Park, K. H., and Kim, D. S. (2009). Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium Channelopathy. J. Clin. Neurol. 5, 186–191.
Lehmann-Horn, F., Kuther, G., Ricker, K., Grafe, P., Ballanyi, K., and Rudel, R. (1987). Adynamia episodica hereditaria with myotonia: a non-inactivating sodium current and the effect of extracellular pH. Muscle Nerve 10, 363–374.
Lehmann-Horn, F., Rudel, R., and Jurkat-Rott, K. (2004). “Nondystrophic myotonias and periodic paralysis,” in Myology, eds C. Franzini-Armstrong, and A. Engel (New York: McGraw-Hill), 1257–1300.
Lehmann-Horn, F., Rudel, R., and Ricker, K. (1993). Non-dystrophic myotonias and periodic paralyses. A European Neuromuscular Center Workshop held 4-6 October 1992, Ulm, Germany. Neuromuscul. Disord. 3, 161–168.
Lerche, H., Heine, R., Pika, U., George, A. L. Jr., Mitrovic, N., Browatzki, M., Weiss, T., Rivet-Bastide, M., Franke, C., Lomonaco, M., Ricker, R., and Lehmann-Horn, F. (1993). Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J. Physiol. (Lond.) 470, 13–22.
Lerche, H., Mitrovic, N., Dubowitz, V., and Lehmann-Horn, F. (1996). Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann. Neurol. 39, 599–608.
Lion-Francois, L., Mignot, C., Vicart, S., Manel, V., Sternberg, D., Landrieu, P., Lesca, G., Broussolle, E., Billette De Villemeur, T., Napuri, S., Des Portes, V., and Fontaine, B. (2010). Severe neonatal episodic laryngospasm due to de novo SCN4A mutations: a new treatable disorder. Neurology 75, 641–645.
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903.
Matthews, E., and Hanna, M. G. (2010). Muscle channelopathies: does the predictedchannelgating pore offer new treatment insights for hypokalaemicperiodicparalysis? J. Physiol. 588, 1879–1886.
Matthews, E., Labrum, R., Sweeney, M. G., Sud, R., Haworth, A., Chinnery, P. F., Meola, G., Schorge, S., Kullmann, D. M., Davis, M. B., and Hanna, M. G. (2009). Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 72, 1544–1547.
Matthews, E., Tan, S. V., Fialho, D., Sweeney, M. G., Sud, R., Haworth, A., Stanley, E., Cea, G., Davis, M. B., and Hanna, M. G. (2008). What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology 70, 50–53.
McClatchey, A. I., Van Den Bergh, P., Pericak-Vance, M. A., Raskind, W., Verellen, C., Mckenna-Yasek, D., Rao, K., Haines, J. L., Bird, T., Brown, R. H. Jr., and Gusella, J. F. (1992). Temperature-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell 68, 769–774.
Miller, T. M., Dias Da Silva, M. R., Miller, H. A., Kwiecinski, H., Mendell, J. R., Tawil, R., Mcmanis, P., Griggs, R. C., Angelini, C., Servidei, S., Petajan, J., Dalakas, M. C., Ranum, L. P., Fu, Y. H., and Ptácek, L. J. (2004). Correlating phenotype and genotype in the periodic paralyses. Neurology 63, 1647–1655.
Mitrovic, N., George, A. L. Jr., Heine, R., Wagner, S., Pika, U., Hartlaub, U., Zhou, M., Lerche, H., Fahlke, C., and Lehmann-Horn, F. (1994). K(+)-aggravated myotonia: destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. J. Physiol. (Lond.) 478, 395–402.
Mitrovic, N., George, A. L. Jr., Lerche, H., Wagner, S., Fahlke, C., and Lehmann-Horn, F. (1995). Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J. Physiol. (Lond.) 487, 107–114.
Noda, M., Shimizu, S., Tanabe, T., Takai, T., Kayano, T., Ikeda, T., Takahashi, H., Nakayama, H., Kanaoka, Y., Minamino, N., Kangawa, K., Matsuo, H., Raftery, M. A., Hirose, T., Inayama, S., Hayashida, H., Miyata, T., and Numa, S. (1984). Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312, 121–127.
Okuda, S., Kanda, F., Nishimoto, K., Sasaki, R., and Chihara, K. (2001). Hyperkalemic periodic paralysis and paramyotonia congenita – a novel sodium channel mutation. J. Neurol. 248, 1003–1004.
Park, Y. H., and Kim, J. B. (2010). An atypical phenotype of hypokalemic periodic paralysis caused by a mutation in the sodium channel gene SCN4A. Korean J. Pediatr. 53, 909–912.
Petitprez, S., Tiab, L., Chen, L., Kappeler, L., Rosler, K. M., Schorderet, D., Abriel, H., and Burgunder, J. M. (2008). A novel dominant mutation of the Nav1.4 alpha-subunit domain I leading to sodium channel myotonia. Neurology 71, 1669–1675.
Plassart, E., Reboul, J., Rime, C. S., Recan, D., Millasseau, P., Eymard, B., Pelletier, J., Thomas, C., Chapon, F., Desnuelle, C., Confavreux, C., Bady, B., Martin, J. J., Lenoir, G., Serratrice, G., Fardeau, M., and Fontaine, B. (1994). Mutations in the muscle sodium channel gene (SCN4A) in 13 French families with hyperkalemic periodic paralysis and paramyotonia congenita: phenotype to genotype correlations and demonstration of the predominance of two mutations. Eur. J. Hum. Genet. 2, 110–124.
Plassart-Schiess, E., Lhuillier, L., George, A. L. Jr., Fontaine, B., and Tabti, N. (1998). Functional expression of the Ile693Thr Na+ channel mutation associated with paramyotonia congenita in a human cell line. J. Physiol. (Lond.) 507, 721–727.
Platt, D., and Griggs, R. (2009). Skeletal muscle channelopathies: new insights into the periodic paralyses and nondystrophic myotonias. Curr. Opin. Neurol. 22, 524–531.
Ptácek, J., and Griggs, R. C. (1996). “Familial periodic paralysis,” in Molecular Biology of Membrane Transport Disorders, ed. T. Andreoli (New York: Plenum Press), 625–642.
Ptácek, L. J., George, A. L. Jr., Barchi, R. L., Griggs, R. C., Riggs, J. E., Robertson, M., and Leppert, M. F. (1992). Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron 8, 891–897.
Ptácek, L. J., Gouw, L., Kwiecinski, H., Mcmanis, P., Mendell, J. R., Barohn, R. J., George, A. L. Jr., Barchi, R. L., Robertson, M., and Leppert, M. F. (1993a). Sodium channel mutations in paramyotonia congenita and hyperkalemic periodic paralysis. Ann. Neurol. 33, 300–307.
Ptácek, L. J., Johnson, K. J., and Griggs, R. C. (1993b). Genetics and physiology of the myotonic muscle disorders. N. Engl. J. Med. 328, 482–489.
Ptácek, L. J., Tawil, R., Griggs, R. C., Engel, A. G., Layzer, R. B., Kwiecinski, H., Mcmanis, P. G., Santiago, L., Moore, M., Fouad, G., Bradely, P., and Leppert, M. F. (1994a). Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77, 863–868.
Ptácek, L. J., Tawil, R., Griggs, R. C., Meola, G., Mcmanis, P., Barohn, R. J., Mendell, J. R., Harris, C., Spitzer, R., Santiago, F., and Leppert, M. F. (1994b). Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology 44, 1500–1503.
Ptácek, L. J., Trimmer, J. S., Agnew, W. S., Roberts, J. W., Petajan, J. H., and Leppert, M. (1991). Paramyotonia congenita and hyperkalemic periodic paralysis map to the same sodium-channel gene locus. Am. J. Hum. Genet. 49, 851–854.
Richmond, J. E., Featherstone, D. E., and Ruben, P. C. (1997). Human Na+ channel fast and slow inactivation in paramyotonia congenita mutants expressed in Xenopus laevis oocytes. J. Physiol. (Lond.) 499, 589–600.
Ricker, K., Bohlen, R., and Rohkamm, R. (1983). Different effectiveness of tocainide and hydrochlorothiazide in paramyotonia congenita with hyperkalemic episodic paralysis. Neurology 33, 1615–1618.
Rios, E., Ma, J. J., and Gonzalez, A. (1991). The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J. Muscle Res. Cell Motil. 12, 127–135.
Rojas, C. V., Neely, A., Velasco-Loyden, G., Palma, V., and Kukuljan, M. (1999). Hyperkalemic periodic paralysis M1592V mutation modifies activation in human skeletal muscle Na+ channel. Am. J. Physiol. 276, C259–C266.
Rossignol, E., Mathieu, J., Thiffault, I., Tetreault, M., Dicaire, M. J., Chrestian, N., Dupre, N., Puymirat, J., and Brais, B. (2007). A novel founder SCN4A mutation causes painful cold-induced myotonia in French-Canadians. Neurology 69, 1937–1941.
Rudel, R., Dengler, R., Ricker, K., Haass, A., and Emser, W. (1980). Improved therapy of myotonia with the lidocaine derivative tocainide. J. Neurol. 222, 275–278.
Ruff, R. L. (1999). Insulin acts in hypokalemic periodic paralysis by reducing inward rectifier K+ current. Neurology 53, 1556–1563.
Ruff, R. L. (2010). Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 74, 269; author reply 169–270.
Ruff, R. L., and Cannon, S. C. (2000). Defective slow inactivation of sodium channels contributes to familial periodic paralysis. Neurology 54, 2190–2192.
Sansone, V., Meola, G., Links, T. P., Panzeri, M., and Rose, M. R. (2008). Treatment for periodic paralysis. Cochrane Database Syst. Rev. CD005045.
Sasaki, R., Takano, H., Kamakura, K., Kaida, K., Hirata, A., Saito, M., Tanaka, H., Kuzuhara, S., and Tsuji, S. (1999). A novel mutation in the gene for the adult skeletal muscle sodium channel alpha-subunit (SCN4A) that causes paramyotonia congenita of von Eulenburg. Arch. Neurol. 56, 692–696.
Sato, C., Ueno, Y., Asai, K., Takahashi, K., Sato, M., Engel, A., and Fujiyoshi, Y. (2001). The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature 409, 1047–1051.
Schoser, B. G., Schroder, J. M., Grimm, T., Sternberg, D., and Kress, W. (2007). A large German kindred with cold-aggravated myotonia and a heterozygous A1481D mutation in the SCN4A gene. Muscle Nerve 35, 599–606.
Simkin, D., Lena, I., Landrieu, P., Lion-Francois, L., Sternberg, D., Fontaine, B., and Bendahhou, S. (2011). Mechanisms underlying a life-threatening skeletal muscle Na+ channel disorder. J. Physiol. (Lond.) 589, 3115–3124.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2005). Ion permeation through a voltage-sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron 47, 183–189.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78.
Sternberg, D., Maisonobe, T., Jurkat-Rott, K., Nicole, S., Launay, E., Chauveau, D., Tabti, N., Lehmann-Horn, F., Hainque, B., and Fontaine, B. (2001). Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 124, 1091–1099.
Struyk, A. F., and Cannon, S. C. (2007). A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J. Gen. Physiol. 130, 11–20.
Struyk, A. F., Markin, V. S., Francis, D., and Cannon, S. C. (2008). Gating pore currents in DIIS4 mutations of NaV1.4 associated with periodic paralysis: saturation of ion flux and implications for disease pathogenesis. J. Gen. Physiol. 132, 447–464.
Struyk, A. F., Scoggan, K. A., Bulman, D. E., and Cannon, S. C. (2000). The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. J. Neurosci. 20, 8610–8617.
Sugiura, Y., Makita, N., Li, L., Noble, P. J., Kimura, J., Kumagai, Y., Soeda, T., and Yamamoto, T. (2003). Cold induces shifts of voltage dependence in mutant SCN4A, causing hypokalemic periodic paralysis. Neurology 61, 914–918.
Takahashi, M. P., and Cannon, S. C. (1999). Enhanced slow inactivation by V445M: a sodium channel mutation associated with myotonia. Biophys. J. 76, 861–868.
Takahashi, M. P., and Cannon, S. C. (2001). Mexiletine block of disease-associated mutations in S6 segments of the human skeletal muscle Na(+) channel. J. Physiol. (Lond.) 537, 701–714.
Tawil, R., Mcdermott, M. P., Brown, R. Jr., Shapiro, B. C., Ptácek, L. J., Mcmanis, P. G., Dalakas, M. C., Spector, S. A., Mendell, J. R., Hahn, A. F., and Griggs, R. C. (2000). Randomized trials of dichlorphenamide in the periodic paralyses. Working Group on Periodic Paralysis. Ann. Neurol. 47, 46–53.
Tombola, F., Pathak, M. M., and Isacoff, E. Y. (2005). Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron 45, 379–388.
Torres, C. F., Griggs, R. C., Moxley, R. T., and Bender, A. N. (1981). Hypokalemic periodic paralysis exacerbated by acetazolamide. Neurology 31, 1423–1428.
Tricarico, D., Pierno, S., Mallamaci, R., Brigiani, G. S., Capriulo, R., Santoro, G., and Camerino, D. C. (1998). The biophysical and pharmacological characteristics of skeletal muscle ATP-sensitive K+ channels are modified in K+-depleted rat, an animal model of hypokalemic periodic paralysis. Mol. Pharmacol. 54, 197–206.
Tricarico, D., Servidei, S., Tonali, P., Jurkat-Rott, K., and Camerino, D. C. (1999). Impairment of skeletal muscle adenosine triphosphate-sensitive K+ channels in patients with hypokalemic periodic paralysis. J. Clin. Invest. 103, 675–682.
Trip, J., Drost, G., Van Engelen, B. G., and Faber, C. G. (2006). Drug treatment for myotonia. Cochrane Database Syst. Rev. CD004762.
Trip, J., Drost, G., Verbove, D. J., Van Der Kooi, A. J., Kuks, J. B., Notermans, N. C., Verschuuren, J. J., De Visser, M., Van Engelen, B. G., Faber, C. G., and Ginjaar, I. B. (2008). In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotonia. Eur. J. Hum. Genet. 16, 921–929.
Tsujino, A., Maertens, C., Ohno, K., Shen, X. M., Fukuda, T., Harper, C. M., Cannon, S. C., and Engel, A. G. (2003). Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc. Natl. Acad. Sci. U.S.A. 100, 7377–7382.
Venance, S. L., Cannon, S. C., Fialho, D., Fontaine, B., Hanna, M. G., Ptácek, L. J., Tristani-Firouzi, M., Tawil, R., and Griggs, R. C. (2006). The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 129, 8–17.
Venance, S. L., Jurkat-Rott, K., Lehmann-Horn, F., and Tawil, R. (2004). SCN4A-associated hypokalemic periodic paralysis merits a trial of acetazolamide. Neurology 63, 1977.
Vicart, S., Sternberg, D., Fontaine, B., and Meola, G. (2005). Human skeletal muscle sodium channelopathies. Neurol. Sci. 26, 194–202.
Vicart, S., Sternberg, D., Fournier, E., Ochsner, F., Laforet, P., Kuntzer, T., Eymard, B., Hainque, B., and Fontaine, B. (2004). New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 63, 2120–2127.
Wagner, S., Lerche, H., Mitrovic, N., Heine, R., George, A. L., and Lehmann-Horn, F. (1997). A novel sodium channel mutation causing a hyperkalemic paralytic and paramyotonic syndrome with variable clinical expressivity. Neurology 49, 1018–1025.
Wang, D. W., Vandecarr, D., Ruben, P. C., George, A. L. Jr., and Bennett, P. B. (1999). Functional consequences of a domain 1/S6 segment sodium channel mutation associated with painful congenital myotonia. FEBS Lett. 448, 231–234.
Wang, G. K., Russell, C., and Wang, S. Y. (2004). Mexiletine block of wild-type and inactivation-deficient human skeletal muscle hNav1.4 Na+ channels. J. Physiol. 554, 621–633.
Webb, J., and Cannon, S. C. (2008). Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia. Neurology 70, 755–761.
Weber, M. A., Nielles-Vallespin, S., Essig, M., Jurkat-Rott, K., Kauczor, H. U., and Lehmann-Horn, F. (2006). Muscle Na+ channelopathies: MRI detects intracellular 23Na accumulation during episodic weakness. Neurology 67, 1151–1158.
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., and Catterall, W. A. (1992). A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914.
Wu, F. F., Gordon, E., Hoffman, E. P., and Cannon, S. C. (2005). A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation. J. Physiol. (Lond.) 565, 371–380.
Wu, F. F., Takahashi, M. P., Pegoraro, E., Angelini, C., Colleselli, P., Cannon, S. C., and Hoffman, E. P. (2001). A new mutation in a family with cold-aggravated myotonia disrupts Na(+) channel inactivation. Neurology 56, 878–884.
Yang, N., George, A. L. Jr., and Horn, R. (1997). Probing the outer vestibule of a sodium channel voltage sensor. Biophys. J. 73, 2260–2268.
Keywords: sodium channel, skeletal muscle, Nav1.4, treatment
Citation: Simkin D and Bendahhou S (2011) Skeletal muscle Na+ channel disorders. Front. Pharmacol. 2:63. doi: 10.3389/fphar.2011.00063
Received: 29 July 2011; Accepted: 28 September 2011;
Published online: 14 October 2011.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Stephen Cannon, University of Texas Southwestern Medical Center, USATheodore R Cummins, Indiana University School of Medicine, USA
Copyright: © 2011 Simkin and Bendahhou. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Saïd Bendahhou, UMR 6097, CNRS, TIANP, University of Nice Sophia Antipolis, Parc Valrose, 06108 Nice, France. e-mail: said.bendahhou@unice.fr