 Fabrice Antigny
Fabrice Antigny- Institut de Physiologie et de Biologie Cellulaires, Université de Poitiers, CNRS, Poitiers, France
Among the diverse physiological functions exerted by calcium signaling in living cells, its role in the regulation of protein biogenesis and trafficking remains incompletely understood. In cystic fibrosis (CF) disease the most common CF transmembrane conductance regulator (CFTR) mutation, F508del-CFTR generates a misprocessed protein that is abnormally retained in the endoplasmic reticulum (ER) compartment, rapidly degraded by the ubiquitin/proteasome pathway and hence absent at the plasma membrane of CF epithelial cells. Recent studies have demonstrated that intracellular calcium signals consequent to activation of apical G-protein-coupled receptors by different agonists are increased in CF airway epithelia. Moreover, the regulation of various intracellular calcium storage compartments, such as ER is also abnormal in CF cells. Although the molecular mechanism at the origin of this increase remains puzzling in epithelial cells, the F508del-CFTR mutation is proposed to be the onset of abnormal Ca2+ influx linking the calcium signaling to CFTR pathobiology. This article reviews the relationships between CFTR and calcium signaling in the context of the genetic disease CF.
Introduction
Cystic fibrosis (CF) is a genetic disease characterized by abnormal ion transport across the apical plasma membrane (PM) of many epithelial tissues, including the airways (Riordan, 1993). The most common CF mutation F508del-CF transmembrane conductance regulator (CFTR) is the deletion of phenylalanine at position 508 (F508del-CFTR) leading to chloride impermeability in many exocrine glands (salivary, airways, pancreatic) associated to reduced volume of the final secretory fluid (Kerem et al., 1989; Riordan et al., 1989; Rommens et al., 1989; Quinton, 1990). CFTR functions as a cyclic AMP-activated and ATP-gated Cl− channel that is expressed at the apical membrane of a wide variety of epithelial cell types. Although the gating of the channel is affected by the F508del-CFTR deletion, the mutant protein retains some functionality as a Cl− channel (Dalemans et al., 1991). Moreover, the protein has a much shorter half-life in the PM than wild-type CFTR (Heda et al., 2001). Therefore, therapeutic efforts have been aimed at overcoming the trafficking defect in order to increase the amount of functional F508del-CFTR proteins present at the cell membrane (Zeitlin, 2000; Becq, 2010). The F508del-CFTR proteins have an improper folding leading to its trapping in the endoplasmic reticulum (ER) by multiple protein chaperone interactions (Norez et al., 2006b) such as calnexin. Calnexin is a Ca2+-sensitive ER-resident protein that interacts with monoglucosylated, trimmed intermediates of the N-linked core glycans on newly synthesized glycoprotein (Helenius and Aebi, 2004). Although CFTR is not a Ca2+-dependent chloride channel, numerous studies have investigated the role of ER Ca2+ on CFTR trafficking and in particular in the context of the abnormal F508del-CFTR trafficking. These studies showed for example that the depletion of ER Ca2+ store by SERCA pump inhibitors (Thapsigargin, CPA, DBHQ…) leads to the partial correction of abnormal F508del-CFTR trafficking by a mechanism, at least in part, dependent on decreasing of calnexin–F508del-CFTR interaction (Egan et al., 2002, 2004; Norez et al., 2006a). Hence, the prevention of F508del-CFTR and calnexin interactions became a valuable target to identify new pharmacological agents able to restore the abnormal F508del-CFTR trafficking. Some of them are also able to modify the Ca2+ homeostasis. These results demonstrated the key role of ER Ca2+ in protein trafficking in epithelial CF and non-CF cells.
Overview of Ca2+ Signaling in Non-Excitable Cells
The diverse physiological functions of Ca2+ are possible because of the existence of a large variety of Ca2+ binding proteins that respond to a rise in [Ca2+]i and transmit the [Ca2+]i information to specific effectors. To ensure high fidelity in reading the Ca2+ signal and to minimize the energy invested in the removal of Ca2+ from the cytoplasm at the end of the Ca2+ signal, cells maintain very low resting [Ca2+]i, usually below 100 nM. The most elemental mechanism of increasing [Ca2+]i in response to extracellular stimuli is an influx into the cell of Ca2+ from the extracellular space, which contains about 2 mM Ca2+.
The ubiquitous mechanism by which external information is translated into a [Ca2+]i signal is by G-protein-coupled receptors (GPCRs). GPCRS are the largest protein family in mammals, and decode stimuli that vary from light to pheromones to peptide hormones. With such diversity, GPRCs regulate virtually any cellular function via multiple second messengers that converge on all known signaling pathways. GPRCs complexes include the receptor, a trimeric G-protein composed of Gα and GβΓ subunits (Gilman, 1987; Freissmuth et al., 1989; Dessauer et al., 1996), an effector (PLCβ in the case of Ca2+ signaling; Rebecchi and Pentyala, 2000), and regulators of G-protein signaling (RGS) proteins (Ishii and Kurachi, 2003; Wieland and Mittmann, 2003). In the case of Ca2+ signaling, the effectors are the different PLCβ isoforms, which cleave the minor lipid PIP2, to yield two different second messengers diacylglycerol (DAG) and IP3. IP3 and DAG act on separate targets. DAG activates protein kinase C (PKC) and some transient receptor potential canonical (TRPC) channels, and IP3 activates the IP3 receptors (IP3Rs) and the Ca2+ release channels in the ER (Berridge, 1993). Activation of IP3Rs by IP3 releases the Ca2+ stored in the ER to the cytoplasm. Since the ER spans the entire cell, such a system can deliver Ca2+ to virtually every domain and every site in the cytoplasm without compromising cellular homeostasis or creating large concentration gradients.
Replenishment of ER Ca2+, by SERCA pump, at the termination of the stimulation state depends on Ca2+ influx channels at the PM that sense the Ca2+ content of the ER and open in response to Ca2+ release from the ER to allow Ca2+ influx into the cytoplasm, which is used to reload the ER with Ca2+ (Parekh et al., 1997; Kiselyov et al., 2003; Parekh and Putney, 2005). Removal of Ca2+ from the cytoplasm depends on the activity of the ER and the PM Ca2+ pumps, SERCA, and PMCA, respectively (Shull, 2000). The Ca2+ signals that can last for many hours, are in the form of Ca2+ oscillations with particular amplitude and frequency.
Airway Epithelia and Calcium Signaling
Changes in cytosolic Ca2+ in non-excitable cells in response to hormone, growth factor, or cytokine that activate C-type phospholipase (PLC) are biphasic. An initial transient rise is followed by less pronounced but sustained elevation (Putney, 1987; Berridge and Irvine, 1989; Berridge, 1993). The initial phase originates from the inositol trisphosphate-induced release Ca2+ from intracellular stores and is transient due to the activity of the membrane Ca2+ pumps ATPase. The sustained phase of the increase in cytosolic Ca2+ requires the continued activation of a Ca2+ influx pathway to maintain sustained response. In airway epithelial cells like numerous cell types, the intracellular Ca2+ increase promotes many cellular processes, as the regulation of Ca2+-activated Cl− conductance (CaCC) in the airway epithelial (Paradiso et al., 1991; Grubb et al., 1994). Furthermore, CaCC conductance plays a role the maintenance of periciliary liquid (PCL) height in CF airway epithelia. In CF epithelia the PCL is more rapidly absorbed from airway surfaces and could not maintain a functional PCL height/volume under basal conditions. To compensate for the absence of CFTR–Cl− secretion, the Ca2+ activated Cl− conductance is thought to play an important role in the regulation of the height of CF epithelium during the motion phase (Tarran et al., 2005). These observations highlighted the role of intracellular Ca2+ and of CaCC conductance in CF physiopathology. Moreover, it has been well established that airway ciliary beat frequency (CBF) is strongly regulated by second messenger, such as Ca2+ (Lansley et al., 1992; Braiman et al., 1998; Evans and Sanderson, 1999). The CBF is a key factor for the regulation of mucociliary transport and thus for the mechanisms of defense of the respiratory tract (Satir and Sleigh, 1990; Wanner et al., 1996). But still, Ribeiro et al. (2005a,b) have demonstrated that Ca2+ mobilization induced by G-protein-coupled specific receptors is increased in human polarized CF epithelial cell compared to control cells and that these differences appear independent of CFTR mutation, but are the consequence of cells exposition to infectious factors.
In CF epithelial cells the presence of external Ca2+ agonists (ATP and histamine) induces a higher Ca2+ mobilization compared to non-CF cells (Antigny et al., 2008a). Several components of Ca2+ signaling pathway are disturbed in human CF cells compared to non-CF cells, and particularly ER Ca2+ release and Ca2+ entry through the PM. At ER level, IP3Rs Ca2+ release is abnormally increased in CF epithelial cells compared to non-CF cells (Antigny et al., 2009). Abnormal increase of IP3R Ca2+ release in CF human epithelial cells is proposed to be the consequence of F508del-CFTR retention in ER compartment (Antigny et al., 2009). In airway epithelial cell lines, the abnormal F508del-CFTR retention provokes concentration of the ER around nucleus followed by an increase of IP3R activity. The abnormal F508del-CFTR ER-retention induces an ER condensation (Ribeiro et al., 2005a; Antigny et al., 2009), which in turn, by causing a likely IP3Rs clustering of the ER membrane could facilitate the formation of highly sensitive Ca2+ release sites.
Ca2+ Signaling in CFTR Protein Chaperoning
The ER is a centrally located organelle, which affects most cellular functions. Its unique luminal environment consists of Ca2+ binding chaperones, which are involved in protein folding, post-translational modification, Ca2+ storage and release, and lipid synthesis and metabolism (Baumann and Walz, 2001; Berridge, 2002). The lumen of the ER contains many proteins which carry out these diverse functions (Bergeron et al., 1994; Meldolesi and Pozzan, 1998; Nicchitta, 1998; Corbett et al., 1999; Corbett and Michalak, 2000; High et al., 2000; Molinari and Helenius, 2000; Baumann and Walz, 2001; Jakob et al., 2001). Many severe diseases result from impaired function of the ER membrane and its protein folding machinery (Brooks, 1999; Jakob et al., 2001; Sherman and Goldberg, 2001). Calreticulin, a major Ca2+ binding chaperone in the ER, is a key component of the calreticulin/calnexin cycle which is responsible for the folding of newly synthesized proteins and glycoproteins and for the quality control pathways in the ER (Michalak et al., 2002; Gelebart et al., 2005). The function of calreticulin, calnexin, and other ER proteins is affected by continuous fluctuations in the concentration of Ca2+ in the ER. Thus, changes in Ca2+ concentration may play a signaling role in the lumen of the ER as well as in the cytosol (Michalak et al., 2002). Of note, the decreasing of calnexin–F508del-CFTR interaction by of ER Ca2+ depletion by SERCA pump inhibitors leads to the partial correction of abnormal F508del-CFTR trafficking (Egan et al., 2002, 2004; Norez et al., 2006a). Recently, Martino et al. (2009) demonstrated that the inflamed CF human bronchial epithelia (HBE), or normal HBE exposed to supernatant from mucopurulent material (SMM) from CF airways, exhibit ER/Ca2+ store expansion and amplified Ca2+-mediated inflammation. HBE inflammation triggers an unfolded protein response (UPR) coupled to mRNA splicing of X-box binding protein-1 (XBP-1). The link between airway epithelial inflammation, XBP-1s, and ER/Ca2+ store expansion was then addressed in murine airways challenged with phosphate-buffered saline or Pseudomonas aeruginosa. P. aeruginosa-challenged mice exhibited airway epithelial ER/Ca2+ store expansion, which correlated with airway inflammation. These findings suggest that, in inflamed HBE, XBP-1s is responsible for the ER/Ca2+ store expansion that confers amplification of Ca2+-dependent inflammatory response.
Ca2+ Signaling and TRPC Channels in CF Cells
It is well established that in secretory epithelial cells, the Ca2+ signal is dependent on the polarity of the cells and is always initiated at the apical pole to propagate to the basal pole (Petersen and Tepikin, 2008). In non-CF cells, there is no evidence in favor of the involvement of apical CFTR activity in the regulation of Ca2+ homeostasis. On the contrary, in CF cells, the different Ca2+ responses observed (versus non-CF cells) are dependent on the presence of CFTR to the PM (Antigny et al., 2008a). How can this be explained? Among others, one possible hypothesis is that TRPC channels constitute a missing link between the abnormal Ca2+ levels observed in CF cells and CFTR dysfunction.
To test for this hypothesis, the involvement of TRPC channels in airway epithelial PM Ca2+ influx has been investigated. TRPC are known to form divalent cation selective and non-selective cation channels (Nilius and Droogmans, 2001; Clapham, 2003). In respiratory diseases, TRPC6 isoform is proposed to contribute to mucus hypersecretion (Li et al., 2003). Interestingly, it was also observed that the store operated Ca2+ entry (SOCE), activated by thapsigargin-ER Ca2+ release in CF cells is similar in non-CF cells, suggesting that STIM1 protein (Stromal interacting molecule 1; Liou et al., 2005; Roos et al., 2005; Zhang et al., 2005) and Orai1 (Feske et al., 2005; Vig et al., 2006; Yeromin et al., 2006) channels are not disrupted in F508del-CFTR expressing cells (Antigny et al., 2011). Thus, TRPC6 channel could be a major actor for supporting the abnormal Ca2+ entry in CF cells. Moreover, recent evidence also suggests that both wt-CFTR and F508del-CFTR PM proteins down-regulate the TRPC6-mediated Ca2+ influx and TRPC6 up-regulates CFTR-dependent Cl− transport (Antigny et al., 2011). This reciprocal coupling has been observed not only in CF and non-CF cell lines but also in freshly isolated ciliated human epithelial cells (Antigny et al., 2011). These observations lead to the emergency of a novel model in which CFTR and TRPC6 are present within the same complex, each channel regulating the other.
However, other molecular entities could also support the abnormal Ca2+ entry in CF cells. For example, a more recent study suggests that F508del-CFTR cells have enhanced SOCE which would be mediated by the ER-resident Ca2+ sensor protein stromal interaction 1 (STIM1) and the Ca2+ release-activated channel Orai1 (Balghi et al., 2011). It is also proposed that the Ca2+ up-regulation results from the absence of CFTR at the PM but would be unrelated to the stimulation of TRPC6 channels because TRPC6 has not been detected in the cell line CF (Balghi et al., 2011) whereas it is present in freshly isolated ciliated human epithelial cells (Antigny et al., 2011).
Thus, further experiments will be needed to clearly understand the molecular mechanisms leading to PM Ca2+ influx in epithelial CF and non-CF cells and the role of TRPCs. Several TRPCs candidates are now knocking at the door such as TRPC6 and TRPC1 as recently shown in isolated pancreatic acini, a tissue in which STIM1 regulates TRPC1 (Hong et al., 2010).
Ca2+ Signaling in CF Inflammation
Cystic fibrosis airways are characterized by persistent infections and an excessive inflammatory response (Muhlebach et al., 1999; Chmiel et al., 2002; Muhlebach and Noah, 2002; Boucher, 2004). In patients with CF, lack of CFTR–Cl− channel function leads to progressive pulmonary damage frequently associated with a severe and persistent neutrophil-dominated endo-bronchial inflammation and bacterial infection (Tirouvanziam, 2006). The molecular mechanisms connecting abnormal CFTR function in airway epithelial cells to excessive lung neutrophilic inflammation have not been fully elucidated.
Abnormal CFTR function activity results in airway surface dehydration and a modification of the properties of mucus clearance that participate to the susceptibility of chronic infection with pathogens such as P. aeruginosa, staphylococcus aureus, and Haemophilus influenza (Pier et al., 1996, 1997; Chmiel and Davis, 2003; Donaldson et al., 2006; Boucher, 2007). In response to bacterial infection, CF airways epithelial cells secrete many inflammatory mediators into the airway lumen (Levine, 1995; Polito and Proud, 1998; Diamond et al., 2000). An exaggerated inflammatory response has been demonstrated in vivo and in in vitro studies. Interleukin-8 (IL-8) levels were increased in bronchial submucosal glands from patients homozygous for F508del-CFTR mutation compared to control subjects, and IL-8 release is 13-fold higher in cultured CF than in normal human bronchial glands (Tabary et al., 2000). Tabary et al. (2000) have also demonstrated that IL-8 secretion is higher in primary human bronchial gland cells derived from F508del-CFTR patients compared to non-CF bronchial gland cells. Then, it has been shown that the intracellular Ca2+ increase is an intermediary step in the signal transduction events linking cell stimulation by inflammatory factors to NF-κB (Nuclear Factor Kappa B) activation (Ribeiro et al., 2005a; Tabary et al., 2006). Moreover, the correction of F508del-CFTR abnormal localization by an incubation of CF cells at 26°C promoted a decrease of NF-κB activity via decrease of Ca2+ release (Tabary et al., 2001).
Ca2+ and Infection in CF
Two major explanations of the mechanisms of dysregulation of intracellular Ca2+ homeostasis in CF are the “infection-driven” and the “mutated CFTR-dependent” mechanistic hypothesis.
Lung mucus production is an adaptive mechanism whose function is to provide a barrier between lung cells and noxious stimuli in the inspired air. These include viruses, bacteria, dust particles, and air pollution. Relevant stimuli for mucus production in CF include bacterial pathogens and inflammation (Oliver et al., 2000). Persistent mucus hypersecretion is often associated with CF. One of the stimulatory mechanisms triggered by bacterial pathogens that contribute to mucin overproduction in CF involves Ca2+ signaling (McNamara and Basbaum, 2001). Indeed, the protein flagellin is a major structural component of bacterial flagella in both Gram positive and Gram negative bacteria and it has been shown that P. aeruginosa flagellin can elicit host cell responses through binding to a glycolipid receptor, asialoGM1 (ASGM1; Feldman et al., 1998). McNamara et al. demonstrated that ASGM1 ligation stimulates transcription of the mucin MUC 2 and this process involves the release of ATP extracellular followed by activation of cell surface ATP receptors and then Ca2+ mobilization (McNamara and Basbaum, 2001). Flagellin increases the association between flagellin receptors ASGM1 and Toll-like receptor 2 as well as 5 (TLR2 and TLR5) to stimulate the release of ATP. ATP binds and activates a G-protein-coupled nucleotide receptor on the cell surface, leading to Ca2+ mobilization (Adamo et al., 2004; McNamara et al., 2006). Finally, it is predicted that the synergistic effects of ATP and other [Ca2+]i-raising agonists to augment activation by flagellin will be larger in CF cells than in non-CF cells, potentially contributing to hyperinflammation in CF airways (Fu et al., 2007). More recently, functional studies performed in HBE cells exposed to P. aeruginosa demonstrate that phospholipase C-β3 (PLCB3), by regulating intracellular calcium transients, play a relevant role in amplifying the expression and release of IL-8, the major chemokine recruiting neutrophils in CF airway lungs (Bezzerri et al., 2011). Balghi et al. (2011) have demonstrated that elevated Ca2+ signaling in CF cells, is caused by an increase in the exocytotic insertion of Orai1 into the PM and the formation of more STIM1/Orai1 complexes during store depletion. This phenomenon induces the increased SOCE in CF cells and enhances IL-8 secretion; therefore it may contribute to the hyperinflammatory state that characterizes CF (Balghi et al., 2011).
Pseudomonas aeruginosa relies on quorum sensing molecules such as the autoinducer N-3-oxododecanoyl homoserine lactone (3O-C12) to drive the expression of numerous genes related to virulence (Erickson et al., 2002), biofilm formation (Singh et al., 2000), and antibiotic resistance (Möker et al., 2010) when colonizing the CF lung. The lactone 3O-C12 has been shown to trigger Ca2+ release from the ER in airway epithelial cells (Schwarzer et al., 2001). The autoinducer 3O-C12 has been demonstrated to induce proinflammatory cytokine production in airway epithelial cells in a calcium-dependent manner, and that dysregulated calcium storage or signaling in CF cells results in an increased production of proinflammatory cytokines (Mayer et al., 2011).
In addition, the expansion of ER observed in CF cells (Ribeiro et al., 2005b; Antigny et al., 2008b) is reversible, in that the removal of the SMM from infected/inflamed bronchi of CF patients normalized the size of the intracellular stores of CF cells and, consistently, non-CF cells show progressive expansion of intracellular calcium stores after long term incubation in vitro with SMM (Ribeiro et al., 2005a). Regarding host defense mechanisms that clear airway surfaces, the raised intracellular Ca2+ release due to ER expansion may provide an adaptive response for both the normal and CF airways. A higher Ca2+ mobilization may be particularly useful to CF patients, who depend solely on Ca2+-dependent Cl− channel to compensate for the absent cAMP-mediated Cl− secretion in CF. However, these data suggested that the increased ER size and Ca2+ storage, following chronic exposure to bacterial factors, is independent of the intrinsic F508del-CFTR defect (Ribeiro et al., 2005a). On contrary, the effect of Cftr genotype on the apoptotic response of airway epithelial cells to P. aeruginosa, indicated that HBE cells expressing F508del-CFTR underwent significantly delayed apoptosis compared to cells expressing wt-CFTR (Cannon et al., 2003). Moreover, mice with wild-type Cftr alleles had apoptotic cell in their lungs after P. aeruginosa infections, whereas mice homozygous for the F508del- or G551D-Cftr alleles showed little apoptosis in response to acute infection (Cannon et al., 2003). Then, CFTR-associated defects in apoptosis may contribute to the pathogenesis of the lung disease in CF.
In conclusion, both major hypothesis are not mutually exclusive, at least since the “mutated CFTR-dependent” mechanism could be worsened in the progression of the disease by the “infection-driven” expansion of the intracellular calcium stores.
Calcium Homeostasis and F508del-CFTR Rescue
At all hot spots of Ca2+ signaling such as global Ca2+ response, IP3R-dependent Ca2+ release, and TRPC6-dependent-Ca2+ influx, a normalization of these Ca2+ responses was observed by the rescue of F508del-CFTR, after pharmacological treatment or after 24 h at low temperature (27°C; Antigny et al., 2008a, 2009, 2011). For example, the daily treatment of CF cells for 2 months with low concentration of the corrector miglustat (N-butyldeoxynojirimycin), an inhibitor of the α-1,2 glucosidase, results in progressive, stable reversible, and sustained correction of F508del-CFTR trafficking (Norez et al., 2009). This progressive correction is also accompanied by a down regulation of Ca2+ homeostasis (Norez et al., 2009). These works demonstrated a good correlation between the presence of F508del-CFTR at the PM and the normalization of global Ca2+ mobilization induced by histamine stimulation.
At the ER level, the IP3R hyper-activity and the ER condensation observed in CF airway were also normalized by the correction of abnormal F508del-CFTR trafficking. Moreover, using CF cells which co-expressed endogenous F508del-CFTR (trapped in the ER) and exogenous wt-CFTR (localized to the PM), the abnormal IP3R activity observed in CF human epithelial cells appears to be the consequence of the retention of endogenous F508del-CFTR in ER compartment (Antigny et al., 2009). In addition to requiring IP3, IP3R are regulated in a biphasic manner by direct interaction with Ca2+, i.e., activation at low concentrations (up to 0.3 μM) and inhibition at higher concentrations (0.5–1 μM; Bezprozvanny et al., 1991). These different regulations of IP3Rs by local Ca2+ concentrations are involved in the complex feedback regulation of the Ca2+ release (De Smedt et al., 1997). According to the results of Ribeiro et al. (2005a,b), even in absence of bacterial infection, the ER network is concentrated around the nucleus and expanded throughout the nucleus in corrected and non-CF epithelial cells. This expansion or condensation of ER network is responsible for the variation in local IP3Rs Ca2+ dependent activity (Antigny et al., 2009). In corrected CF cells, IP3Rs are more distant from each other, leading to reduce propagation of the Ca2+ wave (Figure 1).
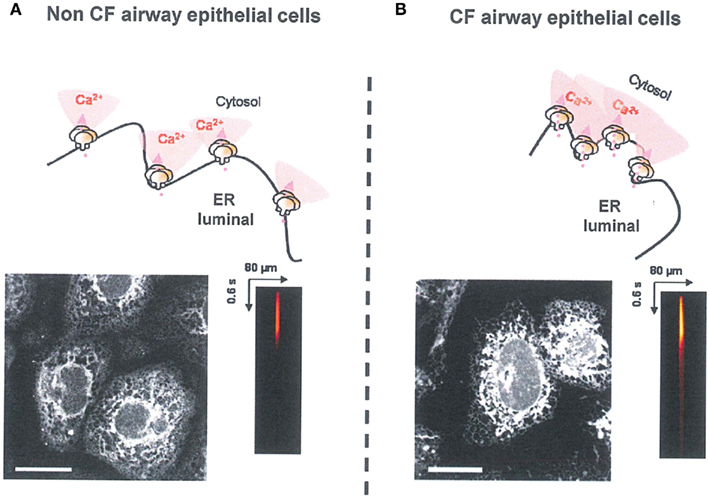
Figure 1. Correlation between ER morphology, IP3R clustering, and IP3R Ca2+ release plasma. (A) In Non-CF cells, the ER was expended at the totality of cell surface, IP3Rs are distant between others and the IP3R ER Ca2+ release was normal (B) In CF cells, the F508del-CFTR was trapped into ER. The ER was concentrated around the nucleus. IP3Rs are more clustered and the Ca2+ propagation wave was abnormally increased. ER staining was performed with ER-tracker (1 μM during 15 min). The IP3R Ca2+ release was measured by using NP-EGTA or IP3-caged techniques.
At the PM level, TRPC6 exacerbated activity in CF epithelial cells could be the result of PM CFTR absence caused by CFTR mutations and not dependent of CFTR activity (Antigny et al., 2011). The down regulation of TRPC6 activity by PM CFTR is reminiscent of the interaction observed for the epithelial Na+ channel (ENaC) and CFTR in airway epithelia (Guggino and Stanton, 2006). Indeed, although still controversial (Itani et al., 2011) CF disease is also characterized by an Na+ hyperabsorption via the apical activity of ENaC channels (Donaldson and Boucher, 2007). CFTR plays a critical role (not fully understood) in the regulation of ENaC activity via intermolecular interaction (Stutts et al., 1995; Donaldson and Boucher, 2007). Moreover, as for Ca2+ signaling (TRPC6 activity for example) the ENaC activity is also normalized by the correction of abnormal trafficking of F508del-CFTR (Noel et al., 2008). Therefore, CFTR seems to control the activity of both ENaC and TRPC6 channels.
The Ca2+ permeable TRPC cation channels are of particular interest in the pathophysiology of CF for several reasons. First, TRPC6 may contribute to mucus hypersecretion, a feature found in many respiratory diseases, including CF (Li et al., 2003). Second, the misregulation of TRPC6 channels may be relevant to CF disease because it might contribute to the increased Na+ absorption described in CF airway epithelial, as described with TRPV4 channel (Arniges et al., 2004).
Conclusion
The relationship between CFTR and Ca2+ signaling in human epithelial cells is far from being fully understood. Some studies showed that the Ca2+ signaling exacerbation in human primary epithelial cells is independent of CFTR and would be the consequence of the presence of infectious factors. But other studies proposed that perturbation of Ca2+ signaling in CF airway epithelia cells is a direct consequence of CFTR mutation and misprocessing. Also, more recent data now suggested a good correlation between CFTR-related defects and abnormal Ca2+ signaling in CF airway epithelial cell.
A novel working hypothesis is now emerging, in which CFTR and TRPC6 channels are both present within a multiprotein membranous complex, each channel down regulating the other; i.e., CFTR down-regulates TRPC6-dependent-Ca2+ influx and TRPC6 up-regulates CFTR-dependent Cl− transport. In addition STIM1, Orai1, and other TRPCs could also contribute to this signaling pathway. In addition STIM1, Orai1, and other TRPCs could also contribute to this signaling pathway. A scheme linking the putative sequence of Ca2+ signal perturbations to F508del-CFTR mutation is proposed in Figure 2.
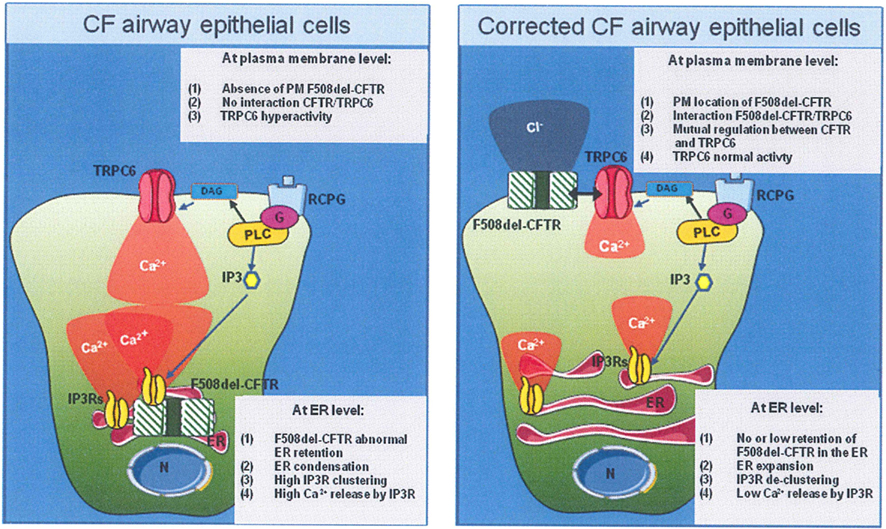
Figure 2. Proposed model linking the F508del-CFTR mutation and Ca2+ homeostasis in airway epithelial cell line. (Left panel) In CF airway epithelial phenotype, in absence of bacterial infection, the F508del-CFTR mutation caused the trapped of mutated CFTR protein into the ER, followed by the ER network condensation, inducing the IP3R clustering. The final consequence was the increased of IP3R activity. Moreover, the CFTR absence of the plasma membrane (PM) induced a hyper-activity of TRPC6 Ca2+ channel. (Right panel) The abnormal F508del-CFTR trafficking correction (pharmacologically or low temperature incubation) induced an ER network expansion. IP3Rs are more distant from each others, leading to reduce Ca2+ release. At PM level, the F508del-CFTR interacted with TRPC6. This interaction seems to induce a normalization of TRPC6 activity.
Despite the progresses made in recent years to understand the role of Ca2+ signaling in CF, further experiments will be needed to dissect and understand the complexity of the regulation of CFTR protein activity and biogenesis by Ca2+ and to precise the molecular interactions between CFTR and TRPC proteins as well as their consequences for the physiopathology of CF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our laboratory is supported by the French associations Vaincre la Mucoviscidose (VLM), ABCF2 Mucoviscidose, and Mucovie.
References
Adamo, R., Sokol, S., Soong, G., Gomez, M., and Prince, A. (2004). Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. Am. J. Respir. Cell Mol. Biol. 30, 627–634.
Antigny, F., Girardin, N., Raveau, D., Frieden, M., Becq, F., and Vandebrouck, C. (2009). Dysfunction of mitochondria Ca2+ uptake in cystic fibrosis airway epithelial cells. Mitochondrion 9, 232–241.
Antigny, F., Norez, C., Becq, F., and Vandebrouck, C. (2008a). Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium 43, 175–183.
Antigny, F., Norez, C., Cantereau, A., Becq, F., and Vandebrouck, C. (2008b). Abnormal spatial diffusion of Ca2+ in F508del-CFTR airway epithelial cells. Respir. Res. 9, 70.
Antigny, F., Norez, C., Dannhoffer, L., Bertrand, J., Raveau, D., Corbi, P., Jayle, C., Becq, F., and Vandebrouck, C. (2011). Transient receptor potential canonical channel 6 links Ca2+ mishandling to cystic fibrosis transmembrane conductance regulator channel dysfunction in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 44, 83–90.
Arniges, M., Vazquez, E., Fernandez-Fernandez, J. M., and Valverde, M. A. (2004). Swelling-activated Ca2+ entry via TRPV4 channel is defective in cystic fibrosis airway epithelia. J. Biol. Chem. 279, 54062–54068.
Balghi, H., Robert, R., Rappaze, B., Zhang, X., Wolhluter-Haddad, A., Evagelidis, A., Luo, Y., Goepp, J., Ferraro, P., Roméo, P., Trebak, M., Wiseman, P., Thomas, D., and Hanrahan, J. (2011). Enhanced Ca2 + entry due to Orai1 plasma membrane insertion increases IL-8 secretion by cystic fibrosis airways. FASEB J. 25. doi: 10.1096/fj.11-187682. [Epub ahead of print].
Baumann, O., and Walz, B. (2001). Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 205, 149–214.
Becq, F. (2010). Cystic fibrosis transmembrane conductance regulator modulators for personalized drug treatment of cystic fibrosis: progress to date. Drugs 70, 241–259.
Bergeron, J. J., Brenner, M. B., Thomas, D. Y., and Williams, D. B. (1994). Calnexin: a membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 19, 124–128.
Berridge, M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249.
Berridge, M. J., and Irvine, R. F. (1989). Inositol phosphates and cell signalling. Nature 341, 197–205.
Bezprozvanny, I., Watras, J., and Ehrlich, B. E. (1991). Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754.
Bezzerri, V., d’Adamo, P., Rimessi, A., Lannzara, C., Crovella, S., Nicolis, A., Tamanini, A., Athanasakis, E., Tebon, M., Bisoffi, G., Drumm, M., Knowles, M., Pinton, P., Gasparini, P., Berton, G., and Cabrini, G. (2011). Phospholipase C-B3 is a key modulator of LI-8 expression in cystic fibrosis bronchial epithelial cells. J. Immunol. 186, 4946–4958.
Boucher, R. C. (2004). New concepts of the pathogenesis of cystic fibrosis lung disease. Eur. Respir. J. 23, 146–158.
Boucher, R. C. (2007). Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol. Med. 13, 231–240.
Braiman, A., Zagoory, O., and Priel, Z. (1998). PKA induces Ca2+ release and enhances ciliary beat frequency in a Ca2+-dependent and -independent manner. Am. J. Physiol. 275, C790–C797.
Brooks, D. A. (1999). Introduction: molecular chaperones of the ER: their role in protein folding and genetic disease. Semin. Cell Dev. Biol. 10, 441–442.
Cannon, C., Kowalski, M., Stopak, K., and Pier, G. (2003). Pseudomonas aeruginosa-induced apoptosis is defective in respiratory epithelial cells expressing mutant cystic fibrosis transmembrane conductance regulator. Am. J. Respir. Cell Mol. Biol. 29, 188–197.
Chmiel, J. F., Berger, M., and Konstan, M. W. (2002). The role of inflammation in the pathophysiology of CF lung disease. Clin. Rev. Allergy Immunol. 23, 5–27.
Chmiel, J. F., and Davis, P. B. (2003). State of the art: why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir. Res. 4, 8.
Corbett, E. F., and Michalak, M. (2000). Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem. Sci. 25, 307–311.
Corbett, E. F., Oikawa, K., Francois, P., Tessier, D. C., Kay, C., Bergeron, J. J., Thomas, D. Y., Krause, K. H., and Michalak, M. (1999). Ca2+ regulation of interactions between endoplasmic reticulum chaperones. J. Biol. Chem. 274, 6203–6211.
Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Crystal, R. G., Pavirani, A., Lecocq, J. P., and Lazdunski, M. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354, 526–528.
De Smedt, H., Missiaen, L., Parys, J. B., Henning, R. H., Sienaert, I., Vanlingen, S., Gijsens, A., Himpens, B., and Casteels, R. (1997). Isoform diversity of the inositol trisphosphate receptor in cell types of mouse origin. Biochem. J. 322(Pt 2), 575–583.
Dessauer, C. W., Posner, B. A., and Gilman, A. G. (1996). Visualizing signal transduction: receptors, G-proteins, and adenylate cyclases. Clin. Sci. (Lond.) 91, 527–537.
Diamond, G., Legarda, D., and Ryan, L. K. (2000). The innate immune response of the respiratory epithelium. Immunol. Rev. 173, 27–38.
Donaldson, S. H., Bennett, W. D., Zeman, K. L., Knowles, M. R., Tarran, R., and Boucher, R. C. (2006). Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 354, 241–250.
Donaldson, S. H., and Boucher, R. C. (2007). Sodium channels and cystic fibrosis. Chest 132, 1631–1636.
Egan, M. E., Glockner-Pagel, J., Ambrose, C., Cahill, P. A., Pappoe, L., Balamuth, N., Cho, E., Canny, S., Wagner, C. A., Geibel, J., and Caplan, M. J. (2002). Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med. 8, 485–492.
Egan, M. E., Pearson, M., Weiner, S. A., Rajendran, V., Rubin, D., Glockner-Pagel, J., Canny, S., Du, K., Lukacs, G. L., and Caplan, M. J. (2004). Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304, 600–602.
Erickson, D., Endersby, R., Kirkham, A., Stuber, K., Vollman, D., Rabin, H., Mitchell, I., and Storey, D. (2002). Pseudomonas aeruginosa quorum-sensing systems may control virulence factor expression in the lungs of patients with cystic fibrosis. Infect. Immun. 70, 1783–1790.
Evans, J. H., and Sanderson, M. J. (1999). Intracellular calcium oscillations regulate ciliary beat frequency of airway epithelial cells. Cell Calcium 26, 103–110.
Feldman, M., Bryan, R., Rajan, S., Scheffler, L., Brunnert, S., Tang, H., and Prince, A. (1998). Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 66, 43–51.
Feske, S., Prakriya, M., Rao, A., and Lewis, R. S. (2005). A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 202, 651–662.
Freissmuth, M., Casey, P. J., and Gilman, A. G. (1989). G proteins control diverse pathways of transmembrane signaling. FASEB J. 3, 2125–2131.
Fu, Z., Bettega, K., Carroll, S., Buchholz, K., and Machen, T. (2007). Role of Ca2+ in responses of airway epithelia to Pseudomonas aeruginosa, flagellin, ATP, and thapsigargin. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L353–L364.
Gelebart, P., Opas, M., and Michalak, M. (2005). Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 37, 260–266.
Gilman, A. G. (1987). G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649.
Grubb, B. R., Vick, R. N., and Boucher, R. C. (1994). Hyperabsorption of Na+ and raised Ca(2+)-mediated Cl- secretion in nasal epithelia of CF mice. Am. J. Physiol. 266, C1478–C1483.
Guggino, W. B., and Stanton, B. A. (2006). New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat. Rev. Mol. Cell Biol. 7, 426–436.
Heda, G. D., Tanwani, M., and Marino, C. R. (2001). The Delta F508 mutation shortens the biochemical half-life of plasma membrane CFTR in polarized epithelial cells. Am. J. Physiol. Cell Physiol. 280, C166–C174.
Helenius, A., and Aebi, M. (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049.
High, S., Lecomte, F. J., Russell, S. J., Abell, B. M., and Oliver, J. D. (2000). Glycoprotein folding in the endoplasmic reticulum: a tale of three chaperones? FEBS Lett. 476, 38–41.
Hong, J. H., Li, Q., Kim, M. S., Shin, D. M., Feske, S., Birnbaumer, L., Cheng, K. T., Ambudkar, I. S., and Muallem, S. (2010). Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12, 232–243.
Ishii, M., and Kurachi, Y. (2003). Physiological actions of regulators of G-protein signaling (RGS) proteins. Life Sci. 74, 163–171.
Itani, O. A., Chen, J. H., Karp, P. H., Ernst, S., Keshavjee, S., Parekh, K., Klesney-Tait, J., Zabner, J., and Welsh, M. J. (2011). Human cystic fibrosis airway epithelia have reduced Cl- conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. U.S.A. 108, 10260–10265.
Jakob, C. A., Chevet, E., Thomas, D. Y., and Bergeron, J. J. (2001). Lectins of the ER quality control machinery. Results Probl. Cell Differ. 33, 1–17.
Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., Buchwald, M., and Tsui, L. C. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080.
Kiselyov, K., Shin, D. M., and Muallem, S. (2003). Signalling specificity in GPCR-dependent Ca2+ signalling. Cell Signal 15, 243–253.
Lansley, A. B., Sanderson, M. J., and Dirksen, E. R. (1992). Control of the beat cycle of respiratory tract cilia by Ca2+ and cAMP. Am. J. Physiol. 263, L232–L242.
Levine, S. J. (1995). Bronchial epithelial cell-cytokine interactions in airway inflammation. J. Investig. Med. 43, 241–249.
Li, S., Westwick, J., and Poll, C. (2003). Transient receptor potential (TRP) channels as potential drug targets in respiratory disease. Cell Calcium 33, 551–558.
Liou, J., Kim, M. L., Heo, W. D., Jones, J. T., Myers, J. W., Ferrell, J. E. Jr., and Meyer, T. (2005). STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241.
Martino, M. E., Olsen, J. C., Fulcher, N. B., Wolfgang, M. C., O’Neal, W. K., and Ribeiro, C. M. (2009). Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. J. Biol. Chem. 284, 14904–14913.
Mayer, M., Sheridan, J., Blohmke, C., Turvey, S., and Hancock, R. (2011). The Pseudomonas aeruginosa autoinducer 3O-C12 homoserine lactone provokes hyperinflammatory responses from cystic fibrosis airway epithelial cells. PLoS ONE 6, e16246. doi:10.1371/journal.pone.0016246
McNamara, N., and Basbaum, C. (2001). Signaling networks controlling mucin production in response to Gram-positive and Gram-negative bacteria. Glycoconj. J. 18, 715–722.
McNamara, N., Gallup, M., Sucher, A., Maltseva, I., McKemy, D., and Basbaum, C. (2006). AsialoGM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate Erk1/2. Am. J. Respir. Cell Mol. Biol. 34, 653–660.
Meldolesi, J., and Pozzan, T. (1998). The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem. Sci. 23, 10–14.
Michalak, M., Robert Parker, J. M., and Opas, M. (2002). Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 32, 269–278.
Möker, N., Dean, C., and Tao, J. (2010). Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J. Bacteriol. 192, 1946–1955.
Molinari, M., and Helenius, A. (2000). Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 288, 331–333.
Muhlebach, M. S., and Noah, T. L. (2002). Endotoxin activity and inflammatory markers in the airways of young patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 165, 911–915.
Muhlebach, M. S., Stewart, P. W., Leigh, M. W., and Noah, T. L. (1999). Quantitation of inflammatory responses to bacteria in young cystic fibrosis and control patients. Am. J. Respir. Crit. Care Med. 160, 186–191.
Nicchitta, C. V. (1998). Biochemical, cell biological and immunological issues surrounding the endoplasmic reticulum chaperone GRP94/gp96. Curr. Opin. Immunol. 10, 103–109.
Nilius, B., and Droogmans, G. (2001). Ion channels and their functional role in vascular endothelium. Physiol. Rev. 81, 1415–1459.
Noel, S., Wilke, M., Bot, A. G., De Jonge, H. R., and Becq, F. (2008). Parallel improvement of sodium and chloride transport defects by miglustat (n-butyldeoxynojyrimicin) in cystic fibrosis epithelial cells. J. Pharmacol. Exp. Ther. 325, 1016–1023.
Norez, C., Antigny, F., Becq, F., and Vandebrouck, C. (2006a). Maintaining low Ca2+ level in the endoplasmic reticulum restores abnormal endogenous F508del-CFTR trafficking in airway epithelial cells. Traffic 7, 562–573.
Norez, C., Noel, S., Wilke, M., Bijvelds, M., Jorna, H., Melin, P., DeJonge, H., and Becq, F. (2006b). Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the alpha-glucosidase inhibitor miglustat. FEBS Lett. 580, 2081–2086.
Norez, C., Antigny, F., Noel, S., Vandebrouck, C., and Becq, F. (2009). A cystic fibrosis respiratory epithelial cell chronically treated by miglustat acquires a non-cystic fibrosis-like phenotype. Am. J. Respir. Cell Mol. Biol. 41, 217–225.
Oliver, A., Canton, R., Campo, P., Baquero, F., and Blazquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lug infection. Science 288, 1251–1254.
Paradiso, A. M., Cheng, E. H., and Boucher, R. C. (1991). Effects of bradykinin on intracellular calcium regulation in human ciliated airway epithelium. Am. J. Physiol. 261, L63–L69.
Parekh, A. B., Fleig, A., and Penner, R. (1997). The store-operated calcium current I(CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell 89, 973–980.
Parekh, A. B., and Putney, J. W. Jr. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810.
Petersen, O. H., and Tepikin, A. V. (2008). Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 70, 273–299.
Pier, G. B., Grout, M., and Zaidi, T. S. (1997). Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc. Natl. Acad. Sci. U.S.A. 94, 12088–12093.
Pier, G. B., Grout, M., Zaidi, T. S., and Goldberg, J. B. (1996). How mutant CFTR may contribute to Pseudomonas aeruginosa infection in cystic fibrosis. Am. J. Respir. Crit. Care Med. 154, S175–S182.
Polito, A. J., and Proud, D. (1998). Epithelia cells as regulators of airway inflammation. J. Allergy Clin. Immunol. 102, 714–718.
Putney, J. W. Jr. (1987). Formation and actions of calcium-mobilizing messenger, inositol 1,4,5-trisphosphate. Am. J. Physiol. 252, G149–G157.
Rebecchi, M. J., and Pentyala, S. N. (2000). Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80, 1291–1335.
Ribeiro, C. M., Paradiso, A. M., Carew, M. A., Shears, S. B., and Boucher, R. C. (2005a). Cystic fibrosis airway epithelial Ca2+ i signaling: the mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia. J. Biol. Chem. 280, 10202–10209.
Ribeiro, C. M., Paradiso, A. M., Schwab, U., Perez-Vilar, J., Jones, L., O’Neal, W., and Boucher, R. C. (2005b). Chronic airway infection/inflammation induces a Ca2+ i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 280, 17798–17806.
Riordan, J. R. (1993). The cystic fibrosis transmembrane conductance regulator. Annu. Rev. Physiol. 55, 609–630.
Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N., Chou, J., Drumm, M. L., Iannuzzi, M. C., Collins, F. S., and Tsui, L. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073.
Rommens, J. M., Iannuzzi, M. C., Kerem, B., Drumm, M. L., Melmer, G., Dean, M., Rozmahel, R., Cole, J. L., Kennedy, D., Hidaka, N., Zsiga, M., Buchwald, M., Riordan, J. R., Tsui, L. C., and Collins, F. S. (1989). Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059–1065.
Roos, J., DiGregorio, P. J., Yeromin, A. V., Ohlsen, K., Lioudyno, M., Zhang, S., Safrina, O., Kozak, J. A., Wagner, S. L., Cahalan, M. D., Velicelebi, G., and Stauderman, K. A. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445.
Satir, P., and Sleigh, M. A. (1990). The physiology of cilia and mucociliary interactions. Annu. Rev. Physiol. 52, 137–155.
Schwarzer, C., Wong, S., Shi, J., Matthes, E., Illek, B., Ianowski, J. P., Arant, R. J., Isacoff, E., Vais, H., Foskett, J. K., Maiellaro, I., Hofer, A. M., and Machen, T. E. (2001). Pseudomonas aeruginosa homoserine lactone activates store-operated cAMP and CFTR-dependent Cl secretion by human airway epithelia. J. Biol. Chem. 285, 34850–34863.
Sherman, M. Y., and Goldberg, A. L. (2001). Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32.
Shull, G. E. (2000). Gene knockout studies of Ca2+-transporting ATPases. Eur. J. Biochem. 267, 5284–5290.
Singh, P., Schaefer, A., Parsek, M., Moninger, T., Welsh, M., and Greenberg, E. (2000). Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilm. Nature 407, 762–764.
Stutts, M. J., Canessa, C. M., Olsen, J. C., Hamrick, M., Cohn, J. A., Rossier, B. C., and Boucher, R. C. (1995). CFTR as a cAMP-dependent regulator of sodium channels. Science 269, 847–850.
Tabary, O., Boncoeur, E., de Martin, R., Pepperkok, R., Clement, A., Schultz, C., and Jacquot, J. (2006). Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cell Signal 18, 652–660.
Tabary, O., Escotte, S., Couetil, J. P., Hubert, D., Dusser, D., Puchelle, E., and Jacquot, J. (2000). High susceptibility for cystic fibrosis human airway gland cells to produce IL-8 through the I kappa B kinase alpha pathway in response to extracellular NaCl content. J. Immunol. 164, 3377–3384.
Tabary, O., Escotte, S., Couetil, J. P., Hubert, D., Dusser, D., Puchelle, E., and Jacquot, J. (2001). Relationship between IkappaBalpha deficiency, NFkappaB activity and interleukin-8 production in CF human airway epithelial cells. Pflugers Arch. 443(Suppl. 1), S40–S44.
Tarran, R., Button, B., Picher, M., Paradiso, A. M., Ribeiro, C. M., Lazarowski, E. R., Zhang, L., Collins, P. L., Pickles, R. J., Fredberg, J. J., and Boucher, R. C. (2005). Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J. Biol. Chem. 280, 35751–35759.
Tirouvanziam, R. (2006). Neutrophilic inflammation as a major determinant in the progression of cystic fibrosis. Drug News Perspect. 19, 609–614.
Vig, M., Peinelt, C., Beck, A., Koomoa, D. L., Rabah, D., Koblan-Huberson, M., Kraft, S., Turner, H., Fleig, A., Penner, R., and Kinet, J. P. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223.
Wanner, A., Salathe, M., and O’Riordan, T. G. (1996). Mucociliary clearance in the airways. Am. J. Respir. Crit. Care Med. 154, 1868–1902.
Wieland, T., and Mittmann, C. (2003). Regulators of G-protein signalling: multifunctional proteins with impact on signalling in the cardiovascular system. Pharmacol. Ther. 97, 95–115.
Yeromin, A. V., Zhang, S. L., Jiang, W., Yu, Y., Safrina, O., and Cahalan, M. D. (2006). Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229.
Zeitlin, P. L. (2000). Pharmacologic restoration of delta F508 CFTR-mediated chloride current. Kidney Int. 57, 832–837.
Keywords: cystic fibrosis, CFTR, trafficking, calcium signaling, calcium stores, pharmacology
Citation: Antigny F, Norez C, Becq F and Vandebrouck C (2011) CFTR and Ca2+ signaling in cystic fibrosis. Front. Pharmacol. 2:67. doi: 10.3389/fphar.2011.00067
Received: 29 July 2011;
Paper pending published: 19 August 2011;
Accepted: 11 October 2011;
Published online: 25 October 2011.
Edited by:
Jean-François Desaphy, University of Bari “Aldo Moro,” ItalyReviewed by:
Michel Vivaudou, Institut de Biologie Structurale, FranceGiulio Cabrini, University Hospital of Verona, Italy
Copyright: © 2011 Antigny, Norez, Becq and Vandebrouck. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Clarisse Vandebrouck, Institut de Physiologie et de Biologie Cellulaires, Université de Poitiers, CNRS, 1 rue Georges Bonnet, Poitiers F-86022, France. e-mail: clarisse.vandebrouck@univ-poitiers.fr