
- 1 Centre de Recherche Université Laval Robert-Giffard, Quebec City, QC, Canada
- 2 Department of Medicine, Université Laval, Quebec City, QC, Canada
- 3 Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA, USA
Voltage-gated sodium Na+ channels are membrane-bound proteins incorporating aqueous conduction pores that are highly selective for sodium Na+ ions. The opening of these channels results in the rapid influx of Na+ ions that depolarize the cell and drive the rapid upstroke of nerve and muscle action potentials. While the concept of a Na+-selective ion channel had been formulated in the 1940s, it was not until the 1980s that the biochemical properties of the 260-kDa and 36-kDa auxiliary β subunits (β1, β2) were first described. Subsequent cloning and heterologous expression studies revealed that the α subunit forms the core of the channel and is responsible for both voltage-dependent gating and ionic selectivity. To date, 10 isoforms of the Na+ channel α subunit have been identified that vary in their primary structures, tissue distribution, biophysical properties, and sensitivity to neurotoxins. Four β subunits (β1–β4) and two splice variants (β1A, β1B) have been identified that modulate the subcellular distribution, cell surface expression, and functional properties of the α subunits. The purpose of this review is to provide a broad overview of β subunit expression and function in peripheral sensory neurons and examine their contributions to neuropathic pain.
Introduction
Ten isoforms of voltage-gated Na+ channels have been identified that vary in tissue distribution, structure, biophysical properties, and sensitivity to neurotoxins (Table 1; Chahine et al., 2005). In standardized nomenclature, the nine confirmed members with >50% common amino acid identity in the transmembrane and extracellular loop regions have been designated as Nav1.1 through Nav1.9. The prefix Nav indicates the chemical symbol of the principal permeating ion (Na) with the principal physiological regulator (voltage; Catterall et al., 2003). The tenth isoform has not yet been fully identified because it has not been functionally expressed. However, this isoform plays an important role in the detection of body fluid Na+ levels and the regulation of salt intake (Watanabe et al., 2000, 2003). At least eight of the mammalian α subunits are expressed in the nervous system: Nav1.1, Nav1.2, Nav1.3, and Nav1.6 are widely expressed in the central nervous system (CNS) while Nav1.7, Nav1.8, and Nav1.9 are preferentially expressed in the peripheral nervous system (PNS; Black et al., 1996).
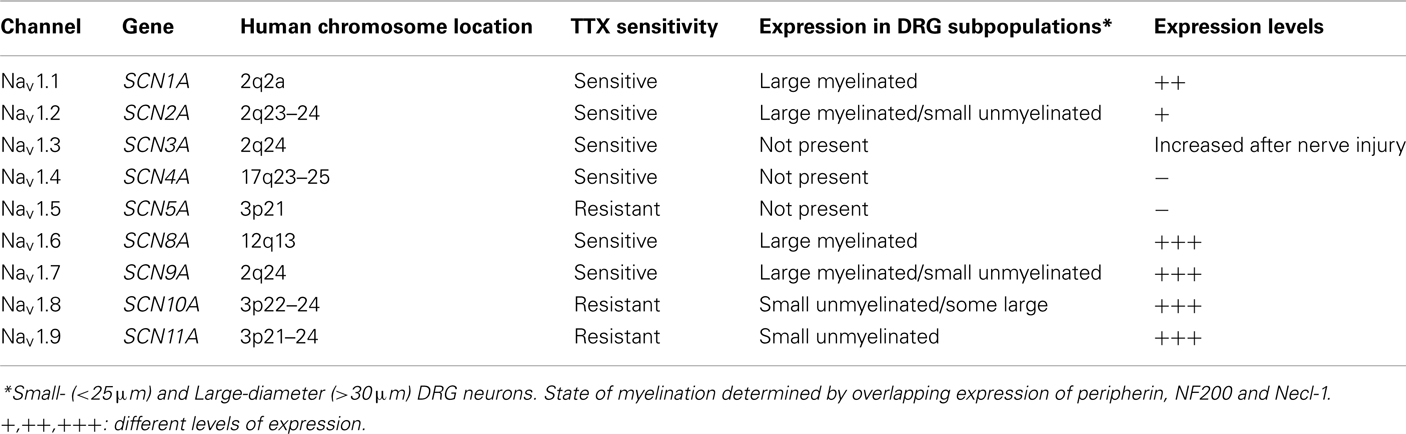
Table 1. Gene location and distribution of Na channels α subunits in subpopulations of DRG sensory neurons.
Primary sensory neurons in the dorsal root ganglia (DRG) give rise to afferent nerve fibers that convey information about thermal, mechanical, and chemical stimulations from peripheral tissues to the CNS. These neurons express a unique combination of tetrodotoxin-sensitive (TTX-S) and tetrodotoxin-resistant (TTX-R) Na+ currents that produce the rapid rising phase of action potentials. Much of what is currently known about the Na+ channels expressed in sensory neurons is derived from electrophysiological studies of cultured DRG neurons (Cummins et al., 2007; Rush et al., 2007). Small-diameter DRG neurons (<25 μm) are the cell bodies of unmyelinated C-fiber nociceptors that preferentially express TTX-R Na+ currents. This contrasts with the myelinated large-diameter (>30 μm) neurons typically associated with low-threshold A-fibers that predominately express TTX-S Na+ currents. Primary sensory neurons express a variety of Na+ channel isoforms that display properties similar to the endogenous TTX-S (Nav1.1, Nav1.2, Nav1.6, Nav1.7) and TTX-R (Nav1.8, Nav1.9) Na+ currents observed in these neurons (Black et al., 1996; Dib-Hajj et al., 1998; Amaya et al., 2000; Ho and O’Leary, 2011).
In vivo, most Na+ channel α subunits are associated with one or more auxiliary β subunits (Isom, 2002). Four distinct isoforms (β1, β4) and two splice variants (β1A, β1B) have been identified (Table 2). They share a common structure (Chahine et al., 2005) consisting of a single membrane spanning domain, a small intracellular C-terminal domain, and a large extracellular N-terminal domain incorporating an immunoglobulin-like fold similar to that of cell adhesion molecules (Figure 1; Isom, 2001; Yu et al., 2003). The β1A and β1B subunits are splice variants of β1. They share an identical N-terminal domain but have a novel C-terminal domain resulting from intron retention (Kazen-Gillespie et al., 2000; Qin et al., 2003). Na+ channel β subunits can be broadly classified based on sequence homology and molecular interactions with α subunits. The β1, β1A–B, and β3 subunits have similar amino acid sequences and form non-covalent interactions with α subunits (Isom et al., 1992; Morgan et al., 2000). This contrasts with the β2 and β4 subunits, which are best characterized as closely related (sharing 35% amino acid sequence), and which are covalently linked to α subunits via a disulfide bridge (Yu et al., 2003). In vivo Na+ channel α subunits are believed to form heteromultimeric complexes consisting of one non-covalently associated (β1, β3) and one covalently (β2, β4) linked β subunit (Catterall et al., 2005). Depending on the composition of the α–β subunit, these interactions have been shown to modulate the gating kinetics, voltage-dependence, and cell surface expression of the associated α subunits (Catterall, 2000). β subunits also function as adhesion molecules that interact with cytoskeleton proteins, the extracellular matrix, and other molecules that regulate cell migration and aggregation (Yu and Catterall, 2003; Brackenbury et al., 2008).
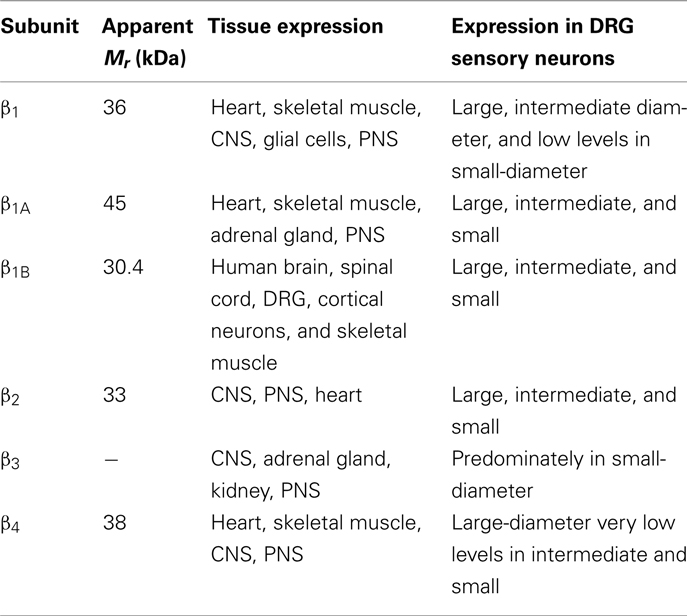
Table 2. Tissue distribution of auxiliary β subunits.
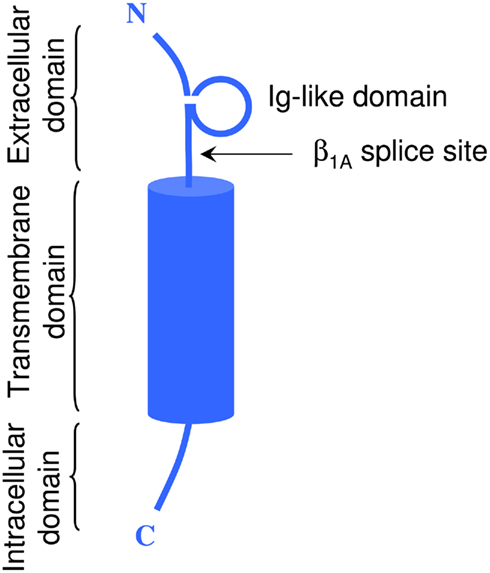
Figure 1. Schematic representation of a typical β subunit, consisting of an NH2-terminal (N) containing an Ig loop and the β1A splice site, a COOH-terminal (C) and a transmembrane-spanning segment.
Voltage-gated Na+ channels are important determinants of sensory neuron excitability, and changes in the expression and gating properties of these channels have been implicated in the development of neuropathic pain (Cummins et al., 2007; Chahine et al., 2008). Immunohistochemistry and in situ hybridization studies have shown that all four isoforms of β subunits and both slice variants are present in DRGs (Kazen-Gillespie et al., 2000; Morgan et al., 2000; Coward et al., 2001; Qin et al., 2003). Given the close physical and functional interactions between α and β subunits, it is not surprising that these auxiliary subunits are also important contributors to pain sensation (Isom, 2001). However, the precise role of these subunits in nociception and neuropathic pain has not been fully elucidated.
The β1 Subunit and its Splice Variants
It has been convincingly demonstrated that the β1 subunit regulates the expression and gating properties of Na+ channels and thereby modulates the electrical excitability of both nerves and muscles (Chahine et al., 2008). Immunohistochemistry and transcript analyses have shown that the β1 subunit is differentially expressed in subpopulations of primary sensory neurons (Oh et al., 1995; Black et al., 1996; Takahashi et al., 2003; Zhao et al., 2011). β1 is abundantly expressed in intermediate- and large-diameter (>30 μm) DRG neurons but is present at comparatively low levels in small-diameter (<25 μm) neurons. The preferential expression of β1 subunits in medium and large neurons suggests that these subunits may contribute significantly to the excitability of low-threshold A-fibers but play a reduced role in small-diameter nociceptors. This is consistent with rodent models of nerve injury where β1 expression is not significantly altered, suggesting that these subunits do not contribute significantly to the development of neuropathic pain (Shah et al., 2001; Takahashi et al., 2003).
The co-expression of β1 subunits with sensory neuron Nav1.7 and Nav1.8 Na+ channels in Xenopus oocytes accelerates current kinetics and produces a hyperpolarizing shift in steady-state inactivation (Vijayaragavan et al., 2001). In addition, β1 selectively increases Nav1.8 current density but has no effect on Nav1.7 expression. These findings indicate that β1 subunits regulate both the gating and cell surface expression of sensory neuron Na+ channels in an isoform-specific manner. More recent work using mammalian cell lines revealed a twofold increase in Nav1.8–β1 peak current density and hyperpolarizing shifts in both activation and inactivation (Zhao et al., 2011). Studies on β subunit chimeras showed that the intracellular C-terminus, but not the membrane spanning or extracellular domains of β1, was critical for retaining the functional regulation of Nav1.8 gating (Zhao et al., 2011).
The role of β1 subunits in sensory neuron excitability has been addressed using SCN1b null mice (Lopez-Santiago et al., 2007). The β1 knockouts exhibit numerous neuronal deficits, including symptoms of epilepsy and ataxia consistent with a broad distribution of this subunit in the CNS (Chen et al., 2004; Lopez-Santiago et al., 2007; Patino et al., 2009). In DRG neurons, the β1 knockout produces a slight reduction in persistent Na+ current associated with small changes in the amplitudes and gating properties of the predominant TTX-S and TTX-R Na+ currents (Lopez-Santiago et al., 2011). Overall, the subtle β1 regulation of DRG Na+ channels coupled with the low level expression in small-diameter neurons and the absence of change in models of nerve injury are inconsistent with the idea that β1 subunits contribute significantly to the development of neuropathic pain.
The β1A and β1B Subunits
There are two splice variants of the β1 subunit, the β1A subunit in the rat and β1B subunit in humans (Kazen-Gillespie et al., 2000; Qin et al., 2003). These variants have N-terminal domains that are identical to that of the β1 subunit, but have novel C-terminals resulting from intron retention. The retained β1B intron codes for a novel membrane spanning and intracellular domain that shares little sequence homology with β1 (17%) or β1A (33%). When co-expressed in oocytes, the β1B subunit increases peak Nav1.2 currents twofold but does not alter the current kinetics or gating properties of the channels (Qin et al., 2003).
The β1A subunit is highly expressed during embryonic development but decreases after birth (Kazen-Gillespie et al., 2000). Western blotting analyses have revealed that β1A is expressed in the heart, brain, spinal cord, and DRGs. When co-expressed in Chinese hamster ovary (CHO) cells, the β1B subunit produces a 2.5-fold increase in Nav1.2 current density and a slight depolarizing shift in activation (<3 mV), but no change in steady-state inactivation or current kinetics. β1A appears to preferentially increase the cell surface expression of Nav1.2 channels, a feature it shares with the parent β1 subunit. These findings suggest that β1B regulation may involve the homologous N-terminal domain that is common to the β1 and β1A variants.
The β2 Subunit
The β2 subunit is widely expressed in DRG neurons of all sizes (Coward et al., 2001; Takahashi et al., 2003) and throughout the CNS, including the spinal cord, cerebral cortex, and cerebellum (Gastaldi et al., 1998). Nav1.2 channels expressed in Xenopus oocytes result in currents that display abnormally slow activation and inactivation kinetics (Auld et al., 1988; Krafte et al., 1988). Co-expressing the β2 subunit induces more rapid activation and inactivation, which is consistent with a shift of Nav1.2 channels from a slow to a fast mode of gating (Isom et al., 1995). The slow gating observed in oocytes contrasts sharply with the properties of Nav1.2 channels expressed in CHO (West et al., 1992) and tsA201(O’Leary, 1998; Qu et al., 2001) cell lines, where rapid kinetics similar to those of native tissues are typically observed. In addition to changes in current kinetics, co-expressing the β2 subunit in oocytes results in a hyperpolarizing shift in Nav1.2 inactivation (2 mV) and a twofold increase in peak current (Isom et al., 1995). Again, this contrasts with results from tsA201 cells, where the β2 subunit produces small depolarizing shifts (3–4 mV) in Nav1.2 activation and inactivation but no changes in current kinetics or recovery from inactivation (Qu et al., 2001). This suggests that β2 regulation of Nav1.2 depends on the host cells used for expression, which may be related to differences in cellular genetic background, post-translational protein modification, or regulation by endogenous signal transduction pathways (West et al., 1992; Qu et al., 2001).
Recent work has focused on β2 subunit regulation of Na+ channel isoforms that are preferentially expressed in sensory neurons. The co-expression of Nav1.8 and β2 subunits in Xenopus oocytes results in a relatively modest depolarizing shift in inactivation (4 mV) but no change in activation, current kinetics, or peak Na+ current (Vijayaragavan et al., 2004). Subsequent studies of Nav1.8–β2, Nav1.6–β2, and Nav1.3–β2 channels expressed in mammalian cells largely confirmed these findings, demonstrating little or no effect of β2 on voltage-dependence, kinetics, or current density (Cummins et al., 2001; Zhao et al., 2011). Similar results have been observed in preliminary studies of heterologously expressed Nav1.7–β2 channels (Ho et al., 2011). Overall, the β2 subunit appears to weakly regulate many of the voltage-gated Na+ channels expressed in sensory neurons.
This contrasts with studies of null mice, where the knockout of the β2 subunit is associated with reductions in TTX-S Na+ current amplitude, mRNA, and protein (Lopez-Santiago et al., 2006). This suggests that β2 expression in DRG neurons increases TTX-S Na+ current amplitude and accelerates current kinetics, effects that are not widely observed in α–β co-expression studies. The underlying cause of this discrepancy is not known. One possibility is that β2 subunits in native DRG neurons interact with endogenous proteins or are the target of signal transduction processes that are not reconstituted in heterologous expression systems. This possibility has gained credence from studies showing that the expression of Nav1.3 in DRG cells results in a depolarizing shift in activation and faster recovery from inactivation compared to Nav1.3 channels expressed in HEK293 cells (Cummins et al., 2001). Interactions with endogenous β subunits or other cell-specific proteins could account for the observed differences in gating properties. Alternatively, the apparent differences in Na+ channel function observed in knockout and heterologous expression studies may stem from the compensatory upregulation of related β subunits and Na+ channel isoforms in null mice (Chen et al., 2004; Yu et al., 2006). Additional studies of the changes in α and β subunit expression that occur in β2 null mice, or the development of conditional β2 knockouts that reduce the opportunity for subunit compensation, may shed light on the apparent discrepancy between the in vivo and in vitro effects of β2 subunit regulation.
Several studies have examined the contribution of β2 subunits to the development of pain behaviors in rodent models of nerve injury. A study investigating β subunit expression using RT-PCR and in situ hybridization found that β2 mRNA levels in DRG neurons are not significantly altered following peripheral nerve injury (Takahashi et al., 2003). However, subsequent studies of β2 protein expression using immunohistochemistry and Western blotting revealed that the β2 protein is upregulated following nerve injuries (Pertin et al., 2005). β2 upregulation has been observed in both injured and uninjured sensory neurons, suggesting that the β2 subunit contributes to the excitability of both these populations. This possibility is supported by studies showing that the β2 knockout decreases the expression of TTX-S Na+ channels in DRG neurons (Lopez-Santiago et al., 2006). Importantly, the mechanical allodynia associated with peripheral nerve injury is attenuated in β2 null mice, which is consistent with a role for this subunit in the development of neuropathic pain (Pertin et al., 2005).
The β3 Subunit
In situ hybridization has shown that β3 subunit mRNA is highly expressed in small- (<25 μm) and medium-diameter (25–45 μm) DRG neurons and to a lesser extent in large-diameter (>45 μm) neurons (Shah et al., 2000, 2001). The cellular distribution of β3 expression extensively overlaps that of TTX-R Nav1.8 and Nav1.9 channels, which are primarily expressed in nociceptors (Akopian et al., 1996; Sangameswaran et al., 1996). In rodent models of neuropathic pain, β3 mRNA increases in C-fiber nociceptors following chronic constriction (Shah et al., 2000), spared nerve ligation, and sciatic nerve transection (Takahashi et al., 2003), and in medium-diameter Aδ fibers in the streptozocin rodent model of diabetes (Shah et al., 2001). The upregulation of β3 observed in animal models of nerve injury is consistent with the increase in β3 protein in human DRG neurons following avulsion injuries (Casula et al., 2004). The preferential expression of β3 subunits in small DRG neurons and their upregulation in models of nerve injury support the idea that β3 is an important contributor to both acute and chronic pain.
β3 Subunits also appear to play a major role in the development of neuropathic pain. Chronic constriction injury and sciatic nerve axotomy have been shown to induce an increase in TTX-S Na+ currents (Cummins and Waxman, 1997) that has been linked to the enhanced expression of Nav1.3 channels in small- and medium-sized DRG neurons (Waxman et al., 1994; Dib-Hajj et al., 1996, 1999; Black et al., 1999; Kim et al., 2002; Takahashi et al., 2003). The injury-induced increase in Nav1.3 expression is paralleled by a similar increase in β3 mRNA and protein levels (Shah et al., 2000; Takahashi et al., 2003). Heterologous expression studies have shown that co-expressing the β3 subunit produces depolarizing shifts in Nav1.3 activation and inactivation, faster recovery from inactivation, and slower current kinetics (Cummins et al., 2001). One possibility is that the upregulation of Nav1.3 channels and β3 subunits may be an attempt by the neurons to compensate for the injury-induced decrease in the expression of Nav1.8 and Nav1.9 channels (Dib-Hajj et al., 1996, 1999; Sleeper et al., 2000). Replacing the slowly gating TTX-R Nav1.8 current with the more rapid TTX-S current of Nav1.3–β3 channels is predicted to reduce the action potential threshold and promote high-frequency firing, thereby contributing to the hyperexcitability of injured DRG neurons (Cummins et al., 2001). However, immunohistochemical analysis suggests that Nav1.3 channels are preferentially upregulated in medium to large size DRG neurons after nerve injury (Kim et al., 2001; Fukuoka et al., 2008) and therefore may not extensively overlap with Nav1.8 channels primarily expressed in small-diameter nociceptors.
Early studies of Nav1.8 channels expressed in Xenopus oocytes found that co-expressing β3 increases Na+ current density and produces a hyperpolarizing shift in activation (Shah et al., 2000). This contrasts with later studies showing that co-expressing β3 in oocytes produces a depolarizing shift in Nav1.8 inactivation but no change in current density (Vijayaragavan et al., 2004). Studies on Nav1.8 expressed in mammalian cells revealed that β3 causes a 31% decrease in peak current density but no change in activation or steady-state inactivation (Zhao et al., 2011). Collectively, these findings suggest that co-expressing the β3 subunit either has no effect or reduces Nav1.8 current density, without altering voltage-dependence or gating kinetics. Similar findings have been reported for the β3 regulation of Nav1.6, a rapidly gating TTX-S Na+ channel that is preferentially expressed at the nodes of Ranvier of peripheral nerve fibers (Krzemien et al., 2000; Tzoumaka et al., 2000; Ulzheimer et al., 2004) and in large-diameter sensory neurons (Black et al., 1996; Fukuoka et al., 2008; Ho and O’Leary, 2011). Heterologous expression studies have indicated that co-expression with the β3 subunit does not alter the peak current density, current kinetics, or voltage-dependence of Nav1.6 channels (Zhao et al., 2011).
The β4 Subunit
The mature β4 subunit protein has a large extracellular Ig-like fold, a single membrane spanning segment, and a short cytoplasmic C-terminal domain that is structurally similar to those of the β1–β3 subunits. β4 shares high amino acid identity (35%) with β2 and includes an extracellular unpaired cysteine that enables β4 to covalently associate with Na+ channel α subunits via disulfide bonds (Yu et al., 2003). The β4 subunit is highly expressed in DRGs and at lower levels in the brain and spinal cord. At the cellular level, β4 is abundantly expressed in large-diameter sensory neurons and at lower levels in intermediate and small neurons (Yu et al., 2003).
The co-expression of β4 with the Nav1.2 channel in tsA201 cells produces a hyperpolarizing shift in activation (−7 mV) but no change in steady-state inactivation (Yu et al., 2003). The effects of β4 on the gating properties of the TTX-S Nav1.6 and TTX-R Nav1.8 channels have also been studied (Chen et al., 2008; Zhao et al., 2011). Co-expressing β4 produces pronounced hyperpolarizing shifts in activation (−17 mV) and steady-state inactivation (−9 mV) of Nav1.8, and a smaller hyperpolarizing shift (−8 mV) in Nav1.6 activation (Zhao et al., 2011). β4 subunits produce similar negative shifts in the activation of the neuronal Nav1.1 and skeletal muscle Nav1.4 channels (Yu et al., 2003; Aman et al., 2009). The consistent hyperpolarizing shift in activation produced by the β4 subunit suggests that this subunit may modulate neuronal excitability by causing Na+ channels to activate at more hyperpolarized voltages.
Resurgent currents were initially described in Purkinje neurons where they were found to promote the discharge of multiple action potentials in response to brief depolarizations (Raman and Bean, 1997, 1999). Subsequent work found that the open-channel block at depolarized voltages coupled with rapid unblocking and slow Na+ channel deactivation at voltages near threshold produce an inward Na+ current (resurgent current) that transiently depolarizes the neurons (Grieco et al., 2005). These resurgent currents increase excitability and are believed to underlie the high-frequency firing of Purkinje neurons (Raman and Bean, 2001). The cytoplasmic C-terminus of the β4 subunit has emerged as a likely candidate for the endogenous blocking particle responsible for resurgent currents (Grieco et al., 2005; Bant and Raman, 2010). This possibility is supported by studies showing that siRNA targeting SCN4b abolishes resurgent currents in cultured cerebellar granule cells and that the exogenous application of synthetic β4 C-terminal peptide 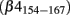 blocks Na+ currents and induces resurgent currents in inactivation-impaired Purkinje neurons. Resurgent currents are substantially reduced in Purkinje neurons isolated from Nav1.6 null mice, indicating that these channels play an important role in the production of resurgent currents (Raman et al., 1997). However, persistent resurgent currents have been reported in the subthalamic nucleus and Purkinje neurons isolated from Nav1.6 null mice, suggesting that other Na+ channel isoforms may also produce these currents (Do and Bean, 2004; Grieco and Raman, 2004).
blocks Na+ currents and induces resurgent currents in inactivation-impaired Purkinje neurons. Resurgent currents are substantially reduced in Purkinje neurons isolated from Nav1.6 null mice, indicating that these channels play an important role in the production of resurgent currents (Raman et al., 1997). However, persistent resurgent currents have been reported in the subthalamic nucleus and Purkinje neurons isolated from Nav1.6 null mice, suggesting that other Na+ channel isoforms may also produce these currents (Do and Bean, 2004; Grieco and Raman, 2004).
The role of β subunits in the generation of resurgent currents has been further investigated in vitro. Co-expressing the β4 subunit does not induce resurgent currents in heterologously expressed Nav1.1 (Aman et al., 2009), Nav1.6 (Zhao et al., 2011), or Nav1.8 (Zhao et al., 2011) channels, indicating that the association with the intact β4 subunit alone is insufficient to produce resurgent current. Additional proteins or post-translational modifications appear to be required to recapitulate the resurgent currents observed in native neurons (Grieco et al., 2002). These endogenous proteins and regulatory pathways may be highly specific to particular cell types and may thus be absent in the mammalian cells lines that are widely used for heterologous expression and cellular electrophysiology studies (Theile and Cummins, 2011). Alternatively, β4-mediated resurgent currents may involve cell-specific enzymatic cleavage by proteases such as β-site amyloid precursor protein cleaving enzyme 1 (BACE1) or other proteases that are required to produce the functionally active blocking peptide (Huth et al., 2011).
Resurgent currents are observed in 40% of large-diameter (35–50 μm) DRG neurons and are substantially reduced in neurons from Nav1.6 null mice (Cummins et al., 2005). Large DRG neurons express both the Nav1.6 (Black et al., 1996; Fukuoka et al., 2008; Ho and O’Leary, 2011) and β4 subunits (Yu et al., 2003), further supporting the idea that Nav1.6–β4 channels may play a role in these currents. This contrasts with small-diameter DRG neurons that do not routinely produce resurgent currents (Cummins et al., 2005) and that express low levels of Nav1.6 (Black et al., 1996; Fukuoka et al., 2008; Ho and O’Leary, 2011) and β4 subunits (Zhao et al., 2011).
Resurgent currents have recently been implicated in the neuronal hyperexcitability and pain associated with paroxysmal extreme pain disorder (PEPD; Jarecki et al., 2010; Theile and Cummins, 2011). In particular, the I1467T mutation in the interdomain III–IV linker of the Nav1.7 channel reduces the rate of inactivation, increases the persistent Na+ current, and induces a depolarizing shift in steady-state inactivation (Fertleman et al., 2006; Jarecki et al., 2008). These changes are consistent with impaired fast inactivation, which increases the probability of open-channel block, a suspected contributor to the generation of resurgent currents (Grieco and Raman, 2004). When heterologously expressed in cultured DRG neurons, the Nav1.7–I1467T mutant channel increases both the percentage of neurons displaying resurgent currents and the peak current amplitude (Theile et al., 2011). Computer simulations further support the idea that PEPD mutations that alter Nav1.7 inactivation induce resurgent currents in DRG neurons that contribute to aberrant action potential firing and increased cellular excitability. The evidence supporting a role for resurgent currents in the development of neuropathic pain is compelling and warrants further investigation.
Summary
All four isoforms (β1–β4) and both splice variants (β1A, β1B) of β subunits are broadly expressed in the PNS. These subunits interact with many of the Na+ channel isoforms in sensory neurons and alter the expression, voltage-dependence, and gating properties of these channels. β subunits are differentially expressed in large-diameter mechanoreceptors (β1, β4) and small-diameter nociceptors (β3). This pattern of β subunit expression suggests that these auxiliary subunits may differentially regulate voltage-gated Na+ currents and the excitability of these neuronal populations. Injury-induced changes in β subunit expression and the altered functional regulation of the Na+ channels expressed in sensory neurons contribute to the hyperexcitability and ectopic firing of sensory neurons. Current evidence suggests that β subunits are important contributors to sensory physiology, nociception, and neuropathic pain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the Heart and Stroke Foundation of Québec and the Canadian Institute of Health Research (CIHR, INO-77909) to M. Chahine; and by a grant from the National Institute of General Medical Sciences (R01GM-078244) to M. E. O’Leary.
References
Akopian, A. N., Sivilotti, L., and Wood, J. N. (1996). A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262.
Aman, T. K., Grieco-Calub, T. M., Chen, C., Rusconi, R., Slat, E. A., Isom, L. L., and Raman, I. M. (2009). Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J. Neurosci. 29, 2027–2042.
Amaya, F., Decosterd, I., Samad, T. A., Plumpton, C., Tate, S., Mannion, R. J., Costigan, M., and Woolf, C. J. (2000). Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol. Cell. Neurosci. 15, 331–342.
Auld, V. J., Goldin, A. L., Krafte, D. S., Marshall, J., Dunn, J. M., Catterall, W. A., Lester, H. A., Davidson, N., and Dunn, R. J. (1988). A rat brain Na+ channel α?subunit with novel gating properties. Neuron 1, 449–461.
Bant, J. S., and Raman, I. M. (2010). Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 107, 12357–12362.
Black, J. A., Cummins, T. R., Plumpton, C., Chen, Y. H., Hormuzdiar, W., Clare, J. J., and Waxman, S. G. (1999). Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J. Neurophysiol. 82, 2776–2785.
Black, J. A., Dib-Hajj, S., McNabola, K., Jeste, S., Rizzo, M. A., Kocsis, J. D., and Waxman, S. G. (1996). Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res. Mol. Brain Res. 43, 117–131.
Brackenbury, W. J., Djamgoz, M. B., and Isom, L. L. (2008). An emerging role for voltage-gated Na+ channels in cellular migration: regulation of central nervous system development and potentiation of invasive cancers. Neuroscientist 14, 571–583.
Casula, M. A., Facer, P., Powell, A. J., Kinghorn, I. J., Plumpton, C., Tate, S. N., Bountra, C., Birch, R., and Anand, P. (2004). Expression of the sodium channel β3 subunit in injured human sensory neurons. Neuroreport 15, 1629–1632.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2003). International Union of Pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol. Rev. 55, 575–578.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Chahine, M., Chatelier, A., Babich, O., and Krupp, J. J. (2008). Voltage-gated sodium channels in neurological disorders. CNS Neurol. Disord. Drug Targets 7, 144–158.
Chahine, M., Ziane, R., Vijayaragavan, K., and Okamura, Y. (2005). Regulation of Na(v) channels in sensory neurons. Trends Pharmacol. Sci. 26, 496–502.
Chen, C., Westenbroek, R. E., Xu, X., Edwards, C. A., Sorenson, D. R., Chen, Y., McEwen, D. P., O’Malley, H. A., Bharucha, V., Meadows, L. S., Knudsen, G. A., Vilaythong, A., Noebels, J. L., Saunders, T. L., Scheuer, T., Shrager, P., Catterall, W. A., and Isom, L. L. (2004). Mice lacking sodium channel β1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J. Neurosci. 24, 4030–4042.
Chen, Y., Yu, F. H., Sharp, E. M., Beacham, D., Scheuer, T., and Catterall, W. A. (2008). Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol. Cell. Neurosci. 38, 607–615.
Coward, K., Jowett, A., Plumpton, C., Powell, A., Birch, R., Tate, S., Bountra, C., and Anand, P. (2001). Sodium channel β1 and β2 subunits parallel SNS/PN3 α-subunit changes in injured human sensory neurons. Neuroreport 12, 483–488.
Cummins, T. R., Aglieco, F., Renganathan, M., Herzog, R. I., Dib-Hajj, S. D., and Waxman, S. G. (2001). Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 21, 5952–5961.
Cummins, T. R., Dib-Hajj, S. D., Herzog, R. I., and Waxman, S. G. (2005). Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 579, 2166–2170.
Cummins, T. R., Sheets, P. L., and Waxman, S. G. (2007). The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 131, 243–257.
Cummins, T. R., and Waxman, S. G. (1997). Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 17, 3503–3514.
Dib-Hajj, S., Black, J. A., Felts, P., and Waxman, S. G. (1996). Down-regulation of transcripts for Na channel α-SNS in spinal sensory neurons following axotomy. Proc. Natl. Acad. Sci. U.S.A. 93, 14950–14954.
Dib-Hajj, S. D., Black, J. A., Cummins, T. R., Kenney, A. M., Kocsis, J. D., and Waxman, S. G. (1998). Rescue of α-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J. Neurophysiol. 79, 2668–2676.
Dib-Hajj, S. D., Fjell, J., Cummins, T. R., Zheng, Z., Fried, K., LaMotte, R., Black, J. A., and Waxman, S. G. (1999). Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 83, 591–600.
Do, M. T., and Bean, B. P. (2004). Sodium currents in subthalamic nucleus neurons from Nav1.6-null mice. J. Neurophysiol. 92, 726–733.
Fertleman, C. R., Baker, M. D., Parker, K. A., Moffatt, S., Elmslie, F. V., Abrahamsen, B., Ostman, J., Klugbauer, N., Wood, J. N., Gardiner, R. M., and Rees, M. (2006). SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774.
Fukuoka, T., Kobayashi, K., Yamanaka, H., Obata, K., Dai, Y., and Noguchi, K. (2008). Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J. Comp. Neurol. 510, 188–206.
Gastaldi, M., Robaglia-Schlupp, A., Massacrier, A., Planells, R., and Cau, P. (1998). mRNA coding for voltage-gated sodium channel beta2 subunit in rat central nervous system: cellular distribution and changes following kainate-induced seizures. Neurosci. Lett. 249, 53–56.
Grieco, T. M., Afshari, F. S., and Raman, I. M. (2002). A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar Purkinje neurons. J. Neurosci. 22, 3100–3107.
Grieco, T. M., Malhotra, J. D., Chen, C., Isom, L. L., and Raman, I. M. (2005). Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron 45, 233–244.
Grieco, T. M., and Raman, I. M. (2004). Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J. Neurosci. 24, 35–42.
Ho, C., and O’Leary, M. E. (2011). Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol. Cell. Neurosci. 46, 159–166.
Ho, C., Zhao, J., Malinowski, S., Chahine, M., and O’Leary, M. E. (2011). Differential expression of sodium channel β subunits and Na channel regulation in subpopulations of dorsal root ganglion sensory neurons. Society of Neurosciences Meeting.
Huth, T., Rittger, A., Saftig, P., and Alzheimer, C. (2011). beta-Site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel beta4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflugers Arch. 461, 355–371.
Isom, L. L. (2002). β subunits: players in neuronal hyperexcitability? Novartis Found. Symp. 241, 124–138.
Isom, L. L., De Jongh, K. S., Patton, D. E., Reber, B. F., Offord, J., Charbonneau, H., Walsh, K., Goldin, A. L., and Catterall, W. A. (1992). Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science 256, 839–842.
Isom, L. L., Ragsdale, D. S., De Jongh, K. S., Westenbroek, R. E., Reber, B. F. X., Scheuer, T., and Catterall, W. A. (1995). Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442.
Jarecki, B. W., Piekarz, A. D., Jackson, J. O., and Cummins, T. R. (2010). Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J. Clin. Invest. 120, 369–378.
Jarecki, B. W., Sheets, P. L., Jackson, J. O., and Cummins, T. R. (2008). Paroxysmal extreme pain disorder mutations within the D3/S4-S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J. Physiol. 586, 4137–4153.
Kazen-Gillespie, K. A., Ragsdale, D. S., D’Andrea, M. R., Mattei, L. N., Rogers, K. E., and Isom, L. L. (2000). Cloning, localization, and functional expression of sodium channel β1A subunits. J. Biol. Chem. 275, 1079–1088.
Kim, C. H., Oh, Y., Chung, J. M., and Chung, K. (2001). The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res. Mol. Brain Res. 95, 153–161.
Kim, C. H., Oh, Y., Chung, J. M., and Chung, K. (2002). Changes in three subtypes of tetrodotoxin sensitive sodium channel expression in the axotomized dorsal root ganglion in the rat. Neurosci. Lett. 323, 125–128.
Krafte, D. S., Snutch, T. P., Leonard, J. P., Davidson, N., and Lester, H. A. (1988). Evidence for the involvement of more than one mRNA species in controlling the inactivation process of rat and rabbit brain Na channels expressed in Xenopus oocytes. J. Neurosci. 8, 2859–2868.
Krzemien, D. M., Schaller, K. L., Levinson, S. R., and Caldwell, J. H. (2000). Immunolocalization of sodium channel isoform NaCh6 in the nervous system. J. Comp. Neurol. 420, 70–83.
Lopez-Santiago, L. F., Brackenbury, W. J., Chen, C., and Isom, L. L. (2011). Na+ channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J. Biol. Chem. 286, 22913–22923.
Lopez-Santiago, L. F., Meadows, L. S., Ernst, S. J., Chen, C., Malhotra, J. D., McEwen, D. P., Speelman, A., Noebels, J. L., Maier, S. K., Lopatin, A. N., and Isom, L. L. (2007). Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J. Mol. Cell. Cardiol. 43, 636–647.
Lopez-Santiago, L. F., Pertin, M., Morisod, X., Chen, C., Hong, S., Wiley, J., Decosterd, I., and Isom, L. L. (2006). Sodium channel β2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J. Neurosci. 26, 7984–7994.
Morgan, K., Stevens, E. B., Shah, B., Cox, P. J., Dixon, A. K., Lee, K., Pinnock, R. D., Hughes, J., Richardson, P. J., Mizuguchi, K., and Jackson, A. P. (2000). β3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. U.S.A. 97, 2308–2313.
Oh, Y., Sashihara, S., Black, J. A., and Waxman, S. G. (1995). Na+ channel β1 subunit mRNA: differential expression in rat spinal sensory neurons. Brain Res. Mol. Brain Res. 30, 357–361.
O’Leary, M. E. (1998). Characterization of the isoform-specific differences in the gating of neuronal and muscle sodium channels. Can. J. Physiol. Pharmacol. 76, 1041–1050.
Patino, G. A., Claes, L. R., Lopez-Santiago, L. F., Slat, E. A., Dondeti, R. S., Chen, C., O’Malley, H. A., Gray, C. B., Miyazaki, H., Nukina, N., Oyama, F., De, J. P., and Isom, L. L. (2009). A functional null mutation of SCN1B in a patient with Dravet syndrome. J. Neurosci. 29, 10764–10778.
Pertin, M., Ji, R. R., Berta, T., Powell, A. J., Karchewski, L., Tate, S. N., Isom, L. L., Woolf, C. J., Gilliard, N., Spahn, D. R., and Decosterd, I. (2005). Upregulation of the voltage-gated sodium channel β2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 25, 10970–10980.
Qin, N., D’Andrea, M. R., Lubin, M. L., Shafaee, N., Codd, E. E., and Correa, A. M. (2003). Molecular cloning and functional expression of the human sodium channel β1B subunit, a novel splicing variant of the β1 subunit. Eur. J. Biochem. 270, 4762–4770.
Qu, Y., Curtis, R., Lawson, D., Gilbride, K., Ge, P., Distefano, P. S., Silos-Santiago, I., Catterall, W. A., and Scheuer, T. (2001). Differential modulation of sodium channel gating and persistent sodium currents by the β1, β2, and β3 subunits. Mol. Cell. Neurosci. 18, 570–580.
Raman, I. M., and Bean, B. P. (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 17, 4517–4526.
Raman, I. M., and Bean, B. P. (1999). Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J. Neurosci. 19, 1663–1674.
Raman, I. M., and Bean, B. P. (2001). Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys. J. 80, 729–737.
Raman, I. M., Sprunger, L. K., Meisler, M. H., and Bean, B. P. (1997). Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19, 881–891.
Rush, A. M., Cummins, T. R., and Waxman, S. G. (2007). Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14.
Sangameswaran, L., Delgado, S. G., Fish, L. M., Koch, B. D., Jakeman, L. B., Stewart, G. R., Sze, P., Hunter, J. C., Eglen, R. M., and Herman, R. C. (1996). Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem. 271, 5953–5956.
Shah, B. S., Stevens, E. B., Gonzalez, M. I., Bramwell, S., Pinnock, R. D., Lee, K., and Dixon, A. K. (2000). beta3, a novel auxiliary subunit for the voltage-gated sodium channel, is expressed preferentially in sensory neurons and is upregulated in the chronic constriction injury model of neuropathic pain. Eur. J. Neurosci. 12, 3985–3990.
Shah, B. S., Stevens, E. B., Pinnock, R. D., Dixon, A. K., and Lee, K. (2001). Developmental expression of the novel voltage-gated sodium channel auxiliary subunit β3, in rat CNS. J. Physiol. 534, 763–776.
Sleeper, A. A., Cummins, T. R., Dib-Hajj, S. D., Hormuzdiar, W., Tyrrell, L., Waxman, S. G., and Black, J. A. (2000). Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J. Neurosci. 20, 7279–7289.
Takahashi, N., Kikuchi, S., Dai, Y., Kobayashi, K., Fukuoka, T., and Noguchi, K. (2003). Expression of auxiliary beta subunits of sodium channels in primary afferent neurons and the effect of nerve injury. Neuroscience 121, 441–450.
Theile, J. W., and Cummins, T. R. (2011). Inhibition of Nav{beta}4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole and anandamide. Mol. Pharmacol. 80, 724–734.
Theile, J. W., Jarecki, B. W., Piekarz, A. D., and Cummins, T. R. (2011). Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J. Physiol. 589, 597–608.
Tzoumaka, E., Tischler, A. C., Sangameswaran, L., Eglen, R. M., Hunter, J. C., and Novakovic, S. D. (2000). Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J. Neurosci. Res. 60, 37–44.
Ulzheimer, J. C., Peles, E., Levinson, S. R., and Martini, R. (2004). Altered expression of ion channel isoforms at the node of Ranvier in P0-deficient myelin mutants. Mol. Cell. Neurosci. 25, 83–94.
Vijayaragavan, K., O’Leary, M. E., and Chahine, M. (2001). Gating properties of Nav1.7 and Nav1.8 peripheral nerve sodium channels. J. Neurosci. 21, 7909–7918.
Vijayaragavan, K., Powell, A. J., Kinghorn, I. J., and Chahine, M. (2004). Role of auxiliary β1-, β2-, and β?-subunits and their interaction with Na(v)1.8 voltage-gated sodium channel. Biochem. Biophys. Res. Commun. 319, 531–540.
Watanabe, E., Fujikawa, A., Matsunaga, H., Yasoshima, Y., Sako, N., Yamamoto, T., Saegusa, C., and Noda, M. (2000). Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J. Neurosci. 20, 7743–7751.
Watanabe, U., Shimura, T., Sako, N., Kitagawa, J., Shingai, T., Watanabe, E., Noda, M., and Yamamoto, T. (2003). A comparison of voluntary salt-intake behavior in Nax-gene deficient and wild-type mice with reference to peripheral taste inputs. Brain Res. 967, 247–256.
Waxman, S. G., Kocsis, J. D., and Black, J. A. (1994). Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 72, 466–470.
West, J. W., Scheuer, T., Maechler, L., and Catterall, W. A. (1992). Efficient expression of rat brain type IIA Na+ channel α subunits in a somatic cell line. Neuron 8, 59–70.
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4, 207.
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., Spain, W. J., McKnight, G. S., Scheuer, T., and Catterall, W. A. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149.
Keywords: voltage-gated sodium channel, pain, β subunit, peripheral nervous system
Citation: Chahine M and O’Leary ME (2011) Regulatory role of voltage-gated Na+ channel β subunits in sensory neurons. Front. Pharmacol. 2:70. doi: 10.3389/fphar.2011.00070
Received: 07 September 2011; Accepted: 19 October 2011;
Published online: 21 November 2011.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Massimo Mantegazza, University of Nice Sophia Antipolis, FranceSaïd Bendahhou, Cantre National de la Recherche Scientifique, France
Copyright: © 2011 Chahine and O’Leary. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Mohamed Chahine, Centre de Recherche Université Laval Robert-Giffard, Local F-6539, 2601, chemin de la Canardière, Quebec City, QC, Canada G1J 2G3. e-mail: mohamed.chahine@phc.ulaval.ca