 Fabrio Sitzia1
Fabrio Sitzia1- 1 Pfizer Applied Neurophysiology Group, School of Physiology and Pharmacology, University of Bristol, Bristol, UK
- 2 Neuroscience Research Unit, Pfizer, Groton, CT, USA
Alpha7 nicotinic acetylcholine receptors (α7 nAChR) are widely distributed throughout the central nervous system and are found at particularly high levels in the hippocampus and cortex. Several lines of evidence indicate that pharmacological enhancement of α7 nAChRs function could be a potential therapeutic route to alleviate disease-related cognitive deficits. A recent pharmacological approach adopted to increase α7 nAChR activity has been to identify selective positive allosteric modulators (PAMs). α7 nAChR PAMs have been divided into two classes: type I PAMs increase agonist potency with only subtle effects on kinetics, whereas type II agents produce additional dramatic effects on desensitization and deactivation kinetics. Here we report novel observations concerning the pharmacology of the canonical type II PAM, PNU120596. Using patch clamp analysis of acetylcholine (ACh)-mediated currents through recombinant rat α7 nAChR we show that positive allosteric modulation measured in two different ways is greatly attenuated when the temperature is raised to near physiological levels. Furthermore, PNU120596 largely removes the strong inward rectification usually exhibited by α7 nAChR-mediated responses.
Introduction
It has long been appreciated that activation of central nervous system (CNS) nicotinic receptors can produce a variety of behavioral changes, including improvements to cognitive function. The most abundant nicotinic acetylcholine receptor (nAChR) subtypes in the mammalian brain are α7-subunit containing homomers (α7 nAChRs) and α4β2 heteromers. Expression of α7 nAChRs receptors is predominantly, but not exclusively, observed in cortex and hippocampus, whereas α4β2 receptors have a somewhat more widespread distribution (Lindstrom, 1996). At the cellular level α7 nAChRs are reported to be found on both neurons and glial cells (Gotti et al., 2006).
Pharmacological and molecular manipulation of both α7- and α4- containing nAChRs has indicated that they both potentially play a role in cognitive processes (Leiser et al., 2009). As a consequence of such findings, both receptors have become enthusiastically pursued drug targets, particularly for those seeking to normalize the cognitive deficits that contribute to the phenotype of devastating CNS diseases, such as schizophrenia and Alzheimer’s disease (Olincy et al., 2006; Leiser et al., 2009; Hajos and Rogers, 2010; Haydar and Dunlop, 2010; Thomsen et al., 2010). These research efforts have produced a growing number of receptor subtype-selective compounds active at either α7 nAChRs or α4β2 nAChRs receptors.
Our own research focus is the α7 nAChR. Evidence specifically implicating this particular nAChR in disease pathophysiology comes from a number of studies. Decreased α7 nAChR expression has been reported in brains from schizophrenia sufferers (Freedman et al., 2001a,b). Genetic linkage studies in schizophrenia implicate the region of the α7 nAChRs gene promoter where polymorphisms result in diminished promoter efficacy to drive receptor expression in vitro (Leonard et al., 2002). Selective α7 nAChRs receptor agonists have also been shown to normalize sensory gating deficits in animal models of schizophrenia (Stevens et al., 1998; Simosky et al., 2001; Hajos et al., 2005), and a recent human study has provided proof-of-concept for the normalization of auditory gating deficits in schizophrenics (Olincy et al., 2006).
The α7 nAChRs is a homomeric Ca2+-permeable ligand-gated channel. It is activated by ACh, choline, and (−)-nicotine and is antagonized by α-bungarotoxin and MLA. Kinetically, α7 nAChRs both activate and deactivate with fast kinetics, they also exhibit a very rapid and profound desensitization that is likely to have important functional consequences. Behavioral experiments suggest that increasing activation of α7 nAChR generates improved cognition in rodents (Arendash et al., 1995a,b; Levin et al., 1999). Recent efforts have lead to the discovery of a number of novel full and partial α7 nAChR agonists that exhibit good selectivity over both other nicotinic receptors and a wide range of other targets (reviewed in Hajos and Rogers, 2010; Haydar and Dunlop, 2010).
An additional pharmacological approach to increasing α7 nAChR activity is through positive allosteric modulation (Bertrand and Gopalakrishnan, 2007). Broad spectrum positive allosteric modulators (PAMs) of nicotinic receptors have been available for some time. More recently agents with good selectivity for α7 nAChRs have been described. Based on their characterization in electrophysiological studies it has recently been proposed that α7 nAChRs PAMs can be subdivided into two classes. Type I PAMs predominantly affect apparent agonist affinity but can also enhance maximum responsiveness, whereas type II PAMs, such as PNU120596 (Hurst et al., 2005), additionally produce profound changes to receptor kinetics, in particular desensitization and deactivation (Gronlien et al., 2007). Recent studies have extended the evaluation of PNU120596 potentiation of α7 nAChRs in some detail to suggest the presence of two distinct desensitization states, one PNU120596 sensitive and the other insensitive to PNU120596 (Williams et al., 2011), although the in vivo significance of such findings remains to be determined. The emerging behavioral properties of α7 nAChRs PAMs in vivo (Hurst et al., 2005; Gronlien et al., 2007; Ng et al., 2007; Timmermann et al., 2007; Dunlop et al., 2009) indicate they share the cognitive-enhancing and normalization of sensory gating properties previously described for α7 nAChR agonists.
In this study we have analyzed in detail the actions at recombinant rat α7 nAChRs of the canonical type II PAM PNU120596. We show for the first time that the pharmacology of these molecules has striking temperature dependence. Furthermore, PNU120596 largely removes the strong inward rectification usually exhibited by α7 nAChRs.
Materials and Methods
Cell Line and Cell Culture
GH4C1 cells stably transfected with rat α7 nAChRs (GH4C1-rα7 cells) were cultured and passaged in standard tissue culture flasks before being transferred to, and cultured on, glass coverslips. The culture medium consisted of Dulbecco’s Modified Eagles Medium (DMEM, Cambrex) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Biosera), 1% penicillin–streptomycin, and 200 μg/ml hygromycin B (Invitrogen) at 37°C in a humidified atmosphere composed of 95% air and 5% CO2.
Electrophysiology
A cell-bearing coverslip was broken into numerous pieces and a single shard placed into a continuously perfused chamber on the stage of an inverted microscope (Nikon Eclipse TE300). The extracellular solution was a standard HEPES-buffered saline (HBSS) consisting of: (in mM) NaCl, 135; KCl, 5; HEPES–NaOH, 10; MgCl2, 1; CaCl2, 2; D-glucose, 30; pH 7.3.
Standard whole-cell patch clamp recordings were made using an Axopatch-200B amplifier (Axon Instruments Inc.) under the control of the pClamp 9.2 software suite (Axon Instruments Inc.). Patch clamp electrodes were of resistance of 3–5 MΩ when filled with the intracellular solution which consisted of (in mM): CsCl, 120; HEPES–CsOH, 10; EGTA, 10; QX314-Br, 5; ATP disodium salt, 4; GTP-disodium salt, 0.3; MgCl2, 4; (pH 7.3). The pairing of this solution with HBSS resulted in a calculated liquid junction potential error of 5 mV, which was arithmetically corrected for in analysis. Whole-cell voltage clamp recordings were established in GH4C1-rα7 cells using standard methods. The holding potential was −75 mV except during the determination of current–voltage relationships where the membrane potential was increased stepwise from −75 in 20 mV increments. To permit rapid solution exchange cells were detached from, and lifted above the underlying coverslip and placed adjacent to the control barrel of a fast-switching multibarrel perfusion system (Warner Instruments). To apply drugs the barrels were translated horizontally so the cell was exposed to the drug-containing solution flow from a different, adjacent barrel. The temperature of the solutions applied from all barrels of this device could be set at a single defined level using a commercially available multichannel temperature controller (Warner Instruments). Data are presented as mean ± SEM.
Drugs
Acetylcholine (ACh), choline, and (−)-nicotine were purchased from Sigma-Aldrich, UK. Methyllycaconitine (MLA), PNU120596 were purchased from Tocris Biosciences, UK.
Results
Patch Clamp Analysis of α7 nAChR-Mediated Responses
Application of ACh, (−)-nicotine, and choline all produced the expected inward currents in GH4C1-rα7 cells voltage-clamped at −75 mV. At high concentrations of each of these agonists, inward currents rapidly appeared and then promptly desensitized (Figure 1A). The remaining work in this manuscript employs only ACh as an agonist, since this is likely to be the major physiological activator of alpha7 receptors in vivo.
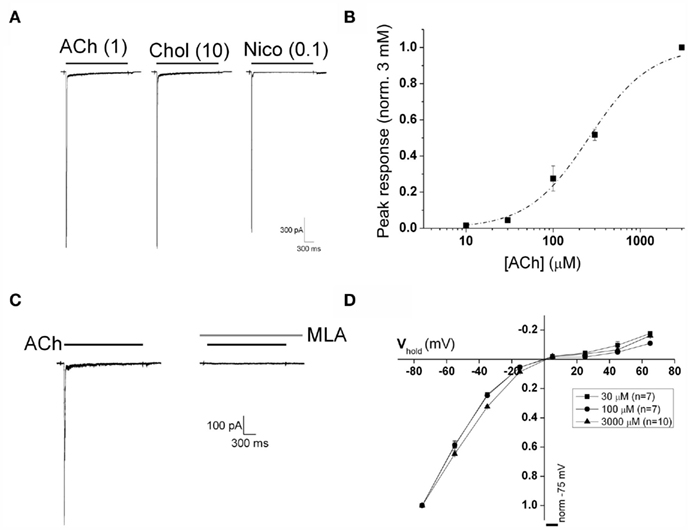
Figure 1. Activation of recombinant α7 nAChR stably expressed in GH4C1 cells. (A) Two seconds duration applications of ACh (1 mM), choline (10 mM), and nicotine (100 μM) elicited α7 nAChR-mediated fast inward currents that rapidly desensitize. (B) Pooled peak current versus concentration data from seven cells. All data from each cell were normalized to the response obtained with 3 mM ACh. The dashed line is a logistic fit to the concentration–response curve of ACh-evoked peak current (EC50 = 260 μM; n = 7). (C) ACh-evoked currents (3 mM) are completely abolished by pre-application of the selective α7 nAChR blocker MLA (100 nM). (D) Current–voltage relationships of currents evoked by three different ACh concentrations [30 μM (n = 7), 100 μM (n = 7), and 3 mM, (n = 10)]. Responses are normalized to the response recorded at a holding potential of −75 mV. Note the strong inward rectification at all agonist concentrations.
The concentration–response behavior of ACh-mediated α7 nAChR activation was determined using 2 s agonist applications at concentrations between 10 and 3000 μM. A plot of peak current versus concentration for such data pooled from seven cells is shown in Figure 1B. The mean EC50 determined from fitting these datasets individually was 260 ± 31 μM, while the mean Hill coefficient was 1.4 ± 0.1. The kinetics of current responses were also strongly concentration-dependent with both activation and desensitization being faster at higher agonist concentrations. Deactivation following removal of 30 μM ACh occurred with a mean time constant of 54.5 ± 9.1 ms, n = 7. Responses to 3 mM ACh were completely eliminated by MLA (100 nM; Figure 1C), confirming currents evoked by ACh in these cells are mediated solely by α7 nAChR. This antagonism was reversed upon MLA washout with a time constant of recovery of approximately 240 s (data not shown). An important caveat to note here is that the estimation of α7 nAChR peak current and kinetics of channel activation and desensitization are impacted profoundly by the kinetics of solution exchange. The α7 nAChR is unique among ligand-gated ion channels with respect to its extremely rapid activation and desensitization kinetics. Although we have employed a rapid perfusion system to activate α7 nAChR channels the estimation of ACh potency and kinetic parameters under such conditions is predominantly governed by the leading edge of the solution application.
Current–voltage relationships for α7 nAChR-mediated currents were obtained by varying the holding potential at which ACh was applied. In line with previous observations, under control conditions the peak current versus voltage plot exhibited strong inward rectification, with little outward current observed at positive potentials (Figure 1D). Similarly strong rectification was observed with all three different ACh concentrations, which produced approximately 10, 30, and 100% levels of peak response amplitude.
Electrophysiological Actions of PNU120596 at Room Temperature
PNU120596 was first described as a PAM at α7-AChR receptors by Hurst et al. (2005). It has more recently been put forward as a canonical example of so-called type II PAMs (Gronlien et al., 2007). In addition to enhancing agonist-induced currents and increasing apparent agonist affinity, type II PAMs produce dramatic effects on receptor kinetics. This behavior is illustrated in Figure 2A where responses to 3 mM ACh from the same cell recorded in either the absence (left) or maintained presence (right) of PNU120596 are compared. In the presence of PNU120596, the maximum current was larger, desensitization was largely eliminated and deactivation was massively slowed. The time constant of deactivation in the presence of PNU120596 (1 μM) was 5211 ± 838 ms (n = 10), close to 100× slower than that seen in the absence of this PAM. Application of PNU120596 produced no agonist effects in its own right.
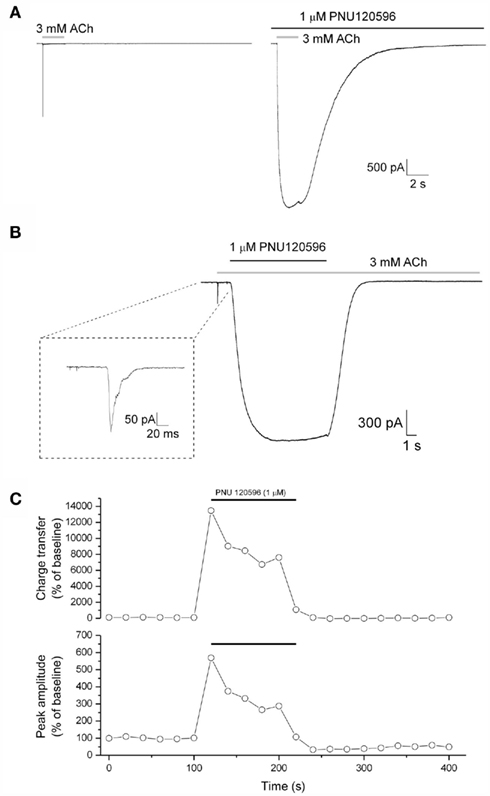
Figure 2. Modulation of ACh-evoked currents by PNU120596. (A) Responses to ACh (3 mM, gray bar; left) are increased in amplitude and kinetically modified in the presence of preapplied PNU120596 (1 μM, black bar; right). (B) An example trace illustrating PNU120596-mediated dedesensitization. Receptors were initially activated and desensitized by ACh (3 mM, gray bar) following which PNU120596 was applied in the maintained presence of ACh. The initial rapidly desensitizing ACh response is shown in the inset box. Following the removal of PNU120596 α7 nAChRs redesensitize over several seconds. (C) The time course of responses to repeated ACh applications (3 mM, 2 s, every 20 s) from a typical recording. Data for both response area (top) and peak amplitude (bottom) are shown, both normalized to the mean baseline value (as 100%). PNU120596 was continuously applied for the time indicated by the black bars.
In addition to slowing desensitization of agonist responses (Figure 2A), PNU120596 was also able to effectively dedesensitize α7 nAChRs previously completely desensitized by application of a high concentration of ACh. An example of this activity is shown in Figure 2B. Here the receptors were first activated and then desensitized by applying 3 mM ACh for 1 s, PNU120596 was applied then in the maintained presence of ACh. This elicited a large non-desensitizing current response that rose to peak with a time constant of 2300 ± 265 ms (n = 17); a rate that is likely to reflect the association kinetics of PNU120596 with its binding site (formally [PNU]. kon + koff). The peak current generated by such dedesensitization was much larger (8.6 ± 2.0-fold; n = 17) than that produced by the initial desensitizing application of 3 mM ACh (an approximately EC99 agonist concentration when applied alone). Removal of PNU120596, in the maintained presence of ACh, caused the receptors to redesensitize with a mean half time of 1456 ± 175 ms (n = 9). This redesensitization rate is >300 times slower than the initial rate of desensitization (which has a half time below 5 ms), and thus the redesensitization kinetics are likely to predominantly reflect the off-rate of PNU120596 from its binding site. In agreement with the rapid redesensitization of α7 nAChR on PNU removal in the presence of ACh (Figure 2B), the effects of preapplied PNU120596 on both the amplitude and kinetics of ACh-evoked responses were rapidly reversed following PNU120596 washout (Figure 2C).
Using the pre-application protocol described above (Figure 2A) we were able to show that PNU120596 also produced a marked change in the voltage-dependence of α7 nAChR-mediated responses (Figures 3A,B). Thus, although current–voltage relationships of agonist responses (3 mM ACh) exhibited marked inward rectification (Figures 1D and 3B), the current–voltage relationship in the presence of PNU120596 was considerably more linear. This was quantified by calculating the rectification index which was 1.38 ± 0.1 for ACh responses in the presence of PNU120596 (n = 5) compared to 3.82 ± 0.3 for responses to 3 mM ACh alone (n = 10, Figure 3C). Consequently, the fold increase in peak current produced by PNU120596 was strongly voltage-dependent. Like other effects of PNU120596 (Figure 2C), the linearization of the I–V relationship was reversed when this agent was removed.
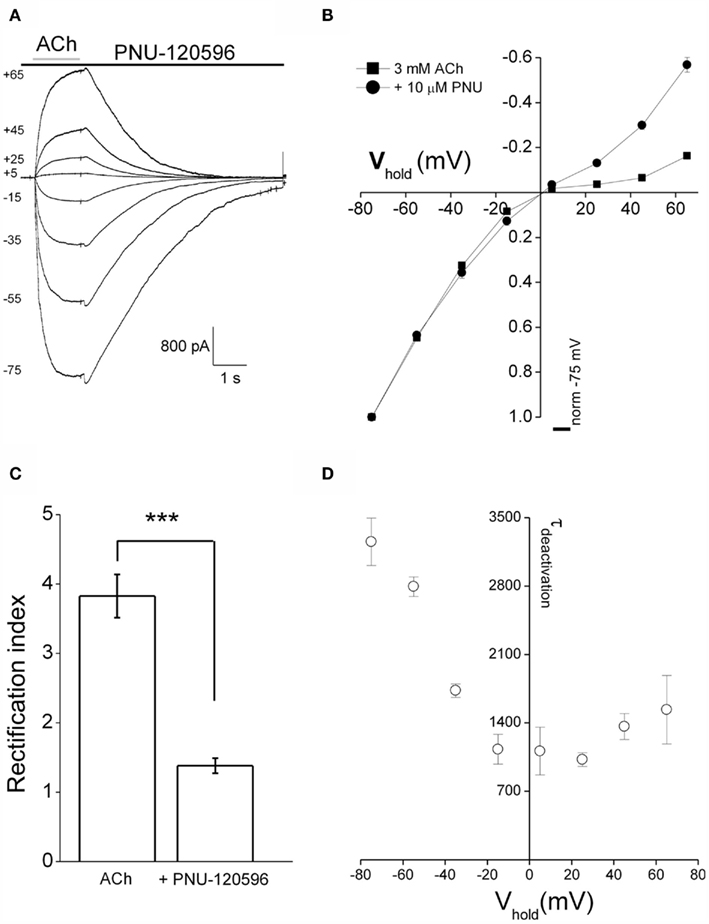
Figure 3. PNU120596 alters voltage-dependence of α7 nAChR-mediated responses. (A) Currents from a standard I–V relationships in response to ACh (3 mM) applied in the continuous presence of 10 μM PNU120596 at room temperature. The numbers to the left of the traces are the holding potential in millivolt. (B) Pooled I–V relationships for ACh alone (3 mM; n = 10, squares) and ACh + PNU120596 (n = 5, circles). PNU120596-modulated ACh-evoked currents have a more linear I–V relationship while currents recorded in ACh alone show classically strong inward rectification. (C) A comparison of rectification index of currents evoked by ACh alone (n = 10) and ACh applied in the presence of PNU120596 (n = 5). Rectification index is defined as the chord conductance between −75 and −55 mV divided by that between +45 and +65 mV. Rectification ratios recorded in PNU120596 were different (***P < 0.001, using Student’s T-test) when compared to currents evoked by ACh (3 mM) alone. (D) A graph plotting the voltage-dependence of the deactivation time constant of ACh-induced currents recorded in the maintained presence of PNU120596. Time constant was determined by making single exponential fits to the current decay following ACh removal. As is visibly apparent in (A), deactivation is considerably faster at less negative membrane potentials.
We also noted that the rate of current deactivation on ACh removal in the presence of PNU120596, as well as being massively slowed, was voltage-dependent. Thus around three times faster deactivation being seen at positive membrane potentials than at a holding potential of −75 mV. This is readily apparent in the traces in Figure 3A as well as the pooled results of exponential fits in Figure 3D.
Temperature Dependence of PNU120596 Actions at α7 nAChRs
The vast majority of patch clamp-based studies of ligand-gated channel pharmacology and biophysics are performed at room temperature, presumably because it is technically much simpler so to do. The findings of such studies are then frequently used to interpret studies in brain slices or in vivo which frequently involve higher temperatures (often around 33°C for brain slices and ~37°C in vivo). PNU120596 has documented in vivo actions and we are ultimately interested in understanding how these actions are generated at a circuit level. Consequently, we were motivated to understand how PNU120596 affected α7 nAChR gating at temperatures around 37°C. To do this we modified our stepping perfusion system such that we could warm the various solution streams, in parallel, to the same temperature.
Increasing the temperature from room temperature (22°C) to physiological temperature (~37°C) produced somewhat variable effects of the amplitude of ACh responses. On average, however, peak amplitude was 20 ± 20% smaller, a non-significant change. Charge transfer over 2 s was significantly reduced by 54 ± 10% (n = 8, Figures 4A,B), due to faster desensitization combined with the slightly reduced peak.
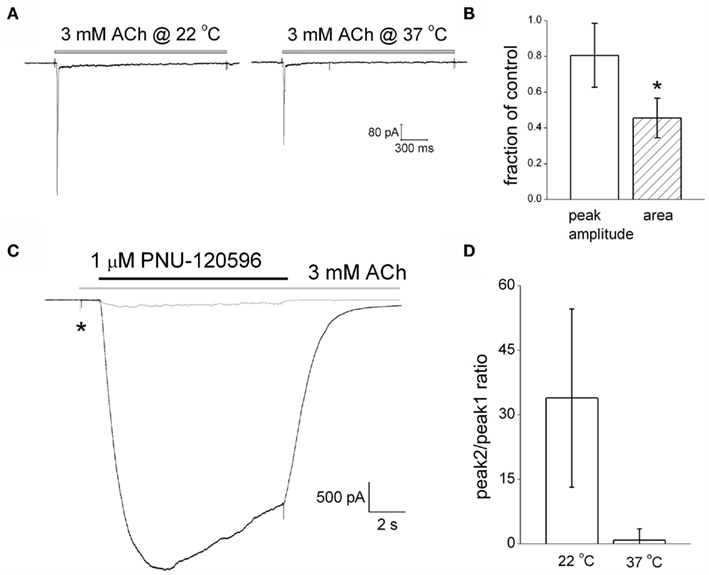
Figure 4. PNU120596-mediated dedesensitization is strongly attenuated at physiological temperatures. (A) ACh-mediated current responses at room temperature and 37°C in the same example cell. (B) A graph plotting the changes in amplitude and charge transfer produced by increasing the recording temperature to 37°C. (C) Example traces showing PNU120596-mediated reversal of ACh-evoked desensitization at room temperature (black trace) and 37°C (gray trace). (D) A histogram showing the ratio between the dedesensitized peak current (P2) evoked by PNU120596 and the initial desensitizing ACh response (P1) at room temperature and 37°C.
We initially studied the temperature dependence of the actions of PNU120596 using the dedesensitization protocol (Figure 4C). As described above (Figure 2B), this involves using a high agonist concentration to completely predesensitize the α7 nAChR population before co-applying PNU120596 to induce recovery (dedesentization) of the receptor response in the maintained presence of agonist. As before, we quantified the effectiveness of a PAM in this assay by plotting the ratio of the dedesensitized response in the presence of the PAM (P2), to that of the initial ACh response prior to desensitization (P1). At room temperature this ratio was 34 ± 20 (n = 4). When we increased the experimental temperature in the same cells it was immediately apparent that PNU120596 became much less effective at dedesensitizing the α7 nAChRs. Thus the P2:P1 ratio fell below 1 (0.86 ± 2; n = 4) at temperature of ~37°C (Figure 4D).
As well as comparing the dedesensitization produced by PNU120596 at room and physiological temperature we also examined the temperature dependence of agonist responses at receptors pre-equilibrated with this PAM. From the example traces shown in Figure 5A, it is clear that the actions of the PNU120597 are greatly curtailed at higher temperatures. This is not only seen in the response amplitude in the presence of PNU120596 but also, for example, in the decreased PAM-mediated slowing of deactivation.
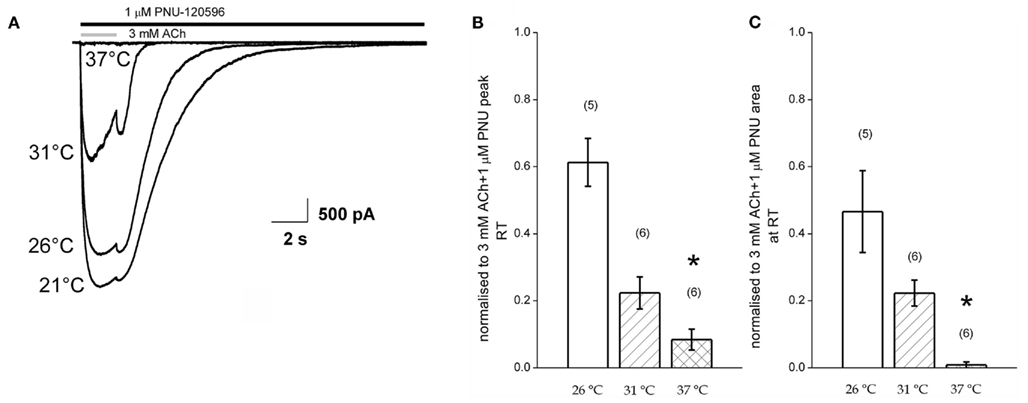
Figure 5. Potentiation of agonist responses at receptors pre-equilibrated with PNU120596 is reduced at higher temperatures. (A) Example responses to 3 mM ACh in the maintained presence of PNU120596 at four different temperatures. (B,C) Pooled data plotting the amplitude (left) and charge transfer (right) of response to ACh in the presence of PNU in three different recording temperature bins. Data are normalized to the corresponding responses recorded at room temperature.
Figures 5B,C plot pooled data from six such experiments. Specifically these graphs plot, for three binned levels of elevated temperature, the relative amplitude (Figure 5B) and charge transfer (Figure 5C) seen in PNU120596 normalized to that seen at room temperature in the same cells. These indicate that the loss of the potentiating effect of the PNU120596 intimately depends on temperature and can be observed with just a few degrees change above room temperature. Together with the dedesensitization measurements in Figure 4 these data provide good evidence that the allosteric actions of PNU120596 are strongly temperature-dependent within the range of standard biological experimentation.
Lastly, we were interested to determine if the temperature dependence of the positive allosteric modulation of α7 nAChR by PNU120596 extended to other structurally diverse α7 nAChR PAMs. We have previously reported on the α7 nAChR PAM activity of SB-206553 (Dunlop et al., 2009), a compound originally characterized as a 5-HT2B/C receptor antagonist. Figure 6 shows the dedesensitization protocol evaluating the effect of SB-206553 on ACh-evoked α7 nAChR currents showing a strongly temperature-dependent effect, similar to PNU120596, with reduced dedesensitization at near physiological temperatures compared to room temperature. Similar to PNU120596, the magnitude of potentiation of α7 nAChR currents in response to ACh in cells pre-equilibrated with SB-206553 was reduced at physiological temperature compared to room temperature (data not shown).
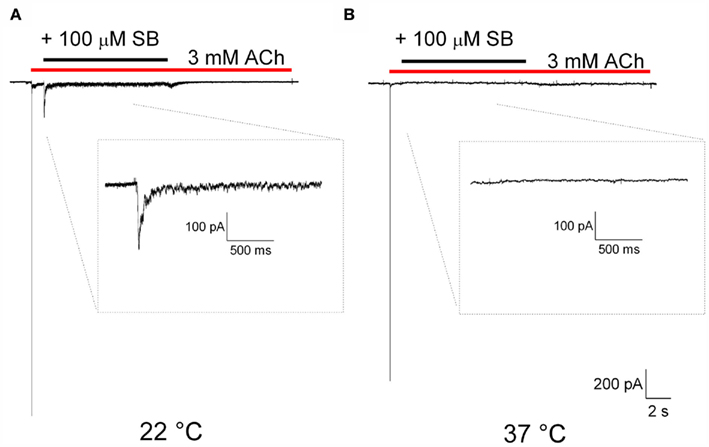
Figure 6. SB-206553-mediated dedesensitization is strongly attenuated at physiological temperatures. (A) Example trace showing SB-206553-mediated reversal of ACh-evoked desensitization at room temperature, and (B) Example trace showing lack of SB-206553-mediated reversal of ACh-evoked desensitization at 37°C, illustrating a strong temperature dependence for this SB-206553 mediated effect.
Discussion
Selective pharmacological manipulation of α7 nicotinic receptors holds considerable promise for the future pharmacotherapy of schizophrenia and other human diseases characterized by significant cognitive deficits (Leiser et al., 2009; Hajos and Rogers, 2010; Haydar and Dunlop, 2010; Thomsen et al., 2010). Furthermore, pharmacological modulation of α7 nAChR also has some potential promise as a neuroprotective strategy (Kihara et al., 2001; Hellstrom-Lindahl et al., 2004; Hu et al., 2007; Roncarati et al., 2009). The current over-arching view is that increasing activity of α7 nAChRs is the required route to improved cognitive function, a supposition that is supported by behavioral data using agonists, and to a lesser extent PAMs. It is this view-point that has shaped the recent extensive drug discovery efforts in many pharmaceutical companies around the world.
At room temperature, the type II PAM PNU120596 produces remarkable effects on gating of α7 receptors. As well as potentiating peak current generated by agonist concentrations that produce near maximal responses in the absence of a PAM, this agent almost eliminates desensitization and slows deactivation by around 100-fold (Figure 2A). Furthermore, this agent can completely dedesensitize α7 nAChRs that have been fully desensitized with a high agonist concentration (Figure 2B). At room temperature these effects are massive compared to the positive allosteric modulation of other neurotransmitter-gated channels by well known drugs, for example, the effects of benzodiazepines on GABAA receptors and the actions of so-called “AMPAkines” on the AMPA sub-class of glutamate receptors. Importantly, all of the effects of PNU120596 we observed in both activation and desensitization assays were substantially reduced as temperature was increased toward physiological levels (Figures 4 and 5). Similarly, the profound temperature dependence for the α7 nAChR PAM activity of PNU120596 was also observed with the structurally distinct PAM SB-206553.
It has been suggested that PAMs like PNU120596 which produce profound effects on the kinetics of α7 nAChRs are unlikely to become useful as clinical therapeutic agents (Ng et al., 2007). The reasoning behind this suggestion is that the elimination of desensitization by such type II PAMs may produce excessive opening of α7 nAChRs leading to cell death through massively enhanced Ca2+ entry (Orrenius et al., 2003). Of course, any such actions will ultimately be dose-dependent. When Ng et al. (2007) compared the cytotoxicity of PNU120596 and a type I PAM, CCMI (also known as Compound 6), they saw significant cell death with the former but not the latter compound. These toxicity assays were, however, performed at room temperature and our work here shows that pharmacological actions of PNU120596 are much greater at room temperature and become substantially attenuated nearer physiological temperature. Notably, another more recent study with PNU120596 revealed no toxic effects of 24 h of PNU120596 treatment on cultured cortical neurons or PC12 cells, although both cell types were clearly shown to express functional α7 nAChRs (Hu et al., 2009). These toxicity studies, unlike those of Ng et al. (2007), were performed at physiological temperatures.
Our recordings also demonstrated that the strong rectification of the I–V relationship that typifies α7 nAChR-mediated currents was considerably reduced in the presence of the PAM. Thus, it seems likely that the presence of the PAM inhibits whatever process generates inward rectification in α7 nAChRs. To our knowledge, however, the molecular basis of the rectification properties of α7 nAChRs is not as well understood as it is for other nicotinic receptors (Haghighiand and Cooper, 1998, 2000).
The loss of strong rectification in the maintained presence of PNU120596 meant it was possible to measure response deactivation kinetics at positive potentials. The combination of two facets of α7 nAChR pharmacology means that such measurements are very difficult to make in the absence of a type II PAM. The first of these is that the strong inward rectification properties seen in the absence of a PAM mean responses at positive potentials are by definition small. Secondly, low agonist concentrations are best for measuring deactivation, because they elicit slower and thus less prior desensitization; but such concentration only elicit small currents. In the presence of PNU120596 it was clear that the rate of deactivation became two to three times faster as the membrane potential was depolarized. Deactivation of many ligand-gated channels reflects agonist unbinding kinetics. Thus our observations may mean ACh unbinding may be more rapid at more positive membrane potentials. The direction of this change suggests that voltage-dependent changes to agonist occupancy are very unlikely to be the cause of the loss of rectification in the presence of PNU120596. In future, it would be informative to examine the details of the voltage-dependence of the concentration-dependence of α7 nAChR activation, i.e., measuring EC50 at different membrane potentials.
Clearly, the temperature dependence we have identified here will have implications for how we need to think about PAM actions at α7 nAChR in vivo. It also means any measures of PAM efficacy generated in vitro at ambient temperatures will need to be considered with caution.
We do not as yet understand the molecular basis of this striking effect of temperature. It is interesting to note that the PNU120596 binding site is located within what is predicted to be the transmembrane region of the α7 nAChR (Young et al., 2008). Consequently, the biophysical or chemical state of the membrane may play a role in determining the actions of PAMs at this site. Raising the temperature the from room temperature to 37°C will have substantial effects on various properties of the plasma membrane not least its fluidity. Lipid interactions are known to affect various aspects of nicotinic receptor function including gating (Barrantes, 2004). With respect to changes in membrane fluidity, it is interesting to note that another key determinant of membrane fluidity, cholesterol content, has a number of effects on nicotinic receptors (Barrantes, 2007). α7 nAChR are reported to be localized in cholesterol rich lipid rafts (Bruses et al., 2001) and changes to membrane cholesterol content seem to cause nAChR to disaggregate from multi-receptor nanoclusters (Kellner et al., 2007). It would be interesting to examine if chemical depletion of cholesterol from cells with agents like methyl-beta-cyclodextrin has any effect on the actions of PAMs such as PNU120596.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arendash, G. W., Sanberg, P. R., and Sengstock, G. J. (1995a). Nicotine enhances the learning and memory of aged rats. Pharmacol. Biochem. Behav. 52, 517–523.
Arendash, G. W., Sengstock, G. J., Sanberg, P. R., and Kem, W. R. (1995b). Improved learning and memory in aged rats with chronic administration of the nicotinic receptor agonist GTS-21. Brain Res. 674, 252–259.
Barrantes, F. J. (2004). Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res. Brain Res. Rev. 47, 71–95.
Barrantes, F. J. (2007). Cholesterol effects on nicotinic acetylcholine receptor. J. Neurochem. 103(Suppl. 1), 72–80.
Bertrand, D., and Gopalakrishnan, M. (2007). Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 74, 1155–1163.
Bruses, J. L., Chauvet, N., and Rutishauser, U. (2001). Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J. Neurosci. 21, 504–512.
Dunlop, J., Lock, T., Jow, B., Sitzia, F., Grauer, S., Jow, F., Kramer, A., Bowlby, M., Randall, A., Kowal, D., Gilbert, A., Comery, T., Laroque, J., Soloveva, V., Brown, J., and Roncarati, R. (2009). Old and new pharmacology: positive allosteric modulation of the alpha7 nicotinic acetylcholine receptor by the 5-HT2B/C receptor antagonist SB-206553. J. Pharmacol. Exp. Ther. 328, 766–776.
Freedman, R., Leonard, S., Gault, J. M., Hopkins, J., Cloninger, C. R., Kaufmann, C. A., Tsuang, M. T., Farone, S. V., Malaspina, D., Svrakic, D. M., Sanders, A., and Gejman, P. (2001a). Linkage disequilibrium for schizophrenia at the chromosome 15q13-14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7). Am. J. Med. Genet. 105, 20–22.
Freedman, R., Leonard, S., Olincy, A., Kaufmann, C. A., Malaspina, D., Cloninger, C. R., Svrakic, D., Faraone, S. V., and Tsuang, M. T. (2001b). Evidence for the multigenic inheritance of schizophrenia. Am. J. Med. Genet. 105, 794–800.
Gotti, C., Moretti, M., Bohr, I., Ziabreva, I., Vailati, S., Longhi, R., Riganti, L., Gaimarri, A., McKeith, I. G., Perry, R. H., Aarsland, D., Larsen, J. P., Sher, E., Beattie, R., Clementi, F., and Court, J. A. (2006). Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer’s disease, Parkinson’s disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol. Dis. 23, 481–489.
Gronlien, J. H., Hakerud, M., Ween, H., Thorin-Hagene, K., Briggs, C. A., Gopalakrishnan, M., and Malysz, J. (2007). Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol. Pharmacol. 72, 715–724.
Haghighiand, A. P., and Cooper, E. (1998). Neuronal nicotinic acetylcholine receptors are blocked by intracellular spermine in a voltage-dependent manner. J. Neurosci. 18, 4050–4062.
Haghighiand, A. P., and Cooper, E. (2000). A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine alpha3beta4 and alpha4beta2 receptors. J. Neurosci. 20, 529–541.
Hajos, M., Hurst, R. S., Hoffmann, W. E., Krause, M., Wall, T. M., Higdon, N. R., and Groppi, V. E. (2005). The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J. Pharmacol. Exp. Ther. 312, 1213–1222.
Hajos, M., and Rogers, B. N. (2010). Targeting alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia. Curr. Pharm. Des. 16, 538–554.
Haydar, S. N., and Dunlop, J. (2010). Neuronal nicotinic acetylcholine receptors – targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr. Top Med. Chem. 10, 144–152.
Hellstrom-Lindahl, E., Court, J., Keverne, J., Svedberg, M., Lee, M., Marutle, A., Thomas, A., Perry, E., Bednar, I., and Nordberg, A. (2004). Nicotine reduces A beta in the brain and cerebral vessels of APPsw mice. Eur. J. Neurosci. 19, 2703–2710.
Hu, M., Gopalakrishnan, M., and Li, J. (2009). Positive allosteric modulation of alpha7 neuronal nicotinic acetylcholine receptors: lack of cytotoxicity in PC12 cells and rat primary cortical neurons. Br. J. Pharmacol. 158, 1857–1864.
Hu, M., Schurdak, M. E., Puttfarcken, P. S., El Kouhen, R., Gopalakrishnan, M., and Li, J. (2007). High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 1151, 227–235.
Hurst, R. S., Hajos, M., Raggenbass, M., Wall, T. M., Higdon, N. R., Lawson, J. A., Rutherford-Root, K. L., Berkenpas, M. B., Hoffmann, W. E., Piotrowski, D. W., Groppi, V. E., Allaman, G., Ogier, R., Bertrand, S., Bertrand, D., and Arneric, S. P. (2005). A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J. Neurosci. 25, 4396–4405.
Kellner, R. R., Baier, C. J., Willig, K. I., Hell, S. W., and Barrantes, F. J. (2007). Nanoscale organization of nicotinic acetylcholine receptors revealed by stimulated emission depletion microscopy. Neuroscience 144, 135–143.
Kihara, T., Shimohama, S., Sawada, H., Honda, K., Nakamizo, T., Shibasaki, H., Kume, T., and Akaike, A. (2001). Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem. 276, 13541–13546.
Leiser, S. C., Bowlby, M. R., Comery, T. A., and Dunlop, J. (2009). A cog in cognition: how the alpha7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol. Ther. 122, 302–311.
Leonard, S., Gault, J., Hopkins, J., Logel, J., Vianzon, R., Short, M., Drebing, C., Berger, R., Venn, D., Sirota, P., Zerbe, G., Olincy, A., Ross, R. G., Adler, L. E., and Freedman, R. (2002). Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch. Gen. Psychiatry 59, 1085–1096.
Levin, E. D., Bettegowda, C., Blosser, J., and Gordon, J. (1999). AR-R17779, and alpha7 nicotinic agonist, improves learning and memory in rats. Behav Pharmacol 10, 675–680.
Ng, H. J., Whittemore, E. R., Tran, M. B., Hogenkamp, D. J., Broide, R. S., Johnstone, T. B., Zheng, L., Stevens, K. E., and Gee, K. W. (2007). Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc. Natl. Acad. Sci. U.S.A. 104, 8059–8064.
Olincy, A., Harris, J. G., Johnson, L. L., Pender, V., Kongs, S., Allensworth, D., Ellis, J., Zerbe, G. O., Leonard, S., Stevens, K. E., Stevens, J. O., Martin, L., Adler, L. E., Soti, F., Kem, W. R., and Freedman, R. (2006). Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch. Gen. Psychiatry 63, 630–638.
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565.
Roncarati, R., Scali, C., Comery, T. A., Grauer, S. M., Aschmi, S., Bothmann, H., Jow, B., Kowal, D., Gianfriddo, M., Kelley, C., Zanelli, U., Ghiron, C., Haydar, S., Dunlop, J., and Terstappen, G. C. (2009). Procognitive and neuroprotective activity of a novel alpha7 nicotinic acetylcholine receptor agonist for treatment of neurodegenerative and cognitive disorders. J. Pharmacol. Exp. Ther. 329, 459–468.
Simosky, J. K., Stevens, K. E., Kem, W. R., and Freedman, R. (2001). Intragastric DMXB-A, an alpha7 nicotinic agonist, improves deficient sensory inhibition in DBA/2 mice. Biol. Psychiatry 50, 493–500.
Stevens, K. E., Kem, W. R., Mahnir, V. M., and Freedman, R. (1998). Selective alpha7-nicotinic agonists normalize inhibition of auditory response in DBA mice. Psychopharmacology (Berl.) 136, 320–327.
Thomsen, M. S., Hansen, H. H., Timmerman, D. B., and Mikkelsen, J. D. (2010). Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr. Pharm. Des. 16, 323–343.
Timmermann, D. B., Gronlien, J. H., Kohlhaas, K. L., Nielsen, E. O., Dam, E., Jorgensen, T. D., Ahring, P. K., Peters, D., Holst, D., Chrsitensen, J. K., Malysz, J., Briggs, C. A., Gopalakrishnan, M., and Olsen, G. M. (2007). An allosteric modulator of the alpha7 nicotinic acetylcholine receptor possessing cognition-enhancing properties in vivo. J. Pharmacol. Exp. Ther. 323, 294–307.
Williams, D. K., Wang, J., and Papke, R. L. (2011). Investigation of the molecular mechanism of the alpha7 nicotinic acetylcholine receptor positive allosteric modulator PNU-120596 provides evidence for two distinct desensitized states. Mol. Pharmacol. 80, 1013–1032.
Keywords: nicotinic receptors, electrophysiology, allosteric modulator, channel, patch clamp
Citation: Sitzia F, Brown JT, Randall AD and Dunlop J (2011) Voltage- and temperature-dependent allosteric modulation of α7 nicotinic receptors by PNU120596. Front. Pharmacol. 2:81. doi: 10.3389/fphar.2011.00081
Received: 09 July 2011; Accepted: 30 November 2011;
Published online: 27 December 2011.
Edited by:
Ralf Franz Kettenhofen, Axiogenesis AG, GermanyReviewed by:
Roger L. Papke, University of Florida, USAAndrea Bruggemann, Nanion Technologies GmbH, Germany
Copyright: © 2011 Sitzia, Brown, Randall and Dunlop. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: John Dunlop, Neuroscience Research Unit, Pfizer, Eastern Point Road, Groton, CT 06340, USA. e-mail: john.dunlop@pfizer.com