 Tanchun Wang1†
Tanchun Wang1†- 1 Departments of Pharmacology and Physiology, Drexel University College of Medicine, Philadelphia, PA, USA
- 2 Pathology and Laboratory Medicine, Drexel University College of Medicine, Philadelphia, PA, USA
Contraction of bladder smooth muscle is predominantly initiated by M3 muscarinic receptor-mediated activation of the Gq/11-phospholipase C β-protein kinase C (PKC) and the G12/13-RhoGEF-Rho kinase (ROCK) pathways. However, these pathways and their downstream effectors are not well understood in bladder smooth muscle. We used phorbol 12,13-dibutyrate (PDBu), and 1,2-dioctanoyl-sn-glycerol (DOG), activators of PKC, in this investigation. We were interested in dissecting the role(s) of PKC and to clarify the signaling pathways in bladder smooth muscle contraction, especially the potential cross-talk with ROCK and their downstream effectors in regulating myosin light chain phosphatase activity and force. To achieve this goal, the study was performed in the presence or absence of the PKC inhibitor bisindolylmaleimide-1 (Bis) or the ROCK inhibitor H-1152. Phosphorylation levels of Thr38-CPI-17 and Thr696/Thr850 myosin phosphatase target subunit (MYPT1) were measured during PDBu or DOG stimulation using site specific antibodies. PDBu-induced contraction in bladder smooth muscle involved both activation of PKC and PKC-dependent activation of ROCK. CPI-17 as a major downstream effector, is phosphorylated by PKC and ROCK during PDBu and DOG stimulation. Our results suggest that Thr696 and Thr850-MYPT1 phosphorylation are not involved in the regulation of a PDBu-induced contraction. The results also demonstrate that bladder smooth muscle contains a constitutively active isoform of ROCK that may play an important role in the regulation of bladder smooth muscle basal tone. Together with the results from our previous study, we developed a working model to describe the complex signaling pathways that regulate contraction of bladder smooth muscle.
Introduction
It is well accepted that phosphorylation of the 20-kDa regulatory myosin light chain (MLC), catalyzed by the Ca2+/calmodulin dependent MLC kinase and dephosphorylation catalyzed by the MLC phosphatase, play a primary role in the regulation of smooth muscle contraction and relaxation (Sobieszek, 1977; for reviews see Kamm and Stull, 1985; Somlyo and Somlyo, 1994). In addition, agonist stimulation of G-protein coupled receptors increases contraction in smooth muscle by both activating the MLC kinase and inhibiting the MLC phosphatase (Kimura et al., 1996; Somlyo and Somlyo, 2003), the result of which is an increase in MLC phosphorylation levels to values greater than those achieved by the simple activation of the MLC kinase alone. Inhibition of the MLC phosphatase can result from either protein kinase C (PKC)-catalyzed phosphorylation of CPI-17 or Rho kinase (ROCK)-catalyzed phosphorylation of the myosin phosphatase target subunit (MYPT1). Both pathways play key roles in the enhancement of myofilament Ca2+ sensitivity (Eto et al., 1995, 2004; Hartshorne, 1998; Kitazawa et al., 2000; Somlyo and Somlyo, 2003). These signaling pathways have been extensively studied in vascular and some non-vascular smooth muscle (Eto et al., 1997; Feng et al., 1999). However, studies in bladder smooth muscle are not as numerous and therefore our understanding of the mechanisms underlying Ca2+ sensitization in bladder is significantly less.
Contraction of bladder smooth muscle during voiding is predominantly initiated by acetylcholine activation of M3 muscarinic receptors (Frazier et al., 2008). We used carbachol in a previous study to initiate contraction of bladder smooth muscle with the goal of studying the potential role(s) of PKC and ROCK pathways in an agonist-induced contraction (Wang et al., 2009). Our results demonstrated that phosphorylation of both CPI-17 and MYPT1, effectors of the PKC and ROCK pathways respectively, are involved in the regulation of carbachol-dependent bladder smooth muscle contraction (Wang et al., 2009). It is unclear, however, what role(s) PKC and ROCK play in the phosphorylation of CPI-17 and MYPT1 and whether or not there is any cross-talk between the two pathways during bladder smooth muscle contraction as has been suggested in other smooth muscles (Niiro et al., 2003). For example, activation of ROCK by PKC, specifically the PKCδ isoform, has been shown to be involved in several cell functions, including vascular smooth muscle migration and contraction (Kandabashi et al., 2003; Ohtsu et al., 2005; Poole and Furness, 2007).
The first goal of this study was to more clearly define the role(s) of PKC and ROCK in bladder smooth muscle contraction and test the hypothesis that both PKC and ROCK regulate contraction through phosphorylation of CPI-17. We were also interested in testing the specific hypothesis that in addition to direct phosphorylation of CPI-17, activation of PKC also increases ROCK activity resulting in an increase in CPI-17 phosphorylation. To test this hypothesis, we used phorbol 12,13-dibutyrate (PDBu), 1,2-dioctanoyl-sn-glycerol (DOG) an analog of the more physiologically relevant diacylglycerol (DAG), and the inactive isomer of PDBu, 4α-phorbol 12,13-dibutyrate (α-PDBu). Using this approach we could, with some assurance, specifically activate only the PKC pathway (Castagna et al., 1982). Therefore any effect of PDBu or DOG on ROCK activity would be through activation of PKC. The second goal of this study was to build upon our earlier report (Wang et al., 2009) to dissect the effect(s) of PKC activation directly via PDBu or DOG from both PKC and ROCK activation via carbachol stimulation. This will then allow us to develop a working hypothesis to help guide future studies on both the normal physiological regulation of bladder smooth muscle and the altered regulation that may occur in pathophysiological states.
Materials and Methods
Intact Bladder Smooth Muscle Preparation
Male New Zealand White rabbits weighing 2–2.5 kg were used in this study. All animal studies and procedures were approved by the Drexel University College of Medicine’s Institutional Animal Care and Use Committee. Rabbits were euthanized by a pentobarbital overdose administered via the ear vein followed by exsanguination. The bladders were then quickly removed and placed in ice cold physiological salt solution (MOPS-buffered PSS). MOPS-buffered PSS contained (in mM) 140 NaCl, 4.7 KCl, 1.2 MgSO4, 1.6 CaCl2, 1.2 Na2HPO4, 2 MOPS (pH 7.4), 5 D-glucose, and 0.02 EDTA. The bladder neck, trigone, and base regions were removed, leaving only the middle detrusor body for experimentation. The detrusor body was dissected in cold MOPS-buffered PSS under a dissecting microscope. The mucosal and serosal layers were carefully removed and strips containing primarily smooth muscle (∼1.5 mm × 6 mm) were cut along the central axis of the bladder in the longitudinal orientation as previously described (Su et al., 2003) and stored in MOPS-buffered PSS at 4°C until used. Our bladder strips, devoid of serosal and mucosal layers and are predominately smooth muscle, do not typically show spontaneous activity such as those preparations containing urothelial and other modulatory cells (Su et al., 2003; Stanton et al., 2006). Storage was never longer than 24 h and storage had no effect on the presence or absence of spontaneous activity or magnitude of contractile force.
Isometric Force Recording
The bladder strips were mounted between a Grass FT.03 force transducer and a stationary clip in water-jacketed muscle organ baths containing MOPS-buffered PSS at 37°C and aerated with 100% O2. The strips were stretched and allowed to stress–relax until a passive force of ∼1 g was achieved. This passive force approximates the optimal length for maximal active stress development (Lo; Su et al., 2003; Wang et al., 2009). The strips were then allowed to equilibrate for at least 40 min until a stable basal force recording was obtained.
After the equilibration period, strips were stimulated with 110 mM KCl–MOPS-buffered PSS (equal-molar substitution of KCl for NaCl) then relaxed with MOPS-buffered PSS; this contraction–relaxation cycle was repeated four times. One hundred ten millimolar KCl–MOPS-buffered PSS was used as the stimulus to decrease diffusion time of the depolarizing K+ into the tissue, thus decreasing the delay from addition of the stimulus to initiation of contraction, and therefore increase the time to peak contraction as is standard procedure in our laboratory (Moreland and Moreland, 1987; Fulginiti et al., 1993; Gorenne et al., 1998; Su et al., 2003, 2004; Stanton et al., 2006). The peak force in each equilibrated muscle strip was used to normalize the force generated with PDBu, α-PDBu, or DOG stimulation. A concentration response curve was generated in response to DOG, demonstrating that 300 μM produced a maximal response. Data therefore are presented as a percentage of the maximal response to 110 mM KCl. Bisindolylmaleimide-1 (Bis, 3 μM) and H-1152 (1 μM) were used to inhibit PKC (Toullec et al., 1991) and ROCK (Sasaki et al., 2002) activity, respectively. Bis and H-1152 concentrations were chosen based on published and unpublished studies from our lab (Stanton et al., 2006; Wang et al., 2009). We previously used 10 μM Bis to inhibit PKC activity, however recent preliminary studies have shown that 3 μM Bis inhibited PDBu-induced contractions but not Ca2+-dependent contractions of Triton X-100 skinned bladder smooth muscle strips suggesting that this concentration of Bis has no non-significant effects on the MLC kinase (data not shown). The inhibitors were added to the muscle bath 20 min prior to the addition of stimulus. Tissues were allowed to contract for 5 min and then relaxed by rinsing with MOPS-buffered PSS.
In studies to examine the calcium-dependence of PDBu-induced contractions we used procedures previously published by our laboratory (Su et al., 2004). To examine the dependence on extracellular calcium, tissues were incubated in calcium-free MOPS-buffered PSS for 15 min followed by stimulation with PDBu. To examine the PDBu response in the absence of both extracellular and intracellular calcium, the tissues were bathed in MOPS-buffered PSS containing 0.1 mM EGTA and then contracted by the addition of 10 μM carbachol for 7 min to release and chelate intracellular calcium. The tissues were then rinsed in calcium-free MOPS-buffered PSS and subjected to a second carbachol stimulation to ensure removal of intracellular calcium. Tissues were then rinsed and bathed in calcium-free MOPS-buffered PSS without EGTA for 15 min followed by stimulation with PDBu. All muscle strips were stimulated with carbachol at the end of each experiment to ensure the procedures to alter calcium source(s) did not affect tissue viability.
Measurement of MLC Phosphorylation
Myosin light chain phosphorylation levels were measured by standard techniques previously published by our laboratory (Moreland and Moreland, 1987; Moreland et al., 1992; Su et al., 2004; Wang et al., 2009). Briefly, strips were rapidly frozen in a dry ice/acetone slurry containing 6% trichloroacetic acid and 10 mM DTT at various time points (0, 1.5, 3, and 5 min) of PDBu stimulation. The strips were then slowly thawed to room temperature, rinsed in acetone, air dried, and subjected to homogenization in a 1% SDS, 10% glycerol, and 1 mM DTT solution using glass/glass homogenizers. Samples were centrifuged at 12,000 rpm for 6 min and subjected to two-dimensional gel electrophoresis and transferred to nitrocellulose membranes for quantification of MLC phosphorylation as previously described (Moreland et al., 1992). Transferred proteins were visualized with Colloidal Gold Stain (Amersham Biosciences, Piscataway, NJ, USA) and digitized by densitometric analysis using a Bio-Rad GS-800 quantitative densitometer (Bio-Rad, Hercules, CA, USA). MLC phosphorylation levels were calculated by dividing the densitometric analysis of the spot corresponding to the phosphorylated MLC by the sum of the densitometric analyses of the spots corresponding to the phosphorylated and unphosphorylated MLC. Values are presented as mole Pi/mole MLC.
Quantification of CPI-17 Phosphorylation
Tissue strips, resting or stimulated, were rapidly frozen and then processed as previously described (Wang et al., 2009). For studies comparing phosphorylation in response to PDBu in the presence or absence of PKC or ROCK inhibitors the membranes were blocked with 5% bovine serum albumin (BSA, Sigma-Aldrich Inc, St. Louis, MO, USA) in a phosphate buffered saline (PBS) solution for 1 h, and incubated with an antibody against Thr38-phospho-CPI-17 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. The membranes were briefly rinsed with double deionized water and incubated with a polyclonal goat anti-rabbit antibody conjugated to horseradish peroxidase (1:5,000; Upstate Cell Signaling, Billerica, MA, USA). Immunoreactive bands were detected using enhanced chemiluminescence (ECL, Amersham Biosciences, Piscataway, NJ, USA). The blots were stripped and re-probed using a primary antibody against total CPI-17 (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by washes in a PBS-0.05% Tween-20 solution. The washed membranes were incubated with a monoclonal sheep anti-mouse antibody conjugated to horseradish peroxidase (1:10,000; Amersham Biosciences, Piscataway, NJ, USA).
For studies comparing CPI-17 phosphorylation in response to PDBu to that in response to α-PDBu or DOG stimulation, membranes were incubated with an antibody against Thr38-phospho-CPI-17 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and an antibody against CPI-17 (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. Membranes were washed in a PBS-0.1% Tween-20 solution and incubated with a goat anti-mouse IRDye 800cw conjugated secondary antibody and a goat anti-rabbit IRDye 680cw conjugated polyclonal secondary antibody for 45 min at room temperature (1:10,000 goat anti-rabbit, 1:10,000 goat anti-mouse, Li-Cor Biosciences, Lincoln, NE, USA) then washed in 0.1% Tween-PBS. Protein was visualized with the Li-Cor Odyssey Infrared Imaging System.
The ratio of the density of the band corresponding to the Thr38-phospho-CPI-17 to that of the band corresponding to total CPI-17 was used to determine CPI-17 phosphorylation levels in response to PDBu in the presence or absence of inhibitors of PKC or ROCK. For blots quantified using the Odyssey imager all ratios of phosphorylated to unphosphorylated proteins were compared to the ratio in resting tissues, which was set at 100%. In most experiments protein loadings were similar, but for all measurements a direct comparison of the levels of phosphorylated protein to total protein were obtained in the same blot by stripping and re-probing as previously demonstrated (Gorenne et al., 1998). When utilizing the Odyssey imager, phosphorylated and total protein levels were measured on the same blot. Any blot demonstrating saturated spots was not used in the analyses. We have previously shown that this probe, stripping, and re-probing protocol accurately quantifies phosphoprotein levels that are linearly related to kinase phosphotransferase activity (Gorenne et al., 1998).
Quantification of MYPT1 Phosphorylation
Tissue strips, resting or stimulated, were rapidly frozen and then homogenized in a buffer including a Protease Inhibitor Cocktail solution (1:100; Sigma-Aldrich Inc, St. Louis, MO, USA), as described above. The samples were subjected to electrophoresis in a 7.5% SDS-acrylamide gel and transferred onto a nitrocellulose membrane. The membranes were blocked with 3% non-fat dry milk in a PBS solution for 40 min and incubated with a primary antibody against either Thr696 or Thr850 phospho-MYPT1 (1:2,000 Upstate Cell Signaling, Billerica, MA, USA) overnight at 4°C. The membranes were briefly washed with double deionized water and incubated with a polyclonal goat anti-rabbit antibody conjugated to horseradish peroxidase (1:5,000; Upstate Cell Signaling, Billerica, MA, USA). Immunoreactive bands were detected using ECL (Amersham Biosciences, Piscataway, NJ, USA). The blots were stripped and re-probed using a primary MYPT1 antibody (1:10,000; Covance Inc, Princeton, NJ, USA) followed by washes in a PBS-0.05% Tween-20 solution and then incubated with a polyclonal goat anti-rabbit antibody conjugated to horseradish peroxidase (1:5,000; Upstate Cell Signaling, Billerica, MA, USA).
For studies comparing phosphorylation in response to PDBu stimulation to that in response to α-PDBu or DOG, membranes were blocked for 1 h in Odyssey Blocking Buffer (Li-Cor Biosciences) and incubated in primary antibody as described above. Membranes were washed in PBS-0.1% Tween-20 followed by incubation with goat anti-rabbit IRDye 680cw conjugated polyclonal secondary antibody for 45 min at room temperature (1:10,000 goat anti-rabbit, Li-Cor Biosciences, Lincoln, NE, USA) and washed with 0.1% Tween-PBS. The membranes were visualized using the Li-Cor Odyssey Infrared Imaging System. The blots were stripped in NewBlot Nitro Stripping Buffer (Li-Cor Biosciences, Lincoln, NE, USA) for 10 min, washed in PBS for 5 min, and re-probed using a primary MYPT1 antibody (1:10,000; Covance Inc., Princeton, NJ, USA) followed by the same washes, secondary antibody incubation, and visualization as described above.
The ratio of the density of the bands corresponding to phospho-Thr696 or phospho-Thr850 to the bands corresponding to total MYPT1 was used to determine MYPT1 phosphorylation levels. For blots quantified using the Odyssey imager all ratios of phosphorylated to total protein were compared to the same ratio in unstimulated tissues, which was set at 100%. In most experiments protein loadings were similar, but in all blots a direct comparison of phosphorylated protein levels to total protein levels was obtained in the same blot by stripping and re-probing.
Materials and Statistics
All reagents, unless otherwise specified below, were purchased from Fisher Scientific (Pittsburgh, PA, USA) and were of analytical grade or better. All electrophoretic and blotting reagents were obtained from Bio-Rad Laboratories (Hercules, CA, USA) and Li-Cor Biosciences (Lincoln, NE, USA). Bis and H-1152 were purchased from Calbiochem (San Diego, CA, USA). Phorbol 12,13-dibutyrate was purchased from Sigma-Aldrich (St. Louis, MO, USA). DOG and α-PDBu were purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA) and dissolved in dimethyl sulfoxide (DMSO). Unless otherwise noted, membrane antibody stripping solutions were purchased from Pierce Biotechnology (Rockford, IL, USA).
Statistical significance between means was determined using one-way ANOVA followed by the Tukey post hoc test. A P value < 0.05 was taken as significant. All “n” values refer to the number of bladder muscle strips; each strip was taken from a different animal.
Results
Isometric Force in Response to PDBu, α-PDBu, or DOG Stimulation
The addition of PDBu results in a dose dependent contraction of bladder smooth muscle (Yoshida et al., 1992; Stanton et al., 2006; Chang et al., 2009). Three millimolar PDBu was used in the present study to induce a maximal PDBu-dependent contraction in rabbit bladder smooth muscle. Representative tracings of a contraction in response to PDBu alone or in the presence of 3 μM Bis or 1 μM H-1152 are shown in Figure 1A. Several such experiments were averaged and are shown in Figure 1B. As shown in Figure 1B, PDBu-dependent isometric force was measured at 0, 1.5, 3, and 5 min of stimulation and expressed as a percentage of the strips’ maximal response to 110 mM KCl. Maximal stress in response to 110 mM KCl in our preparation of bladder smooth muscle averages 6.8 ± 0.4 × 104 N/m2. Force levels earlier than 1.5 min were lower than those measured at 1.5–3 min. Smooth muscle strips produced a slowly developing contraction in response to PDBu. Peak force was attained at ∼1.5 min of contraction with a value of 36.5 ± 2.2% of the maximal force in response to KCl stimulation. The sustained portion of the PDBu-dependent contraction began to decrease at approximately 3 min and returned to values not significantly different from baseline by 5 min of continued stimulation. The addition of α-PDBu, the inactive isomer of PDBu, did not result in contraction of bladder smooth muscle (Figure 1B). Three millimolar α-PDBu was used in the present study to provide a direct comparison to the same concentration of the active phorbol ester, PDBu. Pre-incubation with the PKC inhibitor Bis abolished the PDBu-induced contraction. Pre-incubation with the ROCK inhibitor H-1152 significantly decreased the PDBu-stimulated contraction as well as decreased basal resting tone of the tissue (Figure 1B). The inhibition of PDBu-stimulated force in response to H-1152 was significantly less than that produced by Bis at 1.5 and 3 min of stimulation. These results suggest that PKC is responsible for the PDBu-dependent contraction; however, there also appears to be a significant role for ROCK activation as evidenced by the loss of developed force in the presence of H-1152.
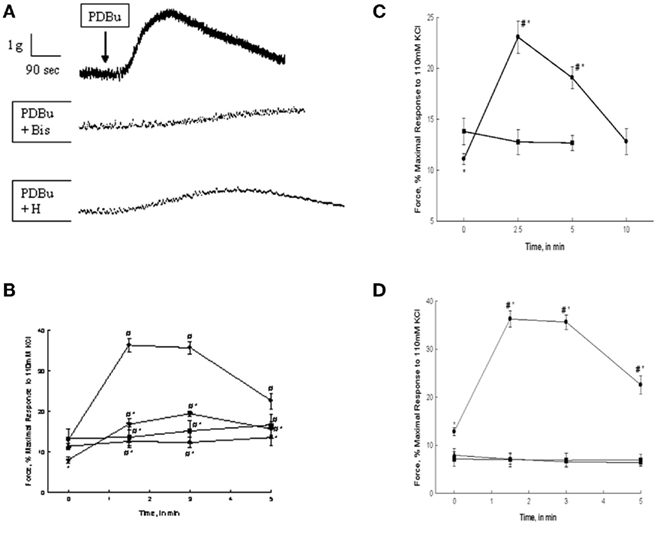
Figure 1. PDBu, α-PDBu, or DOG-stimulated bladder smooth muscle contraction and the effects of PKC or ROCK inhibition or calcium removal. (A) Representative tracing of contractions in response to 3 μM PDBu alone (top tracing), and in the presence of 3 μM Bis (middle tracing), or 1 μM H-1152. Arrow denotes the addition of PDBu, inhibitors were added 20 min prior to stimulation. (B) Intact strips of bladder smooth muscle were contracted for 5 min with PDBu (3 μM) in the presence or absence of Bis (3 μM) or H-1152 (1 μM). Strips contracted in response to PDBu alone (•) generated 36.5 + 2.2% of maximal force at ∼1.5 min and maintained a sustained contraction until ∼3 min. Inhibition with Bis (▲) or H-1152 (▼) significantly decreased force at 1.5 and 3 min. H-1152 also significantly decreased basal force at 0 min. Stimulation with α-PDBu (■) did not elicit an increase in force until 5 min. (C) Intact strips of bladder smooth muscle were contracted for 10 min with DOG (300 μM). DOG (•) generated force significantly greater than basal values at 2.5 and 5 min of stimulation, but not significantly different from basal values at 10 min. Vehicle alone (DMSO, ■) had no effect. (D) Intact strips of bladder smooth muscle were contracted for 5 min with PDBu (3 μM, •) in the presence or absence of extracellular calcium or after depletion of all calcium stores. Strips contracted in the absence of extracellular calcium (■) or following the depletion of all calcium stores (▲) did not respond to PDBu. Values shown are the means ± SE of at least five determinations. *P < 0.05, compared with values in the presence of PDBu alone (B), DMSO alone (C), or the absence of calcium (D). #P < 0.05, compared with values at 0 min of the same treatment.
The addition of DOG (300 μM) resulted in an increase in isometric force in bladder smooth muscle. Preliminary studies demonstrated that 300 μM DOG produced a maximal contractile response. As shown in Figure 1C, isometric force was measured at 0, 2.5, 5, and 10 min of stimulation and was expressed as a percentage of the maximal response to 110 mM KCl. Smooth muscle strips developed a slow contraction in response to DOG qualitatively similar to that seen with PDBu. Peak force was attained at ∼2.5 min of stimulation with a value of 23.1 ± 1.6% of maximal force in response to KCl stimulation and did not return to basal values until 10 min of stimulation. The slower onset of contraction in response to DOG as compared to PDBu is most likely due to slower diffusion rate of DOG through the interstitial space and membrane lipid bilayer. The more rapid decline in force in response to DOG as compared to PDBu is most likely due to the fact that DOG is hydrolyzed, albeit more slowly than diacylglycerol. The vehicle, DMSO, alone had no effect on force.
Bladder smooth muscle strips were subjected to PDBu stimulation after removal of extracellular calcium or removal of all sources of calcium. As shown in Figure 1D, removal of either extracellular calcium or all calcium sources abolished contraction in response to PDBu stimulation. These data suggest that PDBu-induced contractions of bladder smooth muscle are extracellular calcium-dependent.
Myosin Light Chain Phosphorylation in Response to PDBu Stimulation
The mechanism(s) by which PDBu induces contraction of bladder smooth muscle is assumed to be due to the activation of PKC. Our demonstration that DOG produces a contraction similar to PDBu supports this assumption. However, the regulatory steps that are involved beyond activation of PKC are not well defined. In fact the published evidence is not consistent with regards to whether or not MLC phosphorylation is involved in a PDBu-induced contraction (Rembold and Murphy, 1988; Fulginiti et al., 1993; Stanton et al., 2006). In our present study, we addressed this issue by measuring MLC phosphorylation levels in response to PDBu stimulation, in the presence or absence of PKC or ROCK inhibitors. As shown in Figure 2A, stimulation of bladder smooth muscle strips with 3 μM PDBu increased MLC phosphorylation levels with a temporal profile similar to that of force generation in response to PDBu alone; a slow increase followed by a decline to basal values (see force profiles in Figures 1A,B). Considering that maximal PDBu-induced force is approximately 36% of that produced in response to 110 mM KCl, the relatively small increase in MLC phosphorylation levels is not surprising. Pre-incubation with the PKC inhibitor Bis reduced MLC phosphorylation levels, but the reduction only reached levels of significance at 3 min of PDBu stimulation as compared to PDBu-stimulated alone. Similar to PDBu-induced MLC phosphorylation levels at most time points in the presence of Bis, H-1152 also depressed MLC phosphorylation levels, albeit not statistically significant. Interestingly, Bis and H-1152 significantly reduced or abolished PDBu-induced force while the effects on MLC phosphorylation were minimal. In order to determine if a change in basal values of MLC phosphorylation resulting from inhibition of PKC or ROCK affected PDBu-induced increases, we calculated the percent increase in MLC phosphorylation from basal. These calculations are shown in Figure 2B. Although no values were statistically significant due to the variability in a percent change within a very small range, qualitatively the trends are similar in Figure 2B as compared to Figure 2A.
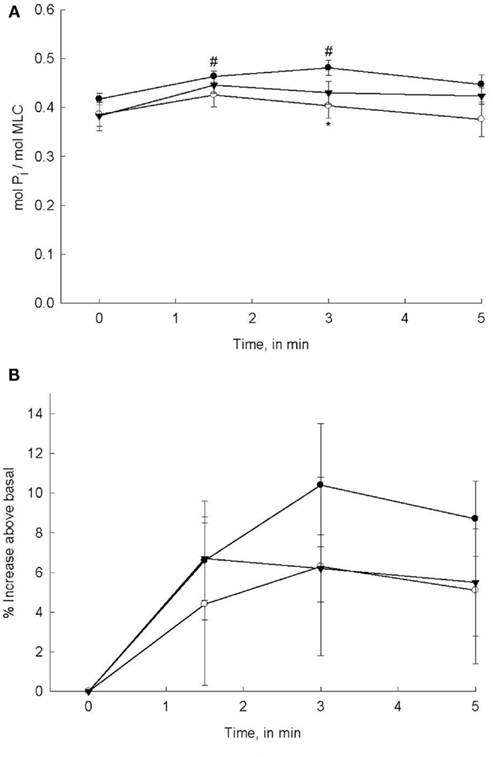
Figure 2. PDBu-stimulated MLC phosphorylation levels in bladder smooth muscle. Intact strips of bladder smooth muscle were contracted for 5 min with PDBu (3 μM) in the presence or absence of Bis (3 μM) or H-1152 (1 μM). Strips were rapidly frozen at 0, 1.5, 3, and 5 min of contraction and then processed for quantification of MLC phosphorylation levels. (A) PDBu stimulation significantly increased MLC phosphorylation levels at 1.5 and 3 min (•). MLC phosphorylation levels in strips incubated with either Bis (o) or H-1152 (▼) prior to PDBu stimulation were not significantly greater than basal values. (B) Percent increase in MLC phosphorylation levels from basal values in response to PDBu alone (•) or after 20 min incubation in Bis (o) or H-1152 (▼). Values shown are the means ± SE of 8–10 determinations. #P < 0.05, as compared with values at 0 min of the same treatment. *P < 0.05, as compared to PDBu alone at the same time point.
Quantification of CPI-17 Phosphorylation
One of the primary mechanisms by which PKC activates or enhances contraction of smooth muscle is via phosphorylation of CPI-17 (Eto et al., 1997, 2004). Phosphorylated CPI-17 binds to the 38-kDa catalytic subunit of the MLC phosphatase which inhibits its activity (Eto et al., 1995; Ito et al., 2004). We have recently shown that carbachol-induced activation of bladder smooth muscle increases CPI-17 phosphorylation and this increase is abolished by Bis and is significantly reduced by H-1152 (Wang et al., 2009). Therefore, we were interested in determining if activation of bladder smooth muscle by PKC alone produced a different sequence of signaling events in terms of PKC and ROCK activity as compared to the numerous signaling pathways activated by carbachol. The first series of experiments to address this question were designed to measure CPI-17 phosphorylation levels during PDBu stimulation in the presence or absence of Bis. Figure 3A shows a representative Western blot of total and phospho-CPI-17 antibody and Figure 3B shows the averaged results of four to eight blots. Levels of Thr38-phospho-CPI-17 were significantly increased at all time points measured during PDBu stimulation. Inhibition of PKC with Bis abolished the PDBu-dependent increase in CPI-17 phosphorylation. Inhibition of ROCK with H-1152 delayed the onset of CPI-17 phosphorylation levels but did not affect the maximal levels attained during the later phases of PDBu stimulation. As expected, these results suggest that CPI-17 phosphorylation is increased by activation of PKC. Moreover, activation of PKC with PDBu also results in ROCK-dependent increases in CPI-17 phosphorylation, at least during the initial phase of a contraction.
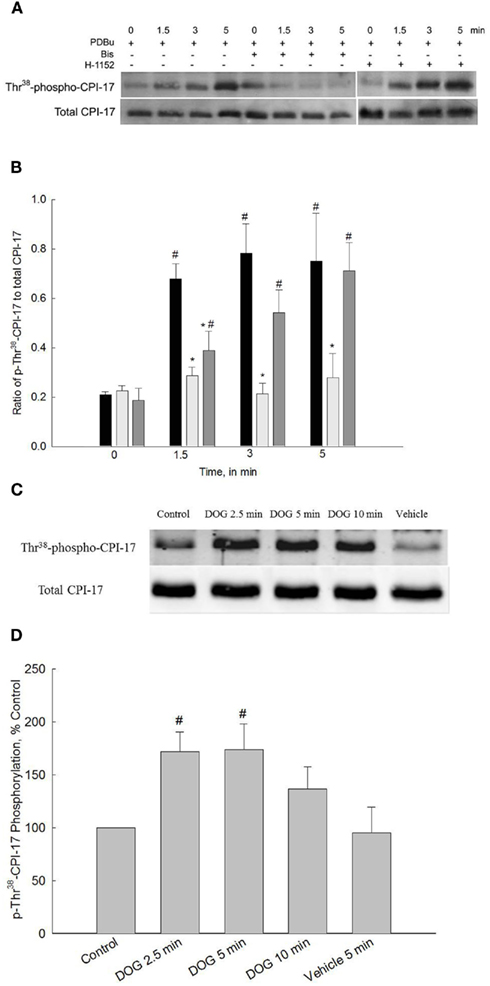
Figure 3. Representative Western blots and quantification of Thr38-CPI-17 phosphorylation levels from bladder smooth muscle contracted with PDBu or DOG. (A) Representative Western blots of strips stimulated with PDBu (3 μM) in the presence or absence of Bis (3 μM) or H-1152 (1 μM). (B) Quantitative results of four to eight blots such as that shown in (A). PDBu stimulation significantly increased phospho-Thr38-CPI-17 levels at all time points (black bars). Inhibition of PKC with Bis (light gray bars) abolished increases in CPI-17 phosphorylation levels at all time points. Inhibition of ROCK with H-1152 (dark gray bars) significantly reduced CPI-17 phosphorylation levels only at 1.5 min. (C) Representative Western blots of bladder smooth muscle strips stimulated with DOG (300 μM) or vehicle (DMSO) alone. (D) Quantitative results of four to eight blots such as that shown in (C). DOG stimulation significantly increased phospho-Thr38-CPI-17 levels at 2.5 and 5 min of stimulation. Values shown are the means ± SE of four to eight determinations. #P < 0.05, compared with values at 0 min or control. *P < 0.05, compared with values in response to PDBu at the same time point.
A second set of experiments were performed to measure CPI-17 phosphorylation levels in response 300 μM DOG. These experiments were performed to provide results using two activators of PKC; the chemical activator PDBu and the analog of the physiological activator DOG. If both PKC activators produce similar results, then this would provide additional support to the assumption that PDBu activates PKC and that our results reflect events that occur during normal physiological stimulation. As shown in Figures 3C,D, Thr38-phospho-CPI-17 levels were significantly increased at 2.5 and 5 min of DOG stimulation. Phosphorylation levels were not significantly different from control values at 10 min of stimulation with DOG or at any time point after the addition of vehicle alone (DMSO, only 5 min is shown). Similar to the finding that the development of force in response to DOG was slower as compared to PDBu-dependent force development, the increase in CPI-17 phosphorylation levels was also slower. CPI-17 phosphorylation levels qualitatively paralleled the increase in force developed in response to both PDBu and DOG (Figures 1B,C as compared to Figures 3B,D).
Quantification of MYPT1 Phosphorylation
ROCK-catalyzed phosphorylation of MYPT1 has been shown to be one of the major mechanisms regulating MLC phosphatase activity (Hartshorne, 1998). Carbachol stimulation of bladder smooth muscle increases Thr850-MYPT1 phosphorylation but not Thr696-MYPT1 phosphorylation (Wang et al., 2009). In the present study, we used PDBu stimulation to address the potential role of PKC in ROCK-catalyzed phosphorylation of MYPT1 at either Thr696 or Thr850 and DOG stimulation to examine ROCK-catalyzed phospho-Thr850-MYPT1. We were also interested in confirming our previous observation that a constitutively active ROCK which phosphorylates Thr850-MYPT1 is functional in resting, unstimulated bladder smooth muscle (Wang et al., 2009).
Figure 4A shows a representative Western blot of PDBu-stimulated phosopho-Thr696-MYPT1 and Figure 4B shows the averaged data of 8–12 blots. Stimulation of bladder smooth muscle with PDBu neither increased nor decreased phosphorylation levels of Thr696-MYPT1. Additionally, neither Bis nor H-1152 had any effect on Thr696-MYPT1 levels at rest or following PDBu stimulation.
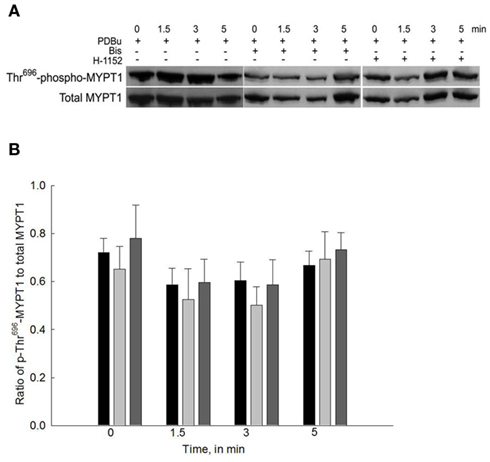
Figure 4. Representative Western blot and quantification of phospho-Thr696-MYPT1 levels from bladder smooth muscle contracted with PDBu in the presence or absence of Bis or H-1152. (A) Representative Western blots of strips stimulated with PDBu (3 μM) in the presence of either Bis (3 μM) or H-1152 (1 μM). (B) Averaged results of 8–12 experiments of the type shown in (A). PDBu stimulation alone did not increase phospho-Thr696-MYPT1 levels (black bars). Inhibition of either PKC (light gray bars) or ROCK (dark gray bars) had no effect on phospho-Thr696-MYPT1 levels in all strips. Values shown are the means ± SE of 8–12 determinations.
Figure 5A shows a representative Western blot of PDBu-stimulated phospho-Thr850-MYPT1 and Figure 5B shows the averaged data of 9–11 blots. Stimulation of bladder smooth muscle with PDBu had no effect on Thr850-MYPT1 phosphorylation levels. Consistent with the lack of effect of PDBu on Thr850-MYPT1 phosphorylation, inhibition of PKC with Bis during PDBu stimulation was also without effect. In contrast to the lack of effect of ROCK inhibition on basal levels of phospho-Thr696-MYPT1, inhibition of ROCK with H-1152 (added 20 min prior to “0” time point) significantly inhibited basal levels as well as levels at all other timed measurements of phospho-Thr850-MYPT1. These results suggest that PKC is not involved in the activation of either the constitutively active ROCK or the ROCK that phosphorylates Thr850-MYPT1 during agonist activation.
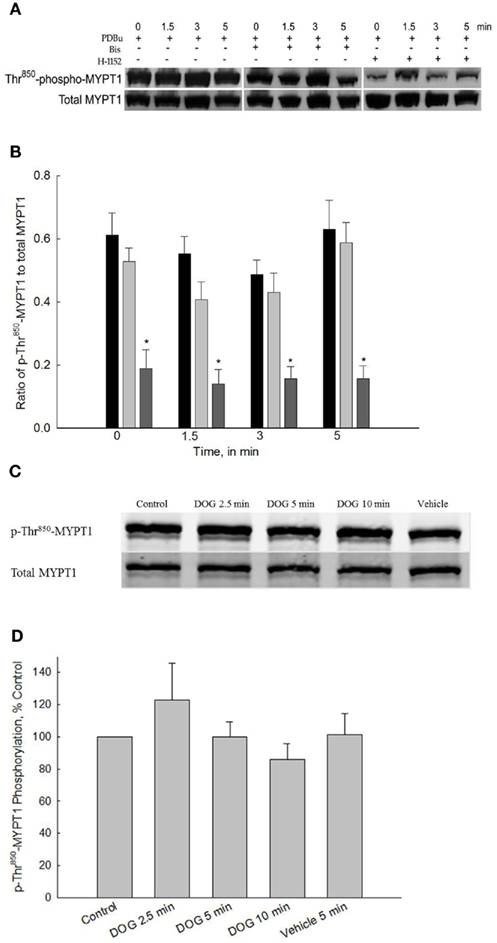
Figure 5. Representative Western blot and quantification of phospho-Thr850-MYPT1 levels from bladder smooth contracted with PDBu or DOG. (A) Representative Western blots of strips stimulated with PDBu (3 μM) in the presence or absence of either Bis (3 μM) or H-1152 (1 μM). (B) Averaged results of 9–11 experiments of the type shown in (A). PDBu stimulation alone did not increase phospho-Thr850-MYPT1 levels (black bars). Inhibition of PKC by Bis (light gray bars) did not affect phospho-Thr850-MYPT1 levels at any time point. Inhibition of ROCK (dark gray bars) significantly decreased phospho-Thr850-MYPT1 levels at all time points measured including basal values. (C) Representative Western blot of strips stimulated with DOG (300 μM). (D) Averaged results of 9–11 experiments of the type shown in (C). Similar to the results with PDBu, DOG stimulation did not increase phospho-Thr850-MYPT1 levels at any time point measured. Values shown are the means ± SE of 9–11 determinations. *P < 0.05, compared with values in strips contracted with PDBu at the same time points.
Figures 5C,D shows the results of 300 μM DOG on phospho-Thr850-MYPT1 levels. DOG stimulation of bladder smooth muscle did not produce any increase in phospho-Thr850-MYPT1 levels as compared to control values. These results further validate the hypothesis that PKC is not involved in activation of ROCK-catalyzed MYPT1 phosphorylation in bladder smooth muscle and that both PDBu and DOG act via similar pathways.
Discussion
We recently presented results aimed at understanding the signaling steps involved in carbachol-induced activation of bladder smooth muscle contraction (Wang et al., 2009). Carbachol stimulation of bladder smooth muscle activates both PKC and ROCK signaling pathways. As such, using carbachol alone, it is difficult to assess the specific role(s) for either PKC or ROCK. Therefore, in this present study we used PDBu and DOG as contractile activators as they are presumed to only activate PKC (Castagna et al., 1982). Any activity consistent with a role for ROCK therefore should be due to activation by PKC or a downstream PKC-dependent mediator. Therefore, using this approach with PDBu and DOG as the stimuli should allow us to dissect which step(s) in the initiation of a contraction is the result of PKC, ROCK, or both.
The major novel findings in this study are: (1) PDBu and DOG-induced contraction of bladder smooth muscle involves activation of PKC directly as well as PKC-dependent activation of ROCK; (2) Consistent with results using vascular smooth muscle (Fulginiti et al., 1993), PDBu-dependent force is not strictly dependent on increased levels of MLC phosphorylation; (3) CPI-17 is phosphorylated directly by PKC and also by PKC-dependent activated ROCK; (4) Although ROCK activation is involved in PDBu and DOG-dependent contractions, phosphorylation of MYPT1 is not; and (5) We confirmed the previously submitted hypothesis that bladder smooth muscle contains a constitutively active isoform of ROCK (Poley et al., 2008; Wang et al., 2009). Our findings coupled with our previous study using carbachol as the contractile stimulus (Wang et al., 2009) have led to the development of a working model for rabbit bladder smooth muscle contraction (shown in Figure 6).
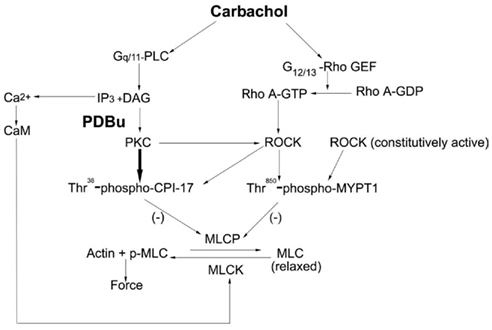
Figure 6. Proposed model for the regulation of bladder smooth muscle contraction. Carbachol stimulation of bladder smooth muscle results in the IP3 dependent release of Ca2+ and MLC kinase-catalyzed phosphorylation of the MLC. Additionally, both PKC and ROCK are activated. PKC directly catalyzes the phosphorylation of CPI-17 while ROCK catalyzes CPI-17 phosphorylation via its activation by PKC. The result is inhibition of the MLC phosphatase and an increase in force. ROCK also catalyzes the phosphorylation of Thr850-MYPT1; however, this is either via a different isoform or pool of ROCK than that which phosphorylates CPI-17. A constitutively active pool of ROCK is present in bladder smooth muscle and is proposed to be responsible for the high basal phosphorylation values of Thr850-MYPT1 due to its inhibition of the MLC phosphatase. PLC, phospholipase C; DAG, diacylglycerol; Rho GEF, Rho guanine nucleotide exchange factor; MLCP, MLC phosphatase; MLCK, MLC kinase; CaM, calmodulin.
PDBu Versus Carbachol-Induced Force and MLC Phosphorylation: Role(s) of PKC and ROCK
The mechanism by which PDBu produces contraction of smooth muscle is unclear. A previous report from our laboratory showed that PDBu induces calcium and MLC phosphorylation-independent contraction of vascular smooth muscle (Fulginiti et al., 1993). In contrast others have observed that PDBu-induced vascular smooth muscle and iris sphincter contractions are at least partly dependent on an increase in intracellular Ca2+ and MLC phosphorylation (Howe and Abdel-Latif, 1987; Singer and Baker, 1987). The apparent discrepancy could be due to the existence of various PKC isoforms in different smooth muscles and species. In our present study, we investigated the mechanism by which PKC induces contraction in bladder smooth muscle, in part, by measuring PDBu as compared to carbachol-dependent increases in MLC phosphorylation.
PDBu stimulation causes a small but significant increase in MLC phosphorylation, which is decreased by inhibition of either PKC or ROCK. Carbachol stimulation produces a robust contraction similar in magnitude to that produced by KCl (Wang et al., 2009). PDBu stimulation, in contrast to carbachol, produces a smaller contraction. Removal of extracellular calcium or depletion of all sources of calcium prevented contraction in response to PDBu, demonstrating the calcium-dependence, and specifically the extracellular calcium-dependence of the contraction. This may be due to inactivation of the MLC kinase in the absence of calcium or the isoform of PKC activated by PDBu resulting in contraction is from the calcium-dependent PKC family. Additionally, stimulation with the inactive phorbol ester, α-PDBu, did not produce a contraction in bladder smooth muscle. This suggests that the increase in force in response to PDBu was not simply phorbol ester-dependent but involved activation of downstream signaling pathways specific to PDBu stimulation, specifically PKC-dependent pathways. This finding coupled with the fact that DOG, a 1,2-diacylgycerol analog, also induced a contraction qualitatively similar to that of PDBu supports the conclusion that PKC is the downstream effector. The patterns of force development and MLC phosphorylation in both carbachol and PDBu-dependent contractions are similar; such that in both cases an early increase in force is followed by a decrease to basal or near basal values. Not surprising, PDBu-dependent increases in both MLC phosphorylation and force were abolished by Bis. However, Bis only affected the carbachol-dependent increase in MLC phosphorylation, and reduced but did not abolish the carbachol-induced increase in force (Wang et al., 2009). This suggests that a carbachol-dependent, but PKC and MLC phosphorylation-independent contractile pathway, is present in bladder smooth muscle. These results are inconsistent with an earlier study using lower urinary tract smooth muscle which suggested that PDBu-induced contractions may be only partially mediated by PKC (Yoshida et al., 1992). This discrepancy may be due, in part, to the use of PKC inhibitors with different specificities as the earlier report used H-7.
The conflicting results in the literature raise the potential limitation inherent in all pharmacological investigations, reliance on the specificity of the chosen inhibitor. These limitations are nicely discussed in work from Cohen’s laboratory (Davies et al., 2000; Bain et al., 2007). In our present study we used Bis for inhibition of PKC and H-1152 for inhibition of ROCK. The inhibitors and concentrations of the inhibitors used were based on the results presented in a previous study from our laboratory (Wang et al., 2009). Specifically, we were interested in directly comparing our current results using PDBu or DOG with the results using carbachol stimulation in our earlier report (Wang et al., 2009). As discussed by Cohen’s group (Davies et al., 2000; Bain et al., 2007), Bis and H-1152, at concentrations used in our study, exhibit non-selective activity against kinases in addition to PKC and ROCK. These studies were preformed in biochemical assays and cells as compared to our present results using tissues which have significantly different responses to both activators and inhibitors of kinases. Moreover, we have found that neither 3 μM Bis nor 1 μM H-1152 inhibited Ca2+-dependent contractions in Triton X-100 skinned or α-toxin permeabilized preparations of bladder smooth muscle (data not shown). This suggests that, at a minimum, the MLC kinase is not inhibited by either Bis or H-1152 at the concentrations used in our current study. Therefore, given this information coupled with the fact that the present study was designed to extend and complement our previous findings (Wang et al., 2009), we believe the use of these inhibitors is appropriate if interpretations are made cautiously.
PDBu Versus Carbachol-Induced CPI-17 Phosphorylation: Role(s) of PKC and ROCK
The fact that PDBu-induced contractions are significantly decreased by a ROCK inhibitor suggests that PKC activates ROCK in bladder smooth muscle (Figure 1). Activation of ROCK by PKC, especially the PKCδ isoform, has been shown to be involved in several cell functions, such as vascular smooth muscle migration and contraction (Kandabashi et al., 2003; Ohtsu et al., 2005; Poole and Furness, 2007). Our laboratory demonstrated that PKCδ and most other novel and classic isoforms of PKC are present in rabbit bladder smooth muscle (unpublished results). Our finding that ROCK inhibition abolishes PDBu-dependent increases in MLC phosphorylation but only partially reduces PDBu-dependent increases in force, suggests a direct role for PKC in addition to an indirect role via ROCK activation in bladder smooth muscle contraction.
Our previous study showed that agonist stimulation of bladder smooth muscle significantly increased Thr38-CPI-17 phosphorylation (Wang et al., 2009), which was primarily caused by PKC during both the phasic and tonic-like phases of contraction. ROCK, however, was also involved in CPI-17 phosphorylation but only during the later tonic-like phase of contraction. PDBu stimulation also increased Thr38-CPI-17 phosphorylation by activation of PKC, which is confirmed by the fact that the PKC inhibitor Bis abolished CPI-17 phosphorylation. Stimulation with DOG also increased Thr38-CPI-17 phosphorylation. Because diacylglycerols are activators of PKC these results support the hypothesis that any increase in CPI-17 phosphorylation is due to activation of PKC. Inhibition of ROCK also decreased CPI-17 phosphorylation. However, in contrast to our study using carbachol as the stimulus which provided a role for ROCK-catalyzed CPI-17 phosphorylation in the later phase of contraction, our present results provide a role for ROCK-catalyzed CPI-17 phosphorylation only during the early phase of the PDBu-induced contraction. This suggests that carbachol and PDBu induce the activation of different isoforms of ROCK and/or induce different temporal profiles of ROCK activation.
PDBu Versus Carbachol-Induced MYPT1 Phosphorylation: Role(s) of PKC and ROCK
MYPT1 is the regulatory subunit of the MLC phosphatase holoenzyme. Our laboratory found that in bladder smooth muscle, Thr850-MYPT1 but not Thr696-MYPT1 phosphorylation increases during agonist stimulation (Wang et al., 2009). Unstimulated resting tissues exhibit significant levels of both Thr696 and Thr850-MYPT1 phosphorylation. Basal phosphorylation levels of Thr850-MYPT1 are sensitive to ROCK inhibition while basal phosphorylation of Thr696-MYPT1 is not. In the present study, we activated only the PKC pathway in bladder smooth muscle by the addition of PDBu or DOG and observed the effect of PKC and potentially PKC activated ROCK on the phosphorylation of MYPT1. Our results demonstrate that neither Thr696 nor Thr850-MYPT1 phosphorylation are significantly increased during PDBu or DOG-induced contraction. These results suggest that although Thr850-MYPT1 is apparently involved in the sustained phase of a carbachol-stimulated contraction, it is not important in a PKC-induced contraction and it is not phosphorylated by the pool of ROCK activated by the PKC pathway.
Consistent with our previous report (Wang et al., 2009), Thr850-MYPT1 phosphorylation is important in the maintenance of resting basal tone of bladder smooth muscle as we demonstrated that inhibition of ROCK decreased both Thr850-MYPT1 phosphorylation and resting levels of force. This conclusion is consistent with work showing that phosphorylated MYPT1 is highly resistant to dephosphorylation, such that this mechanism allows for the long-term regulation of force rather than the acute agonist-stimulated contraction (Takizawa et al., 2002b). Also consistent with our previous finding, basal Thr850-MYPT1 phosphorylation is sensitive to ROCK inhibition. We proposed that a constitutively active pool of ROCK, which phosphorylates only Thr850-MYPT1 at rest, is present in bladder smooth muscle. Our proposal is consistent with work from Poley et al. (2008) who also suggested the existence of a constitutively active pool of ROCK which may contribute to contraction in response to the quick-stretch of bladder smooth muscle.
Several kinases other than ROCK have been proposed to phosphorylate Thr696-MYPT1 in vitro and in vivo, such as ZIP-like kinase, integrin-linked kinase, myotonic dystrophy protein kinase, p21-activated protein kinase, and Raf-1 (Woodsome et al., 2001; Takizawa et al., 2002a; Velasco et al., 2002; Wilson et al., 2005). It is unknown to date which kinase phosphorylates Thr696-MYPT1 in the resting state in bladder smooth muscle, although the lack of effect of H-1152 would suggest it is not ROCK. High basal levels of MYPT1 phosphorylation in bladder smooth muscle may contribute to the inactivation of MLC phosphatase resulting in a high resting tone in the bladder wall. Normally as the bladder fills the low compliance of the bladder wall allows for the maintenance of a low pressure while the volume of the bladder increases. If the bladder smooth muscle has an elevated level of MYPT1 phosphorylation and therefore MLC phosphorylation, the compliance of the bladder wall will be decreased resulting in an increase in bladder pressure with filling. Therefore, any pathophysiological state that resulted in an increase in MYPT1 phosphorylation could have dramatic effects on filling pressure. The phenomenon of high resting MYPT1 phosphorylation and possibly high resting tension is not specific to bladder smooth muscle as Niiro et al. (2003) found a significant level of basal Thr641 (Rat sequence, equals Thr696 in chicken sequence) phosphorylation in rabbit femoral artery.
One of the more interesting findings in this study is the differential effect of ROCK inhibition, with H-1152, on CPI-17 phosphorylation as compared to MYPT1 phosphorylation (Figure 3 versus 4 and 5). Based on the inhibitory effects of H-1152 on PDBu-induced increases in CPI-17 phosphorylation, the logical assumption is that PDBu-induced activation of PKC results in activation of ROCK which in turn phosphorylates CPI-17. However, stimulation with PDBu alone does not produce any increase in phosphorylation of the known substrate for ROCK, MYPT1. This would suggest that either PKC activates an isoform of ROCK that does not phosphorylate MYPT1 or PKC activates a specific pool or compartment of ROCK that is specific to CPI-17. An alternate explanation is that there is a non-specific effect of H-1152 on PKC or a downstream substrate of PKC.
Our study proposes an important role for the so-called Ca2+ sensitizers, MYPT1 and CPI-17, in contraction of bladder smooth muscle. A recent report from Kamm’s laboratory (Ding et al., 2009) would suggest that these proteins may only be important in the slower contraction induced by carbachol as compared to electrical field stimulation or during the later phase of a contractile event. They demonstrated that in the very early phase of mouse bladder smooth muscle contraction, there was a rapid, millisecond time-frame increase in cellular Ca2+ and levels of MLC phosphorylation without any evidence of increases in CPI-17 or MYPT1 phosphorylation. It is possible that this is also the case at time points earlier than those measured in our current study. Our results do, however, suggest that both CPI-17 and MYPT1 phosphorylation are important in the later phase of a rabbit bladder contraction. Consistent with our findings that both PKC and ROCK play an important role in a bladder contraction on the time-frame of minutes is that inhibitors of either PKC or ROCK had a greater effect on the tonic-like as compared to the initial phasic part of contraction of guinea pig bladder smooth muscle (Roosen et al., 2009). Thus, it appears plausible that the very early increase in force in the bladder wall is dependent on Ca2+ and MLC phosphorylation whereas the peak development of force and the more tonic-like phase rely more on PKC and ROCK activation and therefore the Ca2+ sensitization pathway.
In summary, we propose the model shown in Figure 6 for the regulation of bladder smooth muscle contraction. Agonist activation of bladder smooth muscle involves both PKC and ROCK signaling pathways in the development and maintenance of a contraction. CPI-17 is a major downstream regulator of PKC both directly and indirectly via activation of ROCK. Whether the ROCK activated by PKC resulting in the phosphorylation of CPI-17 is a different ROCK isoform or from a different pool of ROCK than that important in catalyzing MYPT1 phosphorylation is not known. Although not directly tested in our studies, but based on a vast literature, CPI-17 phosphorylation results in the inhibition of the MLC phosphatase and an increase in force development. Thr850-MYPT1 is phosphorylated by ROCK during agonist stimulation and inhibits MLC phosphatase activity, but that ROCK isoform or pool is not part of the PKC–ROCK signaling pathway. At rest, a constitutively active pool of ROCK phosphorylates Thr850-MYPT1 and an unknown kinase(s) phosphorylates Thr696-MYPT1. Additionally, there are temporal components to the regulation of ROCK such that PKC activated ROCK is important in the early phase of a bladder smooth muscle contraction while direct ROCK activation via agonist stimulation plays a larger role in the later phase of a contraction. Thus, bladder smooth muscle contraction is regulated by a complex interaction of several signaling pathways exhibiting both cross-activation and temporal components.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported, in part, by funds from NIH grants DK 69898 and DK 85734. D. M. Kendig was supported by a Drexel University College of Medicine Aging Initiative Graduate Fellowship. E. M. Smolock was supported by a Predoctoral Fellowship from the Mid-Atlantic Chapter of the American Heart Association.
References
Bain, J., Plater, L., Elliott, M., Shapiro, N., Hastie, C. J., McLauchlan, H., Klevernic, I., Arthur, J. S. C., Alessi, D. R., and Cohen, P. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315.
Castagna, M., Takai, Y., Kaibuchi, K., Sano, K., Kikkawa, U., and Nishizuka, Y. (1982). Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 257, 7847–7851.
Chang, S., Hypolite, J. A., Mohanan, S., Zderic, S. A., Wein, A. J., and Chacko, S. (2009). Alteration of the PKC-mediated signaling pathway for smooth muscle contraction in obstruction-induced hypertrophy of the urinary bladder. Lab. Invest. 89, 823–832.
Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105.
Ding, H.-L., Ryder, J. W., Stull, J. T., and Kamm, K. E. (2009). Signaling processes for initiating smooth muscle contraction upon neural stimulation. J. Biol. Chem. 284, 15541–15548.
Eto, M., Kitazawa, T., and Brautigan, D. L. (2004). Phosphoprotein inhibitor CPI-17 specificity depends on allosteric regulation of protein phosphatase-1 by regulatory subunits. Proc. Natl. Acad. Sci. U.S.A. 101, 8888–8893.
Eto, M., Ohmori, T., Suzuki, M., Furuya, K., and Morita, F. (1995). A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C isolation from porcine aorta media and characterization. J. Biochem. 118, 1104–1107.
Eto, M., Senba, S., Morita, F., and Yazawa, M. (1997). Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI-17) in smooth muscle: it’s specific localization in smooth muscle. FEBS Lett. 410, 356–360.
Feng, J., Ito, M., Ichikawa, K., Isaka, N., Nishikawa, M., Hartshorne, D. J., and Nakano, T. (1999). Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J. Biol. Chem. 274, 37385–37290.
Frazier, E. P., Peters, S. L., Braverman, A. S., Ruggieri, M. R., Sr., and Michel, M. C. (2008). Signal transduction underlying the control of urinary bladder smooth muscle tone by muscarinic receptors and beta-adrenoceptors. Naunyn Schmiedebergs Arch. Pharmacol. 377, 449–462.
Fulginiti, J., Singer, H. A., and Moreland, R. S. (1993). Phorbol ester-induced contractions of swine carotid artery are supported by slowly cycling cross bridges which are not dependent on calcium or myosin light chain phosphorylation. J. Vasc. Res. 30, 315–322.
Gorenne, I., Su, X., and Moreland, R. S. (1998). Inhibition of p42 and p44 MAP kinase activity does not alter smooth muscle contraction in swine carotid artery. Am. J. Physiol. Heart Circ. Physiol. 275, H131–H138.
Hartshorne, D. J. (1998). Myosin phosphatase: subunits and interactions. Acta Physiol. Scand. 164, 483–493.
Howe, P. H., and Abdel-Latif, A. A. (1987). Phorbol ester-induced protein phosphorylation and contraction in sphincter smooth muscle of rabbit iris. FEBS Lett. 215, 279–284.
Ito, M., Nakano, T., Erdodi, F., and Hartshorne, D. J. (2004). Myosin phosphatase: structure, regulation and function. Mol. Cell. Biochem. 259, 197–209.
Kamm, K. E., and Stull, J. T. (1985). The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu. Rev. Pharmacol. Toxicol. 25, 593–620.
Kandabashi, T., Shimokawa, H., Miyata, K., Kunihiro, I., Eto, Y., Morishige, K., Matsumoto, Y., Obara, K., Nakayama, K., Takahashi, S., and Takeshita, A. (2003). Evidence for protein kinase C-mediated activation of Rho-kinase in a porcine model of coronary artery spasm. Arterioscler. Thromb. Vasc. Biol. 23, 2209–2214.
Kimura, K., Ito, M., Amano, M., Chihara, K., Fukata, Y., Nakafuku, M., Yamamori, B., Feng, J., Nakano, T., Okawa, K., Iwamatsu, A., and Kaibuchi, K. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248.
Kitazawa, T., Eto, M., Woodsome, T. P., and Brautigan, D. L. (2000). Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J. Biol. Chem. 275, 9897–9900.
Moreland, S., and Moreland, R. S. (1987). Effects of dihydropyridines on stress, myosin phosphorylation, and Vo in smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 252, H1049–H1058.
Moreland, S., Nishimura, J., van Breemen, C., Ahn, H.-Y., and Moreland, R. S. (1992). Transient myosin phosphorylation at constant Ca2+ during agonist activation of permeabilized arteries. Am. J. Physiol. 263, C540–C544.
Niiro, N., Koga, Y., and Ikebe, M. (2003). Agonist-induced changes in the phosphorylation of the myosin- binding subunit of myosin light chain phosphatase and CPI17, two regulatory factors of myosin light chain phosphatase, in smooth muscle. Biochem. J. 369, 117–128.
Ohtsu, H., Mifune, M., Frank, G. D., Saito, S., Inagami, T., Kim-Mitsuyama, S., Takuwa, Y., Sasaki, T., Rothstein, J. D., Suzuki, H., Nakashima, H., Woolfolk, E. A., Motley, E. D., and Eguchi, S. (2005). Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 25, 1831–1836.
Poley, R. N., Dosier, C. R., Speich, J. E., Miner, A. S., and Ratz, P. H. (2008). Stimulated calcium entry and constitutive RhoA kinase activity cause stretch-induced detrusor contraction. Eur. J. Pharmacol. 599, 137–145.
Poole, D. P., and Furness, J. B. (2007). PKC delta-isoform translocation and enhancement of tonic contractions of gastrointestinal smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G887–G898.
Rembold, C. M., and Murphy, R. A. (1988). [Ca2+]-dependent myosin phosphorylation in phorbol diester stimulated smooth muscle contraction. Am. J. Physiol. 255, C719–C723.
Roosen, A., Fry, C. H., Guiping, S., and Changhao, W. (2009). Adreno-muscarinic synergy in the bladder trigone: calcium-dependent and -independent mechanisms. Cell Calcium 45, 11–17.
Sasaki, Y., Suzuki, M., and Hidaka, H. (2002). The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]-homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol. Ther. 93, 225–232.
Singer, H. A., and Baker, K. M. (1987). Calcium dependence of phorbol 12,13-dibutyrate-induced force and myosin light chain phosphorylation in arterial smooth muscle. J. Pharmacol. Exp. Ther. 243, 814–821.
Sobieszek, A. (1977). Ca-linked phosphorylation of a light chain of vertebrate smooth-muscle myosin. Eur. J. Biochem. 73, 477–483.
Somlyo, A. P., and Somlyo, A. V. (1994). Signal transduction and regulation in smooth muscle. Nature 372, 231–236.
Somlyo, A. P., and Somlyo, A. V. (2003). Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358.
Stanton, M. C., Austin, J. C., Delaney, D. P., Gosfield, A., Marx, J. O., Zderic, S. A., Chacko, S., and Moreland, R. S. (2006). Partial bladder outlet obstruction selectively abolishes protein kinase C induced contraction of rabbit detrusor smooth muscle. J. Urol. 176, 2716–2721.
Su, X., Smolock, E. M., Marcel, K. N., and Moreland, R. S. (2004). Phosphatidylinositol 3-kinase modulates vascular smooth muscle contraction by calcium and myosin light chain phosphorylation-independent and -dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 286, H657–H666.
Su, X., Stein, R., Stanton, M. C., Zderic, S., and Moreland, R. S. (2003). Effect of partial outlet obstruction on rabbit urinary bladder smooth muscle function. Am. J. Physiol. Renal Physiol. 284, F644–F652.
Takizawa, N., Koga, Y., and Ikebe, M. (2002a). Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem. Biophys. Res. Commun. 297, 773–778.
Takizawa, N., Niiro, N., and Ikebe, M. (2002b). Dephosphorylation of the two regulatory components of myosin phosphatase, MBS and CPI17. FEBS Lett. 515, 127–132.
Toullec, D., Pianetti, P., Coste, H., Bellevergue, P., Grand-Perret, T., Ajakane, M., Baudet, V., Boissin, P., Boursier, E., and Loriolle, F. (1991). The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 266, 15771–15781.
Velasco, G., Armstrong, C., Morrice, N., Frame, S., and Cohen, P. (2002). Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1 M at Thr850 induces its dissociation from myosin. FEBS Lett. 527, 101–104.
Wang, T., Kendig, D. M., Smolock, E. M., and Moreland, R. S. (2009). Carbachol-induced rabbit bladder smooth muscle contraction: roles of protein kinase C and Rho kinase. Am. J. Physiol. Renal Physiol. 297, F1534–F1542.
Wilson, D. P., Susnjar, M., Kiss, E., Sutherland, C., and Walsh, M. P. (2005). Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: rho-associated kinase-mediated phosphorylation of MYPT1 at Thr-855, but not Thr-697. Biochem. J. 389, 763–774.
Woodsome, T. P., Eto, M., Everett, A., Brautigan, D. L., and Kitazawa, T. (2001). Expression of CPI-17 and myosin phosphatase correlates with Ca2+ sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J. Physiol. 535, 553–564.
Keywords: MLC phosphatase, myosin phosphatase target subunit, CPI-17, bisindolylmaleimide-1, H-1152, MLC phosphorylation
Citation: Wang T, Kendig DM, Trappanese DM, Smolock EM and Moreland RS (2012) Phorbol 12,13-dibutyrate-induced, protein kinase C-mediated contraction of rabbit bladder smooth muscle. Front. Pharmacol. 2:83. doi: 10.3389/fphar.2011.00083
Received: 30 June 2011;
Accepted: 06 December 2011;
Published online: 02 January 2012.
Edited by:
Issy Laher, University of British Columbia, CanadaReviewed by:
Hamid Akbarali, Virginia Commonwealth University, USAChun Y. Seow, University of British Columbia, Canada
Copyright: © 2012 Wang, Kendig, Trappanese, Smolock and Moreland. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Robert S. Moreland, Department of Pharmacology and Physiology, Drexel University College of Medicine, 245 N 15th Street, MS #488, Philadelphia, PA 19102, USA. e-mail: robert.moreland@drexelmed.edu
†Tanchun Wang and Derek M. Kendig have contributed equally to this work.