
- School of Biomedical Sciences and Pharmacy, Centre for Translational Neuroscience and Mental Health Research, Hunter Medical Research Institute, University of Newcastle, Callaghan, NSW, Australia
Complex neuroadaptations within key nodes of the brain’s “reward circuitry” are thought to underpin long-term vulnerability to relapse. A more comprehensive understanding of the molecular and cellular signaling events that subserve relapse vulnerability may lead to pharmacological treatments that could improve treatment outcomes for psychostimulant-addicted individuals. Recent advances in this regard include findings that drug-induced perturbations to neurotrophin, metabotropic glutamate receptor, and dopamine receptor signaling pathways perpetuate plasticity impairments at excitatory glutamatergic synapses on ventral tegmental area and nucleus accumbens neurons. In the context of addiction, much previous work, in terms of downstream effectors to these receptor systems, has centered on the extracellular-regulated MAP kinase signaling pathway. The purpose of the present review is to highlight the evidence of an emerging role for another downstream effector of these addiction-relevant receptor systems – the mammalian target of rapamycin complex 1 (mTORC1). mTORC1 functions to regulate synaptic protein translation and is a potential critical link in our understanding of the neurobiological processes that drive addiction and relapse behavior. The precise cellular and molecular changes that are regulated by mTORC1 and contribute to relapse vulnerability are only just coming to light. Therefore, we aim to highlight evidence that mTORC1 signaling may be dysregulated by drug exposure and that these changes may contribute to aberrant translation of synaptic proteins that appear critical to increased relapse vulnerability, including AMPARs. The importance of understanding the role of this signaling pathway in the development of addiction vulnerability is underscored by the fact that the mTORC1 inhibitor rapamycin reduces drug-seeking in pre-clinical models and preliminary evidence indicating that rapamycin suppresses drug craving in humans.
Introduction
Drug addiction is a complex neuropsychiatric condition with the risk of relapse after quitting considered a fundamental obstacle to long-term treatment management (O’Brien, 2005; Kalivas and O’Brien, 2008). Despite significant recent efforts, our understanding of the neurobiological processes that lead to addiction, and subsequently protracted relapse risk, is inadequate to the point that there has been limited translation of pre-clinical findings into pharmacological treatments for this condition. Critically, further research is required to unravel the complex neuroadpatations that cause psychostimulant addiction, in particular the molecular and cellular signaling events that underpin protracted vulnerability to relapse. This review will focus on recent findings relevant to translational efforts to identify targets to treat psychostimulant addiction.
The brain circuitry that controls drug-seeking and relapse is multi-nodal (Koob and Volkow, 2009), however, the two nodes that have received the most attention with regard to functional and molecular correlates of addiction are the ventral tegmental area (VTA) A10 dopamine neurons and the nucleus accumbens (NAC; Kauer and Malenka, 2007; Kalivas and O’Brien, 2008; Luscher and Malenka, 2011). Although other brain regions, such as the prefrontal cortex (PFC), amygdala, and dorsal striatum, are also known to play important roles in addiction, our understanding of the pre- and post-synaptic neuroadaptations that occur within the VTA and NAC is significantly more detailed than these other major nodes in the brain “relapse circuit” (Kalivas and O’Brien, 2008). Furthermore, the NAC and VTA have been ascribed an important role in reward learning (Stuber et al., 2008; Tsai et al., 2009) and drug-induced synaptic plasticity deficits in these brain regions that occur in response to chronic drug taking have been hypothesized to impede the learning of new strategies to overcome addiction (Martin et al., 2006; Kalivas and Volkow, 2011). (We direct readers to several excellent recent reviews describing the effects of chronic cocaine self-administration on synaptic plasticity within the VTA and NAC; Kauer and Malenka, 2007; Kalivas and O’Brien, 2008; Luscher and Malenka, 2011).
Given their importance, significant research efforts have been, and continue to be, directed at the VTA and NAC in order to improve our understanding of the molecular signaling pathways that are responsible for the drug-induced maladaptations. Advances in these areas have highlighted a role for a number of signaling pathways in the development of addiction (Grimm et al., 2003; Lu et al., 2006; Graham et al., 2007; Dietz et al., 2009; Russo et al., 2009; McGinty et al., 2010; Duncan and Lawrence, 2011) including molecules downstream of neurotrophin (NTrk), metabotropic glutamate (mGluR), and dopamine (DR) receptors. Drug-induced perturbations in these signaling pathways are thought to perpetuate plasticity impairments at excitatory glutamatergic synapses on VTA and NAC neurons. The purpose of the present review is to highlight the emerging evidence that the mammalian target of rapamycin complex 1 (mTORC1), a downstream target of these receptor signaling pathways that regulate synaptic protein translation, is a potentially critical link in our understanding of the neurobiological processes that drive addiction and relapse behavior (Neasta et al., 2010; Wang et al., 2010; Brown et al., 2011). Investigations of mTOR’s role in addiction are presently limited to the NAC and VTA, and consequently our review will outline relevant molecular and cellular adaptations within these structures. However, where necessary we will draw mechanistic links derived from studies assessing the role of mTOR in synaptic plasticity in other brain regions. We begin by briefly summarizing mTORC1 signaling in the CNS, however, we alert readers to a number of important reviews that have recently been published on this topic (Klann et al., 2004; Costa-Mattioli et al., 2009; Richter and Klann, 2009; Hoeffer and Klann, 2010; Sharma et al., 2010).
An Overview of mTORC1 Signaling within the Brain
Mammalian target of rapamycin (mTOR) is a 250-kDa serine/threonine protein kinase. Much of what we know about mTOR signaling is based on the effects of the specific inhibitor, rapamycin, a macroclide (sirolimus) originally identified in a soil bacterium. Rapamycin acts by binding to the cytosolic protein FKBP12, which leads to the displacement of Raptor from mTOR (Lorenz and Heitman, 1995; Sabers et al., 1995). Raptor is critical to mTOR function (Kim et al., 2002) and the complex between these two proteins, together with proline-rich AKT substrate 40 kDa (PRAS40), mammalian lethal with Sec13 protein 8 (mLST8), and DEP-domain-containing mTOR interacting protein (Deptor), is referred to as mTOR complex 1 (mTORC1; Hoeffer and Klann, 2010). Rapamycin-induced displacement of Raptor inhibits its capacity to bind to and appropriately locate mTOR substrates and, as such, they can no longer be phosphorylated (Figure 1).
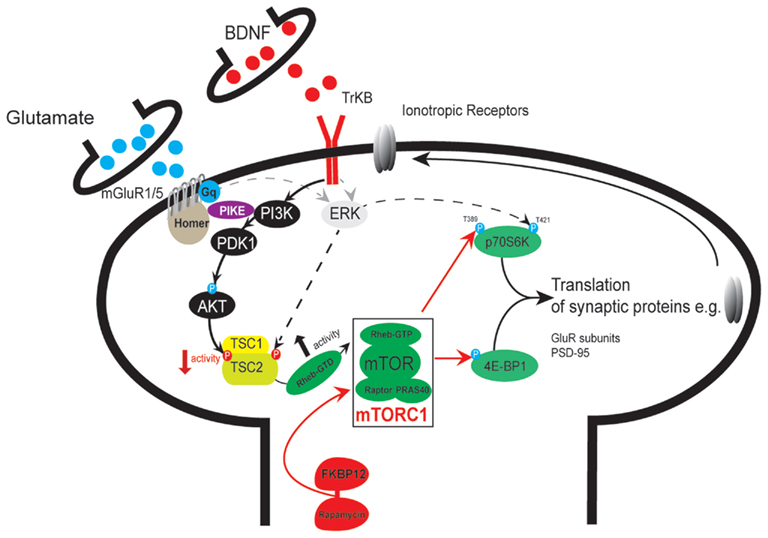
Figure 1. Schematic depicting mTORC1 signaling in a NAC a medium spiny neuron with anterogradely transported BDNF being released from a VTA dopamine neuron terminal and glutamate released from a prefrontal cortex projection neuron. [Note however that BDNF can also be released from PFC projection neurons]. A number of addiction-relevant receptor systems including mGluRs and BDNF (but also dopamine and ionotropic glutamate receptors), can signal via the phosphoinositide-3′ (PI3K) – AKT pathway to regulate mTOR activity. The recruitment of PI3K and the production of phosphatidylinositol 3,4,5-trisphosphate (PIP3) elicits 3-phosphoinositide-dependent protein kinase 1 (PDK1) activity which in turn phosphorylates AKT. AKT activation is then thought to lead to the phosphorylation of tuberin (TSC2). Phosphorylation of TSC2 by AKT reduces TSC1/2 activity and consequently increases mTOR activity; a process that is thought to involve a Rheb-mediated inhibition of FKBP38 (or FKBP12 in the brain, Hoeffer and Klann, 2010) – the latter being an inhibitory binding protein that prevents mTOR complex 1 formation. FKBP12 prevents the binding of the mTOR complex protein regulatory-associated protein with TOR (Raptor) with the FRB binding domain through a process facilitated by rapamycin (FKBP12-rapamycin). mTORC1 phosphorylates 4E-BPs, a family of translational repressor proteins, which leads to their dissociation from eIF4E, enabling it to form the eIF4F complex for cap binding and participation in the initiation of translation of a range of mRNAs. Thus, mTOR-mediated phosphorylation of 4E-BPs and S6Ks leads to increased translation of mRNA – including transcripts that encode proteins required for synaptic plasticity such as GluR1 AMPARs. Akt, also known as protein kinase B; BDNF, brain-derived neurotrophic factor; ERK, extracellular signal-regulated protein kinase; 4E-BPI, eukaryotic initiation factor 4E-binding protein; FKBP12, FK506-binding protein 12; Gq, Gq-protein; mGluR, metabotropic glutamate receptor; mTOR, mammalian target of rapamycin; NMDAR, N-methyl-D-aspartate receptor; PI3K, phosphoinositide-3 kinase; PIKE, PI3K-enhancer; mTORC1, mTOR complex 1; PDK1, phosphoinositide-dependent kinase 1; PRAS40, proline-rich Akt/PKB substrate 40 kDa; Raptor, regulatory-associated protein with TOR; Rheb, Ras homolog enriched in brain; S6K, p70 S6 kinase; TrkB, tyrosine receptor kinase B; TSC1/2, tuberous sclerosis complex 1 and 2; T, threonine; P, phosphorylation.
Studies have implicated mTORC1 signaling in a range of neural functions and neurological conditions including autism, mental retardation, and epilepsy (Ehninger et al., 2008; Bourgeron, 2009), memory formation and brain plasticity (Hoeffer and Klann, 2010), brain metabolism (Potter et al., 2010), feeding behavior (Flier, 2006), as well as age-related neuropathologies (Garelick and Kennedy, 2011). Rapamycin studies have directly led to the finding that mTORC1 plays an important role in activity-dependent translation of proteins required for synaptic plasticity (i.e., long-term potentiation; Cammalleri et al., 2003) and long-term depression (LTD; Mameli et al., 2007) by regulating the translation initiation complex through phosphorylation of p70S6K and 4E-BP1. Indeed, there is strong evidence supporting a role for mTORC1 in regulating the translation of a number of proteins necessary for synaptic plasticity, such as Ca2+/Calmodulin-dependent kinase II alpha (CamKIIα), AMPAR receptor subunits (GluRs), microtubule-associated protein (MAP2), post-synaptic density protein 95 (PSD-95), glutamate receptor interaction protein (GRIP1), and NMDARs, for example, GRIN1 (Hou and Klann, 2004; Mameli et al., 2007).
Regulation of protein synthesis by downstream effectors of mTORC1 generally occurs at the mRNA translation initiation step and involves a complex array of initiation factors and binding proteins working with the ribosomes to ensure that the mRNA start codon is efficiently identified for the initiation of translation (Sonenberg and Hinnebusch, 2007, 2009). An important component of the translation initiation mechanism is the activation of the mRNA by binding of the eIF4F complex to the mRNA’s 5′ cap. mRNA activation through cap recognition is one way translation is regulated and, thus, eIF4F binding constitutes a key regulatory step. Regulation of eIF4F is under either direct or indirect control by mTORC1 (Hay and Sonenberg, 2004; Hoeffer and Klann, 2010). For example, mTORC1 phosphorylates 4E-BPs, a family of translational repressor proteins, which leads to their dissociation from eIF4E, enabling it to form the eIF4F complex for cap binding and participation in the initiation of translation of a range of mRNAs (Gingras et al., 2004; Richter and Klann, 2009). mTORC1 can indirectly influence eIF4F activity through S6K protein kinases that phosphorylate the ribosomal protein S6 and eIF4E, which in turn increases mRNA translation (Raught et al., 2004). Therefore, mTORC1-mediated phosphorylation of 4E-BPs and S6Ks leads to increased translation of mRNA (Hay and Sonenberg, 2004; Richter and Klann, 2009).
There is a second mTOR complex known as mTORC2 that shares only three proteins in common with mTORC1 – that is, mTOR, mLST8, and Deptor. The additional proteins making up the mTORC2 complex include SAPK Interacting protein I (SIN1), rapamycin-insensitive companion of mTOR (Rictor), and protein observed with Rictor (Protor). Unlike mTORC1, mTORC2 is insensitive to acute rapamycin treatment, although there are reports that chronic treatment with the mTOR inhibitor may disrupt mTORC2 complex formation (Sarbassov et al., 2006). Two known targets of mTORC2 include the phosphorylation of AKT (at Serine 473) and protein kinase Cα (Hay and Sonenberg, 2004; Richter and Klann, 2009; Hoeffer and Klann, 2010). While the role of mTORC1 complex in synaptic plasticity is well established, recent studies have identified a potential molecular link between mTORC2/AKT signaling and actin cytoskeletal remodeling, suggesting a possible role in structural plasticity within the brain (Sarbassov et al., 2004; Russo et al., 2009). Consistent with this possibility, there is emerging evidence of an important role for mTORC2 in promoting structural plasticity of VTA dopamine neurons in response to opiates, which we outline in more detail below (Mazei-Robison et al., 2011). Because the roles of mTORC2 in the brain are only beginning to be characterized, our primary focus will be the more established roles of mTORC1 and synaptic plasticity and its relevance to addiction.
Neural Membrane Receptors Upstream of mTORC1
One pathway for mTORC1 activation in neurons involves neurotrophin receptor signaling through receptor tyrosine kinases in response to stimulation by trophic factors, e.g., BDNF. Tyrosine kinase B (TrkB) receptor activation by BDNF leads to the activation of the phosphoinositide-3′ (PI3K)/AKT pathway (Figure 1). Through a phosphorylation cascade, mTORC1 activity is increased by PI3K–AKT activation. The resultant increase in mTOR activity is thought to occur through a Rheb-mediated inhibition of FKBP38 (or FKBP12 in the brain; Hoeffer and Klann, 2010) – the latter being an inhibitory binding protein that prevents mTOR complex formation. It is important to note, however, that mTORC1 is actually at the “crossroads” of a number of addiction-relevant signaling pathways that regulate the translation of synaptic proteins, and thus LTP and LTD in the brain (Figure 1; Hoeffer and Klann, 2010). For example, in addition to neurotrophins, the ionotropic N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors and dopamine D1, D2 receptors also can induce mTORC1 activity (Hay and Sonenberg, 2004; Hoeffer and Klann, 2010). Particularly relevant to the present review, given their well established role in addiction-related behaviors (Bird and Lawrence, 2009; Jupp and Lawrence, 2010; Duncan and Lawrence, 2011), are findings that group I mGluRs can influence mTOR activity and evoke LTD (Rong et al., 2003; Gong et al., 2006; Mameli et al., 2007; Ronesi and Huber, 2008; Hoeffer and Klann, 2010; Sharma et al., 2010). mGluR1/5s are linked to the enhancer of PI3K (PIKE) by the Homer family of scaffold proteins and in the hippocampus, recruitment of this pathway enhances PI3K → AKT phosphorylation and induces mTOR activity and LTD (Rong et al., 2003; Gong et al., 2006; Ronesi and Huber, 2008; Mameli et al., 2009; Hoeffer and Klann, 2010; Sharma et al., 2010). It will be important to determine if a similar mechanism regulates VTA and NAC plasticity in response to addictive drugs.
Metabotropic glutamate receptor, TrkB, and NMDA receptors can also increase the translation of synaptic proteins through the extracellular-regulated kinase MAP kinase (ERK) pathway (Mao et al., 2005; Banko et al., 2006; Gong et al., 2006; Page et al., 2006; Gelinas et al., 2007). In fact, co-incidental activation of mTOR and ERK may be necessary for the precise control of the translational machinery that regulates changes in synaptic efficacy (Gelinas et al., 2007). Importantly though, it does appear that under some circumstances mTOR activation alone is sufficient for synaptic protein translation and synaptic plasticity (Mameli et al., 2007; Ronesi and Huber, 2008). With respect to ERK’s involvement in the control of translation and its interaction with mTOR, ERK can also inhibit tuberin (TSC2), and therefore influence mTOR activity. Further, ERK can independently phosphorylate the same downstream effectors of translation that mTOR targets, e.g., p70S6K and 4E-BP1, an effect that appears to be mediated by the recruitment of MAPK-interacting kinase (Mnk1; Banko et al., 2004; Laplante and Sabatini, 2009; Santini et al., 2009).
mTORC1’s Role in Synaptic Plasticity and Long-Term Memory Formation
This section aims to present a brief overview of the role of mTORC1 in synaptic plasticity and memory formation as a preface to a discussion of the possible links between this role and cocaine-induced plasticity impairments. Translation of synaptic proteins in dendrites is thought to be an important mechanism for altering synapse structure and function (Kandel, 2001; Bramham and Wells, 2007). Early studies first alluded to this by demonstrating that dendrites contain the necessary substrates for protein translation including mRNA, ribosomes, and translation initiation factors (Bodian, 1965; Torre and Steward, 1992; Crino and Eberwine, 1996; Tian et al., 1999; Aakalu et al., 2001; Asaki et al., 2003). Importantly, these initial observations were consistent with functional studies showing that late phase LTP (L-LTP) is dependent on the transcription and synthesis of dendritic proteins, whereas early phase LTP (E-LTP) relies on modifications to existing proteins (Kandel, 2001). These studies reported effects on hippocampal L-LTP and long-term facilitation in Aplysia (Brown et al., 1995; Beretta et al., 1996; Casadio et al., 1999; Tang et al., 2002; Cammalleri et al., 2003). Subsequent studies by Cammalleri and Tang demonstrated a post-synaptic co-localization of mTOR (and other translation regulation factors such as 4E-BP1 and 2) with the PSD-95, in dendrites of cultured hippocampal neurons. These groups also provided evidence of activity-dependent changes in the phosphorylation status of a number of key substrates within this pathway (Tang et al., 2002; Cammalleri et al., 2003), linking synaptic activity with phosphorylation of mTOR substrates.
Similar to L-LTP, long-term memory formation in intact animals also requires new protein synthesis. For example, broad-spectrum protein synthesis inhibitors, such as anisomycin, affect the consolidation and reconsolidation of fear memories (Schafe et al., 1999; Nader et al., 2000; Schafe and LeDoux, 2000; Parsons et al., 2006b). Importantly, behavioral studies employing rapamycin support a role for mTORC1 signaling in long-term memory formation. Accordingly, systemic rapamycin treatment suppresses reconsolidation of fear memory in mice (Blundell et al., 2008), and when injected into the auditory cortex, the mTOR inhibitor was found to block long-term fear memory consolidation (Tischmeyer et al., 2003). Rapamycin delivered into specific brain sites such as the amygdala or hippocampus also suppresses the consolidation and reconsolidation of fear memories (Parsons et al., 2006a), indicating a widespread role for mTORC1 in synapse function. With respect to the specific complement of synaptic proteins that are regulated by mTORC1 and are required for learning in these paradigms, Parsons et al. (2006a) showed that intra-amygdala rapamycin reduced fear learning to a similar extent to anisomysin. However, while anisomycin reduced protein synthesis by an estimated 60–80%, rapamycin reduced protein synthesis by only 10%. These findings were taken to indicate that only a specific subset of proteins, controlled by mTORC1, are required and fundamental to the learning processes responsible for fear memory (Parsons et al., 2006a).
While most animal studies using rapamycin support a role for mTORC1 in long-term memory formation, Panja et al. (2009) found that whereas rapamycin inhibited hippocampal L-LTP in vitro, the mTOR inhibitor failed to block L-LTP in vivo (Panja et al., 2009). Studies using mice possessing genetic manipulations of molecules that increase mTORC1 activity have also demonstrated inconsistent results with respect to synaptic plasticity and long-term memory formation (see Stoica et al., 2011 supplementary data for review). Importantly, in a comprehensive set of experiments confirming the importance of mTORC1 in synaptic plasticity, Stoica et al. (2011) recently reported that mTOR heterozygote mice (mTOR+/−) display normal L-LTP and long-term memory formation (Stoica et al., 2011). However, a sub-threshold dose of rapamycin, ineffective in reducing LTM and L-LTP in wild type mice, significantly reduced these parameters in mTOR+/− animals.
It is also noteworthy, given the now well-characterized role for BDNF in neuroadaptations associated with drug addiction (see further discussion below), that BDNF-induced LTP is blocked by rapamycin in the hippocampus and that this process requires new protein synthesis (Tang et al., 2002). Interestingly, Slipczuk et al. (2009) demonstrated that BDNF activates mTOR and regulates inhibitory avoidance memory formation (Slipczuk et al., 2009). This process was found to involve an mTORC1-dependent upregulation of GluR1 AMPARs. These data are consistent with other studies describing BDNF/mTORC1-dependent increases in synaptic plasticity proteins including PSD-95, GluRs, and other plasticity-associated proteins (Schratt et al., 2004; Page et al., 2006; Mameli et al., 2007). It will be interesting to determine the role of BDNF in driving mTORC1-dependent processes in brain regions known to be involved in addiction.
Putative Role for mTORC1 in Addiction-Relevant Synaptic Plasticity
Maladaptive behaviors that are characteristic of addiction are underpinned by persistent changes in synaptic properties, particularly in the brain reward pathways.
As discussed above, activation of a number of receptor systems that can produce synaptic plasticity, including TrkB, NMDA, Group I mGluRs, and DRs have been shown to activate the mTORC1 pathway and are implicated in the neurobiological processes that lead to the usurpation of the VTA–NAC pathway during the addiction process (Kalivas, 2009; Kalivas and Volkow, 2011).
With respect to drug-induced plasticity in the VTA, Mameli et al. (2007) showed that cocaine-evoked LTP was reversed by pharmacological stimulation of mGluR1 receptors using (RS)-3,5-dihydroxyphenylglycine (DHPG), an mGluR1/5 agonist. Notably, this effect involved mTORC1-dependent synthesis and subsequent insertion of Ca2+ impermeable GluR2 AMPAR subunits. Interestingly, although there is known to be significant crosstalk between ERK and mTORC1 signaling pathways, the ERK inhibitor U0126 had no effect on mGluR1-induced LTD in cocaine-exposed animals. Subsequent studies have implicated an mGluR1–Homer interaction in the LTD that appears protective, at least initially, against cocaine-induced potentiation of excitatory inputs on VTA dopamine neurons. The resultant potentiation of excitatory inputs onto VTA dopamine neurons during the early stages of cocaine self-administration was found to be necessary for subsequent plasticity changes within the NAC (Mameli et al., 2009) and the later expression of addiction behaviors including REINSTATEMENT, i.e., “relapse” (see Box 1 – descriptions). These findings have led to the hypothesis that exhaustion of mGluR and mTOR-dependent LTD in midbrain dopamine neurons may predispose addiction-vulnerable animals to deficits in plasticity within the NAC (Luscher and Huber, 2010).
Box 1. Glossary terms/definitions – cited within text.
Conditioned place preference (CPP): animals are conditioned to sides of an experimental chamber that have different contextual cues – e.g., the walls and or floors are made of different materials. An experimenter injects saline or drug intraperitoneally (i.p.), before placing the mouse or rat into designated sides of the compartment. The animal’s preference for the drug-paired versus saline paired side (CPP) is assessed in a subsequent test, performed under drug free conditions. This procedure is thought to involve Pavlovian conditioning.
Psychomotor sensitization: a progressive increase (reverse tolerance) to a drug’s locomotor activating effects that is usually induced by a single unit dose of drug injected by the experimenter over 5–10 days. The expression of locomotor sensitization is often assessed during withdrawal after an acute injection of the drug (an equivalent dose is used to that given during the induction phase).
Drug self-administration: animals are trained to lever press in operant responding chambers for a unit dose of drug (or natural reward, e.g., sucrose). Using this procedure animals can learn to regulate their own drug intake and depending on the type of schedule, behaviors such as motivation to lever press can be assessed. This is achieved by increasing the ratio, i.e., the number of lever presses required to receive a drug infusion or reward (a progressive ratio schedule).
Reinstatement (relapse-like behavior): a method to gage the propensity to seek and consume drug that employs drug cues that have been associated through classical conditioning procedures during the drug taking phase, stress (most commonly footshock), or small priming doses of drug to “reinstate” lever pressing after a period of extinction training. After training animals to lever press for drug the animals are subjected to a period of forced abstinence during which they are re-exposed to the operant chamber but lever pressing is not reinforced with drug infusions. Once a pre-determined extinction period or an extinction criterion is met, the animals are exposed to drug cues, stress, or priming doses of drug and reinstatement is assessed. Importantly, this tests occurs in the absence of subsequent drug reward. Recovery of responding can also be tested after periods of abstinence without extinction training.
Two-bottle free choice: used to assess alcohol drinking behaviors. Two bottles are place in equivalent areas within the home cage and are filled with water and water combined with the test concentration of alcohol – commonly 5–20%. Alcohol preference is determined by comparing the weight or volume of liquid consumed. Blood alcohol concentrations are often used to verify that the animals are exposed to the drug.
While it is unclear if striatal synaptic plasticity also involves an mTORC1-dependent process (Luscher and Huber, 2010), drug-induced maladaptation of Group I mGluR, and Homer signaling are linked with impairments in the ability to evoke LTD in the NAC (Martin et al., 2006; Moussawi et al., 2009). Indeed, experimenter or self-administered cocaine reduces both mGluR1/5 and Homer1b/c levels in the NAC (Szumlinski et al., 2006) and the recovery of LTD at cortico-accumbal synapses in cocaine-trained rats appears to be dependent on restoring tone to mGluR5 receptors (Moussawi et al., 2009). As highlighted above, an important role for mGluR1/5 receptors and Homer in the induction of synaptic plasticity now exists and there is substantial evidence linking this signaling complex to the regulation of extracellular glutamate levels in the NAC and the disruption of glutamate homeostasis after chronic drug taking (Szumlinski et al., 2004, 2008; Kalivas and Volkow, 2011). Interestingly therefore, Ronesi and Huber (2008) have linked LTD in the hippocampus to a Homer/mGluR1/5 interaction that requires mTOR but not ERK. It will be interesting to determine if the loss of LTD seen at cortico-accumbal synapses after chronic cocaine exposure may involve a dysregulation of Homer/mGluR1/5 → mTOR pathway.
Signaling Molecules Upstream of mTORC1 that are Implicated in Drug-Motivated Behaviors
The first evidence that mTORC1 signaling might play a role in addiction came from the demonstration that rapamycin infused into the NAC of male Sprague Dawley rats prevented psychostimulant-induced SENSITIZATION of methamphetamine place preference – a behavior shown to require structural plasticity in the striatum (Narita et al., 2005). These authors also showed that methamphetamine administration activated the downstream target of mTOR, p70S6K in the NAC (Narita et al., 2005). Consistent with the methamphetamine findings, Szumlinski et al. recently showed that systemic rapamycin (10 mg/kg) suppressed the expression of COCAINE-INDUCED PLACE PREFERENCE (CPP) in C57/B6 mice (see Box 1; Bailey et al., 2011). Intriguingly, while the expression of CPP was found to be rapamycin sensitive, the development of cocaine sensitization or CPP was not affected by prior treatment with the mTOR inhibitor (Bailey et al., 2011). In contrast, Wu et al. (2011) showed that rapamycin dose dependently suppressed the development of cocaine sensitization and CPP in female rats (Wu et al., 2011). It will be important to determine if these differences are related to sex or species differences.
A number of cell membrane receptor signaling molecules that can influence mTOR have been implicated in the neuroadaptations that control psychostimulant-motivated behaviors using experimenter-administered drug exposure procedures (Russo et al., 2007, 2009; McGinty et al., 2008; Shi and McGinty, 2011). Noteworthy is the well-characterized psychostimulant-induced changes to BDNF/TrkB receptor pathway in the VTA and NAC, making this system a strong candidate for drug-induced mTORC1 activation (Narita et al., 2003; Russo et al., 2009; Ghitza et al., 2010; McGinty et al., 2010). It is also important to note that changes in BDNF/TrkB signaling is not limited to psychostimulants, with studies implicating this neurotrophic factor in opiate- (Russo et al., 2007) and alcohol-motivated behaviors (Moonat et al., 2010). With respect to psychostimulant-induced BDNF changes, mRNA and protein levels for this neurotrophic factor are altered in response to experimenter-administered cocaine injections (Zhang et al., 2002; Le Foll et al., 2005) and Bdnf+/1 mice are more hyperactive than wildtypes to psychostimulant injections (Saylor and McGinty, 2008). Furthermore, BDNF protein infused into the NAC or VTA enhances cocaine-induced locomotor activity (Horger et al., 1999; Hall et al., 2003). More recently, Lobo et al. (2010) employed a genetic and optogenetic approach to assess the role of BDNF/TrkB signaling in D1 versus D2 receptor-expressing medium spiny neurons (MSNs) in cocaine-motivated behaviors. To achieve this they used D1 or D2-Cre-floxedTrkB mice, in which the TrkB receptor is selectively deleted in D1 versus D2 MSNs. Interestingly, D1-Cre-floxedTrkB mice showed enhanced cocaine-motivated behaviors, including CPP, whereas the opposite behavioral effects were observed in D2-Cre-floxedTrkB mice (Lobo et al., 2010). These data were further probed by expressing the blue light sensitive (∼470 nm) cation channel, channelrhodopsin-2 (ChR2), in D1 or D2 MSN populations by NAC-directed injections of a Cre-inducible adenovirus DIO-AAV. Optical activation of ChR2, which caused depolarization in D1 or D2 MSNs, recapitulated the cocaine-induced increase or decrease in place preference displayed by D1 or D2-Cre–flTrkB mice respectively, providing complementary support for differential roles for BDNF in D1 versus D2 MSNs. These data highlight the need to better understand the contributions of specific signaling pathways to drug-induced plasticity in these sub-populations of striatal neurons.
Signaling molecules that influence mTOR activity and are downstream of addiction-relevant cell membrane receptors such as TrkB, NMDARs, mGluRs, and DRs (Figure 1), have been directly implicated in drug-induced modification of behavioral output. For example, in the case of PI3K, Izzo et al. (2002) reported that the inhibitor LY294002 suppressed the expression, but not the induction, of cocaine-sensitization (Izzo et al., 2002). Further, p85a/p110 PI3K activity is observed within the shell of the NAC during cocaine sensitization and systemic injections of the dual D1/D2 receptor agonist pergolide suppressed cocaine sensitization and the concomitant increase in PI3K activity (Zhang et al., 2006). In the case of AKT, psychostimulants produce an initial increase in phosphorylation of this kinase through a D1-R-dependent mechanism, however at later time points, AKT activity decreases through an effect that is thought to involve D2-R activation. It should be pointed out that much of this work relates to the dorsal striatum (Brami-Cherrier et al., 2002; Beaulieu et al., 2004, 2005, 2006; Shi and McGinty, 2007, 2011; McGinty et al., 2008). An involvement for AKT in drug-induced neuroadaptations extends to opiates, as Russo and co-workers have also shown that exposure to morphine decreases AKT phosphorylation in the NAC and VTA (Mueller et al., 2004). Furthermore, this group implicated AKT pathway in morphine induced structural adaptations in VTA dopamine neurons, with these effects being linked with opiate tolerance and escalation of drug taking (Russo et al., 2007). Interestingly, a very recent study from this group found that a decrease in mTORC2/AKT signaling was responsible for the opiate-induced neuroadaptations to dopamine neurons. Alterations in mTORC1-associated molecules were also observed in this study, however these changes were in the opposite direction to mTORC2 and independent of the opiate-induced structural changes to VTA dopamine neurons. Understanding the crosstalk between these systems and the relevance of increased mTORC1 signaling to opiate-induced synaptic plasticity would seem highly relevant given the work of Mameli et al. (2007) discussed above.
Much previous work, in terms of downstream effectors of addiction-relevant receptor systems, has centered on the ERK pathway (Valjent et al., 2000, 2004, 2005; Girault et al., 2007). Particularly relevant to the present review, ERK has been shown to contribute to L-DOPA-induced activation of mTORC1, an effect assumed to occur through phosphorylation of TSC2 or Raptor (Santini et al., 2009). It will be for future work to determine which of these systems (see Figure 1) signal to mTOR in response to drug exposure and if co-incidental activation of these pathways is responsible for the full expression of drug-induced behavioral output (Gelinas et al., 2007; Girault et al., 2007). Importantly, the data discussed above demonstrate an important involvement of upstream regulators of mTOR in drug-motivated behaviors.
Evidence Arising from Studies Utilizing Self-Administration Drug Exposure Procedures
Strongly supporting the predicted role for mTORC1 in addiction, Shi et al. (2009) showed that in a small-randomized control study, acute systemic rapamycin (2.5 or 5 mg) suppressed cue-induced drug craving in abstinent heroin addicts (Shi et al., 2009). In light of this finding, it is interesting that several recent reports using drug self-administration models, a method of drug exposure that has significantly greater face validity to human drug taking than experimenter-administered modes, also support a role for mTORC1 in addiction. First, Neasta et al. (2010) showed that phosphorylated mTORC1 and p70S6K (but interestingly not ERK) is increased after binge alcohol exposure using two-bottle free choice and operant alcohol self-administration procedures (see Box 1). The mTORC1 signaling pathway induction observed by Neasta et al. (2010) was accompanied by increases in the synaptic proteins, Homer and GluR1 AMPAR subunits in the NAC. Pre-treatment with rapamycin prevented the increase in Homer. Consistent with systemic rapamycin injections, intra-NAC rapamycin injections also reduced alcohol-motivated behaviors, including self-administration and extinction responding. In a follow-up study, these authors showed that pharmacological inhibition in the NAC of signaling substrates upstream of mTORC1 also suppressed alcohol drinking. Thus, the PI3K inhibitor wortmannin, or triciribine, an AKT inhibitor, reduced alcohol-motivated behaviors in both mice and rats, including binge drinking and operant alcohol self-administration. Notably, these manipulations were found to have no effect on water or sucrose self-administration.
In studies specifically designed to test the role of mTORC1 signaling in cocaine-seeking and relapse-like behavior, Lu et al. (2006) infused rapamycin into the core of the NAC of rats 30 min prior to cue-induced reinstatement testing (Wang et al., 2010). The mTORC1 inhibitor significantly reduced cocaine- but not sucrose-reinstatement. Interestingly, unlike its effects on alcohol drinking in mice, rapamycin had no effect on cocaine self-administration, suggesting a differential role for mTORC1 in psychostimulant versus alcohol reinforcement. These authors also indicated that rapamycin blocked an NMDA receptor-induced potentiation of cocaine-seeking and recruitment of the mTORC1 pathway. These data indicate a potential regulatory pathway initially involving NMDA receptor activation, at least in the processes acutely regulating relapse behavior.
In a recent study aimed at identifying putative molecular substrates that are responsible for the development of addiction and relapse vulnerability, we phenotyped rats using criteria derived from the Diagnostic and Statistical Manual for Substance Dependence (DSM-IV; Brown et al., 2011) – similar to Deroche-Gamonet and colleagues’ behavioral approach first published in 2004. The behavioral tests that were applied at different time points across the addiction cycle (Deroche-Gamonet et al., 2004) included: (i) testing for an animals’ ability to refrain from drug taking during a signaled period of drug non-availability; (ii) testing the motivation to take the drug by progressive ratio testing (Deroche-Gamonet et al., 2004); (iii) testing for relapse vulnerability using a cue-induced reinstatement procedure. To be phenotyped as addiction–vulnerable, animals were required to score within the top 40% of the cohort for reinstatement responding and the top third of the distribution for the two other vulnerability markers that were assessed at earlier points in the addiction cycle, i.e., motivation to lever press on a progressive ratio schedule for cocaine and the ability to refrain from drug taking during a period of signaled non-drug availability (NDA), the latter being a presumptive test of compulsive drug-seeking. Twenty-four hours after relapse testing, animals were sacrificed, and gene expression for synaptic plasticity-associated transcripts was assessed using qPCR in the ventral and dorsal striatum. Interestingly, compared to resistant animals, rats classified as addiction, and relapse vulnerable displayed reduced gene expression for a number of plasticity-associated transcripts, including mTOR, activity-regulated cytoskeletal protein (Arc), Group I mGluRs, GluR subunits, and DRs. Importantly, animals that were separated into vulnerability groups did not differ in terms of their total cocaine intake across the testing period, suggesting that the effects on gene expression are likely to relate to neural processes underpinning the development of addiction, rather than drug-effects alone. In ongoing studies in our laboratory, we have also found that intra-NAC rapamycin reduced progressive ratio break points in cocaine-trained rats and lever pressing during NDA periods. Remarkably, intra-NAC rapamycin infusions also suppressed cue-induced reinstatement 6–8 weeks after treatment and this behavioral effect was associated with a reduction in p70S6K expression at this time point (Manuscript in preparation).
The reduced gene expression we observed for mTOR mRNA in the NAC of addiction and relapse vulnerable rats may initially seem at odds with the effects of systemic or intra-cranial injections of rapamycin on drug-motivated behaviors described above. One possible explanation for this apparent discrepancy is that increased activity within the mTORC1 signaling pathway may have manifested in addiction/relapse vulnerable animals as downregulated mTORC1 transcript expression at the final point of the addiction cycle, i.e., 24 h after relapse test, corresponding with when the brains were harvested. Post-transcriptional repression by microRNAs or other epigenetic regulators may lead to a feedback inhibition of mTORC1 mRNA synthesis in attempt to limit the cocaine-induced increase in mTORC1 activity. In this regard, it is interesting that several recent studies have demonstrated an important role for epigenetic changes which appear to regulate the neural process that control the motivation to consume cocaine (Kumar et al., 2005; Hollander et al., 2010; Im et al., 2010).
The exact cellular and molecular changes that are regulated by mTORC1 and contribute to relapse vulnerability after withdrawal and extinction training are yet to be determined. However, the role of mTORC1 in relapse vulnerability may be related to its involvement in the translation of synaptic proteins during withdrawal. Indeed, one of the most consistently reported post-synaptic changes in the NAC (and VTA) that is linked with elevated relapse vulnerability is increased expression and signaling through glutamatergic, excitatory AMPARs (Mameli et al., 2007, 2009; Anderson et al., 2008; Conrad et al., 2008; Wolf and Ferrario, 2010). Cornish and Kalivas published the first clear evidence pointing to a role for AMPARs in drug-seeking, showing that AMPA receptor antagonists infused in the NAC suppress cocaine reinstatement (Cornish et al., 1999; Cornish and Kalivas, 2000). More recent studies have confirmed and extended these important initial findings by showing that it is an incorporation of GluR1 (Gria1) subunits into AMPARs during withdrawal that triggers relapse-like behavior (Anderson et al., 2008; Conrad et al., 2008). These studies showed that injections into the NAC of a specific antagonist of the GluR1 subunit (1-naphthylacetylsperimine), or a viral vector that prevents the transport of GluR1s for insertion into the synaptic membrane, decreased relapse-like behavior (Anderson et al., 2008; Conrad et al., 2008). Importantly, propensity to relapse was dependent on an increase in surface, intracellular, and total GluR1 levels (and not other GluR subunits). The increase in both “pools” of GluR1s is consistent with a requirement for production/translation of new rather than a redistribution of old/existing GluR1s (Anderson et al., 2008; Conrad et al., 2008). Electrophysiological studies confirmed these biochemical changes in NAC tissue are accompanied by alterations in the firing properties of NAC MSNs. Thus, a “pathological” drive to increase GluR1 AMPA subunits during withdrawal appears to be fundamental to increased vulnerability for relapse. Consistent with these data are reports of impairments in the ability to evoke LTP or LTD in the NAC of animals with a clinically relevant history of cocaine self-administration (Martin et al., 2006; Moussawi et al., 2009; Kasanetz et al., 2010). Indeed, using the behavioral procedure outlined above to identify addiction–vulnerable animals, Kasanetz et al. (2010) showed that impairments in the ability to evoke LTD only persist in animals classified as addiction vulnerable (Kasanetz et al., 2010).
It is interesting in light of evidence presented above that drug self-administration models and relapse procedures have identified an important role for striatal BDNF in addiction and relapse vulnerability (Grimm et al., 2003) and that BDNF drives GluR1 AMPARs through an mTORC1-dependent process (Slipczuk et al., 2009). Indeed, as described above, the BDNF → PI3K → AKT pathway is a “prototypic” regulator of mTORC1 (Laplante and Sabatini, 2009). Thus, it is plausible that BDNF drives mTOR during early phases of the addiction cycle and that this chronically upregulates mTOR activity over time. Somewhat consistent with this proposal, Graham et al. (2007) reported that cocaine self-administration and the effort rats are willing to exert to obtain cocaine is increased after repeated intra-NAC infusions of BDNF. This manipulation also increased relapse vulnerability after extinction training (Graham et al., 2007). Importantly, given the potential supra-physiological effects of this manipulation, anti-sera raised against BDNF produced opposing results. BDNF also appears to drive addiction-relevant maladaptations in the dorsal striatum (Im et al., 2010) and we found a strong trend for reduced mTOR expression in global dissections of this brain region (Brown et al., 2011). Whether BDNF influences striatal mTOR activity in cocaine-trained rats is yet to be assessed.
Is important to note that the role of BDNF in drug-motivated behaviors, particularly drug-seeking, is both drug- and brain region-dependent (McGinty et al., 2010). Thus, infusions of BDNF in the PFC have opposite effects to striatal injections on cocaine-seeking (Berglind et al., 2007, 2009; McGinty et al., 2010), with these effects shown to be linked with normalization of basal extracellular glutamate release in the NAC. Interestingly, the suppressive effect of intra-PFC BDNF on reinstatement appears to depend on MAPK/ERK signaling (and not PI3K), as infusions of a MEK1/2 inhibitor U0126 prevent the suppression of cocaine-seeking produced by BDNF. It will be important for future studies to determine whether the reductions in drug-seeking after intra-PFC BDNF injections are accompanied by changes in PFC mTOR (Whitfield et al., 2011). These studies also highlight the importance of understanding the role for signaling pathways downstream of BDNF at time points across the addiction cycle to determine whether changes in MAPK/ERK or PI3K pathways are brain region- and/or addiction cycle-dependent.
Conclusion: Potential Clinical Utility of mTORC1 Inhibition
In the present review we have summarized the current understanding of the role of the mTORC1 signaling pathway in the development of addiction and relapse vulnerability. The ultimate goal of understanding the complex molecular signaling events that controls protracted addiction and relapse behaviors is to identify possible targets for treatments for this condition. It is interesting in this regard that mTORC1 inhibition reduces craving in heroin addicts; albeit in small study involving acute rapamycin treatment (Shi et al., 2009). Also, chronic rapamycin increased lifespan in both male and female mice (Harrison et al., 2009) and rapamycin is approved for use in humans for the treatment of renal cell carcinoma (Bhatia and Thompson, 2009). It should be mentioned that recent reports indicate that chronic rapamycin can produce weight loss and new onset diabetes (Johnston et al., 2008; Deblon et al., 2011).
Despite the promising pre-clinical data using the animal models of addiction that we have outlined above, a number of important questions remain to be answered with regards to the role of mTOR in the addiction process and CNS processes more generally. Below we will outline a number of knowledge gaps that, when filled, may progress our understanding of the importance of mTORC1 signaling toward this end. It is important to point out that regardless of the potential clinical utility of mTORC1 inhibition, pharmacological agents targeting this pathway are likely to serve as important tools for identifying the critical subset of synaptic proteins that are responsible for neuroadaptations underpinning addiction and relapse vulnerability and brain plasticity more broadly.
A full understanding of the involvement of mTOR in the addiction process will require a comprehensive assessment of the changes within the mTOR signaling pathway across the addiction cycle. Using addiction phenotyping strategies to identify vulnerable animals at early and late stages of the addiction cycle, including into withdrawal, would improve our understanding of how this system becomes engaged and contributes to the processes that increase synaptic proteins critical for relapse. It will also be important to assess changes in the mTOR pathway in other addiction-relevant brain areas such as the PFC and amygdala. This review highlights BDNF as being a possible mediator of drug-induced mTOR activity, however, as has also been made clear throughout this review, other receptor systems (e.g., NMDA, mGluRs, and DRs) can also regulate mTOR activity and synaptic protein translation through parallel signaling pathways. Thus, it will be necessary to determine the likely interactions between PI3K → AKT and alternative signaling pathways that appear to exhibit crosstalk with mTOR in initiating plasticity-associated protein translation (e.g., ERK). In addition, although this review has focused on mTORC1, mTORC2 has recently been identified as playing an important role in opiate-induced structural plasticity (Mazei-Robison et al., 2011). In the striatum, it will be important to determine whether mTOR expression is restricted to specific cell types within the NAC, as appears to be the case for psychostimulant-induced ERK expression (Valjent et al., 2005). Preliminary studies in this regard indicate that dopamine agonists may specifically increase mTOR in MSNs that express dopamine-1 receptors (Santini et al., 2009).
Current data implicating mTORC1 in addiction has relied primarily on acute pharmacological inhibition of mTOR using rapamycin. Determining whether chronic treatment of animals with rapamycin and other specific inhibitors of mTOR can suppress addiction and relapse-like behavior without producing “off-target” effects will be required. Addiction studies would benefit from the use of genetic manipulations of key components of the mTOR pathway. Unfortunately, mTORC1 knockout mice are lethal and genetic manipulations of upstream effectors have produced conflicting results in terms of synaptic plasticity and learning. Use of a pharmacogenetic approach similar to Stoica et al. (2011) could circumvent this problem. It would be interesting to utilize this approach in animal models of addiction to assess behavioral and functional effects, e.g., LTP and LTD in NAC and VTA.
In conclusion, the present review has outlined the recent evidence that mTORC1 signaling is involved in the development and expression of addiction behaviors. To this end we have discussed the role of mTOR in synaptic protein translation in the context of known addiction-associated signaling molecules. Further work will be required to determine the exact role of mTOR in the addiction process and whether members of this signaling pathway will prove to be appropriate targets for medications development to reduce relapse addiction and risk.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Parts of this work were supported by NHMRC and HMRI grants to Christopher V. Dayas. We thank Emily Levi and Morgan James for their editorial assistance.
References
Aakalu, G., Smith, W. B., Nguyen, N., Jiang, C., and Schuman, E. M. (2001). Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30, 489–502.
Anderson, S. M., Famous, K. R., Sadri-Vakili, G., Kumaresan, V., Schmidt, H. D., Bass, C. E., Terwilliger, E. F., Cha, J. H., and Pierce, R. C. (2008). CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci. 11, 344–353.
Asaki, C., Usuda, N., Nakazawa, A., Kametani, K., and Suzuki, T. (2003). Localization of translational components at the ultramicroscopic level at postsynaptic sites of the rat brain. Brain Res. 972, 168–176.
Bailey, J., Ma, D., and Szumlinski, K. K. (2011). Rapamycin attenuates the expression of cocaine-induced place preference and behavioral sensitization. Addict Biol. doi: 10.1111/j.1369-1600.2010.00311.x. [Epub ahead of print].
Banko, J. L., Hou, L., and Klann, E. (2004). NMDA receptor activation results in PKA- and ERK-dependent Mnk1 activation and increased eIF4E phosphorylation in hippocampal area CA1. J. Neurochem. 91, 462–470.
Banko, J. L., Hou, L., Poulin, F., Sonenberg, N., and Klann, E. (2006). Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 26, 2167–2173.
Beaulieu, J. M., Sotnikova, T. D., Gainetdinov, R. R., and Caron, M. G. (2006). Paradoxical striatal cellular signaling responses to psychostimulants in hyperactive mice. J. Biol. Chem. 281, 32072–32080.
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Beretta, L., Gingras, A. C., Svitkin, Y. V., Hall, M. N., and Sonenberg, N. (1996). Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 15, 658–664.
Berglind, W. J., See, R. E., Fuchs, R. A., Ghee, S. M., Whitfield, T. W. Jr., Miller, S. W., and McGinty, J. F. (2007). A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur. J. Neurosci. 26, 757–766.
Berglind, W. J., Whitfield, T. W. Jr., LaLumiere, R. T., Kalivas, P. W., and McGinty, J. F. (2009). A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J. Neurosci. 29, 3715–3719.
Bhatia, S., and Thompson, J. A. (2009). Temsirolimus in patients with advanced renal cell carcinoma: an overview. Adv. Ther. 26, 55–67.
Bird, M. K., and Lawrence, A. J. (2009). The promiscuous mGlu5 receptor – a range of partners for therapeutic possibilities? Trends Pharmacol. Sci. 30, 617–623.
Blundell, J., Kouser, M., and Powell, C. M. (2008). Systemic inhibition of mammalian target of rapamycin inhibits fear memory reconsolidation. Neurobiol. Learn. Mem. 90, 28–35.
Bodian, D. (1965). A suggestive relationship of nerve cell rna with specific synaptic sites. Proc. Natl. Acad. Sci. U.S.A. 53, 418–425.
Bramham, C. R., and Wells, D. G. (2007). Dendritic mRNA: transport, translation and function. Nat. Rev. Neurosci. 8, 776–789.
Brami-Cherrier, K., Valjent, E., Garcia, M., Pages, C., Hipskind, R. A., and Caboche, J. (2002). Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J. Neurosci. 22, 8911–8921.
Brown, A. L., Flynn, J. R., Smith, D. W., and Dayas, C. V. (2011). Down-regulated striatal gene expression for synaptic plasticity-associated proteins in addiction and relapse vulnerable animals. Int. J. Neuropsychopharmacol. 14, 1099–1110.
Brown, E. J., Beal, P. A., Keith, C. T., Chen, J., Shin, T. B., and Schreiber, S. L. (1995). Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature 377, 441–446.
Cammalleri, M., Lutjens, R., Berton, F., King, A. R., Simpson, C., Francesconi, W., and Sanna, P. P. (2003). Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc. Natl. Acad. Sci. U.S.A. 100, 14368–14373.
Casadio, A., Martin, K. C., Giustetto, M., Zhu, H., Chen, M., Bartsch, D., Bailey, C. H., and Kandel, E. R. (1999). A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99, 221–237.
Conrad, K. L., Tseng, K. Y., Uejima, J. L., Reimers, J. M., Heng, L. J., Shaham, Y., Marinelli, M., and Wolf, M. E. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121.
Cornish, J. L., Duffy, P., and Kalivas, P. W. (1999). A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience 93, 1359–1367.
Cornish, J. L., and Kalivas, P. W. (2000). Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J. Neurosci. 20, RC89.
Costa-Mattioli, M., Sossin, W. S., Klann, E., and Sonenberg, N. (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26.
Crino, P. B., and Eberwine, J. (1996). Molecular characterization of the dendritic growth cone: regulated mRNA transport and local protein synthesis. Neuron 17, 1173–1187.
Deblon, N., Bourgoin, L., Veyrat-Durebex, C., Peyrou, M., Vinciguerra, M., Caillon, A., Maeder, C., Fournier, M., Montet, X., Rohner-Jeanrenaud, F., and Foti, M. (2011). Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br. J .Pharmacol. doi: 10.1111/j.1476-5381.2011.01716.x. [Epub ahead of print].
Deroche-Gamonet, V., Belin, D., and Piazza, P. V. (2004). Evidence for addiction-like behavior in the rat. Science 305, 1014–1017.
Dietz, D. M., Dietz, K. C., Nestler, E. J., and Russo, S. J. (2009). Molecular mechanisms of psychostimulant-induced structural plasticity. Pharmacopsychiatry 42(Suppl. 1), S69–S78.
Duncan, J. R., and Lawrence, A. J. (2011). The role of metabotropic glutamate receptors in addiction: Evidence from preclinical models. Pharmacol. Biochem. Behav. 100, 811–824.
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., Ramesh, V., and Silva, A. J. (2008). Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat. Med. 14, 843–848.
Flier, J. S. (2006). Neuroscience. Regulating energy balance: the substrate strikes back. Science 312, 861–864.
Gelinas, J. N., Banko, J. L., Hou, L., Sonenberg, N., Weeber, E. J., Klann, E., and Nguyen, P. V. (2007). ERK and mTOR signaling couple beta-adrenergic receptors to translation initiation machinery to gate induction of protein synthesis-dependent long-term potentiation. J. Biol. Chem. 282, 27527–27535.
Ghitza, U. E., Zhai, H., Wu, P., Airavaara, M., Shaham, Y., and Lu, L. (2010). Role of BDNF and GDNF in drug reward and relapse: a review. Neurosci. Biobehav. Rev. 35, 157–171.
Gingras, A. C., Raught, B., and Sonenberg, N. (2004). mTOR signaling to translation. Curr. Top. Microbiol. Immunol. 279, 169–197.
Girault, J. A., Valjent, E., Caboche, J., and Herve, D. (2007). ERK2: a logical AND gate critical for drug-induced plasticity? Curr. Opin. Pharmacol. 7, 77–85.
Gong, R., Park, C. S., Abbassi, N. R., and Tang, S. J. (2006). Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J. Biol. Chem. 281, 18802–18815.
Graham, D. L., Edwards, S., Bachtell, R. K., DiLeone, R. J., Rios, M., and Self, D. W. (2007). Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat. Neurosci. 10, 1029–1037.
Grimm, J. W., Lu, L., Hayashi, T., Hope, B. T., Su, T. P., and Shaham, Y. (2003). Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J. Neurosci. 23, 742–747.
Hall, F. S., Drgonova, J., Goeb, M., and Uhl, G. R. (2003). Reduced behavioral effects of cocaine in heterozygous brain-derived neurotrophic factor (BDNF) knockout mice. Neuropsychopharmacology 28, 1485–1490.
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., Nadon, N. L., Wilkinson, J. E., Frenkel, K., Carter, C. S., Pahor, M., Javors, M. A., Fernandez, E., and Miller, R. A. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395.
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75.
Hollander, J. A., Im, H. I., Amelio, A. L., Kocerha, J., Bali, P., Lu, Q., Willoughby, D., Wahlestedt, C., Conkright, M. D., and Kenny, P. J. (2010). Striatal microRNA controls cocaine intake through CREB signalling. Nature 466, 197–202.
Horger, B. A., Iyasere, C. A., Berhow, M. T., Messer, C. J., Nestler, E. J., and Taylor, J. R. (1999). Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J. Neurosci. 19, 4110–4122.
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361.
Im, H. I., Hollander, J. A., Bali, P., and Kenny, P. J. (2010). MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat. Neurosci. 13, 1120–1127.
Izzo, E., Martin-Fardon, R., Koob, G. F., Weiss, F., and Sanna, P. P. (2002). Neural plasticity and addiction: PI3-kinase and cocaine behavioral sensitization. Nat. Neurosci. 5, 1263–1264.
Johnston, O., Rose, C. L., Webster, A. C., and Gill, J. S. (2008). Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J. Am. Soc. Nephrol. 19, 1411–1418.
Jupp, B., and Lawrence, A. J. (2010). New horizons for therapeutics in drug and alcohol abuse. Pharmacol. Ther. 125, 138–168.
Kalivas, P. W. (2009). The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 10, 561–572.
Kalivas, P. W., and O’Brien, C. (2008). Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33, 166–180.
Kalivas, P. W., and Volkow, N. D. (2011). New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol. Psychiatry 16, 974–986.
Kandel, E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038.
Kasanetz, F., Deroche-Gamonet, V., Berson, N., Balado, E., Lafourcade, M., Manzoni, O., and Piazza, P. V. (2010). Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328, 1709–1712.
Kauer, J. A., and Malenka, R. C. (2007). Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–858.
Kim, D. H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., Tempst, P., and Sabatini, D. M. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175.
Klann, E., Antion, M. D., Banko, J. L., and Hou, L. (2004). Synaptic plasticity and translation initiation. Learn. Mem. 11, 365–372.
Koob, G. F., and Volkow, N. D. (2009). Neurocircuitry of addiction. Neuropsychopharmacology 35, 217–238.
Kumar, A., Choi, K. H., Renthal, W., Tsankova, N. M., Theobald, D. E., Truong, H. T., Russo, S. J., Laplant, Q., Sasaki, T. S., Whistler, K. N., Neve, R. L, Self, D. W., and Nestler, E. J. (2005). Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48, 303–314.
Le Foll, B., Diaz, J., and Sokoloff, P. (2005). A single cocaine exposure increases BDNF and D3 receptor expression: implications for drug-conditioning. Neuroreport 16, 175–178.
Lobo, M. K., Covington, H. E. III, Chaudhury, D., Friedman, A. K., Sun, H., Damez-Werno, D., Dietz, D. M., Zaman, S., Koo, J. W., Kennedy, P. J., Mouzon, E., Mogri, M., Neve, R. L., Deisseroth, K., Han, M. H., and Nestler, E. J. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390.
Lorenz, M. C., and Heitman, J. (1995). TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J. Biol. Chem. 270, 27531–27537.
Lu, L., Koya, E., Zhai, H., Hope, B. T., and Shaham, Y. (2006). Role of ERK in cocaine addiction. Trends Neurosci. 29, 695–703.
Luscher, C., and Huber, K. M. (2010). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459.
Luscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663.
Mameli, M., Balland, B., Lujan, R., and Luscher, C. (2007). Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317, 530–533.
Mameli, M., Halbout, B., Creton, C., Engblom, D., Parkitna, J. R., Spanagel, R., and Luscher, C. (2009). Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat. Neurosci. 12, 1036–1041.
Mao, L., Yang, L., Tang, Q., Samdani, S., Zhang, G., and Wang, J. Q. (2005). The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J. Neurosci. 25, 2741–2752.
Martin, M., Chen, B. T., Hopf, F. W., Bowers, M. S., and Bonci, A. (2006). Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat. Neurosci. 9, 868–869.
Mazei-Robison, M. S., Koo, J. W., Friedman, A. K., Lansink, C. S., Robison, A. J., Vinish, M., Krishnan, V., Kim, S., Siuta, M. A., Galli, A., Niswender, K. D., Appasani, R., Horvath, M. C., Neve, R. L., Worley, P. F., Snyder, S. H., Hurd, Y. L., Cheer, J. F., Han, M. H., Russo, S. J., and Nestler, E. J. (2011). Role for mTOR signaling and neuronal activity in morphine-induced adaptations in ventral tegmental area dopamine neurons. Neuron 72, 977–990.
McGinty, J. F., Shi, X. D., Schwendt, M., Saylor, A., and Toda, S. (2008). Regulation of psychostimulant-induced signaling and gene expression in the striatum. J. Neurochem. 104, 1440–1449.
McGinty, J. F., Whitfield, T. W. Jr., and Berglind, W. J. (2010). Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 1314, 183–193.
Moonat, S., Starkman, B. G., Sakharkar, A., and Pandey, S. C. (2010). Neuroscience of alcoholism: molecular and cellular mechanisms. Cell. Mol. Life Sci. 67, 73–88.
Moussawi, K., Pacchioni, A., Moran, M., Olive, M. F., Gass, J. T., Lavin, A., and Kalivas, P. W. (2009). N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci. 12, 182–189.
Mueller, O., Lightfoot, S., and Schroeder, A. (2004). RNA Integrity Number (RIN) – Standardization of RNA Quality Control. Waldbronn: Agilent Application Note Publication 5989-1165, 1–8.
Nader, K., Schafe, G. E., and Le Doux, J. E. (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406, 722–726.
Narita, M., Akai, H., Kita, T., Nagumo, Y., Narita, M., Sunagawa, N., Hara, C., Hasebe, K., Nagase, H., and Suzuki, T. (2005). Involvement of mitogen-stimulated p70-S6 kinase in the development of sensitization to the methamphetamine-induced rewarding effect in rats. Neuroscience 132, 553–560.
Narita, M., Aoki, K., Takagi, M., Yajima, Y., and Suzuki, T. (2003). Implication of brain-derived neurotrophic factor in the release of dopamine and dopamine-related behaviors induced by methamphetamine. Neuroscience 119, 767–775.
Neasta, J., Ben Hamida, S., Yowell, Q., Carnicella, S., and Ron, D. (2010). Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc. Natl. Acad. Sci. U.S.A. 107, 20093–20098.
O’Brien, C. P. (2005). Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am. J. Psychiatry 162, 1423–1431.
Page, G., Khidir, F. A., Pain, S., Barrier, L., Fauconneau, B., Guillard, O., Piriou, A., and Hugon, J. (2006). Group I metabotropic glutamate receptors activate the p70S6 kinase via both mammalian target of rapamycin (mTOR) and extracellular signal-regulated kinase (ERK 1/2) signaling pathways in rat striatal and hippocampal synaptoneurosomes. Neurochem. Int. 49, 413–421.
Panja, D., Dagyte, G., Bidinosti, M., Wibrand, K., Kristiansen, A. M., Sonenberg, N., and Bramham, C. R. (2009). Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J. Biol. Chem. 284, 31498–31511.
Parsons, R. G., Gafford, G. M., and Helmstetter, F. J. (2006a). Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J. Neurosci. 26, 12977–12983.
Parsons, R. G., Riedner, B. A., Gafford, G. M., and Helmstetter, F. J. (2006b). The formation of auditory fear memory requires the synthesis of protein and mRNA in the auditory thalamus. Neuroscience 141, 1163–1170.
Potter, W. B., O’Riordan, K. J., Barnett, D., Osting, S. M., Wagoner, M., Burger, C., and Roopra, A. (2010). Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS ONE 5, e8996. doi:10.1371/journal.pone.0008996
Raught, B., Peiretti, F., Gingras, A. C., Livingstone, M., Shahbazian, D., Mayeur, G. L., Polakiewicz, R. D., Sonenberg, N., and Hershey, J. W. (2004). Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23, 1761–1769.
Richter, J. D., and Klann, E. (2009). Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev. 23, 1–11.
Ronesi, J. A., and Huber, K. M. (2008). Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J. Neurosci. 28, 543–547.
Rong, R., Ahn, J. Y., Huang, H., Nagata, E., Kalman, D., Kapp, J. A., Tu, J., Worley, P. F., Snyder, S. H., and Ye, K. (2003). PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat. Neurosci. 6, 1153–1161.
Russo, S. J., Bolanos, C. A., Theobald, D. E., DeCarolis, N. A., Renthal, W., Kumar, A., Winstanley, C. A., Renthal, N. E., Wiley, M. D., Self, D. W., Russell, D. S., Neve, R. L., Eisch, A. J., and Nestler, E. J. (2007). IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat. Neurosci. 10, 93–99.
Russo, S. J., Mazei-Robison, M. S., Ables, J. L., and Nestler, E. J. (2009). Neurotrophic factors and structural plasticity in addiction. Neuropharmacology 56(Suppl. 1), 73–82.
Sabers, C. J., Martin, M. M., Brunn, G. J., Williams, J. M., Dumont, F. J., Wiederrecht, G., and Abraham, R. T. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270, 815–822.
Santini, E., Heiman, M., Greengard, P., Valjent, E., and Fisone, G. (2009). Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2, ra36.
Sarbassov, D. D., Ali, S. M., Kim, D. H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., Tempst, P., and Sabatini, D. M. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302.
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., Markhard, A. L., and Sabatini, D. M. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168.
Saylor, A. J., and McGinty, J. F. (2008). Amphetamine-induced locomotion and gene expression are altered in BDNF heterozygous mice. Genes Brain Behav. 7, 906–914.
Schafe, G. E., and LeDoux, J. E. (2000). Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J. Neurosci. 20, RC96.
Schafe, G. E., Nadel, N. V., Sullivan, G. M., Harris, A., and LeDoux, J. E. (1999). Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn. Mem. 6, 97–110.
Schratt, G. M., Nigh, E. A., Chen, W. G., Hu, L., and Greenberg, M. E. (2004). BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J. Neurosci. 24, 7366–7377.
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., and Zukin, R. S. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702.
Shi, J., Jun, W., Zhao, L. Y., Xue, Y. X., Zhang, X. Y., Kosten, T. R., and Lu, L. (2009). Effect of rapamycin on cue-induced drug craving in abstinent heroin addicts. Eur. J. Pharmacol. 615, 108–112.
Shi, X., and McGinty, J. F. (2007). Repeated amphetamine treatment increases phosphorylation of extracellular signal-regulated kinase, protein kinase B, and cyclase response element-binding protein in the rat striatum. J. Neurochem. 103, 706–713.
Shi, X., and McGinty, J. F. (2011). D1 and D2 dopamine receptors differentially mediate the activation of phosphoproteins in the striatum of amphetamine-sensitized rats. Psychopharmacology (Berl.) 214, 653–663.
Slipczuk, L., Bekinschtein, P., Katche, C., Cammarota, M., Izquierdo, I., and Medina, J. H. (2009). BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS ONE 4, e6007. doi:10.1371/journal.pone.0006007
Sonenberg, N., and Hinnebusch, A. G. (2007). New modes of translational control in development, behavior, and disease. Mol. Cell 28, 721–729.
Sonenberg, N., and Hinnebusch, A. G. (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745.
Stoica, L., Zhu, P. J., Huang, W., Zhou, H., Kozma, S. C., and Costa-Mattioli, M. (2011). Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. U.S.A. 108, 3791–3796.
Stuber, G. D., Klanker, M., de Ridder, B., Bowers, M. S., Joosten, R. N., Feenstra, M. G., and Bonci, A. (2008). Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science 321, 1690–1692.
Szumlinski, K. K., Abernathy, K. E., Oleson, E. B., Klugmann, M., Lominac, K. D., He, D. Y., Ron, D., During, M., and Kalivas, P. W. (2006). Homer isoforms differentially regulate cocaine-induced neuroplasticity. Neuropsychopharmacology 31, 768–777.
Szumlinski, K. K., Ary, A. W., and Lominac, K. D. (2008). Homers regulate drug-induced neuroplasticity: implications for addiction. Biochem. Pharmacol. 75, 112–133.
Szumlinski, K. K., Dehoff, M. H., Kang, S. H., Frys, K. A., Lominac, K. D., Klugmann, M., Rohrer, J., Griffin, W. III, Toda, S., Champtiaux, N. P., Berry, T., Tu, J. C., Shealy, S. E., During, M. J., Middaugh, L. D., Worley, P. F., and Kalivas, P. W. (2004). Homer proteins regulate sensitivity to cocaine. Neuron 43, 401–413.
Tang, S. J., Reis, G., Kang, H., Gingras, A. C., Sonenberg, N., and Schuman, E. M. (2002). A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 99, 467–472.
Tian, Q. B., Nakayama, K., Okano, A., and Suzuki, T. (1999). Identification of mRNAs localizing in the postsynaptic region. Brain Res. Mol. Brain Res. 72, 147–157.
Tischmeyer, W., Schicknick, H., Kraus, M., Seidenbecher, C. I., Staak, S., Scheich, H., and Gundelfinger, E. D. (2003). Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur. J. Neurosci. 18, 942–950.
Torre, E. R., and Steward, O. (1992). Demonstration of local protein synthesis within dendrites using a new cell culture system that permits the isolation of living axons and dendrites from their cell bodies. J. Neurosci. 12, 762–772.
Tsai, H. C., Zhang, F., Adamantidis, A., Stuber, G. D., Bonci, A., de Lecea, L., and Deisseroth, K. (2009). Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science 324, 1080–1084.
Valjent, E., Corvol, J. C., Pages, C., Besson, M. J., Maldonado, R., and Caboche, J. (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709.
Valjent, E., Pages, C., Herve, D., Girault, J. A., and Caboche, J. (2004). Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur. J. Neurosci. 19, 1826–1836.
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., Stipanovich, A., Caboche, J., Lombroso, P. J., Nairn, A. C., Greengard, P., Hervé, D., and Girault, J. A. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496.
Wang, X., Luo, Y. X., He, Y. Y., Li, F. Q., Shi, H. S., Xue, L. F., Xue, Y. X., and Lu, L. (2010). Nucleus accumbens core mammalian target of rapamycin signaling pathway is critical for cue-induced reinstatement of cocaine seeking in rats. J. Neurosci. 30, 12632–12641.
Whitfield, T. W. Jr., Shi, X., Sun, W. L., and McGinty, J. F. (2011). The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J. Neurosci. 31, 834–842.
Wolf, M. E., and Ferrario, C. R. (2010). AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci. Biobehav. Rev. 35, 185–211.
Wu, J., McCallum, S. E., Glick, S. D., and Huang, Y. (2011). Inhibition of the mammalian target of rapamycin pathway by rapamycin blocks cocaine-induced locomotor sensitization. Neuroscience 172, 104–109.
Zhang, D., Zhang, L., Lou, D. W., Nakabeppu, Y., Zhang, J., and Xu, M. (2002). The dopamine D1 receptor is a critical mediator for cocaine-induced gene expression. J. Neurochem. 82, 1453–1464.
Keywords: mTOR, addiction, relapse, cocaine, mGluR, BDNF, plasticity, drug
Citation: Dayas CV, Smith DW and Dunkley PR (2012) An emerging role for the mammalian target of rapamycin in “pathological” protein translation: relevance to cocaine addiction. Front. Pharmacol. 3:13. doi: 10.3389/fphar.2012.00013
Received: 11 November 2011;
Paper pending published: 30 November 2011;
Accepted: 20 January 2012;
Published online: 06 February 2012.
Edited by:
Andrew Lawrence, Florey Neuroscience Institutes, AustraliaReviewed by:
Jason B. Wu, Cedars-Sinai Medical Center, USAJacqueline McGinty, University of South Carolina, USA
Copyright: © 2012 Dayas, Smith and Dunkley. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christopher V. Dayas, School of Biomedical Sciences and Pharmacy, The University of Newcastle, Callaghan, NSW 2308, Australia. e-mail: christopher.dayas@newcastle.edu.au