
- 1 Neurosciences, Ottawa Hospital Research Institute, Ottawa, ON, Canada
- 2 Institut de Physique Ottawa-Carleton, Campus de l’Université d’Ottawa, Ottawa, ON, Canada
Mechanical, ischemic, and inflammatory injuries to voltage-gated sodium channel (Nav)-rich membranes of axon initial segments and nodes of Ranvier render Nav channels dangerously leaky. By what means? The behavior of recombinant Nav1.6 (Wang et al., 2009) leads us to postulate that, in neuropathologic conditions, structural degradation of axolemmal bilayer fosters chronically left-shifted Nav channel operation, resulting in ENa rundown. This “sick excitable cell Nav-leak” would encompass left-shifted fast- and slow-mode based persistent INa (i.e., Iwindow and slow-inactivating INa). Bilayer-damage-induced electrophysiological dysfunctions of native-Nav channels, and effects on inhibitors on those channels, should, we suggest, be studied in myelinated axons, exploiting INa(V,t) hysteresis data from sawtooth ramp clamp. We hypothesize that (like dihydropyridines for Ca channels), protective lipophilic Nav antagonists would partition more avidly into disorderly bilayers than into the well-packed bilayers characteristic of undamaged, healthy plasma membrane. Whereas inhibitors using aqueous routes would access all Navs equally, differential partitioning into “sick bilayer” would co-localize lipophilic antagonists with “sick-Nav channels,” allowing for more specific targeting of impaired cells. Molecular fine-tuning of Nav antagonists to favor more avid partitioning into damaged than into intact bilayers could reduce side effects. In potentially salvageable neurons of traumatic and/or ischemic penumbras, in inflammatory neuropathies, in muscular dystrophy, in myocytes of cardiac infarct borders, Nav-leak driven excitotoxicity overwhelms cellular repair mechanisms. Precision-tuning of a lipophilic Nav antagonist for greatest efficacy in mildly damaged membranes could render it suitable for the prolonged continuous administration needed to allow for the remodeling of the excitable membranes, and thus functional recovery.
Nav Channel Gating Mode Terminology
As originally described by Hodgkin and Huxley (1952) voltage-gated sodium channels (Nav) have a predominant fast mode of gating. However, for any population of Nav channels, at any given time, a small fraction will be switching reversibly between fast-mode and slow mode (Figure 1). Terminology for Nav kinetics has evolved along with the structure/function studies propelling the voltage-gated channel field and it is sometimes unclear what is meant by “persistent sodium current.” Thus, before embarking on a discussion of how membrane injury alters Nav channel behavior and what this might signify for drug therapies, we show (Figure 1) how we use kinetic terms. The simplified schematic is co-labeled with conventional (current-based) and newer (structure/function-based) terms: Nav channel conformations can be either Active mode (= fast) or Relaxed mode (= slow; Villalba-Galea et al., 2008). For transitions between modes, we prefer “mode-switch” to “mode-shift” (see Lin et al., 2007), since “switch” better evokes a protein conformation change than does “shift,” which in any case is easily confounded with the graphical relocation of Boltzmann curves along a voltage axis, a routine occurrence for both Active mode and Relaxed mode channels (this will be seen, e.g., in Figure 3C and Figures 4C,D). The mode-switch conformation changes, though not well understood in Nav channels (Webb et al., 2009), are not dependent on voltage (Villalba-Galea et al., 2008).
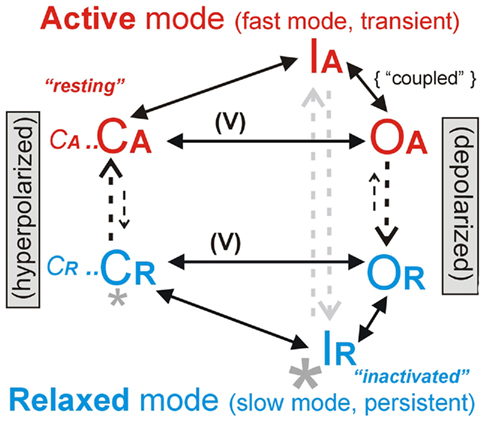
Figure 1. Two major gating modes. A Nav channel has four voltage sensor domains and complex gating kinetics; this simplified scheme emphasizes voltage-dependent (V) activation [closed, open (C,O)], plus inactivation (I) to an open-like but occluded state in each of the two major modes (Active and Relaxed). The smaller letters (CA… and CR…) serve as a reminder that a full kinetic scheme (e.g., Taddese and Bean, 2002; Lenkey et al., 2011) would have many more states. Structural changes underlying mode switching, a feature of all voltage-gated proteins, are not yet understood (Villalba-Galea et al., 2008). During prolonged depolarizations and hyperpolarizations, inter-mode transitions (vertical arrows), which are voltage-independent (Villalba-Galea et al., 2008), would drive the Nav system toward IR and CA (“inactivated” and “resting” in the parlance of Lenkey et al., 2011), respectively. Macroscopic INa(t) includes INa-fast (fast, transient, or broadly speaking, Hodgkin–Huxley type) plus INa-slow (also variously called “persistent,” “slowly inactivating,” “non-inactivating”), which flow through open Active and Relaxed mode Nav channels (OA, OR), respectively. Depolarization-induced repacking of voltage sensor domains (largely within the trans-bilayer electric field) yields net outward gating charge movement and facilitates C → O transitions. Activation curves, i.e., gNa(V) associated with CA–OA or with CR–OR, yield the “Boltzmann” sigmoid curves that summarize voltage-dependent fast or slow-gating energetics. Hyperpolarizing and depolarizing Boltzmann shifts are called “left shifts” or “right-shifts” respectively. The process by which the Nav “inactivation particle” binds (OA → IA), though not inherently voltage dependent, is rate-limited by (hence coupled to) the voltage-dependent process of fast activation (Wang et al., 2009; Boucher et al., 2012). Thus, when activation [gNa-fast(V)] left shifts due to bilayer damage, steady-state inactivation left shifts by the same number of millivolts, as depicted in Figure 3. This gNa-fast(V) phenomenon we term coupled-left shift (CLS). Importantly, the “subthreshold persistent current” of pacemaker neurons (Taddese and Bean, 2002) and of first-nodes (Kole, 2011) is a gNa-fast(V) based phenomenon, as is CLS. So-called “persistent current inhibitors” (e.g., riluzole, ranolazine) principally act by stabilizing non-conducting Relaxed mode states (large asterisk), but they also cause use-dependent ↓ INa-fast, perhaps through binding to Active mode channels (Lenkey et al., 2011; though inhibitor-bound CR states (small asterisk) might contribute to ↓ [CA] and thence to ↓ [OA]).
The Problem: Nav Channels Get Leaky in Sick Excitable Cells
As summarized in Table 1, Nav channels are dangerously leaky in a multitude of “sick-cell excitable cell” conditions. Mechanical trauma and/or ischemia and/or inflammation, and/or various genetic diseases result in damaged excitable cell membranes. More specifically, at Nav-rich membranes, bilayer structure degradation renders the Nav channels leaky. A CNS trauma example is depicted in Figure 2A: stretch trauma has caused axolemmal blebbing at the node of Ranvier. The fully blebbed bilayer, though detached from the specialized nodal spectrin skeleton (Figure 2C), would still be well-endowed with Nav1.6 channels. Even in such egregiously bleb-damaged plasma membrane, Nav channels are capable of voltage-dependent gating (Milton and Caldwell, 1990). In fact they gate “too well” (Figure 2B) in that they activate at inappropriately hyperpolarized (left-shifted) voltages (Shcherbatko et al., 1999; Tabarean et al., 1999; Wang et al., 2009; Beyder et al., 2010). A Nav channel whose operation becomes left-shifted in this manner is, in effect, leaky.
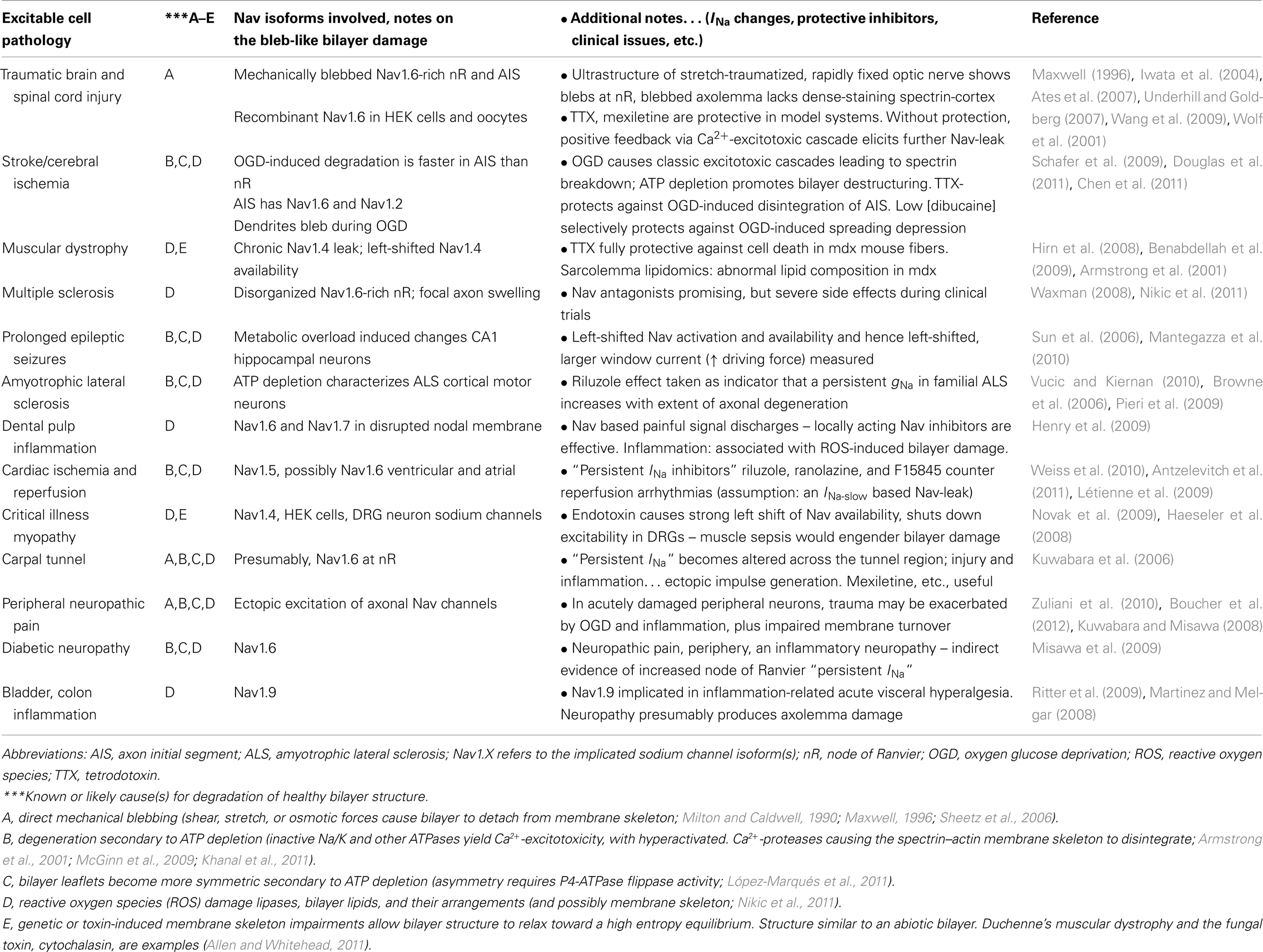
Table 1. Sick excitable cells and their leaky, hyperactive, lethal Nav channels.
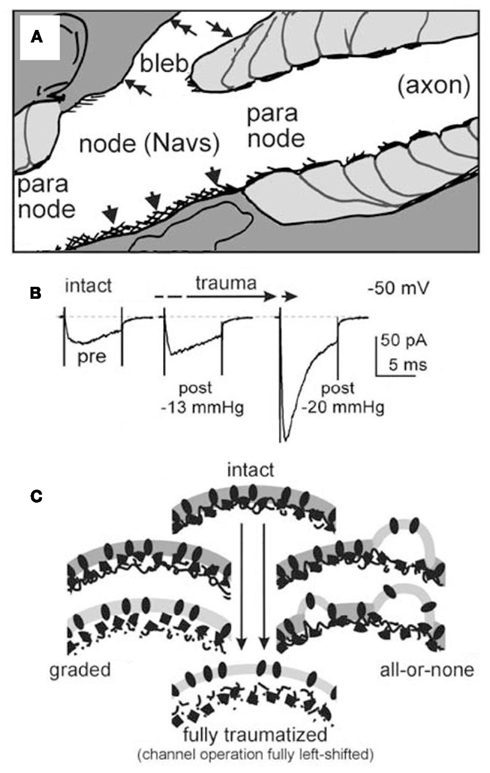
Figure 2. (A) From a stretch-traumatized optic nerve, the nodal and paranodal regions of a node of Ranvier (cartooned by tracing an electron micrograph, Figure 2A of Maxwell, 1996) showing intact (arrowheads) and severely blebbed yet unruptured (double-headed arrows) nodal (hence Nav-rich) axolemma. (B) INa(t) from recombinant αNav1.6/β1 in a cell-attached oocyte patch, before (intact) then again after bouts of pipette aspiration (experimental conditions as in Wang et al., 2009). The changing INa(t) (at −50 mV) is consistent with increasingly strong coupled-left shift, the mechanism outlined in Figure 3. (C) In sick excitable cells, Nav channels that become “leaky,” we assume, experience nano-structural environmental changes; here we depict a Nav-rich membrane going from intact (top) to blebbed (bottom). If INa changes such as seen in B are due to increasing bilayer entropy, then graded structural changes of each channel’s nano-environments (left) would be expected. If, instead, as suggested by some (Shcherbatko et al., 1999; but see Morris et al., 2006), pipette aspiration induced-blebbing alters Nav kinetics by disrupting discrete channel/cytoskeleton interactions, then each Nav channel will see an all-or-none structural change, such as depicted at right.
By virtue of molecular coupling between the fast-mode activation and inactivation (availability) processes in a Nav channel (Banderali et al., 2010), these two processes left shift in synchrony: if the activation Boltzmann [“m3∞(V)” in Hodgkin–Huxley parlance] left shifts by, say, 7 (or 11 or 20…) mV, then availability [“h∞(V)”] left shifts by 7 (or 11 or 20…) mV as well. This behavior of fast-mode Nav channels, shown for recombinant Nav1.6 channels in Figures 3A,B we term “coupled-left shift” (CLS). Crucially, as shown in Figure 3C, Nav-CLS yields a left-shifted steady-state window conductance [“m3h(V)”]. Here, m3h(V) is shown before (black) and after a 20-mV CLS (gray). Note that the Nav-CLS version of m3h(V) now peaks at what would normally be a subthreshold voltage range (i.e., below the normal Vrest). Even if only a fraction (say 10%) of the nodal membrane suffered damage, that fraction, by virtue of its left-shifted m3h(V), should generate a “subthreshold persistent current” that would depolarize the adjacent (intact) Nav-rich membrane, causing the axon to fire ectopically. Because it facilitates rhythmic firing, a subthreshold persistent INa is used by pacemaker neurons (Taddese and Bean, 2002) and by the first node (beyond the axon initial segment) to elicit rhythmic firing (Kole, 2011), but injury-induced Nav-CLS based subthreshold persistent INa triggers pathological ectopic activity. We have modeled the consequences of Nav-CLS for axonal ion homeostasis and excitability (Boucher et al., 2012). In a nutshell, Nav-CLS causes [Na+] gradients to run down, it overtaxes the Na/K-ATPase and, depending on CLS pervasiveness and severity, produces subthreshold voltage oscillations, bursting, and diverse other manifestations of hyper -and hypo-excitability.
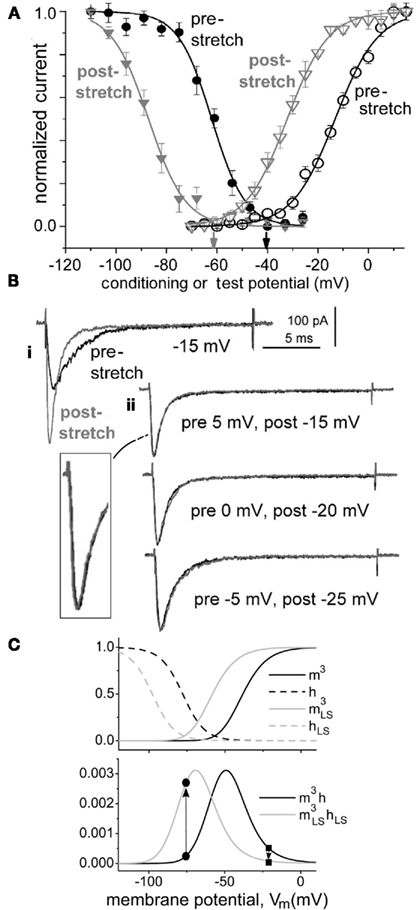
Figure 3. Coupled-left shift (Nav-CLS). (A) Traumatic membrane stretch causes irreversible hyperpolarizing shifts of activation and steady-state fast inactivation (availability) for recombinant Nav1.6 (aspirated cell-attached oocyte patches; likewise in B). (B) CLS as seen from INa traces: i, INa(t) during a step to −15 mV before and after stretch-induced membrane damage – the change is irreversible. ii Nav channels in this patch (same as i) underwent a membrane-injury-induced CLS found to be of magnitude 20 mV, as evidenced by the fact that amplitude-normalized pre/post INa(t) traces measured at Vm steps 20 mV apart, as listed, have indistinguishable time courses (expanded section: pre 5 mV, post −15 mV traces). CLS like this is observed whether left shifts are small or large (see Figure 3 of Wang et al., 2009). This justifies co-left-shifting of HH Boltzmanns, 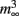 (V) and h∞(V) (= availability), along the voltage axis (Figure 1 legend) to model axonal Nav channels in trauma-injured membrane (Boucher et al., 2012). (C) Activation [m3(V)] and inactivation [h(V)] variables at t = ∞ for regular HH-Nav channels and after each function is left-shifted 20 mV (mimicking intact versus “strongly traumatized”). The product (lower panel) of overlapping activation and availability Boltzmann’s, m3h(V) = steady-state open probability or non-dimensional gwindow. Arrows: a 20-mV left-shift
(V) and h∞(V) (= availability), along the voltage axis (Figure 1 legend) to model axonal Nav channels in trauma-injured membrane (Boucher et al., 2012). (C) Activation [m3(V)] and inactivation [h(V)] variables at t = ∞ for regular HH-Nav channels and after each function is left-shifted 20 mV (mimicking intact versus “strongly traumatized”). The product (lower panel) of overlapping activation and availability Boltzmann’s, m3h(V) = steady-state open probability or non-dimensional gwindow. Arrows: a 20-mV left-shift 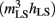 increases gwindow near typical Vrest while decreasing it nearer 0 mV.
increases gwindow near typical Vrest while decreasing it nearer 0 mV.
After finding that reversible bilayer stretch elicits reversible Nav-CLS in recombinant Nav1.5 (Morris and Juranka, 2007), we turned to recombinant Nav1.6 (Wang et al., 2009). There, irreversible bilayer damage due to “membrane stretch” (cell-attached patch clamp, with stretch- and bleb-inducing pipette aspiration) yields irreversible CLS (Figures 2B,C) whether or not Nav1.6 channels are co-expressed with β-subunits. Once the irreversible damage has “saturated” (typically producing >20 mV of irreversible CLS), further stretch of the bilayer elicits reversible Nav-CLS because now, pipette aspiration is indeed causing reversible thinning/disordering (i.e., stretching) of the bilayer. For Nav1.6, as for Nav1.5 (Morris and Juranka, 2007; Banderali et al., 2010) for comfortably non-lytic stretch stimuli, reversible Nav-CLS (Figure 5 in Wang et al., 2009) is small (typically <5 mV). When studied in fast-mode, Nav1.4 and Nav1.2 channels (co-expressed with β-subunits) behave the same way as fast-mode Nav1.6 (Catherine E. Morris and P. F. Juranka, unpublished observations). It is now known, however, that expressed in HEK cells Nav1.5 channels exhibit large substantially irreversible Nav-CLS (Beyder et al., 2010). An interesting possible explanation for the isoform differences in oocytes is an idea that would be tested by immuno-biochemistry, namely that in oocytes, Nav1.5 traffics to bilayer subdomains with a high degree of disorderliness whereas Nav1.2, Nav1.4, and Nav1.6 traffic to highly ordered cholesterol-rich domains. It would, in fact, be worth exploiting this stark difference between Nav1.5 and the other Nav isoforms as a way to correlate channel-lipid interactions and kinetic behavior.
What appears to be irreversible Nav-CLS arising from metabolic (not mechanical) injury has been reported for the Nav channels of hippocampal neuronal somata subjected to prolonged epileptic discharge: the somatic Nav channels (presumably Nav1.2) show a measurably left-shifted Iwindow associated with left-shifted (fast-mode) activation and availability (Sun et al., 2006). Presumably these overworked neurons are ATP-depleted and their plasma membranes are beset by the usual chemical insults of ischemia. Skeletal muscles subject to inflammatory conditions appear to have a comparable type of Nav-leak (Haeseler et al., 2008; Nikic et al., 2011). A recurring theme here is that it will be necessary to revisit native-Nav channels linked to disease states in Table 1 to establish whether CLS correlates broadly with increasing Nav-leak.
Both Fast and Slow Mode Nav Currents Contribute to Nav-Leak
In injured membrane, as will be discussed further in Figure 4 (see also Figure 2Ciii in Wang et al., 2009), Nav channels gating in slow mode exhibit left-shifted activation. However, whether slow inactivation is kinetically coupled to slow activation is unknown (in parallel to the coupling between fast activation and fast inactivation marked in between OA and IA in Figure 1). Typically, in a population of Nav channels, ∼1% would be in slow mode. Though slow mode current (INa-slow) is a kinetically distinct entity (see Figure 1) from any fast-mode based persistent currents (the coupled-“m3h” or subthreshold gwindow of Taddese and Bean, 2002), it too is called persistent current. In computational models, INa-slow is typically simplified to a small stand-alone sub-population of Nav channels with no inactivation process (e.g., Coggan et al., 2010). In reality, slow and fast modes interconnect (Figure 1) and the simplest assumption is that any injured membrane Nav channel (Figure 3) would show CLS when in fast mode and left-shifted activation when in slow mode. Thus, when we refer to “Nav-CLS”-injury, an implicit assumption is that the affected Nav channels “leak” in both fast and slow modes.
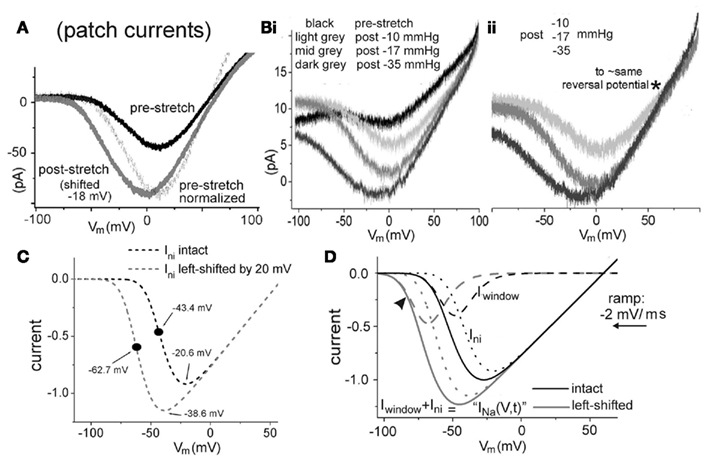
Figure 4. Hyperpolarizing ramps. (A) Recombinant Nav1.6, oocyte patch, INa elicited by hyperpolarizing ramps from +100 to −100 mV, −2 mV/ms, before and after membrane damage. (Bi). Similar data set before stretch then after increasingly strong ∼20 s bouts of pipette aspiration (conditions as in (A); further explanation in text). (Bii) Damaged-membrane traces, y-axis adjusted, as described in text. (C) Simulated INa-ni(V,t) for “intact” and 20 mV left-shifted non-inactivating HH-type Nav channels (ramps: −2 mV/ms). The INa midpoint shifts ∼−18 mV, as expected for the 20-mV shift imposed on m3(V) [equal-sized INa-ni(V) and m3 shifts would correspond to infinitely steep voltage-dependence in mni(V); see Boucher, 2011]. (D) Simulated INa(V,t) = (INa-fast + INa-ni) for hyperpolarizing ramps from +150 to −150 mV at −2 mV/ms, gni/gNa-fast = 0.0075; for control (black) and −20 mV shifted (gray). Total INa(V,t) (solid lines) mimic experimental traces, given such a gni/gNa-fast mix; they would be comprised of the two components, as labeled.
Increased Entropy and Nav-CLS
Formally, plasma membrane blebs induced by pipette aspiration (Sheetz et al., 2006) or other shear forces in the mechanically traumatized CNS (Maxwell, 1996) are defined as regions of high fluidity bilayer dissociated from the cortical cytoskeleton. Clearly, the provenance of such injury-induced blebs differs radically from that of transient cell-mediated blebs used for ameboid-crawling (Charras et al., 2009); there, the blebbing-membrane loses its F-actin (due to precision cortical acto-myosin contractions) while retaining those elements of the spectrin skeleton needed for re-adhesion to the cortex. In these motility-blebs, the bilayer, we expect, would retain much of its healthy, far-from-equilibrium structure. By contrast, in excitable cell injury (Maxwell, 1996), as in pipette aspiration induced-blebbing (Morris et al., 2006; Sheetz et al., 2006), bleb formation, being an externally initiated unregulated process, is governed largely by raw physics. Bilayer structure in a blebbing-membrane region of a sick excitable cell will, thus, approach equilibrium, exhibiting increased disorder both vertically (between leaflets) and horizontally (within plane). By virtue of non-specific electrostatic binding of polar lipid headgroups to filamentous cortical proteins (Sheetz et al., 2006) plus the actions of lipid-ATPases (Shevchenko and Simons, 2010), certain phospholipids like phosphatidylserine are normally disproportionately high in the inner leaflet. As blebs develop, however, this vertical asymmetry dissipates: artificial bilayers and dead bilayers are symmetrical (Musters et al., 1993; Schlegel and Williamson, 2001; van Genderen et al., 2008). Simultaneously, lateral phase separation patterns found among the bilayer lipids will diminish in complexity (Maxwell, 1996; Kaiser et al., 2009; Lingwood and Simons, 2010; Shevchenko and Simons, 2010; Levental et al., 2011): within-plane organization will become more random, more disordered. In summary, in the plasma membrane of sick excitable cells, bilayer structure will slowly relax toward the minimum energy, maximum entropy situation of a self-organized bilayer. It is still a bilayer, but one with a radically lower information content than the healthy far-from-equilibrium bilayer that various particular Nav channel isoforms have come (via evolution) to expect. Additionally, once it has areas denuded of cortical cytoskeleton, plasma membrane bilayer should be more prone to inappropriate insertion of intracellular membrane vesicles and to excessive uptake of free fatty acids (see Figure 1 of Milton and Caldwell, 1990; Morris and Homann, 2001). The expectation is that damaged Nav-rich bilayer will be disorderly and that its lipid-species composition too will render it abnormally fluidized.
Thus, while certain mutant variants of Nav channels are dangerously “leaky” due to structurally defective proteins (Catterall et al., 2008), Nav-CLS posits that genetically normal Nav channels in sick excitable cells become dangerously leaky when they find themselves embedded in structurally defective bilayers.
Positive Feedback: From Small Nav-Rich Bleb to Large Nav-Rich Bleb
Consider, for a Nav-rich region of an excitable cell membrane (an axon initial segment, say, or the post-synaptic membrane of a skeletal muscle end-plate), an initially minor Nav-CLS leak due to a small bleb. Or, an entire Nav-rich membrane could be mildly damaged and have a similarly small Nav-CLS leak due, say, to inflammation-induced reactive oxygen species (ROS). A mild mechanically or chemically induced Nav-CLS leak should grow via a vicious cycle: the external event (trauma, infection, ischemia, etc.) degrades plasma membrane structural integrity. As detailed elsewhere (Morris, 2011a,b,c), abnormally fluidized, more easily thinned bilayers, like depolarization, lower the energy barrier between a voltage sensor’s “down” and “up” conformations, and so will engender left-shifted Nav gating, i.e., Nav-CLS. The resulting Na+-leak puts excess demands on Na+/Ca2+ exchangers and on a cell’s various ATP-driven pumps. When the demands can no longer be met an excitotoxic cascade occurs (Wolf et al., 2001; Yuen et al., 2009). Elevated [Ca2+]intracellular hyperactivates Ca2+-protease-mediated cleavage of cortical cytoskeleton elements, enlarging the extent of bleb-like membrane. Via Na-CLS, the initially minor mechanically induced bleb has subverted cellular processes so that those processes proceed to damage the Nav-rich membrane’s bilayer structure. The process is irreversible once Ca2+-protease dependent spectrin fragments become detectable (McGinn et al., 2009). In cerebral ischemia (Schafer et al., 2009) and in cardiomyocytes (Weiss et al., 2010) during post-ischemia/reperfusion, the initial insults that bring on Nav-leak are chemical. Dystrophic muscle cells are genetically bleb-prone and have left-shifted Nav availability linked to cell-lethal Nav-leaks (Hirn et al., 2008; Allen and Whitehead, 2011).
In the hours and days following traumatic or ischemic injury, mildly damaged excitable cells in “penumbra” zones abutting the primary insult zones succumb to deepening, self-induced (and, evidently, tetrodotoxin-sensitive) secondary injury. Thus, for various model systems there is a tetrodotoxin-sensitive axon loss in white matter, cell death in gray matter, loss of dystrophic skeletal muscle fibers, and, at cardiac infarct border-zones, loss of cardiomyocytes. Table 1 signifies, we think, that sick excitable cells that suffer an initially minor Nav-CLS can expect excitotoxic demise if Nav-CLS based Na+-leak is not stopped either by membrane remodeling that rids the cells of the damaged Nav-rich bilayer, or by therapeutic drugs that staunch the leak.
Protein Partners
For Nav1.6 channels expressed in oocytes, injury-induced CLS phenomena described here occur with or without co-expressed β subunits (Wang et al., 2009). For that reason, and since virtually nothing is known about in situ modulatory interactions of β subunits (or ankyrin-G, or other of Nav channels’ many putative protein partners, Dib-Hajj and Waxman, 2010), we do not explicitly deal with them here. On the other hand, our insistence on how crucial it is to study sick-cell Nav-leak and its attendant Nav-pharmacology in native and not just recombinant systems, acknowledges that native lipid structures in conjunction with diverse protein partners in the immediate vicinity of native-Nav channels, are likely to determine the specifics of sick-cell Nav-leak in different types of excitable cells.
Nav Inhibitors
Tetrodotoxin, being a pore blocker, inhibits both fast (Active) and slow (Relaxed) mode Nav channels. Its exclusive selectivity for Nav channels has made it a powerful tool in cell/tissue models of disease, as just described. Like many Nav inhibitors, tetrodotoxin is powerfully protective in cellular models of injury to Nav-rich excitable membranes (Table 1) but it is a universal Nav-pore blocker and as such, lethal upon systemic administration. Nav inhibitors with more appropriate clinical traits include heterocyclic molecules like ranolazine and riluzole (Antzelevitch et al., 2011; Cadotte and Fehlings, 2011). These lipophilic compounds preferentially bind and stabilize Nav channels in non-conducting slow-gating states (Song et al., 1997; Antzelevitch et al., 2011). Often called “persistent current” blockers, these drug molecules are especially effective at stabilizing slow mode Nav in non-conducting states and at higher concentrations they inhibit fast-mode channels (Jo and Bean, 2011; Lenkey et al., 2011). Because of severe side effects (Waxman, 2008), however, none of the available Nav antagonists is routinely used to counter the devastating, slow-developing consequences of traumatic brain injury described at the neurological level as diffuse axonal injury (Wolf et al., 2001; Iwata et al., 2004) though for spinal injury, riluzole trial are underway (Cadotte and Fehlings, 2011).
Lipophilicity and Nav Inhibitor Efficacy
Although it is recognized that clinically effective Nav inhibitors are lipophiles (or strongly lipophilic amphiphiles), what explains the importance of lipophilicity is unclear (Jo and Bean, 2011; Lenkey et al., 2011; Nesterenko et al., 2011). We formulate, below, a two-part hypothesis in which, for sick excitable cells, the known requirement for lipophilicity in effective Nav antagonists (Lenkey et al., 2011) correlates with the elevated bilayer-fluidity origin of Nav-CLS. Before doing so, we direct the reader to Box 1 which itemizes some physiological, pharmacological, physico-chemical, and computational findings that bear on the idea.
Box 1. A brief history: bilayer partitioning and intra-bilayer orientation of lipophilic/amphiphilic molecules that bind voltage-gated channels** and other membrane proteins•.
**Herbette et al. (1989) partitioning of dihydropyridines (DHPs) into lipid bilayer could precede binding to voltage-gated Ca2+ channels. Sarcolemma/buffer partition coefficients: 5,000–150,000 range.
**Mason et al. (1992) voltage-gated Ca2+ channel antagonists and cholesterol. X-ray diffraction and equilibrium binding techniques: ↑membrane cholesterol →marked decrease in DHP partition coefficients (likewise verapamil, diltiazem).
**Mason (1993) Ca2+ channel DHP type antagonists – interactions with bilayers. Lipid composition (e.g., cholesterol content, acyl chain saturation) effects on membrane partitioning of antagonists should affect bioavailability under normal versus pathological conditions with altered membrane lipids. Specifics of bilayer composition may help concentrate and orient drug molecules relative to a hydrophobic binding site at channel/bilayer interface. For desirable pharmacokinetics, ↑’d efficacy, ↓’d side effects, drug design should anticipate contributions from membrane lipid compartment.
**Lee and MacKinnon (2004) amphiphilic voltage sensor toxins of arachnid venoms reach their target by partitioning into the lipid bilayer. Accumulation of toxin where voltage sensors reside and exploiting the free energy of partitioning of appropriately oriented amphiphilic toxins → high-affinity inhibition.
•Zhang et al. (2007) tetracaine/vesicle interactions: partitioning into solid-gel membrane depends mostly on steric accommodation between lipids, whereas in liquid-crystalline membrane (larger inter-lipid distances, lower steric hindrance), hydrophobic and ionic interactions between tetracaine and lipid molecules predominate. Bilayer partition coefficients ↓’d by cholesterol.
•Baenziger et al. (2008) bilayer lipid composition alters tetracaine action at nicotinic AChRs.
•Eckford and Sharom (2008) cholesterol-modulation of P-glycoprotein-mediated drug transport appears to operate via effects on drug partitioning into the bilayer and by changes in the protein’s local lipid environment.
•Chisari et al. (2009) GABA-R both specific (e.g., enantiomer-dependent) and non-specific (e.g., bilayer partitioning) properties contribute to potency and longevity of steroid action.
•Lombardi et al. (2009) β2 agonist, indacaterol fluidizes membranes less than salmeterol and yields faster-onset, longer-duration therapeutic effects, perhaps because of synergy between indacaterol’s better partitioning into raft micro domains and its faster membrane permeation.
**Schmidt and MacKinnon (2008) the mechanical state of bilayer lipids in the plasma membrane is crucial to the efficacy of amphiphilic peptide toxins that have evolved to bind to and right-shift voltage sensors.
**Milescu et al. (2009) conversion of sphingomyelin to ceramide shows that binding efficacy of an amphiphilic voltage sensor toxin to the membrane-embedded voltage sensors of Kv channels depends on the bilayer’s lipid composition.
•Ali et al. (2010) loading of drugs (diazepam, ibuprofen, midazolam, propofol) into liposome bilayers is sensitive to cholesterol content: ↑’d cholesterol = ↓’d drug incorporation.
•Tejwani et al. (2011) all-atoms molecular dynamics simulations: a bilayer’s hydrocarbon chain interior is the region most selective to the chemical structures of drug-type solutes. Multiple discrete polar groups (same solute) contribute additively to bilayer partitioning. Significant orientational preferences for the drugs are evident in specific bilayer regions.
•Jastrzebska et al. (2011) bilayer composition/organization regulates classic G protein-coupled receptor, rhodopsin.
**Lenkey et al. (2011) lipophilicity is a key factor in Nav antagonist potency, allowing inhibitor molecules to bind at two distinct sites, one most accessible at hyperpolarized potentials (“resting site”), the other at depolarized potentials (“inactivated site”). In addition, steric/orientation features involving aromatic rings, hydrogen acceptors, and sheer molecular bulk are important determinants of potency at the inactivated site.
Relevant to this Box, Muddana et al. (2012) recently showed differential partitioning of non-lipid amphiphiles (vitamin-E, benzyl alcohol, Triton-X 100) into bilayers depending on mechanical properties of different lipid subdomains.
To probe riluzole inhibition, Narahashi and colleagues clamped INa in DRG neurons (Song et al., 1997). Noting the drug’s enhanced effectiveness in damaged neurons, they invoked high-affinity binding to the slow-inactivated Nav channels of damage-depolarized cells. In such situations, the Nav channels of interest are not only in damaged cells, we suggest, but damaged bilayer. This should affect the working concentration of lipophilic Nav antagonists (like riluzole) if they partition differently into damaged versus intact membrane. Further, riluzole efficacy might be higher in damaged than intact bilayer if its ability to orient appropriately with respect to intra-bilayer binding sites on the DRG Nav channels (see Lenkey et al., 2011) is better in less structured bilayers.
What little we could find in the literature relative to these issues of the partitioning and orientation of lipophilic drugs in different classes of bilayer structure is summarized in Box 1. Of particular note, highly ordered, cholesterol-rich membrane subdomains accumulate lipophilic drugs more poorly than cholesterol-poor fluidized membranes. Strikingly, the drugs in question are clinically important voltage-gated calcium channel antagonists, the dihydropyridines. The collection of findings sketched in Box 1 suggest that at ones peril would one assume bilayer order/disorder to be irrelevant for the interactions of lipophilic drugs with dynamic membrane proteins (see also Benabdellah et al., 2009; Rajendran et al., 2010; Morris, 2011b). To our knowledge, however, these issues have not been investigated experimentally for Nav inhibitors.
Lenkey et al. (2011) showed that simple lipophilicity (measured from oil/water partition coefficients) correlates strongly with Nav inhibitor efficacy, and, beyond that, that molecular orientation and steric constraints are particularly important for inhibition of INa-slow (Lenkey et al., 2011). The possibility that, for sick excitable cells, clinically useful Nav inhibitors could selectively target the malfunctioning Nav channels by (1) accumulating preferentially in disordered bilayers, and (2) exhibiting orientation/steric properties appropriate for within-bilayer binding to their targets (see Schmidt and MacKinnon, 2008; Morris, 2011a,b) directly echoes Mason’s (1993) ideas regarding the dihydropyridines (see Box 1). It contrasts strikingly, however, with a recent model for the differential atrium/ventricle efficacy of ranolazine for (healthy) cardiac myocyte Nav channels. There, it is explicitly assumed that drug access to binding sites (Active mode and Relaxed mode conformations) is via hydrophilic pathways (Nesterenko et al., 2011).
Hypothesis: In Sick Excitable Cells, Bilayer Entropy Engenders Nav-CLS But Simultaneously Enhances the Efficacy of Lipophilic Nav Antagonists
Against the above background material, we now restate some of the ideas formally in a two-part hypothesis. The hypothesis is grounded in our work on recombinant axon-isoform Nav channels in traumatized membranes (Wang et al., 2009), on Schmidt and MacKinnon’s (2008) observations on Kv channel activation and drug interactions in intact versus mechanically disturbed plasma membranes, and on the Krepkiy et al. (2009) finding that isolated voltage sensors in “S4-up-states” locally thin/disorder the bilayer (explained in Morris, 2011a,b,c). In conjunction with the meta-analysis of Lenkey et al. (2011) for Nav-antagonist physical chemistry and efficacy, which postulates bilayer-embedded binding sites for fast and slow mode Nav channel conformations, these findings lead us to hypothesize:
H1: Mechanical and/or chemical insult causes the far-from-equilibrium order of a healthy bilayer to decay toward equilibrium, i.e., toward the more disordered dynamically self-organized arrangement of abiotic bilayers. In injured Nav-rich excitable cells including neurons, the Nav channels that leak are irreversibly “left-shifted” due to irreversibly increased bilayer disorder, with the shift proportionate to the extent of disorder in the Nav-embedding bilayer;
H2: Because complex lipophilic Nav inhibitors in aqueous solution partition into and may also orient more appropriately in damaged (disordered) bilayers than in well-packed bilayers of healthy Nav-rich membranes, lipophilic Nav inhibitors achieve their best global efficacy precisely at the loci of the leaky Nav channels.
Our title combines items H1, H2 as a question–hypothesis, and the question is not rhetorical. The hypothesis moreover, needs to be tested on native-Nav channels in their native-bilayer settings. The kinetics of native-Nav channels expressed and maintained at high-density (Kole et al., 2008) remain poorly understood (Fu et al., 2011); add trauma (or other sick excitable cell conditions) and this is even more true. We re-iterate: there is an unmet need to study the electrophysiological and pharmacological behavior of native-Nav/bilayer/membrane-skeleton assemblages before, during, and after imposing assorted types of membrane damage (see “A–E” column, Table 1 and Box 1).
Nav-Rich Native Membranes Need to be Investigated
Myelinated axon INa is carried by Nav1.6 channels (see Chatelier et al., 2010). Nodal INa has fast and slow components (Stys et al., 1993), but the discrete responses of these components to trauma or ischemia or other bilayer-damaging pathological states have not, to our knowledge, been studied electrophysiologically. Assuming fast and slow axonal INa arises from one Nav population (Figure 1), damaged axolemma should have co-existing left-shifted INa-fast (hence left-shifted Iwindow) and left-shifted INa-slow. Carefully devised (though currently little-used) techniques exist, fortunately, to study drug actions on INa at voltage clamped nodes of Ranvier. An understanding of the pharmacological idiosyncrasies of nodal Nav1.6 channels in both modes is needed. Voltage clamp ramps (see Box 2) would be the most feasible way to simultaneously monitor fast and slow INa in intact versus damaged nodal membrane. Based on recombinant Nav1.6 ramp data and on simulations, the next three sections suggest what to watch for from nodal Nav1.6 current.
Box 2. Vaseline-gap voltage clamp of a node of Ranvier.
This technique (Conti et al., 1976; Benoit and Escande, 1991; Schwarz et al., 1995, 2006), though challenging, was the major electrophysiological approach used for Nav channel pharmacology several decades ago, before INa-slow was recognized as a pharmacological target. It would be worth resurrecting the method to study native-Nav1.6 channels in damaged nodes of Ranvier. The specific aim would be to perform sawtooth ramp clamp of the nodal axolemma to concurrently detect INa-fast and INa-slow and their responses to membrane damage and to antagonists. Standard step voltage clamp would be used as well for purposes of technical, biophysical, and pharmacological benchmarking. In amphibians, nodes yield ∼50 nA of INa-fast and 1 nA traces show good signal-to-noise (Conti et al., 1976). Given a gNa-slow/ gNa-fast of ∼1%, peak ramp INa(V) for INa-slow should be ∼0.5 nA. Mechanically traumatizing a node after recording control data is not feasible. Nodal bilayer damage could, alternatively, be imposed by ATP depletion and/or Ca2+-protease hyperactivation and/or generation of reactive oxygen species (Dong and Hare, 2005). Cyclodextrin extraction of membrane cholesterol could used to disrupt healthy bilayer structure. It is possible that accidental stretch, a recognized hazard while mounting axons for vaseline-gap clamp (see Methods section in Schwarz et al., 1995), could be exploited, or abrupt nerve stretch (Maxwell, 1996) prior to axon dissection. High resolution video-recording at all stages would allow for blinded post hoc classification of trauma (unintentional and intentional) intensity. Whatever the methods used to damage the nodal bilayer, ancillary imaging, and/or chemical evidence (Rajendran et al., 2010; Shevchenko and Simons, 2010) that initially intact nodal membranes had incurred structural/compositional damage would needed. Changes to nodal INa-fast and INa-slow would be most usefully tracked via the hysteresis information from sawtooth clamp. To illustrate, in Figures 4 and 5, we summarize what little information is available for ramp-elicited total INa(V,t) from recombinant Nav1.6 channels before and after mechanical membrane damage, then we simulate some ramp clamp outcomes for a 0.0075 mixture of HH-type Nav channels without and with inactivation.
Ramp Clamp, Nav1.6 Current, and Membrane Damage to Date
In our recordings of recombinant Nav1.6 current in oocyte patches, INa-slow was hard to study since usually it was unmeasurable against background noise (see also Chatelier et al., 2010). Nevertheless, Figures 4A,B shows ramp-elicited INa traces from cell-attached oocyte patches. To minimize INa-fast in favor of slow-gating current, hyperpolarizing ramps were used. The Nav-bearing membrane patches were injured via pipette aspiration induced-blebbing. Figure 3A is from Wang et al. (2009) while the Figure 4B data are from the same group of experiments, included here to help illustrate Nav1.6 ramp INa(V,t) during progressively deepening membrane damage. The ramp-elicited INa(V,t) progressively left shifts with stronger damage, its maximal amplitude increases and, in the gmax region, its slope conductance steepens. Offsets and drift (though largely absent from Figure 4A) are the norm for INa from ramp clamp, as in Figure 4Bi [in Figure 4Bii, current offsets in the three post-trauma traces were handled by repositioning traces on the y-axis to a common reversal potential (asterisk, ∼−55 mV, ENa), making more evident the post-trauma left shifts and the steepening slope conductance]. Steeper “gmax” slopes signify increased numbers of maximally activated Nav channels and/or greater maximal Popen values. Either way, these INa(V,t) traces would be consistent with post-trauma “Nav-leak” through left-shifted Relaxed mode channels. In these experiments, strong patch-trauma left-shifted fast INa ≥20 mV (Wang et al., 2009), but whether slow and fast INa in a given patch shifted the same amount was not established.
Rudimentary Simulations for Ramp Clamp – the Simplest Model
To concurrently monitor co-existing fast and slow INa in Nav-rich excitable cells is not straightforward. Simulations can help with experimental design. For slow INa we simply used gni, a non-inactivating version of fast gNa [i.e., the usual Hodgkin–Huxley gNa but with h(V) = 1], with gni/gNa-fast = 0.0075. Eventually, more detailed modeling of INa (with slow mode inactivation and mode-switch transitions, Figure 1) will be needed, but even our rudimentary simulation illustrates some important points.
In Figure 4C, with fast-mode gNa zeroed, INa-ni(V,t) is depicted alone to illustrate the consequence of a left shift. As there is no inactivation or mode switching, the slope conductance in the gmax region is fixed. Maximal INa-ni(V,t) increases because shifted channels activate where Vm − ENa is greater. In Figure 4D the gNa-fast and gni components are combined (black, control; gray, 20 mV left-shifted). Even though a hyperpolarizing ramp was used and even though the two-component INa(V,t) elicited has the look of “just non-inactivating current,” it is not. Encroaching on the Vm range near typical Vrest (arrowhead), the major component is, in fact, “window current” (Iwindow). Though gni is modeled as having precisely the same activation characteristics as gNa-fast, it yields insignificant Ini in the “subthreshold” Vm range where Iwindow flows, simply because of gNa-fast:gni ratio (>100:1). The dominance of an Iwindow-based subthreshold Nav-leak could be consequential, especially if putative therapeutic inhibitors for mild injury were chosen to target gNa-slow.
It is instructive to consider this scenario in light of Nav inhibitors and trauma. If a mild CLS-type injury depolarized an axon by a few millivolts, the key Nav-leak for the axon would be the shifted Iwindow. Accelerated Na/K pumping would be required to sustain a “resting” Vm below the zone ∼10 mV more depolarized (Figure 4D) where Ini-leak starts to take over. The pharmacological goal, therefore: target the damaged-membrane’s Nav-CLS channels with lipophilic inhibitors that, at voltages near Vrest, stabilize CR or CA conformations (see Figure 1). Strongly lipophilic antagonists that readily cross the blood–brain-barrier (e.g., Weston et al., 2009) would be ideal. While these may be relatively poorer inhibitors of slow INa (see Lenkey et al., 2011) staunching the Nav-CLS leak in mildly damaged cells before they depolarize is the “first responder” goal.
With this as the target, it becomes important (in pharmacologically relevant settings) to better understand what modulates the ratios of Active to Relaxed channels. This is a question of mode switching and it is unexplored territory. Inhibiting OA → OR (Figure 1A) or promoting CR → CA would minimize slow mode and maximize fast-mode channels. Based on Nav1.4 behavior in oocyte membranes (Tabarean et al., 1999) we know that in some cell types, membrane damage can dramatically modulate Nav channel mode switch transitions: conversion to fast-mode gating occurs. When examining native-Nav channels’ responses to injury, the possibility of mode switch modulation should be borne in mind.
Exploiting the Hysteresis of Sawtooth Ramps
Figure 5A emphasizes how ramp rate and direction strongly influence the “shape” of INa(V,t) = [INa-fast + Ini](V,t). For either direction and for all speeds, INa(V,t) is identical above ∼−25 mV since there, only gni is active [in its Popen(V) ∼ 1 range]. For real Nav channels (which undergo slow inactivation), both ramp speed and direction will affect INa(V,t) in this range, yielding information on Relaxed mode (slow) and mode-switch transitions. Relaxed and Active mode channels are from a single population dominated by Active mode, and sawtooth protocols allow for concurrent semi-quantitative monitoring of both. In membrane-damaged excitable cells, on-going left shift will be expected, making the system response a “moving target.” Moreover, when following electrophysiologically the consequences of irreversible membrane damage, damage intensity, and its rate of change will not be tightly controlled. Within-preparation comparisons and time course data are therefore imperative (Tabarean et al., 1999; Morris et al., 2006; Wang et al., 2009).
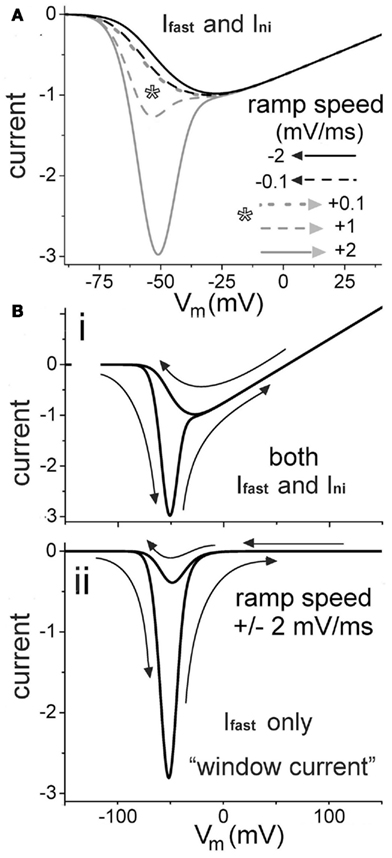
Figure 5. Modeling how to dissect fast and slow INa using sawtooth hysteresis. (A) For gni/gNa-fast = 0.0075 (also in B), total INa(V,t) during negative (black) and positive-going (gray) voltage ramps of various speeds, normalized to peak INa for the negative 0.1 mV/ms ramp. Positive to ∼−25 mV, all plots overlap. Asterisk: speed range of commonly used depolarizing ramp protocols. (Bi,ii) Current hysteresis (arrows show direction of ramps) during sawtooth ramp clamp, as labeled, for both INa-fast and INa-ni [i.e., the total INa(V,t) hysteresis trace as expected in a control experiment] and for INa-fast only (expected outcome if a drug inhibited INa-slow and not INa-fast).
With relatively fast ramps, obtaining larger within-preparation data sets is easier, but as our simulations illustrate, faster ramps increase the “contamination” of INa(V,t) by INa-fast (i.e., Iwindow) even though traces appear to be exclusively “INa-slow” (or, in the simulated form, Ini). Standard practice in most labs is to use depolarizing ramps at ∼+0.1–1.0 mV/ms, but only at the slowest end of this range would Iwindow contamination be minimal. INa(V,t) is essentially identical in either direction at +0.1 mV/ms, but fortunately, −2 mV/ms is almost as good (Figure 5A). As in Figure 5Bi, a sawtooth ramp with a fairly speedy depolarizing limb (i.e., ∼−2 mV/ms) can locate the INa-fast(V) window current. The hyperpolarizing counterpart will then reveal the only slightly “contaminated” slow current. Ramps need not, of course, be mirror images; the empirical choice is for whatever yields informative and resolvable hysteresis traces. As membrane damage deepened, quick sawtooths like this could monitor both components’ changes over time. Time course data over minutes could show whether, at minimal concentrations, lipophilic antagonists show evidence of accumulating in the bilayer, and if, say, they were acting more powerfully on slow or fast components. Depending on how rapidly injury-related membrane characteristics were changing, quick sawtooths could make it possible to capture multiple runs for better signal-to-noise. Intermittently obtained step protocol I/V data would, of course, be a useful supplement to the hysteresis data.
In Figure 5Bii, below the two-component hysteresis trace, is the INa-fast only component. To a first approximation, these hysteresis sets would mimic start and end points of a notional experiment in which a “persistent current blocker” was added to the system, fully inhibiting slow mode current while having no effect on fast mode current. During Nav-antagonist time course experiments, changes of this ilk, in combination with damage-induced left-shifting (Figure 3) would be expected. It seems unlikely that all this could be captured in a timely fashion via the usual Nav voltage clamp approach of steps-for-INa-fast, slow-ramps-for-INa-slow.
To re-iterate, continual monitoring of nodal INa(V,t) hysteresis via empirically worked-out sawtooth protocols could give a picture of Active and Relaxed mode Nav protein behavior in injured axons. For each mode, it could show if/how injury affects susceptibility to Nav antagonists. If Nav agonists showed lower efficacy in intact versus damaged membrane, this would raise a multitude of ancillary questions regarding the avidity of lipophilic drug partitioning into intact versus damaged membranes. Comparing inhibitor efficacy time courses in intact (cholesterol-packed) against cholesterol-extracted membranes (a recognized form of membrane damage) could, for example, be informative.
Old Idea, New Wrinkle
Differential partitioning of lipophilic drugs into different membrane subdomains (i.e., intact versus damaged) is a new idea for Nav channels, but a related concept is now general currency for G-proteins (Lombardi et al., 2009; Rajendran et al., 2010). Our proposal for Nav channels has a different twist, since we envisage disease states, not normal cell-mediated membrane modeling, as the factor underlying the therapeutically relevant structural heterogeneity in excitable Nav-rich membranes. Data have, interestingly, been available on membrane partitioning of the lipophilic antagonists of voltage-gated Ca channels for ∼20 years. Mason and colleagues (Box 1, first three entries) suggested, moreover, that these drugs would tend to partition differently into healthy membranes versus ones whose composition was altered due to pathological conditions. At about the same time, however, molecular-level descriptions of protein structure and of hydrophobic drug binding sites became increasingly dominant in pharmacology. The idea that bilayer structure must influence the activities of membrane proteins (Phillips et al., 2009) was largely put aside (see discussion in Finol-Urdaneta et al., 2010).
A recent finding from Andersen’s group (Rusinova et al., 2011), that initially may seem tangential, is, on closer inspection, entirely germane to this issue: membrane-fluidizing insulin receptor sensitizers (insulin receptors are G-proteins) at clinically used levels cause “off-target” left shift of Nav channel gating. In effect, Nav left shift due to increased bilayer fluidity is such a robust phenomenon that it can be used as a bioassay for side effects! This, these authors point out, “underscores the importance of exploring bilayer effects of candidate drugs early in drug development.” Framed in similar terms, we stress here the need to establish experimentally whether bilayer damage in sick excitable cells constitutes a “membrane-fluidizing Nav-antagonist sensitizer” for Nav channels. If so, it may be possible to modify antagonists to operate optimally (thus more selectively) on Nav channels in damaged membranes: this would be a “silver bullet” scenario – a “bullet” targeting only the “the bad guys.” MacKinnon and colleagues (Schmidt and MacKinnon, 2008) among others (Lee and MacKinnon, 2004; Milescu et al., 2009, see Box 1) reported such an case for an amphiphilic voltage sensor toxin that targets Kv channels in damaged but not intact membrane (described in Morris, 2011a,b,c).
If lipophilic Nav antagonists can be fine-tuned for maximal efficacy in precisely the pathologically fluidized bilayers where they are most needed, the hoped-for result would be: hyper-accumulation of a drug that shows ultra-Nav-affinity in “sick” high entropy bilayers (see Table 1). Given the diversity of Nav channel isoforms and the multitude of bilayer-structural features that differentiate intact from damaged Nav-rich membranes, silver bullet antagonists might be possible.
High throughput recombinant channel techniques make superb preliminary drug screens and could be adapted for first round studies of damaged Nav-bearing membrane. But to design optimally neuroprotective Nav agents for sick axons, the “retro” approach of the node of Ranvier vaseline-gap voltage clamp (Box 2) would be better.
Concluding Overview
In diverse sick excitable cells, Nav channel leak causes hyperexcitability and excitotoxicity-induced cell demise (Table 1). Pharmacological mitigation is poor and side effects too severe (Waxman, 2008). There is uncertainty about the nature and subcellular location of the offending Nav channels and how they are accessed by clinically promising lipophilic antagonists. We argue that for pathologies featuring membrane damage, Nav channel operation, and antagonist binding in damaged versus intact membranes will be pivotal.
Native-Nav channel kinetics can differ appreciably from kinetics for the same isoform expressed in HEK cells or oocytes (Tabarean et al., 1999; Fu et al., 2011). The sick-Nav/sick-bilayer hypothesis presented here will need testing in native settings. The axolemma of a Xenopus sciatic nerve axon would, for instance, more closely resemble a human myelinated axon than does the plasma membrane of an undifferentiated human embryonic kidney cell. Too little is known about how, in cell-specific bilayer environments, fast and slow-gating native-Nav channels (plus whatever protein partners may accompany them) respond to sick-cell type membrane damage and then to lipophilic inhibitors. To illustrate how INa-hysteresis data (from sawtooth ramp clamp of nodes of Ranvier) could facilitate the concurrent study of fast and slow mode INa during damage and antagonist application, we simulated INa for various ramped voltage stimuli.
Our global hypothesis about sick excitable cells posits that the primary drug target for lipophilic antagonists is not “axonal Nav channels” or “cardiac channels” or “skeletal muscle Nav channels” but sick-bilayer Nav channels. Ignoring the physico-chemical consequences of bilayer damage could result in sub-optimal protective antagonists for sick excitable cells. Information on native-Nav/native-bilayer/Nav-antagonist interactions could underpin the design of safer neuroprotective drugs, i.e., ones that could be administered at low enough systemic concentrations to generate few side effects, while selectively accumulating in the high entropy bilayer locale of leaky Nav channels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Support for this work is from NSERC and CIHR, Canada.
References
Ali, M. H., Kirby, D. J., Mohammed, A. R., and Perrie, Y. (2010). Solubilisation of drugs within liposomal bilayers: alternatives to cholesterol as a membrane stabilising agent. J. Pharm. Pharmacol. 62, 1646–1655.
Allen, D. G., and Whitehead, N. P. (2011). Duchenne muscular dystrophy – what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 43, 290–294.
Antzelevitch, C., Burashnikov, A., Sicouri, S., and Belardinelli, L. (2011). Electrophysiological basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 8, 1281–1290.
Armstrong, S. C., Latham, C. A., Shivell, C. L., and Ganote, C. E. (2001). Ischemic loss of sarcolemmal dystrophin and spectrin: correlation with myocardial injury. J. Mol. Cell. Cardiol. 33, 1165–1179.
Ates, O., Cayli, S. R., Gurses, I., Turkoz, Y., Tarim, O., Cakir, C. O., and Kocak, A. (2007). Comparative neuroprotective effect of sodium channel blockers after experimental spinal cord injury. J. Clin. Neurosci. 14, 658–665.
Baenziger, J. E., Ryan, S. E., Goodreid, M. M., Vuong, N. Q., Sturgeon, R. M., and daCosta, C. J. (2008). Lipid composition alters drug action at the nicotinic acetylcholine receptor. Mol. Pharmacol. 73, 880–890.
Banderali, U., Juranka, P. F., Clark, R. B., Giles, W. R., and Morris, C. E. (2010). Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin) 4, 12–21.
Benabdellah, F., Yu, H., Brunelle, A., Laprévote, O., and De La Porte, S. (2009). MALDI reveals membrane lipid profile reversion in MDX mice. Neurobiol. Dis. 36, 252–258.
Benoit, E., and Escande, D. (1991). Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflugers Arch. 419, 603–609.
Beyder, A., Rae, J. L., Bernard, C., Strege, P. R., Sachs, F., and Farrugia, G. (2010). Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J. Physiol. 588, 4969–4985.
Boucher, P.-A. (2011). Stress Driven Changes in the Kinetics of Bilayer Embedded Proteins: A Membrane Spandex and a Voltage-Gated Sodium Channel. 2011. Ph.D. thesis, University of Ottawa (downloadable at UO Research, search “Boucher spandex”), Ottawa, ON.
Boucher, P. A., Joós, B., and Morris, C. E. (2012). Coupled left-shift of Nav channels: modeling the Na+-loading and dysfunctional excitability of damaged axons. J. Comput. Neurosci. (in press).
Browne, S. E., Yang, L., DiMauro, J. P., Fuller, S. W., Licata, S. C., and Beal, M. F. (2006). Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 22, 599–610.
Cadotte, D. W., and Fehlings, M. G. (2011). Spinal cord injury: a systematic review of current treatment options. Clin. Orthop. Relat. Res. 469, 732–741.
Catterall, W. A., Dib-Hajj, S., Meisler, M. H., and Pietrobon, D. (2008). Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J. Neurosci. 28, 11768–11777.
Charras, G. T., Mitchison, T. J., and Mahadevan, L. (2009). Animal cell hydraulics. J. Cell. Sci. 122, 3233–3241.
Chatelier, A., Zhao, J., Bois, P., and Chahine, M. (2010). Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: a single-channel analysis. Pflügers Arch. 460, 1–10.
Chen, S., Tran, S., Sigler, A., and Murphy, T. H. (2011). Automated and quantitative image analysis of ischemic dendritic blebbing using in vivo 2-photon microscopy data. J. Neurosci. Methods 195, 222–231.
Chisari, M., Eisenman, L. N., Krishnan, K., Bandyopadhyaya, A. K., Wang, C., Taylor, A., Benz, A., Covey, D. F., Zorumski, C. F., and Mennerick, S. (2009). The influence of neuroactive steroid lipophilicity on GABAA receptor modulation: evidence for a low-affinity interaction. J. Neurophysiol. 102, 1254–1264.
Coggan, J. S., Prescott, S. A., Bartol, T. M., and Sejnowski, T. J. (2010). Imbalance of ionic conductances contributes to diverse symptoms of demyelination. Proc. Natl. Acad. Sci. U.S.A. 107, 20602–20609.
Conti, F., Hille, B., Neumcke, B., Nonner, W., and Stämpfli, R. (1976). Conductance of the sodium channel in myelinated nerve fibres with modified sodium inactivation. J. Physiol. (Lond.) 262, 729–742.
Dib-Hajj, S. D., and Waxman, S. G. (2010). Isoform-specific and pan-channel partners regulate trafficking and plasma membrane stability; and alter sodium channel gating properties. Neurosci. Lett. 486, 84–91.
Dong, C. J., and Hare, W. A. (2005). Contribution to ischemic injury of rat optic nerves by intracellular sodium overload. Doc. Ophthalmol. 110, 15–23.
Douglas, H. A., Callaway, J. K., Sword, J., Kirov, S. A., and Andrew, R. D. (2011). Potent inhibition of anoxic depolarization by the sodium channel blocker dibucaine. J. Neurophysiol. 105, 1482–1494.
Eckford, P. D., and Sharom, F. J. (2008). Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: effects on ATPase activity, drug binding and transport. Biochemistry 47, 13686–13698.
Finol-Urdaneta, R. K., McArthur, J. R., Juranka, P. F., French, R. J., and Morris, C. E. (2010). Modulation of KvAP unitary conductance and gating by 1-alkanols and other surface active agents. Biophys. J. 98, 762–772.
Fu, Y., Struyk, A., Markin, V., and Cannon, S. (2011). Gating behaviour of sodium currents in adult mouse muscle recorded with an improved two-electrode voltage clamp. J. Physiol. (Lond.) 589, 525–546.
Haeseler, G., Foadi, N., Wiegand, E., Ahrens, J., Krampfl, K., Dengler, R., and Leuwer, M. (2008). Endotoxin reduces availability of voltage-gated human skeletal muscle sodium channels at depolarized membrane potentials. Crit. Care Med. 36, 1239–1247.
Henry, M. A., Luo, S., Foley, B. D., Rzasa, R. S., Johnson, L. R., and Levinson, S. R. (2009). Sodium channel expression and localization at demyelinated sites in painful human dental pulp. J. Pain. 10, 750–758.
Herbette, L. G., Vant Erve, Y. M., and Rhodes, D. G. (1989). Interaction of 1,4 dihydropyridine calcium channel antagonists with biological membranes: lipid bilayer partitioning could occur before drug binding to receptors. J. Mol. Cell. Cardiol. 21, 187–201.
Hirn, C., Shapovalov, G., Petermann, O., Roulet, E., and Ruegg, U. T. (2008). Nav1.4 deregulation in dystrophic skeletal muscle leads to Na+ overload and enhanced cell death. J. Gen. Physiol. 132, 199–208.
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. (Lond.) 117, 500–544.
Iwata, A., Stys, P. K., Wolf, J. A., Chen, X. H., Taylor, A. G., Meaney, D. F., and Smith, D. H. (2004). Traumatic axonal injury induces proteolytic cleavage of the voltage gated sodium channels modulated by tetrodotoxin and protease inhibitors. J. Neurosci. 24, 4605–4613.
Jastrzebska, B., Debinski, A., Filipek, S., and Palczewski, K. (2011). Role of membrane integrity on G protein-coupled receptors: rhodopsin stability and function. Prog. Lipid Res. 50, 267–277.
Jo, S., and Bean, B. P. (2011). Inhibition of neuronal voltage-gated sodium channels by brilliant blue G. Mol. Pharmacol. 80, 247–257.
Kaiser, H. J., Lingwood, D., Levental, I., Sampaio, J. L., Kalvodova, L., Rajendran, L., and Simons, K. (2009). Order of lipid phases in model and plasma membranes. Proc. Natl. Acad. Sci. U.S.A. 106, 16645–16650.
Khanal, G., Chung, K., Solis-Wever, X., Johnson, B., and Pappas, D. (2011). Ischemia/reperfusion injury of primary porcine cardiomyocytes in a low-shear microfluidic culture and analysis device. Analyst 136, 3519–3526.
Kole, M. H. (2011). First node of Ranvier facilitates high-frequency burst encoding. Neuron 71, 671–682.
Kole, M. H., Ilschner, S. U., Kampa, B. M., Williams, S. R., Ruben, P. C., and Stuart, G. J. (2008). Action potential generation requires a high sodium channel density in the axon initial segment. Nat. Neurosci. 11, 178–186.
Krepkiy, D., Mihailescu, M., Freites, J. A., Schow, E. V., Worcester, D. L., Gawrisch, K., Tobias, D. J., White, S. H., and Swartz, K. J. (2009). Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462, 473–479.
Kuwabara, S., and Misawa, S. (2008). Pharmacologic intervention in axonal excitability: in vivo assessment of nodal persistent sodium currents in human neuropathies. Curr. Mol. Pharmacol. 1, 61–67.
Kuwabara, S., Misawa, S., Tamura, N., Nakata, M., Kanai, K., Sawai, S., Ogawara, K., and Hattori, T. (2006). Latent addition in human motor and sensory axons: different site-dependent changes across the carpal tunnel related to persistent Na+ currents. Clin. Neurophysiol. 117, 810–814.
Lee, S. Y., and MacKinnon, R. (2004). A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 430, 232–235.
Lenkey, N., Karoly, R., Epresi, N., Vizi, E., and Mike, A. (2011). Binding of sodium channel inhibitors to hyperpolarized and depolarized conformations of the channel. Neuropharmacology 60, 191–200.
Létienne, R., Bel, L., Bessac, A. M., Vacher, B., and Le Grand, B. (2009). Myocardial protection by F 15845, a persistent sodium current blocker, in an ischemia-reperfusion model in the pig. Eur. J. Pharmacol. 624, 16–22.
Levental, I., Grzybek, M., and Simons, K. (2011). Raft domains of variable properties and compositions in plasma membrane vesicles. Proc. Natl. Acad. Sci. U.S.A. 108, 11411–11416.
Lin, W., Laitko, U., Juranka, P. F., and Morris, C. E. (2007). Dual stretch responses of mHCN2 pacemaker channels: accelerated activation, accelerated deactivation. Biophys. J. 92, 1559–1572.
Lingwood, D., and Simons, K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46–50.
Lombardi, D., Cuenoud, B., and Krämer, S. D. (2009). Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur. J. Pharm. Sci. 38, 533–547.
López-Marqués, R. L., Holthuis, J. C., and Pomorski, T. G. (2011). Pumping lipids with P4-ATPases. Biol. Chem. 392, 67–76.
Mantegazza, M., Curia, G., Biagini, G., Ragsdale, D. S., and Avoli, M. (2010). Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 9, 413–424.
Martinez, V., and Melgar, S. (2008). Lack of colonic-inflammation-induced acute visceral hypersensitivity to colorectal distension in Na(v)1.9 knockout mice. Eur. J. Pain 12, 934–944.
Mason, R. P. (1993). Membrane interaction of calcium channel antagonists modulated by cholesterol. Implications for drug activity. Biochem. Pharmacol. 45, 2173–2183.
Mason, R. P., Moisey, D. M., and Shajenko, L. (1992). Cholesterol alters the binding of Ca2+ channel blockers to the membrane lipid bilayer. Mol. Pharmacol. 41, 315–321.
Maxwell, W. L. (1996). Histopathological changes at central nodes of Ranvier after stretch-injury. Microsc. Res. Tech. 34, 522–535.
McGinn, M. J., Kelley, B. J., Akinyi, L., Oli, M. W., Liu, M. C., Hayes, R. L., Wang, K. K., and Povlishock, J. T. (2009). Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J. Neuropathol. Exp. Neurol. 68, 241–249.
Milescu, M., Bosmans, F., Lee, S., Alabi, A. A., Kim, J. I., and Swartz, K. J. (2009). Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16, 1080–1085.
Milton, R. L., and Caldwell, J. H. (1990). Na current in membrane blebs: implications for channel mobility and patch clamp recording. J. Neurosci. 10, 885–893.
Misawa, S., Sakurai, K., Shibuya, K., Isose, S., Kanai, K., Ogino, J., Ishikawa, K., and Kuwabara, S. (2009). Neuropathic pain is associated with increased nodal persistent Na(+) currents in human diabetic neuropathy. J. Peripher. Nerv. Syst. 14, 279–284.
Morris, C. E. (2011a). Voltage-gated channel mechanosensitivity: fact or friction? Front. Physiol. 2:25. doi: 10.3389/fphys.2011.00025
Morris, C. E. (2011b). “Why are so many channels mechanosensitive?” in Cell Physiology Source Book, 4th Edn, ed. N. Sperelakis (Amsterdam: Elsevier), 493–505.
Morris, C. E. (2011c). “Pacemaker, potassium, calcium, sodium: stretch modulation of the voltage-gated channels,” in Cardiac Mechano-Electric Coupling and Arrhythmias: from Pipette to Patient, 2nd Edn, eds P. Kohl, F. Sachs, and M. Franz (Elsevier Saunders), 43–49.
Morris, C. E., and Homann, U. (2001). Cell surface area regulation and membrane tension. J. Membr. Biol. 179, 79–102.
Morris, C. E., and Juranka, P. F. (2007). Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys. J. 93, 822–833.
Morris, C. E., Juranka, P. F., Lin, W., Morris, T. J., and Laitko, U. (2006). Studying the mechanosensitivity of voltage-gated channels using oocyte patches. Methods Mol. Biol. 322, 315–329.
Muddana, H. S., Chiang, H. H., and Butler, P. J. (2012). Tuning membrane phase separation using nonlipid amphiphiles. Biophys. J. 102, 489–497.
Musters, R. J., Otten, E., Biegelmann, E., Bijvelt, J., Keijzer, J. J., Post, J. A., Op den Kamp, J. A., and Verkleij, A. J. (1993). Loss of asymmetric distribution of sarcolemmal phosphatidylethanolamine during simulated ischemia in the isolated neonatal rat cardiomyocyte. Circ. Res. 73, 514–523.
Nesterenko, V. V., Zygmunt, A. C., Rajamani, S., Belardinelli, L., and Antzelevitch, C. (2011). Mechanisms of atrial-selective block of sodium channel by ranolazine II. Insights from a mathematical model. Am. J. Physiol. Heart Circ. Physiol. 301, H1615–H1624.
Nikic, I., Merkler, D., Sorbara, C., Brinkoetter, M., Kreutzfeldt, M., Bareyre, F. M., Brück, W., Bishop, D., Misgeld, T., and Kerschensteiner, M. A. (2011). Reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 17, 495–499.
Novak, K. R., Nardelli, P., Cope, T. C., Filatov, G., Glass, J. D., Khan, J., and Rich, M. M. (2009). Inactivation of sodium channels underlies reversible neuropathy during critical illness in rats. J. Clin. Invest. 119, 1150–1158.
Phillips, R., Ursell, T., Wiggins, P., and Sens, P. (2009). Emerging roles for lipids in shaping membrane-protein function. Nature 459, 379–385.
Pieri, M., Carunchio, I., Curcio, L., Mercuri, N. B., and Zona, C. (2009). Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp. Neurol. 215, 368–379.
Rajendran, L., Knölker, H. J., and Simons, K. (2010). Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 9, 29–42.
Ritter, A. M., Martin, W. J., and Thorneloe, K. S. (2009). The voltage-gated sodium channel Nav1.9 is required for inflammation-based urinary bladder dysfunction. Neurosci. Lett. 452, 28–32.
Rusinova, R., Herold, K. F., Sanford, R. L., Greathouse, D. V., Hemmings, H. C. Jr., and Andersen, O. S. (2011). Thiazolidinedione insulin sensitizers alter lipid bilayer properties and voltage-dependent sodium channel function: implications for drug discovery. J. Gen. Physiol. 138, 249–270.
Schafer, D. P., Jha, S., Liu, F., Akella, T., McCullough, L. D., and Rasband, M. N. (2009). Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J. Neurosci. 29, 13242–13254.
Schlegel, R. A., and Williamson, P. (2001). Phosphatidylserine, a death knell. Cell Death Differ. 8, 551–563.
Schmidt, D., and MacKinnon, R. (2008). Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 19276–19281.
Schwarz, J. R., Glassmeier, G., Cooper, E. C., Kao, T. C., Nodera, H., Tabuena, D., Kaji, R., and Bostock, H. (2006). KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J. Physiol. (Lond.) 573, 17–34.
Schwarz, J. R., Reid, G., and Bostock, H. (1995). Action potentials and membrane currents in the human node of Ranvier. Pflügers Arch. 430, 283–292.
Shcherbatko, A., Ono, F., Mandel, G., and Brehm, P. (1999). Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys. J. 77, 1945–1959.
Sheetz, M. P., Sable, J. E., and Döbereiner, H. G. (2006). Continuous membrane cytoskeleton adhesion requires continuous accommodation to lipid and cytoskeleton dynamics. Annu. Rev. Biophys. Biomol. Struct. 35, 417–434.
Shevchenko, A., and Simons, K. (2010). Lipidomics: coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 11, 593–598.
Song, J. H., Huang, C. S., Nagata, K., Yeh, J. Z., and Narahashi, T. (1997). Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 282, 707–714.
Stys, P. K., Sontheimer, H., Ransom, B. R., and Waxman, S. G. (1993). Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc. Natl. Acad. Sci. U.S.A. 90, 6976–6980.
Sun, G. C., Werkman, T. R., and Wadman, W. J. (2006). Kinetic changes and modulation by carbamazepine on voltage-gated sodium channels in rat CA1 neurons after epilepsy. Acta Pharmacol. Sin. 27, 1537–1546.
Tabarean, I. V., Juranka, P., and Morris, C. E. (1999). Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys. J. 77, 758–774.
Taddese, A., and Bean, B. P. (2002). Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron 33, 587–600.
Tejwani, R. W., Davis, M. E., Anderson, B. D., and Stouch, T. R. (2011). Functional group dependence of solute partitioning to various locations within a DOPC bilayer: a comparison of molecular dynamics simulations with experiment. J. Pharm. Sci. 100, 2136–2146.
Underhill, S. M., and Goldberg, M. P. (2007). Hypoxic injury of isolated axons is independent of ionotropic glutamate receptors. Neurobiol. Dis. 25, 284–290.
van Genderen, H. O., Kenis, H., Hofstra, L., Narula, J., and Reutelingsperger, C. P. (2008). Extracellular annexin A5: functions of phosphatidylserine-binding and two-dimensional crystallization. Biochim. Biophys. Acta 1783, 953–963.
Villalba-Galea, C. A., Sandtner, W., Starace, D. M., and Bezanilla, F. (2008). S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. U.S.A. 105, 17600–17607.
Vucic, S., and Kiernan, M. C. (2010). Upregulation of persistent sodium conductances in familial ALS. J. Neurol. Neurosurg. Psychiatr. 81, 222–227.
Wang, J. A., Lin, W., Morris, T., Banderali, U., Juranka, P. F., and Morris, C. E. (2009). Membrane trauma and Na+ leak from Nav1.6 channels. Am. J. Physiol. Cell Physiol. 297, C823–C834.
Waxman, S. G. (2008). Mechanisms of disease: sodium channels and neuroprotection in multiple sclerosis-current status. Nat. Clin. Pract. Neurol. 4, 159–169.
Webb, J., Wu, F. F., and Cannon, S. C. (2009). Slow inactivation of the NaV1.4 sodium channel in mammalian cells is impeded by co-expression of the beta1 subunit. Pflugers Arch. 457, 1253–1263.
Weiss, S., Benoist, D., White, E., Teng, W., and Saint, D. A. (2010). Riluzole protects against cardiac ischaemia and reperfusion damage via block of the persistent sodium current. Br. J. Pharmacol. 160, 1072–1082.
Weston, R. M., Subasinghe, K. R., Staikopoulos, V., and Jarrott, B. (2009). Design and assessment of a potent sodium channel blocking derivative of mexiletine for minimizing experimental neuropathic pain in several rat models. Neurochem. Res. 34, 1816–1823.
Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D., and Smith, D. H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 21, 1923–1930.
Yuen, T. J., Browne, K. D., Iwata, A., and Smith, D. H. (2009). Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J. Neurosci. Res. 87, 3620–3625.
Zhang, J., Hadlock, T., Gent, A., and Strichartz, G. R. (2007). Tetracaine-membrane interactions: effects of lipid composition and phase on drug partitioning, location, and ionization. Biophys. J. 92, 3988–4001.
Keywords: traumatic brain injury, spinal, riluzole, ranolazine, simulation, modeling
Citation: Morris CE, Boucher P-A and Joós B (2012) Left-shifted Nav channels in injured bilayer: primary targets for neuroprotective Nav antagonists? Front. Pharmacol. 3:19. doi: 10.3389/fphar.2012.00019
Received: 03 October 2011; Accepted: 06 February 2012;
Published online: 23 February 2012.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Mirela Milescu, University of Missouri–Columbia, USAMark Rich, Wright State University, USA
Copyright: © 2012 Morris, Boucher and Joós. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Catherine E. Morris, Neurosciences, Ottawa Hospital Research Institute, 450 Smyth Rd, Ottawa, ON, K1H 8M5, Canada. e-mail: cmorris@uottawa.ca