 Jesmond Dalli
Jesmond Dalli- William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, London, UK
It is postulated that peptides derived from the N-terminal region of Annexin A1, a glucocorticoid-regulated 37-kDa protein, could act as biomimetics of the parent protein. However, recent evidence, amongst which the ability to interact with distinct receptors other then that described for Annexin A1, suggest that these peptides might fulfill other functions at variance to those reported for the parent protein. Here we tested the ability of peptide Ac2-26 to induce chemotaxis of human neutrophils, showing that this peptide can elicit responses comparable to those produced by the canonical activator formyl-Met-Leu-Phe (or FMLP). However, whilst disruption of the chemical gradient abolished the FMLP response, addition of peptide Ac2-26 in the top well of the chemotaxis chamber did not affect (10 μM) or augmented (at 30 μM) the neutrophil locomotion to the bottom well, as elicited by 10 μM peptide Ac2-26. Intriguingly, the sole addition of peptide Ac2-26 in the top wells produced a marked migration of neutrophils. A similar behavior was observed when human primary monocytes were used. Thus, peptide Ac2-26 is a genuine chemokinetic agent toward human blood leukocytes. Neutralization strategies indicated that engagement of either the GPCR termed FPR1 or its cognate receptor FPR2/ALX was sufficient to sustain peptide Ac2-26 induced neutrophil migration. Similarly, application of pharmacological inhibitors showed that cell locomotion to peptide Ac2-26 was mediated primarily by the ERK, but not the JNK and p38 pathways. In conclusion, we report here novel in vitro properties for peptide Ac2-26, promoting neutrophil and monocyte chemokinesis; a process that may contribute to accelerate the resolution phase of inflammation. We postulate that the generation of Annexin A1 N-terminal peptides at the site of inflammation may expedite the egress of migrated leukocytes thus promoting the return to homeostasis.
Introduction
Annexin A1 (AnxA1) is a glucocorticoid-regulated 37-kDa protein that exerts important actions on fundamental processes in inflammation. Highly abundant in myeloid cells, AnxA1 is rapidly mobilized upon cell activation (non-genomic externalization) or subsequent to de novo synthesis (genomic activation, e.g., after glucocorticoid treatment or pro-inflammatory cytokine application; Perretti and D’Acquisto, 2009). Once on the cell surface the protein is exposed to extracellular fluids and the in the presence of calcium undergoes structural re-organization, consequent to interaction with phospholipids via the core region of the protein (∼280 amino acid long), which leads to the exposure of the N-terminal region (∼50 amino acid long; Gerke et al., 2005). This conformational change is thought to lead to the interaction of the AnxA1 N-terminus with specific receptors (Hu et al., 2008). It is worth recalling here that both human recombinant AnxA1 and the peptide Ac2-26 exert anti-inflammatory and pro-resolving effects in a variety of experimental models (Perretti and Dalli, 2009). Moreover, AnxA1 null mice are viable and do not have an appreciable phenotype unless challenged with inflammatory stimuli whereby a stronger and often prolonged reaction is then observed (Yang et al., 2004; Damazo et al., 2006; Babbin et al., 2008).
In addition to representing the pharmacophore of the protein affording interaction with counter-ligands, the N-terminus is also a highly regulated region. It can undergo phosphorylation on specific Tyrosine or Serine sites, a pre-requisite for secretion in certain cell types, or can be cleaved by serine proteases (Solito et al., 2006; Vong et al., 2007; D’Acquisto et al., 2008). In fact, both elastase and proteinase 3 have been shown to cleave at specific sites within the AnxA1 N-terminal region (Rescher et al., 2006; Vong et al., 2007) and it is plausible that AnxA1 can be a substrate for many other proteases. Cleavage of this protein has also been reported in human inflammatory samples including bronchoalveolar lavage fluids (Tsao et al., 1998) and blister exudates (Perretti et al., 1999) suggesting that this process is not an in vitro artifact but of biological significance.
Gerke and colleagues published a break-through study showing that peptides derived from the AnxA1 N-terminus activate the formyl peptide receptor type 1 (FPR1; Walther et al., 2000). Subsequently, we showed that full-length AnxA1 can bind and activate a related receptor termed FPR2/ALX (the lipoxin A4 receptor). This interaction was of physiological relevance since a direct association between AnxA1 and FPR2/ALX could be shown in human and mouse activated neutrophils (Perretti et al., 2002). Subsequent observations indicated that peptide Ac2-26 activated all three of the human formyl peptide receptors (Ernst et al., 2004). Parallel studies from our group showed that whilst peptide Ac2-26 could bind both FPR1 and FPR2/ALX, the full-length protein displayed specific binding only toward FPR2/ALX (Hayhoe et al., 2006).
It is believed that AnxA1 cleavage can represent a catabolic event, terminating the “AnxA1 mediated anti-inflammatory tone” (Vong et al., 2007). This hypothesis is backed by the observation that a cleavage-resistant species of the protein afforded higher potency in inflammatory settings (Pederzoli-Ribeil et al., 2010). In addition to this, however, it is also possible that AnxA1 cleavage could release N-terminal derived sequences that would then interact with FPR1 eliciting chemotactic responses. This could be particularly true outside the vasculature were inflammatory exudates rich in serine proteases can cleave AnxA1 generating such peptides.
To address this hypothesis in the present study we assessed (i) the chemotactic response of human neutrophils (PMN) elicited by peptide Ac2-26, (ii) the involvement of FPR1 and/or FPR2/ALX in the observed effects, and (iii) the intracellular pathways engaged by this peptide.
Materials and Methods
Neutrophil and Monocyte Isolation
Experiments using healthy volunteers were approved by the local research ethics committee (P/00/029 East London and The City Local Research Ethics Committee 1). Informed written consent was provided according to the Declaration of Helsinki. Blood was collected into 3.2% sodium citrate and diluted 1:1 in RPMI-1640 before separation through a double-density gradient using Histopaque 10771 and 11191 (Sigma-Aldrich, Poole, UK). After centrifugation at 1340 rpm for 30 min polymorphonuclear (PMN) and mononuclear cells were collected from lower and upper layers respectively. Contaminating erythrocytes in the PMN cell suspension were removed by hypotonic lysis. Cells were finally washed and resuspended in RPMI-1640. Monocytes were isolated from the mononuclear cell suspension by immunomagnetic positive selection using the EasySep® Human CD14 Positive Selection Kit (STEMCELL Technologies, Grenoble, France).
Chemotaxis Assay
For neutrophil chemotaxis, cells were resuspended at a concentration of 4 × 106 cells/ml in RPMI-1640 containing 0.1% BSA. The assay was performed using 3-μm pore size ChemoTx™ 96 well plates (Neuro Probe Inc., Gaithersburg, USA) by adding chemotactic stimuli to the bottom wells, and placing 25 μl of cell suspension on top of the filter. Plates were incubated for 90 min at 37°C with 5% CO2. After this period filters were washed with PBS and plates centrifuged at 1200 rpm for 30 s. Migrated cells were analyzed by taking 20-μl from the bottom wells and incubating with Alamar Blue (Invitrogen Ltd. Paisley, UK) and comparing with a standard curve constructed with known cell numbers. Plates were read after 3 h in a fluorescence spectrophotometer at EX560-EM5 90 nm. For monocyte chemotaxis, the assay was performed following the same protocol outlined above incubating cells for 120 min to allow migration.
In some cases, compounds (see below) were added to freshly prepared neutrophils for 10 min at 37°C prior to cell addition to the top well of the chemotactic plates. Anti-FPR1 or anti-FPR2 antibodies were handled in a similar manner.
Pharmacological Tools
Peptide Ac2-26 (Ac-AMVSEFLKQAWFIENEEQEYVQTVK; Tocris, Bristol, UK) or FMLP (Sigma-Aldrich, Dorset, UK) were used as chemoattractants. In initial experiments, also serum amyloid protein A (SAA; Peprotech, London, UK) was used, since it induces chemotaxis via FPR2 (He et al., 2003; Ye et al., 2009; Dufton et al., 2010). Anti-FPR1 or anti-FPR2 monoclonal antibodies (10 μg/ml final concentration in either case) were obtained from R&D System (Abingdon, UK) or Genovac (Brussels, Belgium), respectively).
Inhibitors of mitogen-activated phosphokinase were tested to learn about the signaling pathway(s) activated by peptide Ac2-26 in these experimental settings. The compounds PD98059 (ERK inhibitor; Maiti et al., 2008), SB203580 (p38 inhibitor; Maiti et al., 2008), and SP600125 (JNK Inhibitor – SP600125; Tokuda et al., 2003) were obtained from Cell Signaling Technologies (Hertfordshire, UK) and used in the concentration range of 3–30 μM as based on published data (Gallicchio et al., 2009). In selected experiments the ERK inhibitor (10 μM) was tested against peptide Ac2-26 validating blockade of phosphor-ERK accumulation by Western blotting using a described methodology (Hayhoe et al., 2006).
Statistics
Results are presented as Mean ± SEM of n experiments performed in triplicate or quadruplicate. Statistical differences were determined by analysis-of-variance followed by Dunnet’s or Bonferroni’s post hoc tests, taking a P-value less than 0.05 as significant.
Results
In initial experiments we tested the effect of peptide Ac2-26 on human PMN chemotaxis in comparison to the FPR1 agonist FMLP and the FPR2/ALX agonist SAA. Figure 1 demonstrates that peptide Ac2-26 can elicit a chemotactic response in the same order as FMLP, with a near maximal effect at 10 μM, which corresponds to 3 μg/ml final concentration. The positive control FMLP induced a marked response between 1 and 100 nM concentration range, and similarly SAA, applied in the bottom well of the chamber at 1–30 μM range (Figure 1). Thus, peptide Ac2-26 causes PMN chemotaxis to a comparable level as produced by FMLP or SAA.
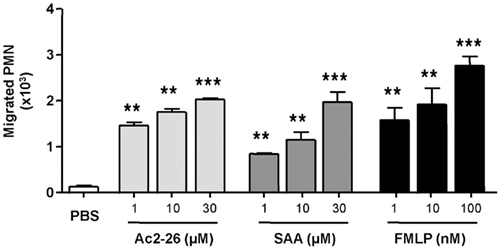
Figure 1. Peptide Ac2-26 provokes migration of human PMN: comparison to FMLP and SAA. Neutrophils isolated from peripheral blood of healthy volunteers were resuspended at 4 × 106 cells/ml and chemotaxis to peptide Ac2-26 (1–30 μM), SAA (1–30 μM), or FMLP (1–100 nM) was assessed over a 90-min period. Results are ±SEM from three to four cell preparations (**P < 0.01, ***P < 0.001 vs. PBS group).
Next we tested whether peptide Ac2-26 elicited genuine PMN chemotaxis or a chemokinetic response. To this end, we used 10 μM peptide Ac2-26 as the chemoattractant concentration, and added in the top wells, vehicle, or Ac2-26 from 1 to 30 μM. Figure 2 illustrates these results, where Ac2-26 provoked ∼4-fold increase in PMN migration above control wells, and this effect was not modified by adding 1 μM Ac2-26 in the top well. However, when 10 or 30 μM peptide Ac2-26 were added to the top wells, the overall PMN migration was markedly augmented yielding, in essence, a doubling effect as compared to 10 μM Ac2-26 in the bottom well alone (Figure 2A). Of interest, the chemotactic response elicited by 1 nM FMLP was abrogated by adding a higher concentration of the tri-peptide in the top well (Figure 2B). Finally, when peptide Ac2-26 was tested in the top well only, it did not alter the extent of PMN migration above control responses, except for the 30-μM concentration that produced a remarkable neutrophil mobilization (Figure 2C).
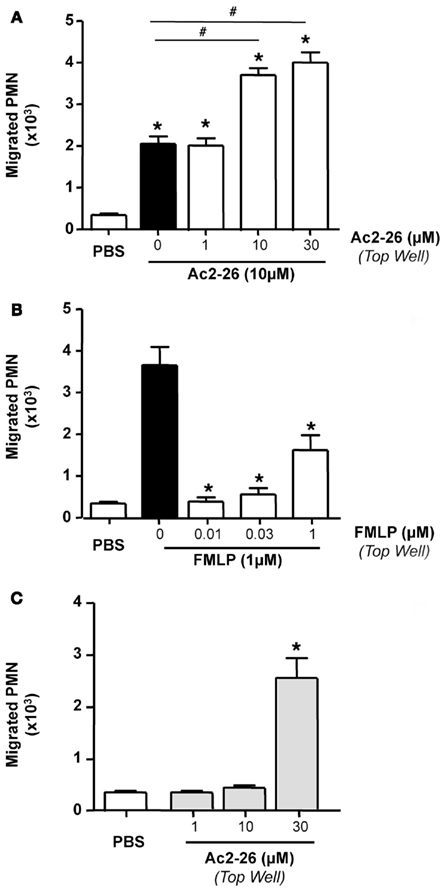
Figure 2. Peptide Ac2-26, but not FMLP, induces PMN chemokinesis. Peripheral blood neutrophils were incubated with either (A) peptide Ac2-26 (1–30 μM) or PBS prior to loading on to 96 well ChemoTx™ plate where the bottom wells were either loaded with peptide Ac2–26 (10 μM) or (B) FMLP (0.01–1 μM) prior to loading on to a chemotaxis plate in the presence of 1 nM FMLP. (C) Neutrophils were incubated with peptide Ac2-26 (1–30 μM) prior to loading on to the chemotaxis plate. Results are ±SEM from three to four cell preparations [(A,B) *P < 0.05 vs. stimulus alone (dose 0 group); (C) *P < 0.05 vs. PBS].
The concentrations of peptide Ac2-26 used in the experiments are congruent with its apparent binding affinities for human FPR1 and FPR2/ALX (Walther et al., 2000; Perretti et al., 2002; Hayhoe et al., 2006). Next, we tested if FPR1 or FPR2/ALX was responsible for the observed effect, using again 10 μM peptide Ac2-26 to induce human PMN migration. To this end, we used specific antibodies against either of the human receptors. Here the cells were incubated with 10 μg/ml (final concentration) of either antibody. Blockade of either formyl peptide receptor was able to abrogate the chemotactic effect elicited by the Ac2-26 (Figure 3A).
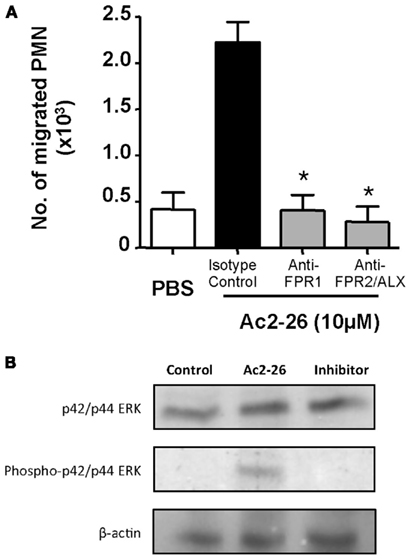
Figure 3. Both FPR1 and FPR2/ALX mediate peptide Ac2-26 induced PMN migration. (A) Peripheral blood neutrophils were pre-incubated for 5 min at RT with anti-FPR1, FPR2/ALX, or an Isotype control antibody, prior to loading on to a 96 well ChemoTx™ plate in the presence of 10 μM peptide Ac2-26. Chemotaxis was evaluated after a 90-min incubation at 37°C and 5% CO2. Results are ±SEM from three distinct cell preparations (*P < 0.05 vs. isotype control). (B) Ac2-26 (10 μM) was added to peripheral blood neutrophils in the presence of absence of PD98059 (ERK inhibitor; 10 μM) for 5 min prior to protein extraction and assessment of phospho- and total ERK isoforms, as well as beta-actin. Blots are representative of triplicate analyses conducted with two distinct neutrophil preparations.
Peptide Ac2-26 engagement of human FPR1 and FPR2/ALX has been shown to activate the MAPK signaling in an uneven way, with phosphorylation of ERK but not JNK or p38 (Walther et al., 2000; Ernst et al., 2004; Hayhoe et al., 2006). Thus, we next evaluated whether this observation also held true in our experimental conditions showing that incubation of freshly prepared PMN with Ac2-26 led to ERK phosphorylation, an effect that could be effectively blocked upon incubation with an ERK specific inhibitor (Figure 3B). We next assessed the whether this pathway was also involved in mediating the chemokinetic effects exerted by the Ac2-26 peptide. Here we treated freshly prepared neutrophils with selective pharmacological inhibitors over an extended concentration range in order to account for any unspecific effects of the inhibitors. Pre-treatment of neutrophils with the ERK inhibitor, compound PD98059, abolished the cellular response to peptide Ac2-26, even at the lowest concentration tested of 3 μM (Figure 4A). Intermediate effects were achieved with the p38 inhibitor which was mildly effective at concentrations of 3 and 10 μM, yet it only significantly attenuated peptide Ac2-26 mediated effects at the highest concentration of 30 μM (Figure 4B). On the other hand pre-treatment of PMN with the JNK inhibitor, did not have any influence on the extent of PMN migration (Figure 4C).
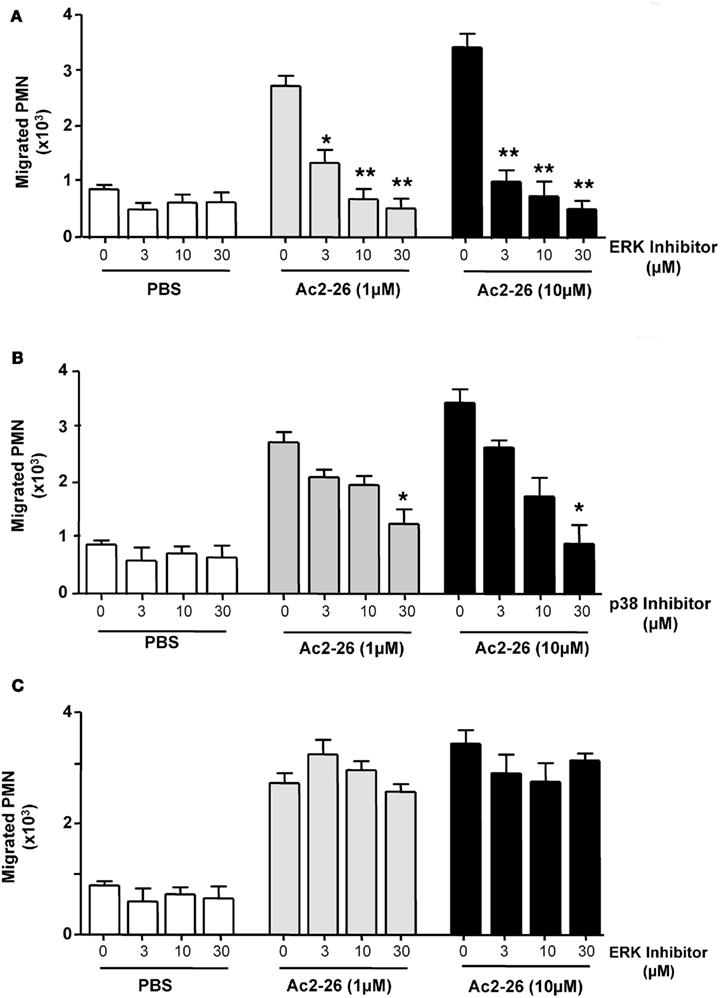
Figure 4. Peptide Ac2-26 induced PMN migration relies primarily on the ERK signaling pathway. Neutrophils were pre-incubated for 15 min at 37°C with either (A) PD98059 (ERK inhibitor; 3–30 μM), (B) SB203580 (p38 inhibitor; 3–30 μM), or (C) SP600125 (JNK inhibitor; 3–30 μM) prior to loading on to a 96 well ChemoTx™ plate in the presence of PBS or peptide Ac2-26 (3–30 μM). Chemotaxis was evaluated after a 90-min incubation at 37°C and 5% CO2. Results are ±SEM from three distinct cell preparations [*P < 0.05, **P < 0.01 vs. stimulus alone (dose 0 group)].
We next extended our observations to another cell type that is also important in the regulation of the inflammatory response, the monocyte. Here we tested whether peptide Ac2-26 elicited chemotactic/chemokinetic responses in this cell type, using a concentration range of 1–30 μM and adding peptide Ac2-26 either to the bottom wells of the chemotactic plates, or in the top wells. As seen in Figure 5, addition of the peptide to either the top or bottom compartment of the chemotaxis plate elicited a marked response as measured by number of cells mobilized to the bottom well. We observed an increment of ∼10-fold over controls when either the chemotactic (Figure 5A-addition of the peptide to the bottom well) or the chemokinetic (Figure 5B-addition of the peptide to the top well) effects were tested.
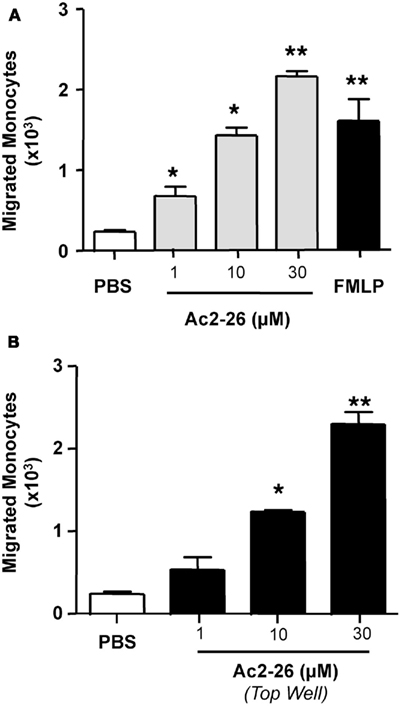
Figure 5. Peptide Ac2-26 induced human monocytes chemokinesis. Monocytes were isolated from peripheral blood of healthy volunteers as indicated in the Section “Materials and Methods” and resuspended at 4 × 106 cells/ml. (A) Chemotaxis toward peptide Ac2-26 (1–30 μM) was evaluated after a 90-min incubation at 37°C and 5% CO2. FMLP (1 nM) was used as positive control. (B) Chemokinesis induced by peptide Ac2-26 was assessed by incubating the cells with the peptide prior to loading on to the top well of the ChemoTx™ plate, the number of migrated cells to the bottom chamber was evaluated after 90 min incubation at 37°C and 5% CO2. Results are ±SEM of three to four distinct monocyte preparations. (*P < 0.05, **P < 0.01 vs. PBS group).
Discussion
We demonstrate here that the AnxA1-derived peptide Ac2-26 exerts chemokinetic, rather than chemotactic, effects on human PMN by acting through either FPR1 or FPR2/ALX and engaging primarily the ERK signaling pathway.
The seminal work of Gerke’s lab over a decade ago indicated that peptides derived from the AnxA1 N-terminal could activate human PMN by engaging the FMLP receptor and in this manner attenuate cell responses such as trans-endothelial migration (Walther et al., 2000). For a long time peptide Ac2-26 has been used as an AnxA1 surrogate in view of its ability to replicate the anti-inflammatory (Perretti and Dalli, 2009) and pro-resolving properties (Maderna et al., 2005, 2010; Scannell et al., 2007) of the native protein. Therefore, clarification of the receptor mechanism engaged by peptide Ac2-26 is of importance as it may have bearing on the pharmacological exploitation of this line of research for the development of novel anti-inflammatory therapeutics.
The model that emerges is that peptide Ac2-26 can activate all three human formyl peptide receptors, promoting calcium fluxes, and cell locomotion (Ernst et al., 2004). Such an effect may be part of a reparatory process as demonstrated with intestinal epithelial cells (Babbin et al., 2006) where addition of the peptide engaged FPRs to activate the cytoskeletal machinery favoring cell migration and repair of the epithelial monolayer. It was therefore surprising when we conducted binding studies and demonstrated that whilst full-length AnxA1 bound, and activated, the human FPR2/ALX, this protein was unable to bind human FPR1 (Hayhoe et al., 2006). In the same study we confirmed the activation of mitogen-activated phosphate kinases as an early post-receptor event, with phosphorylation of ERK but not p38 or JNK, at least within the time frame of our analyses (30 min). It is now clear that in human monocytes peptide Ac2-26 can activate the JAK/STAT/SOCS signaling requiring ≥60 min incubation (Pupjalis et al., 2011). How can we explain this disparate receptor engagement between AnxA1 and its N-terminal pharmacophore?
One reason behind this dichotomy of behavior could be that AnxA1 exerts its inhibitory role in the inflamed vasculature exclusively through FPR2/ALX, whilst the peptide – which might be generated at the site of inflammation or tissue damage – could exert chemokinetics or chemoattractant properties via FPR1. We discussed earlier the generation of N-terminal derived peptides from AnxA1 in the inflammatory exudates by the action of serine proteases and other proteolytic activities. In particular, PMN-derived elastase and proteinase 3 have been advocated as pivotal enzymes that could truncate the N-terminal region of AnxA1, which is exposed following calcium binding to the protein (Gerke and Moss, 2002; Gerke et al., 2005), leading to the release of biologically active peptides. Formation or release of AnxA1-derived peptides in an inflammatory exudate could contribute to resolution (Perretti and D’Acquisto, 2009) by promoting phagocytosis of apoptotic PMN by macrophages (Scannell et al., 2007; Maderna et al., 2010) and dampening inflammatory monocyte activation (Pupjalis et al., 2011). We propose that the data presented here, would argue for another pro-resolving property of peptide Ac2-26; the removal of immune cells (PMN and monocytes) from the site of inflammation favoring their exit back into the blood stream or through the lymphatic circulation and thus promoting tissue restoration and regain of its pre-inflammatory status. If confirmed in in vivo settings, this biological property of peptide Ac2-26 would explain: (i) the apparent discrepancy between AnxA1 and peptide Ac2-26 binding to human FPR1 and FPR2/ALX and (ii) the need for generating these short peptides, whereby AnxA1 would act as a pro-drug. In this respect recent observations made using a super-AnxA1, resistant to serine protease cleavage, would seem to argue against the second hypothesis, since this protein displayed higher anti-inflammatory activity then native AnxA1 (Pederzoli-Ribeil et al., 2010). However, here one needs to make a distinction between the anti-inflammatory properties exerted by AnxA1 (and super-AnxA1) in the vasculature and/or in the initial phases of acute inflammation, and those that may be operative during the onset of resolution, especially at the level of the exudate/tissue site, where these peptides could be generated.
Walther et al. (2000) reported that ∼2 nM circulating AnxA1 can be measured in normal plasma that could augment by 10- to 100-fold in inflammatory settings, yielding concentration that can activate FPR2/ALX (Perretti et al., 2002; Hayhoe et al., 2006). At very high concentrations, (100–500 μM), likely not physiological, peptide Ac2-26 activates human PMN with production of reactive oxygen species (Walther et al., 2000) whilst inhibitory effects are predominant within the 10-μM range (Perretti et al., 1995; Walther et al., 2000). Therefore in our chemotaxis experiments, we selected concentrations of peptide Ac2-26 in the 1- to 30-μM range since we deem that concentrations higher that these would be of little biological significance. This especially in consideration of the fact that the molar ratio AnxA1:peptide Ac2-26 is 1:1, therefore it is really impossible to envisage pathophysiological settings where AnxA1 concentrations ≥30 μM could be reached.
Peptide Ac2-26 promoted a clear chemokinetic response in human PMN being, in this way, clearly distinguishable from FMLP – which produced the expected chemotactic effect (Ye et al., 2009) hence abolished when the chemical gradient was disrupted. In line with our initial data on early signaling responses (Hayhoe et al., 2006), peptide Ac2-26 induced PMN migration was highly reliant on ERK phosphorylation but not p38 or JNK phosphorylation. We reason that the mild effect of the p38 inhibitor, though significant, was solely detected at 30 μM concentration alluding to a possible non-selective response since at concentrations above 10 μM this inhibitor has been shown to also inhibit ERK phosphorylation (Lian et al., 1999). Conversely, the ERK specific inhibitor markedly affected peptide Ac2-26 elicited PMN migration even at the 3-μM concentration. The JNK inhibitor acted as negative control, since it was unable to modulate this cellular response to peptide Ac2-26 to any appreciable extent, at any of the concentration tested, in line with previous observations (Walther et al., 2000; Ernst et al., 2004; Hayhoe et al., 2006).
Finally, the issue of the receptor engaged by peptide Ac2-26 in these experimental settings was addressed. Human PMN express FPR1 and FPR2/ALX but not FPR3, which is more relevant to macrophages and dendritic cells (Ye et al., 2009). Neutralizing antibodies to FPR1 (Yazid et al., 2010) or FPR2/ALX (Hayhoe et al., 2006) abrogated the cellular response to peptide Ac2-26. This is reminiscent of the data produced in human PMN under flow upon incubation with cromones, though in that study each antibody produced a partial effect and the combination gave total abrogation (Yazid et al., 2010). In the current experiments peptide Ac2-26 could engage either FPR1 or FPR2/ALX to promote PMN migration and each receptor seemed sufficient to govern this biological response. There are no doubts that human FPR1 is a chemotactic receptor and the same applies to human FPR2/ALX (Le et al., 2005). Our data suggests that simultaneous functional activation is important in eliciting the observed responses, an observation that favors a mechanism involving cross-talk between the two receptors, potentially resulting from heterodimerization, upon peptide Ac2-26 application. Future studies will clarify the potential molecular interlink between FPR1 and FPR2/ALX that these results indicate. It is noteworthy however that peptide Ac2-26 can bind and activate FPR1 and FPR2/ALX with similar affinities (Ernst et al., 2004; Hayhoe et al., 2006).
In conclusion, we provide in vitro evidence that peptide Ac2-26 can promote locomotion of human primary cells, PMN, and monocytes. Such an effect is at least in part is due to a chemokinetic rather than chemotactic effect elicited by this peptide. This suggests that generation of AnxA1 N-terminal derived peptides during the resolution phase of the inflammatory response may play an important role in expediting the removal of blood-borne cells from the tissue. This in turn would favor the restoration of homeostasis. As often the case, these novel results incite new questions that must be addressed in future studies, both in terms of in vivo relevance as well as in the remit of molecular pharmacology, hence the potential modulation of FPR1 and FPR2/ALX localization upon peptide Ac2-26 application.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by the Wellcome Trust (programme 086867/Z/08). This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institutes of Health Research.
References
Babbin, B. A., Laukoetter, M. G., Nava, P., Koch, S., Lee, W. Y., Capaldo, C. T., Peatman, E., Severson, E. A., Flower, R. J., Perretti, M., Parkos, C. A., and Nusrat, A. (2008). Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J. Immunol. 181, 5035–5044.
Babbin, B. A., Lee, W. Y., Parkos, C. A., Winfree, L. M., Akyildiz, A., Perretti, M., and Nusrat, A. (2006). Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J. Biol. Chem. 281, 19588–19599.
D’Acquisto, F., Perretti, M., and Flower, R. J. (2008). Annexin-A1: a pivotal regulator of the innate and adaptive immune systems. Br. J. Pharmacol. 155, 152–169.
Damazo, A. S., Yona, S., Flower, R. J., Perretti, M., and Oliani, S. M. (2006). Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J. Immunol. 176, 4410–4418.
Dufton, N., Hannon, R., Brancaleone, V., Dalli, J., Patel, H. B., Gray, M., D’Acquisto, F., Buckingham, J. C., Perretti, M., and Flower, R. J. (2010). Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 184, 2611–2619.
Ernst, S., Lange, C., Wilbers, A., Goebeler, V., Gerke, V., and Rescher, U. (2004). An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. 172, 7669–7676.
Gallicchio, M., Benetti, E., Rosa, A. C., and Fantozzi, R. (2009). Tachykinin receptor modulation of cyclooxygenase-2 expression in human polymorphonuclear leucocytes. Br. J. Pharmacol. 156, 486–496.
Gerke, V., Creutz, C. E., and Moss, S. E. (2005). Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6, 449–461.
Hayhoe, R. P., Kamal, A. M., Solito, E., Flower, R. J., Cooper, D., and Perretti, M. (2006). Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood 107, 2123–2130.
He, R., Sang, H., and Ye, R. D. (2003). Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 101, 1572–1581.
Hu, N. J., Bradshaw, J., Lauter, H., Buckingham, J., Solito, E., and Hofmann, A. (2008). Membrane-induced folding and structure of membrane-bound annexin A1 N-terminal peptides: implications for annexin-induced membrane aggregation. Biophys. J. 94, 1773–1781.
Le, Y., Ye, R. D., Gong, W., Li, J., Iribarren, P., and Wang, J. M. (2005). Identification of functional domains in the formyl peptide receptor-like 1 for agonist-induced cell chemotaxis. FEBS J. 272, 769–778.
Lian, J. P., Huang, R., Robinson, D., and Badwey, J. A. (1999). Activation of p90RSK and cAMP response element binding protein in stimulated neutrophils: novel effects of the pyridinyl imidazole SB 203580 on activation of the extracellular signal-regulated kinase cascade. J. Immunol. 163, 4527–4536.
Maderna, P., Cottell, D. C., Toivonen, T., Dufton, N., Dalli, J., Perretti, M., and Godson, C. (2010). FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 24, 4240–4249.
Maderna, P., Yona, S., Perretti, M., and Godson, C. (2005). Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2-26. J. Immunol. 174, 3727–3733.
Maiti, A., Hait, N. C., and Beckman, M. J. (2008). Extracellular calcium-sensing receptor activation induces vitamin D receptor levels in proximal kidney HK-2G cells by a mechanism that requires phosphorylation of p38alpha MAPK. J. Biol. Chem. 283, 175–183.
Pederzoli-Ribeil, M., Maione, F., Cooper, D., Al-Kashi, A., Dalli, J., Perretti, M., and D’Acquisto, F. (2010). Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood 116, 4288–4296.
Perretti, M., Chiang, N., La, M., Fierro, I. M., Marullo, S., Getting, S. J., Solito, E., and Serhan, C. N. (2002). Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A(4) receptor. Nat. Med. 8, 1296–1302.
Perretti, M., and D’Acquisto, F. (2009). Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 9, 62–70.
Perretti, M., and Dalli, J. (2009). Exploiting the Annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br. J. Pharmacol. 158, 936–946.
Perretti, M., Wheller, S. K., Choudhry, Q., Croxtall, J. C., and Flower, R. J. (1995). Selective inhibition of neutrophil function by a peptide derived from lipocortin 1 N-terminus. Biochem. Pharmacol. 50, 1037–1042.
Perretti, M., Wheller, S. K., Flower, R. J., Wahid, S., and Pitzalis, C. (1999). Modulation of cellular annexin I in human leukocytes infiltrating DTH skin reactions. J. Leukoc. Biol. 65, 583–589.
Pupjalis, D., Goetsch, J., Kottas, D. J., Gerke, V., and Rescher, U. (2011). Annexin A1 released from apoptotic cells acts through formyl peptide receptors to dampen inflammatory monocyte activation via JAK/STAT/SOCS signalling. EMBO Mol. Med. 3, 102–114.
Rescher, U., Goebeler, V., Wilbers, A., and Gerke, V. (2006). Proteolytic cleavage of annexin 1 by human leukocyte elastase. Biochim. Biophys. Acta 1763, 1320–1324.
Scannell, M., Flanagan, M. B., deStefani, A., Wynne, K. J., Cagney, G., Godson, C., and Maderna, P. (2007). Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 178, 4595–4605.
Solito, E., Christian, H. C., Festa, M., Mulla, A., Tierney, T., Flower, R. J., and Buckingham, J. C. (2006). Post-translational modification plays an essential role in the translocation of annexin A1 from the cytoplasm to the cell surface. FASEB J. 20, 1498–1500.
Tokuda, H., Niwa, M., Ito, H., Oiso, Y., Kato, K., and Kozawa, O. (2003). Involvement of stress-activated protein kinase/c-Jun N-terminal kinase in endothelin-1-induced heat shock protein 27 in osteoblasts. Eur. J. Endocrinol. 149, 239–245.
Tsao, F. H., Meyer, K. C., Chen, X., Rosenthal, N. S., and Hu, J. (1998). Degradation of annexin I in bronchoalveolar lavage fluid from patients with cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 18, 120–128.
Vong, L., D’Acquisto, F., Pederzoli-Ribeil, M., Lavagno, L., Flower, R. J., Witko-Sarsat, V., and Perretti, M. (2007). Annexin 1 cleavage in activated neutrophils: a pivotal role for proteinase 3. J. Biol. Chem. 282, 29998–30004.
Walther, A., Riehemann, K., and Gerke, V. (2000). A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 5, 831–840.
Yang, Y. H., Morand, E. F., Getting, S. J., Paul-Clark, M. J., Liu, D. L., Yona, S., Hannon, R., Buckingham, J. C., Perretti, M., and Flower, R. J. (2004). Modulation of inflammation and response to dexamethasone by annexin-1 in antigen-induced arthritis. Arthritis Rheum. 50, 976–984.
Yazid, S., Leoni, G., Getting, S. J., Cooper, D., Solito, E., Perretti, M., and Flower, R. J. (2010). Antiallergic cromones inhibit neutrophil recruitment onto vascular endothelium via annexin-A1 mobilization. Arterioscler. Thromb. Vasc. Biol. 30, 1718–1724.
Keywords: inflammation, annexin A1, resolution, neutrophil, migration
Citation: Dalli J, Montero-Melendez T, McArthur S and Perretti M (2012) Annexin A1 N-terminal derived peptide Ac2-26 exerts chemokinetic effects on human neutrophils. Front. Pharmacol. 3:28. doi: 10.3389/fphar.2012.00028
Received: 24 November 2011; Paper pending published: 14 December 2011;
Accepted: 13 February 2012; Published online: 28 February 2012.
Edited by:
Orina Belton, Universtiy College Dublin, IrelandReviewed by:
Derek W. Gilroy, University College London, UKDerek Brazil, Queen’s University Belfast, UK
Copyright: © 2012 Dalli, Montero-Melendez, McArthur and Perretti. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mauro Perretti, William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. e-mail: m.perretti@qmul.ac.uk