 Xianglan Yao1
Xianglan Yao1
- 1 Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, Bethesda, MD, USA
- 2 Cognosci, Inc., Research Triangle Park, NC, USA
New treatments are needed for severe asthmatics to improve disease control and avoid severe toxicities associated with oral corticosteroids. We have used a murine model of house dust mite (HDM)-induced asthma to identify steroid-unresponsive genes that might represent targets for new therapeutic approaches for severe asthma. This strategy identified apolipoprotein E as a steroid-unresponsive gene with increased mRNA expression in the lungs of HDM-challenged mice. Furthermore, apolipoprotein E functioned as an endogenous negative regulator of airway hyperreactivity and goblet cell hyperplasia in experimental HDM-induced asthma. The ability of apolipoprotein E, which is expressed by lung macrophages, to attenuate AHR, and goblet cell hyperplasia is mediated by low density lipoprotein (LDL) receptors expressed by airway epithelial cells. Consistent with this, administration of an apolipoprotein E mimetic peptide, corresponding to amino acids 130–149 of the LDL receptor-binding domain of the holo-apoE protein, significantly reduced AHR and goblet cell hyperplasia in HDM-challenged apoE−/− mice. These findings identified the apolipoprotein E – LDL receptor pathway as a new druggable target for asthma that can be activated by administration of apoE-mimetic peptides. Similarly, apolipoprotein A-I may have therapeutic potential in asthma based upon its anti-inflammatory, anti-oxidative, and anti-fibrotic properties. Furthermore, administration of apolipoprotein A-I mimetic peptides has attenuated airway inflammation, airway remodeling, and airway hyperreactivity in murine models of experimental asthma. Thus, site-directed delivery of inhaled apolipoprotein E or apolipoprotein A-I mimetic peptides may represent novel treatment approaches that can be developed for asthma, including severe disease.
Why are New Treatments Needed for Patients with Severe Asthma?
Severe asthma is a major public health and economic problem. The World Health Organization1 estimates that approximately 235 million people worldwide suffer from asthma. Although less than 5% of asthmatics have severe disease, they account for approximately 50% of all the direct and indirect costs associated with asthma (Braman, 2006). The definition of severe, refractory asthma, as determined by the American Thoracic Society, is the need for either continuous or near continuous oral corticosteroids for more than 50% of the year and/or high-dose inhaled corticosteroids to achieve control to a level similar to patients with mild or moderate persistent asthma (American Thoracic Society, 2000). The definition of severe, refractory asthma also requires two or more of the following; daily treatment with a controller medication in addition to inhaled corticosteroids, daily or near daily need for short-acting β-agonist to control asthma symptoms, persistent airflow obstruction (FEV1 < 80% predicted), one or more urgent care visits for asthma per year, three of more oral steroid “bursts” per year, prompt decline in asthma control with ≤25% reduction in oral or inhaled corticosteroid dose, or near fatal asthma event in past (American Thoracic Society, 2000). Consistent with this definition, severe asthmatics, as compared to those with mild or moderate asthma, are more likely to be older with a longer duration of disease, have more daily symptoms, have increased rates of urgent health care utilization, have more sinusitis and pneumonia, are less atopic, and have lower lung function with increased gas trapping and bronchodilator reversibility (Moore et al., 2007; Sorkness et al., 2008).
Limited treatment options are available for severe asthmatics. The most recent NHLBI National Asthma Education and Prevention Expert Panel 3 Report recommends that severe asthmatics be treated with high doses of an inhaled corticosteroid plus a long-acting beta2-agonist (LABA) and oral corticosteroids. Omalizumab, an anti-IgE monoclonal antibody, can reduce the frequency of asthma exacerbations and the dose of inhaled corticosteroids in severe, allergic asthmatics whose serum IgE levels fall within the target range (Fanta, 2009). Limitations regarding omalizumab are that it requires parenteral administration, is expensive and can infrequently be associated with anaphylaxis in 0.1% of patients (Strunk and Bloomberg, 2006). Bronchial thermoplasty is a new treatment option that requires several bronchoscopies to deliver thermal energy that ablates airway smooth muscle (Castro et al., 2010). The U.S. Food and Drug Administration has recently approved this procedure for adult asthmatics who have persistent symptoms despite inhaled corticosteroids plus LABAs. There, however, remains an unfulfilled need for the development of additional new treatment options for severe asthma that improve disease control, are cost–effective and can spare patients from the potentially devastating side-effects related to chronic corticosteroid therapy, but do not require parenteral administration or an invasive procedure.
Bench-to-Beside: Using a Murine Model of House Dust Mite-Induced Asthma to Identify Steroid-Unresponsive Genes that may Modulate Asthma Pathogenesis and Disease Severity
Although corticosteroids can improve symptom control in severe asthmatics, chronic administration can be associated with debilitating side-effects, which include diabetes, aseptic bone necrosis, weight gain, hypertension, and impaired host defense. Therefore, severe asthmatics who require chronic oral corticosteroids frequently seek an alternative medication that will allow a reduction, or ideally a discontinuation, of chronic oral corticosteroid therapy.
Furthermore, patients with severe asthma or asthmatics who smoke cigarettes may be poorly responsive or completely resistant to corticosteroids based upon the presence of glucocorticoid resistance due to a decrease in histone deacetylase 2 activity or reduced nuclear translocation of glucocorticoid receptors (Barnes, 2011). We reasoned that there might exist differentially expressed genes in asthmatic lungs that are up-regulated in the setting of asthma and remain persistently up-regulated despite treatment with corticosteroids. Furthermore, we considered that these steroid-unresponsive genes might modulate the pathogenesis of asthma and represent novel therapeutic targets for the development of new asthma medications.
To identify these steroid-unresponsive genes, we utilized a murine model of house dust mite (HDM)-induced airway inflammation, airway remodeling, and airway hyperreactivity. We selected HDM because it is a common aeroallergen that is an environmental trigger and risk factor for the development of persistent asthma (Sears et al., 2003; Johnson et al., 2004). HDM is a complex mixture of proteins and lipopolysaccharide (LPS) that induces airway inflammation via allergic and non-allergic mechanisms (Thomas et al., 2002; De Alba et al., 2009; Lambrecht and Hammad, 2009). We proposed that genes could be identified whose expression is up-regulated in the lung in response to HDM challenge and remain greater than twofold up-regulated despite treatment with corticosteroids as compared to control saline-challenged mice. This scenario simulates a patient who presents to their health care provider with persistent asthma symptoms despite treatment with high doses of inhaled or oral corticosteroids. Furthermore, we reasoned that these corticosteroid–unresponsive genes might provide mechanistic insights into the pathogenesis of asthma and potentially represent new therapeutic targets that could be relevant for the treatment of severe asthma.
Wild-type A/J mice, which have enhanced susceptibility to the induction of asthma and allergen-mediated airway hyperreactivity, received saline, or HDM by nasal administration 5 days per week for 6 weeks (Levitt and Mitzner, 1988; Wills-Karp and Ewart, 1997; Finkelman et al., 2010). Dexamethasone (1 mg/kg) or saline was administered via an osmotic mini-pump during weeks four through six to simulate treatment of established asthma (Yao et al., 2010). We found that corticosteroid treatment dissociated airway hyperreactivity from airway inflammation and goblet cell hyperplasia in this experimental murine model of HDM-induced asthma (Yao et al., 2010). While corticosteroid administration was very effective at suppressing airway inflammation and goblet cell hyperplasia, it resulted in only a small, but statistically significant, decrease in airway hyperreactivity that remained increased as compared to control mice. This finding is consistent with prior murine and human studies, which showed that airway inflammation and airway hyperreactivity, are mediated by distinct pathways that may be uncoupled (Crimi et al., 1998; Swedin et al., 2009). Thus, administration of dexamethasone to HDM-challenged mice generated a steroid-unresponsive model of airway hyperreactivity, which we reasoned could be utilized to identify novel genes and pathways that modulate AHR in asthma.
Using this model system, genome-wide expression profiling of the asthmatic lung transcriptome identified 68 candidate steroid-unresponsive genes, defined as having a greater than twofold increase in expression in response to HDM challenge and remained greater than twofold increased despite treatment with corticosteroids. From these candidate genes, we used real-time quantitative RT-PCR to confirm that lung mRNA levels of apolipoprotein E (APOE) were significantly increased in HDM-challenged mice (Yao et al., 2010). Furthermore, APOE mRNA levels remained greater than twofold elevated in the lungs of HDM-challenged mice that received corticosteroid treatment as compared to control mice, thereby identifying APOE as a steroid-unresponsive gene. Therefore, we hypothesized that apolipoprotein E might modulate the pathogenesis of asthma.
Why Investigate the Role of Apolipoprotein E in the Pathogenesis of Asthma?
Apolipoprotein E (apoE) is a 34-kDa protein that plays a key role in cholesterol transport and lipoprotein metabolism. An important function of apolipoprotein E is to facilitate the high-affinity binding of lipoproteins, such as very low density lipoproteins (VLDL), a subclass of high density lipoproteins (HDL) and chylomicron remnants, to the low density lipoprotein (LDL) receptor (Mahley, 1988). Binding of lipoprotein particles to the LDL receptor facilitates the transport of cholesterol into cells via the internalization of lipoprotein particles. This is termed receptor-mediated endocytosis (Goldstein and Brown, 2009). Consistent with this function, elevations in plasma cholesterol levels are seen in individuals who lack either apoE (familial apoE deficiency) or have genetic defects in the LDL receptor (familial hypercholesterolemia; Schaefer et al., 1986; Goldstein and Brown, 2009; Pendse et al., 2009).
Apolipoprotein E is synthesized by most organs, including the lung. Macrophages, including CD68+ alveolar macrophages, express apoE (Mahley, 1988; Yao et al., 2010). Apolipoprotein E has been shown to modulate lung development in mice, as apoE knockout mice have diminished developmental alveologenesis, high airways resistance and more rapid loss of lung recoil with aging (Massaro and Massaro, 2008).
In the liver, hepatic parenchymal cells are the primary cellular source of apoE, where it is secreted as a component of VLDL particles (Mahley, 1988). ApoE is also expressed by astrocytes in the brain and is found in cerebrospinal fluid, as well as in smooth muscle cells where it may modulate cellular proliferation and differentiation (Mahley, 1988). Alveolar type I cells in the rat have also been reported to express apolipoprotein E (Chen et al., 2006).
Although the primary function of apoE is to facilitate lipid transport into cells by receptor-mediated endocytosis mediated by the LDL receptor, it is has been recognized that apoE modulates a variety of additional important biological functions. The human APOE gene has three common polymorphic alleles that result in proteins that have different amino acid sequences at two sites. ApoE3 has a cysteine at amino acid 112 and an arginine at amino acid 158, whereas apoE2 has cysteines at both sites while apoE4 has arginines at both sites (Mahley et al., 2009). ε3 is the most frequent APOE allele (65–70%), while ε2 (5–10%) and ε4 (15–20%) are less common. The APOE ε4 allele has been identified a major genetic risk factor for the development of late-onset Alzheimer’s disease (AD) via its role in modulating the production, clearance or aggregation of amyloid-β peptides or, alternatively, by directly modulating lipid metabolism in the brain or by regulating synaptic plasticity or repair (Bu, 2009). The APOE ε4 allele has also been implicated as a risk factor for other neurological disorders, including Parkinson’s disease and multiple sclerosis, which suggests that it may facilitate accelerated neurodegeneration (Bu, 2009; Mahley et al., 2009).
Apolipoprotein E also modulates adaptive immune responses and host susceptibility to infection. Apolipoprotein E facilitates the presentation of lipid antigens to antigen presenting cells, such as dendritic cells and B cells via a process that involves the endocytosis of apoE–lipid–antigen complexes via the LDLR (van den Elzen et al., 2005; Allan et al., 2009). Individuals with the APOE ε4/ε4 genotype have an accelerated disease course with progression to death as compared to individuals with the APOE ε3/ε3 genotype in the setting of human immunodeficiency virus (HIV) infection. In particular, the APOE ε4/ε4 genotype may serve as a determinant of HIV–AIDS progression as a consequence of enhanced HIV fusion and cell entry (Burt et al., 2008). ApoE is also required for the assembly of hepatitis C virions (HCV) and subsequent cellular infection, which occurs via an interaction with the HCV non-structural protein, 5A (NS5A; Chang et al., 2007; Cun et al., 2010). ApoE has also been implicated in modulating the pathogenesis of ocular infections caused by herpes simplex virus-1 (Hill et al., 2007).
Apolipoprotein E Functions as an Endogenous Negative Regulator of Airway Hyperreactivity and Goblet Cell Hyperplasia in Experimental House Dust Mite-Induced Asthma
Based upon the finding that apolipoprotein E has multiple functions, we hypothesized that it may modulate the pathogenesis or disease severity in asthma. To investigate the role of apoE in asthma, we compared the asthmatic phenotype of HDM-challenged apoE knockout mice (apoE−/−) with that of wild-type mice (Yao et al., 2010). Experimental asthma was induced by nasal administration of HDM (25 μg), 5 days per week for 5 weeks. HDM-challenged apoE−/− had significant increases in both AHR, goblet cell hyperplasia, and mRNA levels of the MUC5AC mucin gene and CLCA3 (Gob-5), a calcium-activated chloride channel that has been implicated in the development of goblet cell hyperplasia and mucus production (Long et al., 2006). This demonstrates that apolipoprotein E functions as an endogenous negative regulator of airway hyperreactivity and goblet cell hyperplasia in experimental HDM-induced asthma.
We next investigated whether the LDL receptor, which is the major receptor for apoE, is required for apoE to negatively regulate AHR and goblet cell hyperplasia in experimental HDM-induced asthma (Yao et al., 2010). We used confocal immunofluorescence microscopy to demonstrate that the LDL receptor is expressed on ciliated airway epithelial cells where it may interact with apoE that is released from alveolar macrophages. Next, we hypothesized that if the LDL receptor functions as the relevant receptor in the lung for apoE, then the phenotype of HDM-challenged LDLR−/− mice should recapitulate that of HDM-challenged apoE−/− mice. Consistent with this hypothesis, we found that HDM-challenged LDLR−/− mice had a phenotype of enhanced AHR and goblet cell hyperplasia. Interestingly, type 2 rhinovirus has also been found to bind to LDL receptors expressed by human tracheal epithelial cells, which up-regulates the expression of both pro-inflammatory cytokines and the LDL receptor (Suzuki et al., 2001). This suggests a potential role for LDL receptors in the regulation of airway inflammatory responses to viral infections, which are the most common environmental trigger for asthma exacerbations (Wark and Gibson, 2006).
The Therapeutic Potential of Apolipoprotein E Mimetic Peptides
The finding that apoE functions as an endogenous negative regulator of airway hyperreactivity and goblet cell hyperplasia, suggested that apoE-mimetic peptides might be utilized experimentally to rescue the phenotype of HDM-challenged apoE−/− mice. Furthermore, we hypothesized that apoE-mimetic peptides might be considered as a new asthma therapy that targets a pathway that is distinct from current asthma treatments. The development of apoE-mimetic peptides for treatment of disease has its origins in research aimed at defining the biology of APOE genotypes, which are best known to alter the course and severity of AD (Corder et al., 1993). The precise mechanism by which proteins encoded by the APOE4 gene increase susceptibility to inflammation is being studied. Part of the challenge is to understand whether the APOE4 gene and its apoE4 protein products are providing a dominant gain of negative function or, alternatively, that they represent the disease-associated loss of a protective function. Human clinical data shows heterozygous APOE3/4 patients have onset of disease and severities of AD that are intermediate between the most severe APOE4/4 homozygous patients and those with homozygous APOE3/3 genotypes (Corder et al., 1993). Since these findings are consistent with either gain of negative functions or loss of protective functions, we employed APOE knockout mice and transgenic, targeted replacement mice in which the protein-encoding exons for the mouse apoE gene were replaced with those encoding human APOE3 or human APOE4 genes as tools to decipher the influence of apolipoprotein E protein isoforms on inflammation (Lynch et al., 2003). Using LPS treatment of peritoneal macrophages to stimulate release of TNF as a biomarker of inflammation, we found that cells from APOE4/4 animals had significantly greater TNF levels than their APOE3/3 counterparts. However, the TNF levels from APOE 0/0 (knockout) macrophages were not significantly different from APOE4/4 TNF levels. These findings suggest that apoE4 proteins had significantly reduced abilities to suppress inflammation. To further characterize these anti-inflammatory activities of apoE proteins, we mated APOE3/3 and APOE0/0 (knockout) mice to generate APOE3/0 heterozygous mice and found their LPS-stimulated TNF levels to be intermediate between those from APOE3/3 and APOE4/4 mice. This finding strongly supports the concept that the apoE3 protein directly functions as an anti-inflammatory agent, that the anti-inflammatory effect depends upon the concentration of apoE protein available, and that the apoE4 protein is a much less potent anti-inflammatory agent.
The development of apoE-mimetic peptides was initially driven by early studies that mapped the region necessary for receptor-binding to residues 130–160 of the holo-apoE-protein (Lalazar et al., 1988; Mahley et al., 2009). Reasoning that peptides spanning this region might also bind to apoE receptors, peptides were synthesized that corresponded to residues 130–149 of the holo-apoE protein (aka. COG130 = acetyl-TEELRVRLASHLRKLRKRLL-amide; Figure 1; Laskowitz et al., 2001). Similar to the holo-apoE protein, COG130 and COG133, an amino-terminally truncated peptide spanning residues 133–149 (acetyl-LRVRLASHLRKLRKRLL-amide), displayed significant anti-inflammatory activity as measured by reduced TNF release from LPS-stimulated BV2 microglial cells. Subsequent studies showed that intravenous administration of COG133 significantly reduced TNF and IL-6 levels in the blood and brains of LPS-stimulated wild-type mice (Lynch et al., 2003). More recent unpublished studies show that COG133 reduces TNF release in wild-type (mouse apoE alone), APOE0/0 (knockout) mice, transgenic replacement APOE3/3 and APOE4/4 mice. From these results, we hypothesized that holo-apoE proteins and apoE-mimetic peptides, such as COG133, must stimulate an anti-inflammatory mechanism within macrophages and other cells that display apoE receptors.
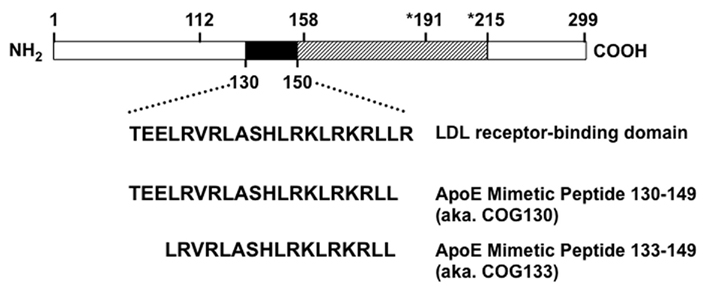
Figure 1. Structure of apolipoprotein E and apoE-mimetic peptides. Mature holo-apoE is a 299 amino acid protein with polymorphisms at residues 112 and 158 that define the different protein isotypes: apoE2 (cys112, cys158), apoE3 (cys112, arg158), and apoE4 (arg112, arg158). The LDL receptor-binding domain includes residues 130–160. ApoE-mimetic peptides derive from the region including amino acids 130–150 as shown by the filled box. The hatched box extends past the thrombin cleavage site at position 191 to the beginning of the lipid-binding domain at position 215. The amino-terminal portion through residues 1–191 forms a 4-helix anti-parallel bundle followed by a hinge region that extends to residue 215. The carboxy terminal region from 215 to 299 defines the largely unstructured lipid-binding domain with a major lipid-binding region from residues 244 to 272. The apoE-mimetic peptide spanning residues 130–149 is also known as COG130, while the peptide spanning residues 133–149 is also known as COG133.
To define the mechanism of this anti-inflammatory action, we performed a series of studies aimed at finding binding partners for apoE and apoE-mimetic peptides (Christensen et al., 2011). While it is well known that the holo-apoE protein and COG130 can bind to one or more of a family of structurally related apoE receptor proteins and extracellular matrix molecules, no studies have reported intracellular binding partners for them. To find these binding partners, we modified COG133 to contain an amino-terminal biotin moiety and used this biotinylated-COG133 as a hook to catch proteins that bound to the biotin–apoE–mimetic peptide. After addition of biotin-COG133 to mouse microglial BV2 cells for 30 min, lysates containing putative COG133-binding protein complexes were isolated by affinity chromatography on streptavidin–agarose columns. After extensive washing of the column, the resin with bound peptides and protein complexes was boiled in Laemmli sample buffer and electrophoresed on polyacrylamide gels. Silver staining revealed two distinct proteins that eluted from the resin that were submitted for characterization by mass spectrometry (ms). Results of the ms/ms characterization were compared to the protein database and a match was found with the protein known as SET, which is also known as inhibitor number 2 of protein phosphatase 2A (I2PP2A). Additional affinity purifications followed by Western blotting showed that the 39- and 25-kDa proteins were immunoreactive with antibodies directed to the SET protein. Similar binding studies showed that recombinant SET and purified holo-apoE also co-immunoprecipitated. Based upon these results, we hypothesized that COG133 could bind and antagonize SET, thereby permitting re-activation of PP2A to increase cellular phosphatase activity levels. Proof supporting this hypothesis was shown by measurements of phosphorylated-p38 MAP kinase (p-p38) where levels of p-p38 increased with LPS treatments, but then decreased with LPS plus COG133 treatments. Since LPS-mediated activation of toll-Like receptor 4 (TLR4) leads to increased cellular levels of p-p38 and eventually to increased release of TNF and nitric oxide, COG133-mediated reduction of p-p38 levels should lead to decreased levels of TNF and nitric oxide, a result which was experimentally verified in numerous cell-based and whole animal studies (unpublished observation; Christensen et al., 2011). These data support a novel mechanism where apoE-mimetic peptides or holo-apoE bind to SET, thereby permitting reactivation of PP2A that then leads to decreased signal transduction in pathways associated with generation of an inflammatory response.
Administration of an Apolipoprotein E Mimetic Peptide Attenuates the Induction of Experimental House Dust Mite-Induced Asthma
Based upon the anti-inflammatory properties of apoE-mimetic peptides, we hypothesized that they might be utilized to modify disease severity in asthma. Furthermore, we reasoned that apoE-mimetic peptides could be used to rescue the phenotype of enhanced airway hyperreactivity and goblet cell hyperplasia in HDM-challenged apoE−/− mice and thereby confirm the role of apoE in the pathogenesis of asthma. We elected to utilize an apoE(130–149) mimetic peptide that corresponded to amino acids 130–149 of the LDL receptor-binding domain of the native apoE holoprotein. Systemic administration of the apoE(130–149) mimetic peptide coincident with daily nasal HDM challenges successfully attenuated the increased AHR and goblet cell hyperplasia, as well as the increased expression of MUC5AC and CLCA3 genes (Yao et al., 2010). Furthermore, systemic administration of the apoE(130–149) mimetic peptide also attenuated multiple indices of airway inflammation, such as bronchoalveolar lavage fluid eosinophilia, total serum IgE levels, and mRNA levels of Th2 (IL-4, IL-13) and Th17 (IL-17A) cytokines, as well as chemokines for eosinophils (CCL7, CCL11, and CCL17). Expression of products of alternatively activated macrophages, such as chitinase 3-like 3, arginase 1, and Fizz1 (resistin-like α) were also reduced.
In addition, we reasoned that if apoE negatively regulates HDM-induced increases in AHR and goblet cell hyperplasia via the LDL receptor, then HDM-challenged LDLR−/− mice should be resistant to rescue by the apoE(130–149) mimetic peptide. Consistent with this, systemic administration of the apoE(130–149) mimetic peptide to HDM-challenged LDLR−/− mice did not attenuate AHR, airway inflammation or goblet cell hyperplasia (Yao et al., 2010). Therefore, we showed that systemic administration of the apoE(130–149) mimetic peptide attenuated airway inflammation, AHR and goblet cell hyperplasia in HDM-challenged apoE−/−, but not LDLR−/− mice, which is consistent with the conclusion that the effects of apoE in murine HDM-induced experimental asthma are mediated via the LDL receptor.
These findings demonstrate that an apoE – LDL receptor pathway in the lung functions as an endogenous negative regulator of HDM-induced airway hyperreactivity and goblet cell hyperplasia in a murine model of experimental asthma (Yao et al., 2010). Lung apoE mRNA levels are increased following HDM challenge and remain persistently elevated despite corticosteroid treatment (Yao et al., 2010). Apolipoprotein E may be released from alveolar macrophages into epithelial lining fluid, where it can interact with LDL receptors, which are expressed by airway epithelial cells, to attenuate airway hyperreactivity and goblet cell hyperplasia (Figure 2). Our laboratory is currently actively investigating the mechanism by which this is mediated. Furthermore, administration of an apoE-mimetic peptide [apoE(130–149)] to HDM-challenged apoE−/− mice not only rescued the phenotype of enhanced AHR and goblet cell hyperplasia, but also attenuated the induction of HDM-induced eosinophilic airway inflammation. This suggests that the apoE – LDL receptor pathway represents a novel druggable target for the development of new treatment approaches for asthma, especially those with severe disease. This pathway can be activated by administration of apoE-mimetic peptides that correspond to the LDL receptor-binding domain of holo-apolipoprotein E. Thus, our data from an experimental murine model of HDM-induced asthma support the concept of developing inhaled apoE-mimetic peptides, such as the apoE(130–149) peptide, as a potential new therapeutic approach for asthma.
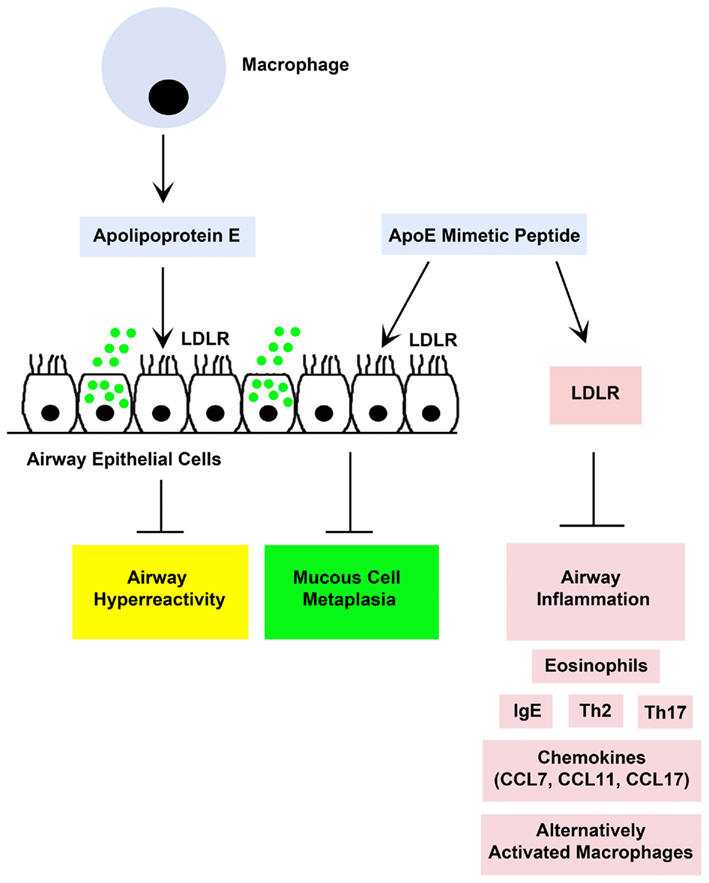
Figure 2. A proposed model by which endogenous holo-apolipoprotein E (apoE) and exogenously administered apolipoprotein E mimetic peptides [e.g., apoE(130–149)] modulate experimental house dust mite (HDM)-induced asthma (Yao et al., 2010). Both endogenous apoE, which is produced by lung macrophages, and an exogenously administered apoE(130–49) mimetic peptide attenuated airway hyperreactivity and mucous cell metaplasia via an interaction with low density lipoprotein receptors (LDLR), such as those expressed by ciliated airway epithelial cells. Although endogenous apoE did not modulate HDM-induced airway inflammation, administration of the apoE(130–149) mimetic peptide attenuated airway inflammation in apoE knockout mice, but not in LDLR knockout mice, which suggests that the anti-inflammatory effects of the apoE(130–149) mimetic peptide are mediated via a LDLR-dependent mechanism.
The Therapeutic Potential of Apolipoprotein A-I Mimetic Peptides
The success of the apoE(130–149) mimetic peptide suggested to us that apolipoprotein A-I mimetic peptides might be similarly effective at attenuating the manifestations of asthma. Apolipoprotein A-I (apoA-I) accounts for approximately 70% of the protein component of HDL (Navab et al., 2011). The main function of HDL is to mediate reverse cholesterol transport out of peripheral cells, such as macrophages, and thereby protect against atherosclerosis (Navab et al., 2011; Osei-Hwedieh et al., 2011). Cholesterol is transported out of cells via the ATP-binding cassette transporter 1 (ABCA1), as well as ABCG1 and SR-BI (Osei-Hwedieh et al., 2011). HDL, however, has additional functions in addition to reverse cholesterol transport, which include anti-inflammatory, anti-oxidant, anti-fibrotic, and anti-thrombotic properties (Navab et al., 2011). HDL is also protective against the effects of bacterial LPS and increases nitric oxide production. Consistent with this, apoA-I has been found to have protective effects in the lung as apoA-I knockout mice under basal conditions have an increase in lung inflammation, oxidative stress, and collagen deposition as compared to wild-type mice (Wang et al., 2010).
ApoA-I mimetic peptides have been developed as a therapeutic approach based upon their ability to recapitulate the key biologic functions of holo-apolipoprotein A-I and HDL, which include cholesterol transport as well as anti-inflammatory and anti-oxidative properties (Navab et al., 2011; Osei-Hwedieh et al., 2011). ApoA-I mimetic peptides were first designed to understand the structure of apolipoproteins. One of the first such peptides developed, 18A (DWLKAFYDKVAEKLKEAF), was synthesized to have a similar distribution of polar and hydrophobic amino acids as apoA-I and was found to form an amphipathic helix, which is now known to be a common structural motif of apolipoproteins (Anantharamaiah et al., 1985; Segrest et al., 1994). Apolipoproteins typically have a tandem array of amphipathic helices, which enable them when on the surface of lipoproteins to bind to lipids. These helices are oriented parallel to the surface of the phospholipid monolayer on lipoproteins, with the hydrophobic side facing inward toward the lipids, whereas the polar side faces outward toward the aqueous environment (Segrest et al., 1994). ApoA-I mimetic peptides were first considered as possible therapeutic agents when it was shown in clinical trials that four to five weekly intravenous infusion of the full-length apoA-I protein complexed with phospholipid, which was designed to simulate the structure and composition of nascent HDL, can significantly reduce atherosclerotic plaque in patients with acute coronary syndrome (Nissen et al., 2003; Tardif et al., 2007). One difficulty with this approach, however, is the relatively large doses of apoA-I needed, approximately 40 mg/Kg, which makes this type of treatment prohibitively expensive and has generated interest in the use of short synthetic apoA-I mimetic peptides as an alternative (Remaley et al., 2008).
Besides serving as tools for understanding the structure of apoA-I, synthetic peptide mimics of apoA-I have also been shown to share some of the same biological properties of the full-length protein (D’Souza et al., 2010). One of the best studied apoA-I mimetic peptides is the 4F peptide (Navab et al., 2002). It is similar in sequence to the 18A peptide but contains 4 phenylalanine substitutions, and hence its name, in its hydrophobic face. Because of its relatively high hydrophobicity, the increase in phenylalanine content was found to increase the ability of this peptide to bind to oxidized lipids, a known anti-atherogenic function of HDL (Anantharamaiah et al., 2007). A version of the peptide when synthesized with D-amino acids called D-4F was found to be partially orally available and to reduce atherosclerosis in animal models (Navab et al., 2002). Both oral D-4F and an intravenous preparation of the peptide made with L-amino acids, L4F, have been tested in Phase I clinical trials and have been shown to be safe, although there was limited effect on the functional properties of HDL from the treated patients (Bloedon et al., 2008; Watson et al., 2011).
The ability of HDL to remove excess cholesterol from peripheral cells and deliver it to the liver for excretion by the reverse cholesterol transport pathway is believed to be one of the main anti-atherogenic functions of HDL (Kellner-Weibel and de la Llera-Moya, 2011). This process begins when apoA-I, by acting like a protein detergent, removes phospholipid and cholesterol from a lipid microdomain in the plasma membrane created by the ABCA1 transporter (Nofer and Remaley, 2005). The ABCA1 transporter is believed to be a phospholipid flippase and is the defective gene in Tangier disease, which is a disorder associated with very low HDL-C levels due to the hypercatabolism of apoA-I. Normally when apoA-I acquires phospholipid by the ABCA1 transporter, it becomes too large to be catabolized by the kidney and also becomes competent for effluxing cholesterol from cells by additional mechanisms, such as by the ABCG1 transporter and by SR-BI, as well as by a passive aqueous diffusion process (Kellner-Weibel and de la Llera-Moya, 2011).
Short synthetic peptides containing amphipathic helices can also promote lipid efflux by the ABCA1 transporter (Remaley et al., 2003). One such peptide is the 5A peptide (Figure 3; Sethi et al., 2008). It is a bihelical peptide (DWLKAFYDKVAEKLKEAF-P-DWAKAAYDKAAEKAKEAA); the first helix is identical to 18A, whereas the second helix is a modified 18A peptide, which contains 5 alanine substitutions and hence its name. The two helices are joined together by proline, which confers a beta turn and is often found between the 10 different helices in the full-length apoA-I protein. Alanine, which is only slightly hydrophobic, was substituted for more hydrophobic amino acids in the hydrophobic face of the second helix of 5A to reduce its lipid-binding affinity, which increases the specificity of the peptide for cholesterol efflux by the ABCA1 transporter (Sethi et al., 2008). The 5A peptide and several other apolipoprotein mimetic peptides that promote cholesterol efflux are now actively being investigated as possible therapeutic agents for the treatment of atherosclerosis (Sethi et al., 2007; Murphy et al., 2008; Amar et al., 2010; Tabet et al., 2010; Osei-Hwedieh et al., 2011).
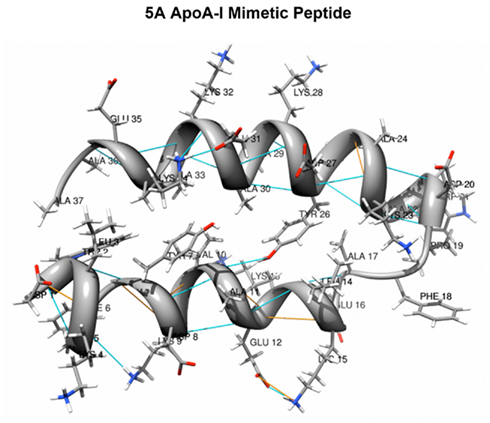
Figure 3. 5A Apolipoprotein A-I mimetic peptide. The 5A peptide is a 37 amino acid long bihelical peptide, containing a proline that links two amphipathic helices. 5 Alanine substitutions are made in the hydrophobic face of the second helix to reduce its lipid-binding affinity (courtesy of Alexander Andrianov, Ph.D. and adapted with permission from Yao et al., 2011b).
The 5A Apolipoprotein A-I Mimetic Peptide Attenuates the Induction of Experimental House Dust Mite-Induced Asthma
We hypothesized that the anti-inflammatory, anti-oxidant, and anti-fibrotic properties of apoA-I might be utilized therapeutically for the treatment of asthma. Therefore, we assessed whether systemic administration of the 5A apoA-I mimetic peptide could attenuate the induction of HDM-induced asthma in wild-type A/J mice. Systemic administration of the 5A apoA-I mimetic to HDM-challenged wild-type A/J mice inhibited the cardinal manifestations of asthma, including airway inflammation, airway remodeling, and airway hyperreactivity (Figure 4; Yao et al., 2011a). In particular, HDM-challenged mice that received the 5A apoA-I mimetic peptide had a significant reduction in the numbers of bronchoalveolar lavage fluid inflammatory cells, which included eosinophils, lymphocytes, and neutrophils, as well as the number of lung alternatively activated macrophages and the expression of Th2 (IL-4, IL-5, IL-13) and Th17 (IL-17A) cytokines. The expression of lung CC chemokines (CCL7, CCL11, CCL17, and CCL24) that promote T cell and eosinophil chemotaxis were also reduced. Administration of the 5A apoA-I mimetic peptide also attenuated airway remodeling responses, including goblet cell hyperplasia and expression of collagen genes, Cola1a1 and Col3a1.
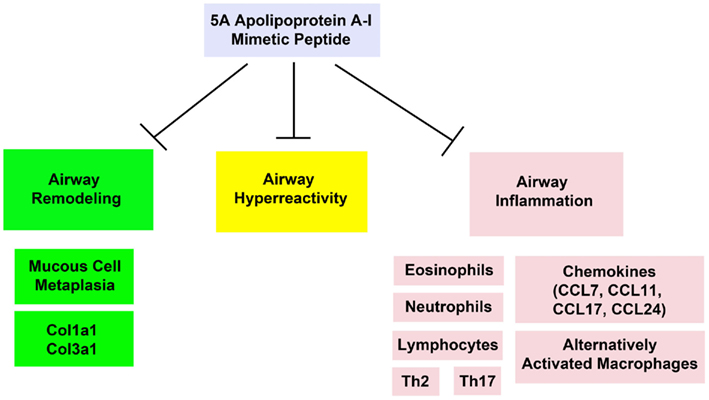
Figure 4. Exogenous administration of the 5A apolipoprotein A-I mimetic peptide attenuates the induction of airway remodeling, airway hyperreactivity, and airway inflammation in a murine model of house dust mite-induced asthma (Yao et al., 2011a).
D-4F, another amphipathic, α-helical apoA-I mimetic peptide has also been reported to attenuate the manifestations of ovalbumin (OVA)-induced asthma in an experimental murine model (Nandedkar et al., 2011). In this study, nasal administration of the D-4F apoA-I mimetic peptide to treat OVA-challenged mice decreased airway inflammation, as evidenced by a reduction in bronchoalveolar lavage fluid eosinophils and eosinophil peroxidase activity. Total serum IgE and levels of plasma pro-inflammatory HDL were decreased in mice that received D-4F treatment. D-4F also reduced airway hyperreactivity and airway remodeling responses as indicated by a reduction in peri-alveolar and peri-vascular collagen deposition. The beneficial effects of D-4F in experimental OVA-induced asthma may be mediated by a reduction in oxidative stress (Nandedkar et al., 2011).
Taken together, the ability of the 5A and D-4F apoA-I mimetic peptides to attenuate the manifestations of experimental HDM- and OVA-induced asthma in murine models suggests that administration of apoA-I mimetic peptides might represent a novel treatment approach for asthmatic patients.
Can Inhaled Apolipoprotein Mimetic Peptides be Developed into a Novel Treatment Approach for Asthma?
Data generated from pre-clinical murine models suggest that site-directed delivery of inhaled apoE or apoA-I mimetic peptides may represent novel therapeutic approaches for asthma. This concept is based upon the ability of apoE and apoA-I mimetic peptides to prevent the induction of experimental asthma in murine models. The effectiveness of this approach, however, will ultimately require confirmation in future clinical studies of asthmatic patients, including those with severe disease. Furthermore, we hypothesize that if efficacious, the beneficial effects of apoE and apoA-I mimetic peptides might be most helpful for patients with severe disease, especially if the apoE or apoA-I mimetic peptides can be shown to be synergistic with corticosteroids and thereby have a steroid-sparing effect, or ideally, permit the discontinuation of corticosteroids. For example, administration of apolipoprotein mimetic peptides might be used synergistically with corticosteroids to attenuate persistent airway hyperreactivity, especially when airway hyperreactivity does not respond to corticosteroid treatment. The clinical setting where this could be especially relevant is in individuals with a pauci-granulocytic sub-phenotype of severe asthma, based upon the absence of elevated eosinophils or neutrophils in induced sputum samples. Patients with a pauci-granulocytic phenotype comprise approximately 35% of severe asthmatics and would be anticipated to be poorly responsive to the anti-inflammatory effects of corticosteroids (Simpson et al., 2008). In addition, apolipoprotein mimetic peptides might also be effective in attenuating the induction of eosinophilic and/or neutrophilic airway inflammation. Establishing whether apoE or apoA-I mimetic peptides are effective for the treatment of severe asthma or specific severe asthma phenotypes, however, will require data from future clinical trials of asthmatic patients.
Based upon the data generated in our murine studies, we propose that the apolipoprotein mimetic peptides be investigated in clinical trials of human subjects to assess its efficacy and safety. Furthermore, we propose that the apolipoprotein mimetic peptides be administered via an inhalational approach, which has the advantage of directly targeting the peptide to the airway epithelium, where, in the case of the apoE(130–149) mimetic peptide, may facilitate its interaction with LDL receptors to thereby attenuate the pathogenesis of asthma. Similarly, an inhaled apolipoprotein mimetic peptide would be easy to administer and thereby obviate the need for alternative treatment options that require either parenteral administration or an invasive procedure. If shown to be safe and efficacious in asthmatic patients, apolipoprotein mimetic peptides could potentially enter clinical practice as novel treatment for asthma that has a mechanism of action that is distinct from currently available therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, NIH, Bethesda, MD, USA.
Footnote
References
Allan, L. L., Hoefl, K., Zheng, D. J., Chung, B. K., Kozak, F. K., Tan, R., and van den Elzen, P. (2009). Apolipoprotein-mediated lipid antigen presentation in B cells provides a pathway for innate help by NKT cells. Blood 114, 2411–2416.
Amar, M. J., D’Souza, W., Turner, S., Demosky, S., Sviridov, D., Stonik, J., Luchoomun, J., Voogt, J., Hellerstein, M., and Remaley, A. T. (2010). 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J. Pharmacol. Exp. Ther. 334, 634–641.
American Thoracic Society. (2000). Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. Am. J. Respir. Crit. Care Med. 162, 2341–2351.
Anantharamaiah, G. M., Jones, J. L., Brouillette, C. G., Schmidt, C. F., Chung, B. H., Hughes, T. A., Bhown, A. S., and Segrest, J. P. (1985). Studies of synthetic peptide analogs of the amphipathic helix. Structure of complexes with dimyristoyl phosphatidylcholine. J. Biol. Chem. 260, 10248–10255.
Anantharamaiah, G. M., Mishra, V. K., Garber, D. W., Datta, G., Handattu, S. P., Palgunachari, M. N., Chaddha, M., Navab, M., Reddy, S. T., Segrest, J. P., and Fogelman, A. M. (2007). Structural requirements for antioxidative and anti-inflammatory properties of apolipoprotein A-I mimetic peptides. J. Lipid Res. 48, 1915–1923.
Bloedon, L. T., Dunbar, R., Duffy, D., Pinell-Salles, P., Norris, R., DeGroot, B. J., Movva, R., Navab, M., Fogelman, A. M., and Rader, D. J. (2008). Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J. Lipid Res. 49, 1344–1352.
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344.
Burt, T. D., Agan, B. K., Marconi, V. C., He, W., Kulkarni, H., Mold, J. E., Cavrois, M., Huang, Y., Mahley, R. W., Dolan, M. J., McCune, J. M., and Ahuja, S. K. (2008). Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc. Natl. Acad. Sci. U.S.A. 105, 8718–8723.
Castro, M., Rubin, A. S., Laviolette, M., Fiterman, J., De Andrade Lima, M., Shah, P. L., Fiss, E., Olivenstein, R., Thomson, N. C., Niven, R. M., Pavord, I. D., Simoff, M., Duhamel, D. R., McEvoy, C., Barbers, R., Ten Hacken, N. H., Wechsler, M. E., Holmes, M., Phillips, M. J., Erzurum, S., Lunn, W., Israel, E., Jarjour, N., Kraft, M., Shargill, N. S., Quiring, J., Berry, S. M., and Cox, G. (2010). Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am. J. Respir. Crit. Care Med. 181, 116–124.
Chang, K. S., Jiang, J., Cai, Z., and Luo, G. (2007). Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81, 13783–13793.
Chen, J., Chen, Z., Chintagari, N. R., Bhaskaran, M., Jin, N., Narasaraju, T., and Liu, L. (2006). Alveolar type I cells protect rat lung epithelium from oxidative injury. J. Physiol. (Lond.) 572(Pt 3), 625–638.
Christensen, D. J., Ohkubo, N., Oddo, J., Van Kanegan, M. J., Neil, J., Li, F., Colton, C. A., and Vitek, M. P. (2011). Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J. Immunol. 186, 2535–2542.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., Roses, A. D., Haines, J. L., and Pericak-Vance, M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923.
Crimi, E., Spanevello, A., Neri, M., Ind, P. W., Rossi, G. A., and Brusasco, V. (1998). Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am. J. Respir. Crit. Care Med. 157, 4–9.
Cun, W., Jiang, J., and Luo, G. (2010). The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 84, 11532–11541.
De Alba, J., Raemdonck, K., Dekkak, A., Collins, M., Wong, S., Nials, A. T., Knowles, R. G., Belvisi, M. G., and Birrell, M. A. (2009). HDM induces direct airway inflammation in vivo: implications for future disease therapy? Eur. Respir. J. 35, 1377–1387.
D’Souza, W., Stonik, J. A., Murphy, A., Demosky, S. J., Sethi, A. A., Moore, X. L., Chin-Dusting, J., Remaley, A. T., and Sviridov, D. (2010). Structure/function relationships of apolipoprotein a-I mimetic peptides: implications for antiatherogenic activities of high-density lipoprotein. Circ. Res. 107, 217–227.
Finkelman, F. D., Hogan, S. P., Hershey, G. K., Rothenberg, M. E., and Wills-Karp, M. (2010). Importance of cytokines in murine allergic airway disease and human asthma. J. Immunol. 184, 1663–1674.
Goldstein, J. L., and Brown, M. S. (2009). The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29, 431–438.
Hill, J. M., Bhattacharjee, P. S., and Neumann, D. M. (2007). Apolipoprotein E alleles can contribute to the pathogenesis of numerous clinical conditions including HSV-1 corneal disease. Exp. Eye Res. 84, 801–811.
Johnson, J. R., Wiley, R. E., Fattouh, R., Swirski, F. K., Gajewska, B. U., Coyle, A. J., Gutierrez-Ramos, J. C., Ellis, R., Inman, M. D., and Jordana, M. (2004). Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am. J. Respir. Crit. Care Med. 169, 378–385.
Kellner-Weibel, G., and de la Llera-Moya, M. (2011). Update on HDL receptors and cellular cholesterol transport. Curr. Atheroscler. Rep. 13, 233–241.
Lalazar, A., Weisgraber, K. H., Rall, S. C. Jr., Giladi, H., Innerarity, T. L., Levanon, A. Z., Boyles, J. K., Amit, B., Gorecki, M., Mahley, R. W., and Vogel, T. (1988). Site-specific mutagenesis of human apolipoprotein E. Receptor binding activity of variants with single amino acid substitutions. J. Biol. Chem. 263, 3542–3545.
Lambrecht, B. N., and Hammad, H. (2009). Biology of lung dendritic cells at the origin of asthma. Immunity 31, 412–424.
Laskowitz, D. T., Thekdi, A. D., Thekdi, S. D., Han, S. K., Myers, J. K., Pizzo, S. V., and Bennett, E. R. (2001). Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 167, 74–85.
Levitt, R. C., and Mitzner, W. (1988). Expression of airway hyperreactivity to acetylcholine as a simple autosomal recessive trait in mice. FASEB J. 2, 2605–2608.
Long, A. J., Sypek, J. P., Askew, R., Fish, S. C., Mason, L. E., Williams, C. M., and Goldman, S. J. (2006). Gob-5 contributes to goblet cell hyperplasia and modulates pulmonary tissue inflammation. Am. J. Respir. Cell Mol. Biol. 35, 357–365.
Lynch, J. R., Tang, W., Wang, H., Vitek, M. P., Bennett, E. R., Sullivan, P. M., Warner, D. S., and Laskowitz, D. T. (2003). APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278, 48529–48533.
Mahley, R. W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240, 622–630.
Mahley, R. W., Weisgraber, K. H., and Huang, Y. (2009). Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 50(Suppl.), S183–S188.
Massaro, D., and Massaro, G. D. (2008). Apoetm1Unc mice have impaired alveologenesis, low lung function, and rapid loss of lung function. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L991–L997.
Moore, W. C., Bleecker, E. R., Curran-Everett, D., Erzurum, S. C., Ameredes, B. T., Bacharier, L., Calhoun, W. J., Castro, M., Chung, K. F., Clark, M. P., Dweik, R. A., Fitzpatrick, A. M., Gaston, B., Hew, M., Hussain, I., Jarjour, N. N., Israel, E., Levy, B. D., Murphy, J. R., Peters, S. P., Teague, W. G., Meyers, D. A., Busse, W. W., and Wenzel, S. E. (2007). Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J. Allergy Clin. Immunol. 119, 405–413.
Murphy, A. J., Woollard, K. J., Hoang, A., Mukhamedova, N., Stirzaker, R. A., McCormick, S. P., Remaley, A. T., Sviridov, D., and Chin-Dusting, J. (2008). High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler. Thromb. Vasc. Biol. 28, 2071–2077.
Nandedkar, S. D., Weihrauch, D., Xu, H., Shi, Y., Feroah, T., Hutchins, W., Rickaby, D. A., Duzgunes, N., Hillery, C. A., Konduri, K. S., and Pritchard, K. A. Jr. (2011). D-4F, an ApoA-1 mimetic, decreases airway hyperresponsiveness, inflammation and oxidative stress in a murine model of asthma. J. Lipid Res. 52, 499–508.
Navab, M., Anantharamaiah, G. M., Hama, S., Garber, D. W., Chaddha, M., Hough, G., Lallone, R., and Fogelman, A. M. (2002). Oral administration of an Apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation 105, 290–292.
Navab, M., Reddy, S. T., Van Lenten, B. J., and Fogelman, A. M. (2011). HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 8, 222–232.
Nissen, S. E., Tsunoda, T., Tuzcu, E. M., Schoenhagen, P., Cooper, C. J., Yasin, M., Eaton, G. M., Lauer, M. A., Sheldon, W. S., Grines, C. L., Halpern, S., Crowe, T., Blankenship, J. C., and Kerensky, R. (2003). Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA 290, 2292–2300.
Nofer, J. R., and Remaley, A. T. (2005). Tangier disease: still more questions than answers. Cell. Mol. Life Sci. 62, 2150–2160.
Osei-Hwedieh, D. O., Amar, M., Sviridov, D., and Remaley, A. T. (2011). Apolipoprotein mimetic peptides: Mechanisms of action as anti-atherogenic agents. Pharmacol. Ther. 130, 83–91.
Pendse, A. A., Arbones-Mainar, J. M., Johnson, L. A., Altenburg, M. K., and Maeda, N. (2009). Apolipoprotein E knock-out and knock-in mice: atherosclerosis, metabolic syndrome, and beyond. J. Lipid Res. 50(Suppl.), S178–S182.
Remaley, A. T., Amar, M., and Sviridov, D. (2008). HDL-replacement therapy: mechanism of action, types of agents and potential clinical indications. Expert Rev. Cardiovasc. Ther. 6, 1203–1215.
Remaley, A. T., Thomas, F., Stonik, J. A., Demosky, S. J., Bark, S. E., Neufeld, E. B., Bocharov, A. V., Vishnyakova, T. G., Patterson, A. P., Eggerman, T. L., Santamarina-Fojo, S., and Brewer, H. B. (2003). Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J. Lipid Res. 44, 828–836.
Schaefer, E. J., Gregg, R. E., Ghiselli, G., Forte, T. M., Ordovas, J. M., Zech, L. A., and Brewer, H. B. Jr. (1986). Familial apolipoprotein E deficiency. J. Clin. Invest. 78, 1206–1219.
Sears, M. R., Greene, J. M., Willan, A. R., Wiecek, E. M., Taylor, D. R., Flannery, E. M., Cowan, J. O., Herbison, G. P., Silva, P. A., and Poulton, R. (2003). A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N. Engl. J. Med. 349, 1414–1422.
Segrest, J. P., Garber, D. W., Brouillette, C. G., Harvey, S. C., and Anantharamaiah, G. M. (1994). The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv. Protein Chem. 45, 303–369.
Sethi, A. A., Amar, M., Shamburek, R. D., and Remaley, A. T. (2007). Apolipoprotein AI mimetic peptides: possible new agents for the treatment of atherosclerosis. Curr. Opin. Investig. Drugs 8, 201–212.
Sethi, A. A., Stonik, J. A., Thomas, F., Demosky, S. J., Amar, M., Neufeld, E., Brewer, H. B., Davidson, W. S., D’Souza, W., Sviridov, D., and Remaley, A. T. (2008). Asymmetry in the lipid affinity of bihelical amphipathic peptides. A structural determinant for the specificity of ABCA1-dependent cholesterol efflux by peptides. J. Biol. Chem. 283, 32273–32282.
Simpson, J. L., Powell, H., Boyle, M. J., Scott, R. J., and Gibson, P. G. (2008). Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am. J. Respir. Crit. Care Med. 177, 148–155.
Sorkness, R. L., Bleecker, E. R., Busse, W. W., Calhoun, W. J., Castro, M., Chung, K. F., Curran-Everett, D., Erzurum, S. C., Gaston, B. M., Israel, E., Jarjour, N. N., Moore, W. C., Peters, S. P., Teague, W. G., and Wenzel, S. E. (2008). Lung function in adults with stable but severe asthma: air trapping and incomplete reversal of obstruction with bronchodilation. J. Appl. Physiol. 104, 394–403.
Suzuki, T., Yamaya, M., Kamanaka, M., Jia, Y. X., Nakayama, K., Hosoda, M., Yamada, N., Nishimura, H., Sekizawa, K., and Sasaki, H. (2001). Type 2 rhinovirus infection of cultured human tracheal epithelial cells: role of LDL receptor. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L409–L420.
Swedin, L., Neimert-Andersson, T., Hjoberg, J., Jonasson, S., van Hage, M., Adner, M., Ryrfeldt, A., and Dahlen, S. E. (2009). Dissociation of airway inflammation and hyperresponsiveness by cyclooxygenase inhibition in allergen challenged mice. Eur. Respir. J. 34, 200–208.
Tabet, F., Remaley, A. T., Segaliny, A. I., Millet, J., Yan, L., Nakhla, S., Barter, P. J., Rye, K. A., and Lambert, G. (2010). The 5A apolipoprotein A-I mimetic peptide displays antiinflammatory and antioxidant properties in vivo and in vitro. Arterioscler. Thromb. Vasc. Biol. 30, 246–252.
Tardif, J. C., Gregoire, J., L’Allier, P. L., Ibrahim, R., Lesperance, J., Heinonen, T. M., Kouz, S., Berry, C., Basser, R., Lavoie, M. A., Guertin, M. C., and Rodes-Cabau, J. (2007). Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA 297, 1675–1682.
Thomas, W. R., Smith, W. A., Hales, B. J., Mills, K. L., and O’Brien, R. M. (2002). Characterization and immunobiology of house dust mite allergens. Int. Arch. Allergy Immunol. 129, 1–18.
van den Elzen, P., Garg, S., Leon, L., Brigl, M., Leadbetter, E. A., Gumperz, J. E., Dascher, C. C., Cheng, T. Y., Sacks, F. M., Illarionov, P. A., Besra, G. S., Kent, S. C., Moody, D. B., and Brenner, M. B. (2005). Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 437, 906–910.
Wang, W., Xu, H., Shi, Y., Nandedkar, S., Zhang, H., Gao, H., Feroah, T., Weihrauch, D., Schulte, M. L., Jones, D. W., Jarzembowski, J., Sorci-Thomas, M., and Pritchard, K. A. Jr. (2010). Genetic deletion of apolipoprotein A-I increases airway hyperresponsiveness, inflammation, and collagen deposition in the lung. J. Lipid Res. 51, 2560–2570.
Watson, C. E., Weissbach, N., Kjems, L., Ayalasomayajula, S., Zhang, Y., Chang, I., Navab, M., Hama, S., Hough, G., Reddy, S. T., Soffer, D., Rader, D. J., Fogelman, A. M., and Schecter, A. (2011). Treatment of patients with cardiovascular disease with L-4F, an Apo-A1 mimetic, did not improve select biomarkers of HDL function. J. Lipid Res. 52, 361–373.
Wills-Karp, M., and Ewart, S. L. (1997). The genetics of allergen-induced airway hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 156(4 Pt 2), S89–S96.
Yao, X., Dai, C., Fredriksson, K., Dagur, P. K., Mccoy, J. P., Qu, X., Yu, Z. X., Keeran, K. J., Zywicke, G. J., Amar, M. J. A., Remaley, A. T., and Levine, S. J. (2011a). 5A, an apolipoprotein A-I mimetic peptide, attenuates the induction of house dust mite-induced asthma. J. Immunol. 186, 576–583.
Yao, X., Remaley, A. T., and Levine, S. J. (2011b). New kids on the block: the emerging role of apolipoproteins in the pathogenesis and treatment of asthma. Chest 140, 1048–1054.
Yao, X., Fredriksson, K., Yu, Z. X., Xu, X., Raghavachari, N., Keeran, K. J., Zywicke, G. J., Kwak, M., Amar, M. J., Remaley, A. T., and Levine, S. J. (2010). Apolipoprotein E negatively regulates house dust mite-induced asthma via a LDL receptor-mediated Pathway. Am. J. Respir. Crit. Care Med. 182, 1228–1238.
Keywords: asthma, apolipoprotein E, apolipoprotein A-I, apolipoprotein E mimetic peptide, apolipoprotein A-I mimetic peptide
Citation: Yao X, Vitek MP, Remaley AT and Levine SJ (2012) Apolipoprotein mimetic peptides: a new approach for the treatment of asthma. Front. Pharmacol. 3:37. doi: 10.3389/fphar.2012.00037
Received: 15 November 2011; Paper pending published: 02 December 2011;
Accepted: 20 February 2012; Published online: 09 March 2012.
Edited by:
Gerda Fouche, Council for Scientific and Industrial Research, South AfricaReviewed by:
Adam Antczak, Medical University of Lodz, PolandArmin Braun, Fraunhofer Institute for Toxicology and Experimental Medicine, Germany
Gary Peter Anderson, University of Melbourne, Australia
Copyright: © 2012 Yao, Vitek, Remaley and Levine. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Stewart J. Levine, Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Building 10, Room 6D03, MSC 1590, Bethesda, MD 20892-1590, USA. e-mail:bGV2aW5lc0BuaGxiaS5uaWguZ292