
- 1 Research Center of Computational and Biotechnological Sciences, Second University of Naples, Naples, Italy
- 2 INT Pascale – Cancer Research Center of Mercogliano, Mercogliano, Italy
- 3 Institute for Research in Biomedicine, Molecular Modelling and Bioinformatics Group, Barcelona, Spain
- 4 Department of Biochemistry and Biophysics, Second University of Naples, Naples, Italy
Sirt-1 is defined as a nuclear protein involved in the molecular mechanisms of inflammation and neurodegeneration through the de-acetylation of many different substrates even if experimental data in mouse suggest both its cytoplasmatic presence and nucleo-cytoplasmic shuttling upon oxidative stress. Since the experimental structure of human Sirt-1 has not yet been reported, we have modeled its 3D structure, highlighted that it is composed by four different structural regions: N-terminal region, allosteric site, catalytic core and C-terminal region, and underlined that the two terminal regions have high intrinsic disorder propensity and numerous putative phosphorylation sites. Many different papers report experimental studies related to its functional activators because Sirt-1 is implicated in various diseases and cancers. The aim of this article is (i) to present interactomic studies based human Sirt-1 to understand its most important functional relationships in the light of the gene–protein interactions that control major metabolic pathways and (ii) to show by docking studies how this protein binds some activator molecules in order to evidence structural determinants, physico-chemical features and those residues involved in the formation of complexes.
Introduction
In complex biological systems the protein–gene interactions operate under protein–protein or gene–gene interaction maps where they have specific functional roles (Barabási and Oltvai, 2004). In this context well-connected hubs are of high functional importance (Jeong et al., 2001; He and Zhang, 2006). Consequently, studies based on protein–protein interaction (PPI) networks can be inferred from centrality statistics of proteins associated with disease and biological processes associated with genes and proteins. Genes associated with a particular phenotype or function are not randomly positioned in the PPI network, but tend to exhibit high connectivity; they may cluster together and can occur in central network locations (Goh et al., 2006; Oti and Brunner, 2006). Seven different homologous proteins compose Sirtuin family, and in particular Sirt-1 exhibits a high degree of structural disorder as demonstrated in a recent work of our group (Autiero et al., 2009). In general it has been already that the protein disorder plays a crucial role in PPIs and in regulatory processes for understanding the phenomenon of interactome (Tompa and Fuxreiter, 2008). Therefore, it is important to focus the attention on Sirtuins because they are involved in numerous processes and implicated in different diseases. Importantly the second-degree interaction maps related to these family present 5786 neighbors with average number of neighbors equal to 84.22. However some sirtuins have not yet been well studied and not much information are known in regard to their interaction with other proteins (data not shown) in second order interactome. In particular, Sirt-1 is defined as a nuclear protein even if experimental data suggest also its cytoplasmatic presence and indicate that it is involved into nucleo-cytoplasmic shuttling upon oxidative stress (Autiero et al., 2009). Sirt-1 is a NAD+ dependent histone deacetylates that play important functional roles in many biological processes causing various modifications of histone/protein acetylation status by several class I and II histone deacetylase (HDAC) inhibitors (Kyrylenko et al., 2003). In literature it is reported that Sirt-1 regulates gene silencing, cell cycle, DNA-damage repair and life span. In specific diseased conditions, Sirt-1 regulates or interacts with many proteins: TP53, NEDD8, SMAD4, DYNC1H1, TUBULIN, NUDC, DYNACTIN, HDAC4, POLR2H, and BRCA1. For example, Sirt-1 interacts with TP53 which is a very short lived protein involved in the acetylation processes and gene activation as consequent target (Appella and Anderson, 2001). In fact, the inactivation of HIC1 leads to an up-regulation of Sirt-1 which deacetylates and deactivates TP53. This allows the cells to bypass apoptosis and survive DNA damage (Chen et al., 2005). It is also known that Sirt-1 is involved in inflammatory processes and in neurodegenerative diseases like Huntington (Pallkes et al., 2008). Moreover, in literature it is reported that Sirt-1 interacts also with HDAC2, HDAC4, MEF2, SUMO, and UBIQUITIN and that HDAC4 might function to integrate sumoylation and deacetylation signals via its interaction with UBC9 and Sirt-1 and that acetylation and sumoylation occur on the same lysine residue (Zhao et al., 2005). This evidences the reason for which the analysis of the Sirt-1 interactome is of great interest in order to find the relationships between nodes (i.e., genes, proteins) and their positions as well as the overall relationships in the entire system along with structural inferences of activators associated with it.
Since the 3D structure of Sirt-1 has not yet been obtained experimentally, we have recently modeled this protein by computational methods and highlighted that it is composed by four different regions: N-terminal region, allosteric site, catalytic core and C-terminal region and underlined that the two extended terminal regions of about 250 residues each are highly disordered (Autiero et al., 2009). Sirt-1 is implicated in numerous diseases and cancers and many different papers report experimental studies related to the effects of its activation. In fact, Sirt-1 activation by natural activators seems to show a wide spectrum of beneficial effects in cardiovascular, metabolic, and neurodegenerative diseases and, hence, interest is increasing in testing more potent Sirt-1 activators for the treatment of these aging associated diseases. The natural activator resveratrol has been largely studied because of its low toxicity in humans and its anti-aging properties (Orallo, 2006; Harikumar and Agarwal, 2008). In particular, it is an important constituent of red wine (Zhuang et al., 2003) that increases the cell survival in several animals by stimulating the Sirt-1 dependent deacetylation of TP53 (Howitz et al., 2003). Since natural compounds failed to induce an increased activity of Sirt-1 (Yang et al., 2007), new activators (SRT1460, SRT1720, SRT2183) with a good affinity for Sirt-1 have been synthesized. Recently, a pharmaceutical biotechnology company, starting from these activators, discovered novel selective Sirt-1 activators using a high-throughput screening methodology (Smith et al., 2009; Vu et al., 2009; Yamazaki et al., 2009). In this article we will report studies on the Sirt-1 interactome and on molecular complexes between Sirt-1 and four different activators, i.e., SRT1460, SRT1720, SRT2183, and resveratrol, by molecular docking (Camins et al., 2010). Since the human sirtuin is proving to be a multifunctional protein with a large spectrum of biological activities and partners, the analysis of its interactome is an important step to define which biological process is directly or indirectly controlled by this molecule. This information is preliminary to understand the structural characteristics of complexes between sirtuin and those ligands that have been shown to regulate its biological activity. Starting from this knowledge we can design new molecules in a targeted way to control specific biological functions dependent on sirtuin.
Materials and Methods
Interactomic Studies
Cytoscape software (Kohl et al., 2011) is used to visualize the network of Sirt-1 family. The experimentally evidenced interactions of Sirt family proteins were filtered from Bio grid, HPRD, MINT, and Pathway Interaction Database which are curated from both high-throughput data sets and individual focused studies along with interaction published in peer reviewed journals (Watts and Strogatz, 1998; Stark et al., 2006; Chatr-Aryamontri et al., 2007; Keshava Prasad et al., 2009; Schaefer et al., 2009). Further more the manually curated PPI network is obtained from Center for BioMedical Computing (CBMC) at University of Verona. Centrality statistics of the protein network are vitals for attaining properties of the network (Assenov et al., 2008; Scardoni et al., 2009). In particular, we focused most of our attention on central vertices in complex networks since they might play the role of organizational hubs. Betweenness centrality (BC; Freeman, 1977; Joy et al., 2005) and closeness centrality (CC; Wuchty and Stadler, 2003) are based on the calculation of shortest paths. Przulj et al. showed bottleneck’s importance in protein interaction networks and their correlation with gene essentiality (Przulj et al., 2004; Yu et al., 2004). Lin et al. (2008) proposed two characteristic analysis algorithms: maximum neighborhood component (MNC) and density of maximum neighborhood component (DMNC) for exploring essential proteins (Hub proteins) from protein interaction networks (Lin et al., 2008). Most of these different methods for identifying essential nodes from the network have been stated in literature (Mason and Verwoerd, 2007). We utilized Maximal Clique Centrality (MCC), MNC, and DMNC, EPC, and other centrality based measure are taken into account for exploring the potential hubs in interaction maps of Sirt-1. Gene ontological data were mapped to nodes (Proteins) in the network. Gene Ontological study of a network infers about biological process, molecular function, and cellular location of the interactants present in the interactome. Significant clustering of genes, mapped with proteins, are layered into Graphs of the Gene Ontology and they are identified using the GO enrichment analysis plugin BiNGO (Maere et al., 2005).
Molecular Docking Studies
Most cellular processes are carried out by PPIs. Predicting the 3D structures of protein–protein complexes by docking, it can shed light on their functional mechanisms and roles in cell. Docking can assist in predicting PPIs, in understanding signaling pathways and in evaluating the affinity of complexes (Andrusier et al., 2008). In this work, docking studies were done both to get structural models of those Sirt-1 complexes suggested by the interactome analysis and to understand the structural determinants underlying the interaction of Sirt-1 with small molecules that have the function of effectors. Automated docking is widely used for modeling biomolecular complexes in structure/function analysis and in molecular design. There are several effective methods available, incorporating different parameters such as algorithm and scoring function to provide reasonably good predictions. AutoDock4 is resulted a very useful tool for predicting the complexes conformation and the related binding energies of ligands with proteins. The basic algorithm used for conformational searching in AutoDock4 is Lamarckian genetic algorithm (LGA; Morris et al., 1998). This algorithm works on the basis of the stepwise generation selection. In fact, during the docking simulation a test population of docking conformation is created and in subsequent stepwise generations these individual conformations are selected for the next generation and in this way the best conformation is obtained. LGA has an additional feature called “Lamarckian” that allows to the individual conformation of searching the local conformation space, of finding the local minima and, then, of passing this to next generation. In particular, AutoDock4 uses a semi empirical free energy force field to predict binding free energy of small molecules and macromolecules, presents other traditional features such as Simulated Annealing and genetic algorithm and uses a force field that refers to the form and parameters of mathematical function used to describe the potential energy of a system of particles and leads to calculate the intermolecular energies for predicting free energy of binding. AutoDock 4 is composed by two software packages, i.e., AutoDock and AutoGrid, and consists of Rigid Docking and Flexible Docking modules. Rigid Docking (called also Grid-based approach) allows the ligand to have a large and a fixed conformational space around protein. In this approach the target macromolecule is embedded in the grid, the interaction energy between the probe and the target is computed and stored in this grid and is used as input for docking simulation. In this case, the relative orientation of molecules interacting with each other are allowed to change whereas the internal geometry of the target molecule is kept fixed. On the other hand, Flexible Docking module includes the side chain flexibility. In fact, in this approach a specific part of the target molecule is made flexible and, during the docking time, these flexible parts are treated explicitly allowing rotations of bond angles around torsion degrees of freedom.
The most important part in docking is the selection of the correct active binding site. In certain cases the binding site area on the surface of the protein is found with the help of specific software but the selection is also addressed on the basis of prior knowledge of the protein. Before setting up the docking run, ligands and receptor or target molecule were prepared by adding charges, torsions, and hydrogen atoms by specific tools. This preparation is important to mimic the “in vivo” conditions of molecular interaction (see Figure A1 in Appendix). After the preparation of parameter and map files, AutoDock suite was launched for the process of docking that generates as output a log file (DLG) containing all the information of docked complexes (see Figure A2 in Appendix). The description of AutoDock procedure used to simulate the complexes between Sirt-1 and the four activators is shown in Figure 1. The first docking methods or rigid docking treated proteins as rigid bodies (means the internal geometry of the molecule are kept fixed) in order to reduce the search space for optimal structure of complexes (Wodak and Janin, 1978; Halperin et al., 2002). However ignoring flexibility could prevent docking algorithms from recovering native associations (Andrusier et al., 2008) and specially in the case of unordered proteins or highly flexible proteins one cannot ignore the importance of flexible docking. Moreover, flexibility in docking should be taken into account if docked structures were determined by homology modeling (Marti-Renom et al., 2000) or if loop conformations were modeled (Soto et al., 2008) and this scenario implies in our case the presence of two unordered loops/regions, i.e., N-terminus and C-terminus (Autiero et al., 2009). The benefit of rigid docking procedure is relatively low in computational time and is less complex (Andrusier et al., 2008) but we cannot ignore the structural characteristic of Sirt-1. Therefore we have used this peculiar protocol that use steps of rigid docking followed by steps of flexible docking to generate near native models of complexes made with flexible Sirt-1 protein.
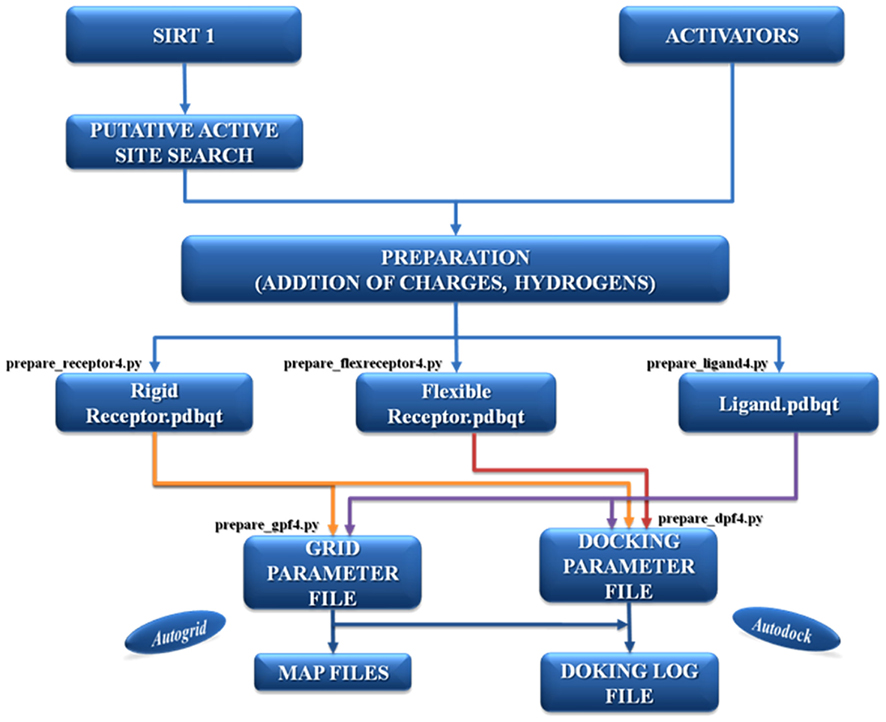
Figure 1. The description of the AutoDock protocol used to simulate the complexes between Sirt-1 and its four activators.
Results
Centrality Statistics of First Order Interaction of Sirt-1
Sirt family first order interaction maps, obtained concerning experimental data reported in protein databases (see Methods section), have 228 nodes and 3769 edges (interactions). The extraction of first order interaction map of Sirt-1 has 136 nodes and 1503 edges with Sirt-1 as a central node of the network (Figure 2). A statistic analysis of first order interaction map of Sirt-1 was performed. In particular, given undirected networks, the clustering coefficient Cn of a node n is defined as Cn = 2en/[kn(kn − 1)], where kn is the number of neighbors of n and en is the number of connected pairs between all neighbors of n (Barabási and Oltvai, 2004). In directed networks, the definition is slightly different: Cn = en/[kn(kn − 1)]. The evaluation of the average clustering coefficient distribution gives the average of the clustering coefficients for all nodes n with k neighbors and identifies a modular organization of networks. The clustering coefficient Cn for undirected network of the Sirt-1 interaction map is 0.717. The mean shortest path length between any two proteins is 1.836 (Figure 3A). The top 30 best-connected nodes obtained by average path length, as calculated by Centiscape (see Table 1), have relatively lesser average path length in respect to TP53 Interactome (Dartnell et al., 2005). The node distribution degree of Sirt-1 interactome gives information of the protein interactions with the k other proteins (Figure 3B). In details, it tends to decrease slowly complying with the power law y = axb where “a” is 4.971 and “b” is −0.232 with a correlation coefficient of 0.113. This value indicates a scale free network (Barabási and Oltvai, 2004) and in general these are very robust against failure, such as removal of arbitrary network elements. This evaluation suggests that the Sirt-1 interaction map is assortative and has a low value of vertices. In this network the average number of interacting partners was evaluated and it resulted equal to 22.10. Moreover, since Jeong et al. (2001) showed that a protein acting as hub is more important than those sparsely connected with a small number of interactions (Jeong et al., 2001), we calculated the putative hub proteins present in our network by using different algorithms like MCC, DMNC, MNC and Edge Percolated component and different centrality based measures. In Table 2 are reported the top 10 hub proteins obtained by these analysis but only five of them (SLC25A3, Sirt-1, JUN, MCF2L2, and EP300) were selected as hub by all used different algorithms.
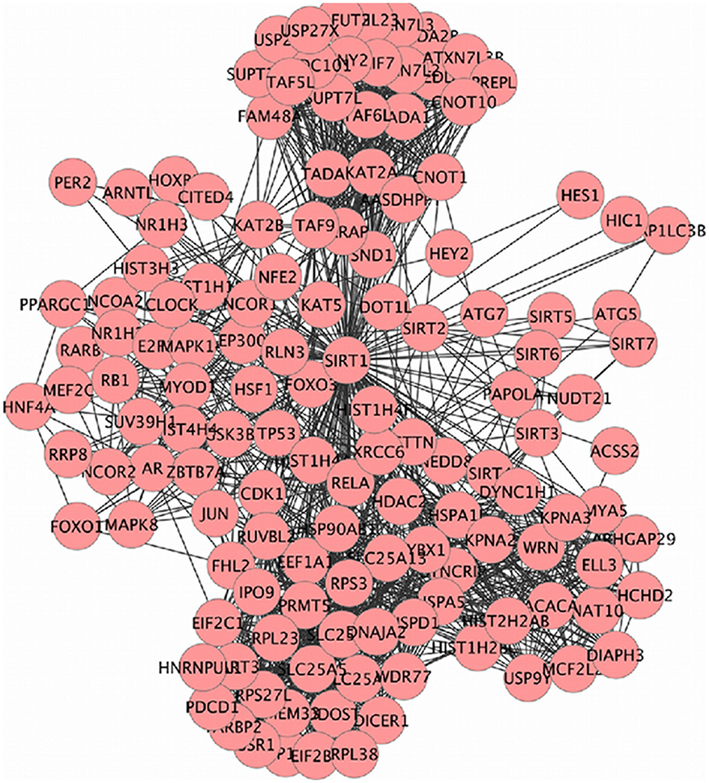
Figure 2. SIRT family interaction maps containing 136 nodes and 1503 edges. Black lines are interactions and nodes (proteins) are represented by Circles.
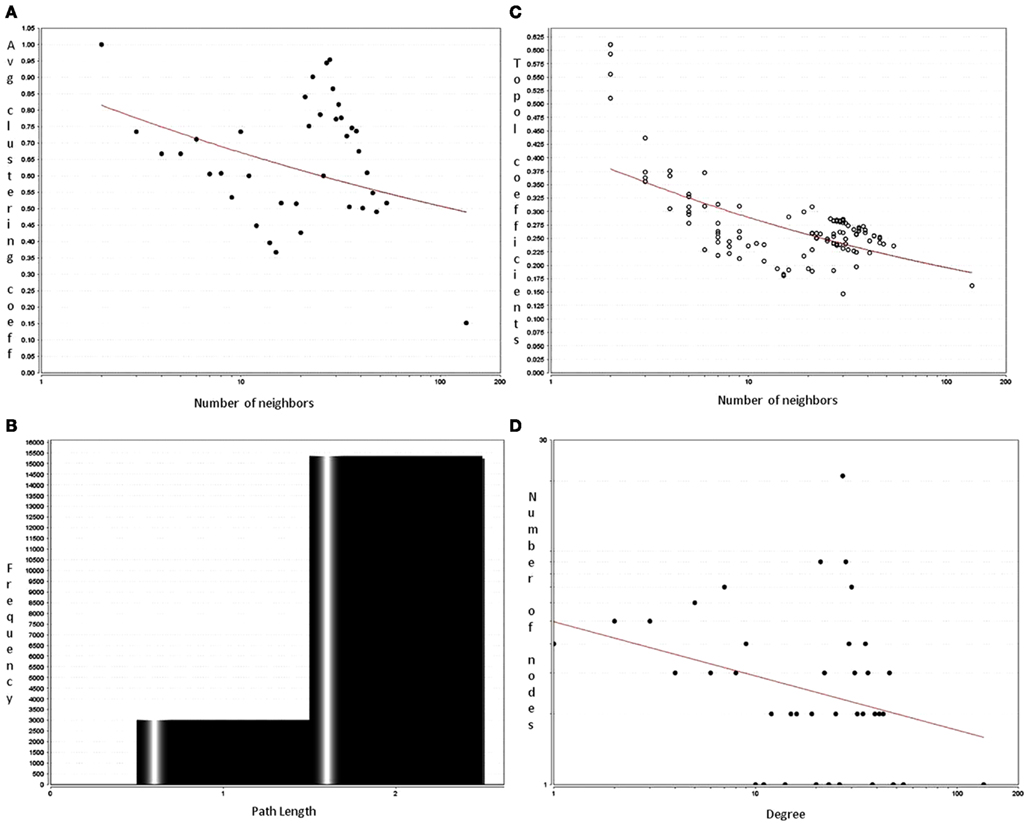
Figure 3. Analysis of first order interactome for Sirt-1 composed by 136 nodes and 1504 edges. The shortest path length distribution (A) indicates that the network possesses small-world property. The node degree distribution (B) shows a scale free network property. (C) Average clustering coefficient of Sirt-1 network showing constant decrease (D) topological coefficient giving insights on the measure of extent to which a protein in the network shares interaction partners with other proteins.
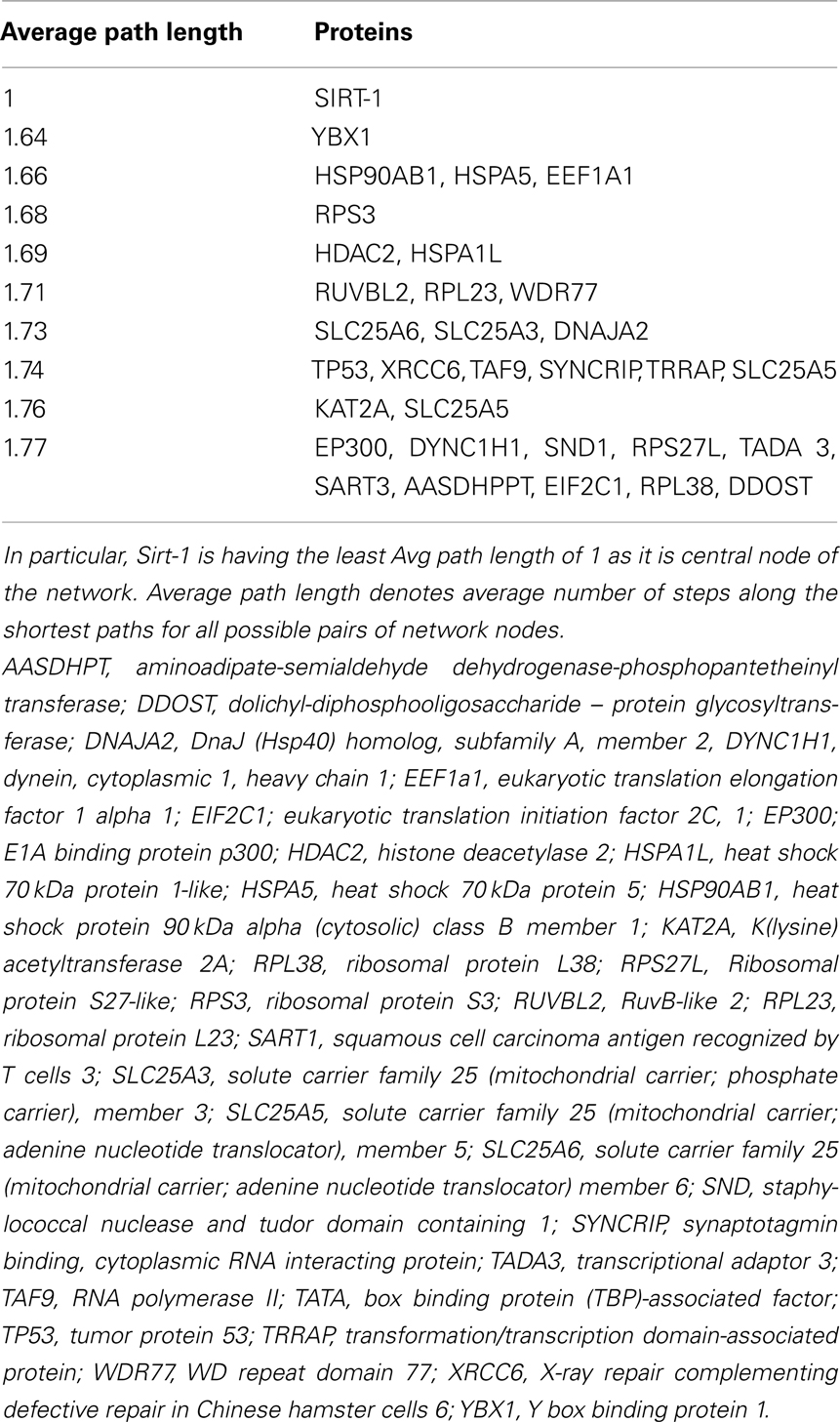
Table 1. Top 30 neighbors based on smallest average path length.
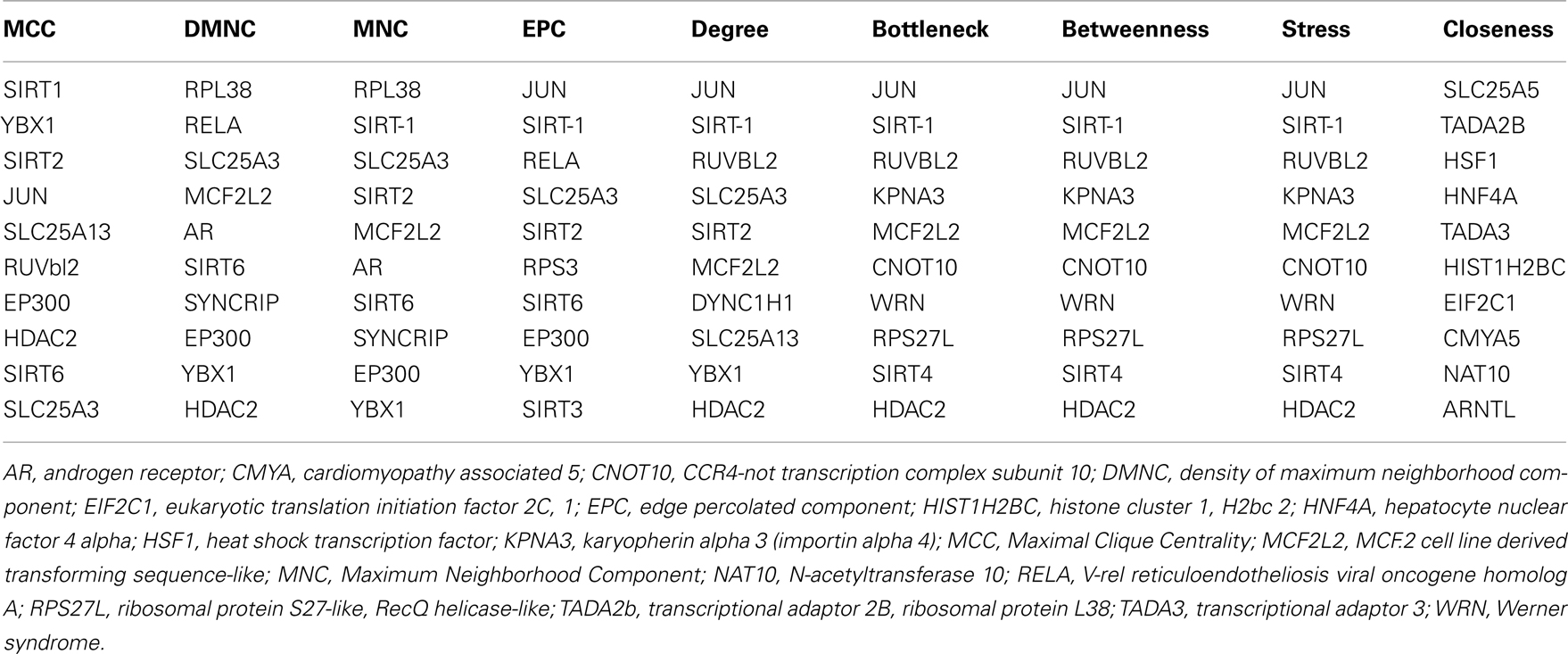
Table 2. The 10 hub proteins present in Sirt-1 first order interaction maps obtained by different algorithms and centrality measures.
Parameters related to topological aspects of Sirt-1 neighbors are acquired by calculating the average clustering coefficient of proteins that shows tendency to form clusters or groups (Barabási and Oltvai, 2004). Sirt-1 network has a constant decrease in clustering coefficients due to the higher number of interaction of each protein (Figure 3C). This suggests that it is a small-world network having hierarchical modularity. Ravasz et al. (2002) showed that highly connected regions connect sparsely connected nodes. In fact they classified networks into two modular organizations: local clustering and global networks. Local networks are considered to have functionality similar to biological processes whereas global connectivity is related to hub proteins present in the network connecting high-end nodes (higher order communication points between protein complexes; Han et al., 2004). Sirt-1 network is showing a tendency to connected global networks.
The decrease of the topological coefficient with the number of interacting partners gives information regarding interaction of proteins with common neighbor (Figure 3D). This shows that hub proteins (except SLC25 protein family) share fewer common neighbors then sparsely connected nodes and it also proves that the early inference of modular organization of Sirt-1 network is correct. A stressed node in the network is Sirt-1 having the highest number of distribution degrees (see supplementary material for details in Table A1 in Appendix). BC has been evaluated as the amount of traffic that a vertex or edge has to handle in a network. In Sirt-1 interactome, the number of nodes has a high degree of BC and this is reported in (see supplementary material for details in Table A1 in Appendix). It has been shown that high degree of connectivity correlates well with pleiotropic effects (Tyler et al., 2009). This indicates also that the most part of Proteins in Sirt 1 interactome map are involved in many different biological processes with different cellular localizations, more precisely AR, RELA, and SYNCRIP are present in nucleus as well as cytoplasm whereas SLC25A5 is in inner mitochondrial membrane as well as cytoplasm. Sirt 1 is found to interact with proteins involved in numerous pathways like Foxo Signaling, Regulation of Androgen receptor activity (Table A3 in Appendix).
Gene Ontological Studies of Sirt-1
GO studies on the hub proteins inferred from our analysis suggest that they are involved in important biological processes related to gene regulation, Metabolism and proton co-transport (Table A2 in Appendix). In details, SLC25A3 is responsible for the inorganic phosphate transport into the mitochondrial matrix, either by proton co-transport or in exchange for hydroxyl ions (k, Entrez Gene description), while JUN interacts directly with specific target DNA sequences to regulate gene expression. The centrality analysis based on hub proteins showed SLC25A3, JUN, Sirt-1, RUVBL2, and MCF2L2 as important proteins of the network. Other Methods based on MCC, DMNC, MNC, and EPC evidenced the same proteins as hub nodes along with EP300, YBX1, RPL38, AR, and Sirt-2.
Genes associated with proteins and found significant in the interactome were analyzed by the BiNGO package in Cytoscape. Sirt-1 first order interacting partners are involved into numerous biological processes. Sirt-1 interactome is significantly involved Metabolism modulation related processes (Figure 4). Sirt-2 in chromatin silencing at rDNA, RPS27L, and RTN4 in regulating anti-apoptotic phenomena.
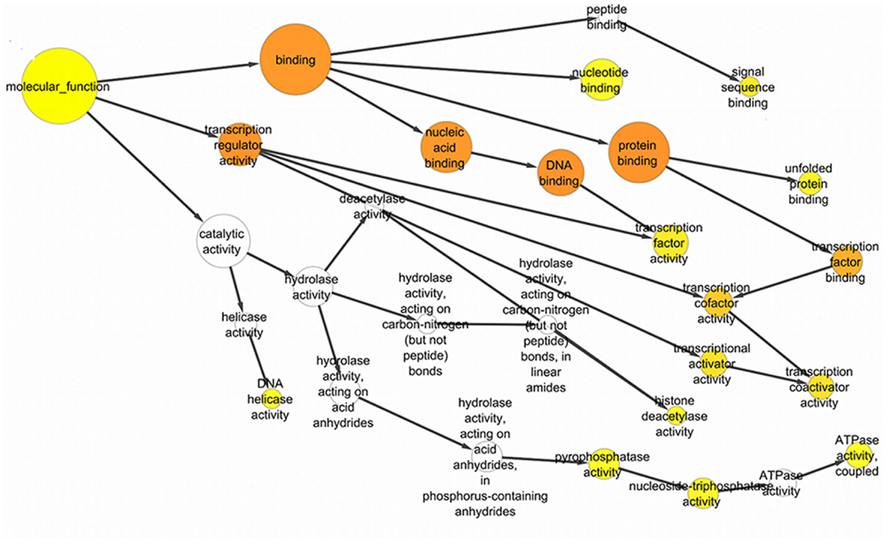
Figure 4. The significant GO ontological data related to molecular function with the GO nodes are listed in circles connected by black arrows to the GO nodes. These yellow and orange color nodes correspond to the statistically significant nodes.
Certain processes, like chromatin remodeling and modification, involve many important proteins of the network like KAT2B, NCOR1, HDAC6, RRP8, HDAC2, and KAT2A. Moreover, TP53, Sirt-2, PPARGC1, CPS1, and JUN are responsible for the processes related to the response to starvation whereas the response to stress is regulated by NCOR1, MYOD1, KRT1, SIRT2, HDAC2, RPS3, RELA, FOXO1, HDAC6, and other proteins involved in ncRNA metabolic processes and in negative regulation of signaling pathways. In particular, the important processes like DNA binding activity transcription factor regulation and DNA repair are shown to have an involvement with proteins like Sirt-1, Sirt-2, TP53, PPARGC1A, JUN, EP300, HDAC2, HDAC6, KAT2A, Kat 2B, RELA, RB1, WRN, XRCC5, and XRCC6 (Figure A3 in Appendix).
In particular, the sirtuin network shows that Sirt-2, HDAC6, HDAC2, Sirt-1, PPARGc1A, TRRAP are implicated in histone modification and histone deacetylation whereas SUV39H1 and DICER1 are involved in gene silencing phenomenon.
The proteins in Sirt 1 interaction maps showed also different cellular localization and molecular function (Figure 4 and Table A4 in Appendix). In details, Sirt family, ARNTL, WRN, EP300, SYNCRIP, JUN, RPS3 are proteins showing pleiotropicity in biological as well as in the cellular localization in the GO analysis maps. In fact, Sirt-1 interacts with cytoplasmic, nuclear, extracellular and mitochondrial proteins as found with a significant p value, i.e., p < 0.05 to p > 0.0000005 that measures the statistical significance of the different essentialities of proteins implicated in the biological processes. RELA and JUN show interactions with mitochondrial proteins, Sirt-1 interacts with other cellular proteins in activating DNA repair and stress protection mechanisms.
Second Order Interaction of Sirt-1
The Sirt-1 second-degree interaction map is composed of 4691 nodes. These nodes correspond to different partners interacting by 221595 edges (Interactions). The second order network of Sirt-1 is scale free and small-world network interacting with numerous proteins implicated in transcription and metabolism related processes. Sirt 1 has a high degree of interactions in second order interaction maps as it is having interactions with high number of proteins like PARP1 (inhibits Sirt activity) and NAMPT (regulates NAD+ levels; Yang et al., 2006). Analyzing centrality statistics and pattern of rearrangement of interacting nodes in Second order interactome of Sirt-1 will provide further insights on the variability in functionality, cellular localization, and pleiotropicity nature of the SIRT interaction map.
Molecular Docking Studies
Dai et al. (2010) suggested that Sirtuin activating compounds (STACS) interact directly with Sirt-1 activating the deacetylation through an allosteric mechanism. This mechanism requires the presence of an allosteric site on the protein; therefore, we have used for modeling the same structural site on which we have recently found that binds AROS, the allosteric effector of Sirt-1 known as endogenous activator (Autiero et al., 2009). So we acquired from the structural model of the complex AROS–SIRT1, obtained after docking and molecular dynamics, the putative residues of interaction (see Table 3). Hence, our docking studies have focused on the interactions between the allosteric site found on the native modeled structure of Sirt-1 and STACs like SRT1720, SRT2183, SRT1460, and resveratrol (Figure A4 in Appendix). The best docking results were obtained by implementing flexible docking in AutoDock4. In particular, the reason for the selection of this site depends from the fact that many experimental data have suggested that the modulation of the catalytic activity of Sirt-1 is exerted through the adjustment implemented by the allosteric site. Recent works also show that the interaction of Sirt-1 with small effectors has a functional relevance for its activation (Zhao et al., 2004; Milne et al., 2007; Bemis et al., 2009). However, the modeled structure of Sirt-1 shows that the allosteric site selected as binding area for activators is near to N-terminal region predicted as unordered (Autiero et al., 2009).

Table 3. Sirt-1 residues resulted at the interaction interface with AROS (Autiero et al., 2009) and used during the docking studies.
We have also focused our attention on the disordered residues flanking the allosteric site (see Table 3) considering them as flexible during the process of docking. This structural region is close to the highly disordered N-terminal segment and involved into the regulation of the enzyme activity (Tanno et al., 2007; Ford et al., 2008; Sasaki et al., 2008).
The grid-based approach was implemented defining a rigid box (of dimension 4.14 × 19.56 × −24.21 Angstroms) on the surface of the protein and around the residues of the allosteric site (see Table 3 and Figure A6 in Appendix) to specify the docking area for the activators. Parametric details of the grid parameters such as “number of spacing,” “number of grid points,” and “center grid box” in all three directions are given in Figure A5 in Appendix.
Figures 5 and 6 and Figure A7 in Appendix show the best docking models computed for all the four activators against Sirt-1. The complexes present negative values of the binding free energy, which indicates that the models between Sirt-1 and its activators are reliable. Their analysis in terms of interaction residues confirms that the binding region is conserved with the involvement of charged and aromatic residues which suggests complexes stabilized by electrostatic and stacking interactions. Moreover, the complex between Sirt-1 and resveratrol resulted not so stable in respect to the complexes of the other three ligands. This can be inferred from the absence of H-bonds and relatively lesser number of charged interaction residues (Table 4). However, the EC1.5 values related to the Sirt-1 activity, and reported in the literature, supports the observation that the resveratrol is a less potent activator (Milne et al., 2007; Bemis et al., 2009). A remarkable observation that supports our models is that the experimental EC1.5 values are linearly correlated with the binding energy values found by AutoDock for the Sirt-1 complexes with the four activators. In fact, a correlation coefficient of 0.73 demonstrates the good agreement between functional data and computational results.
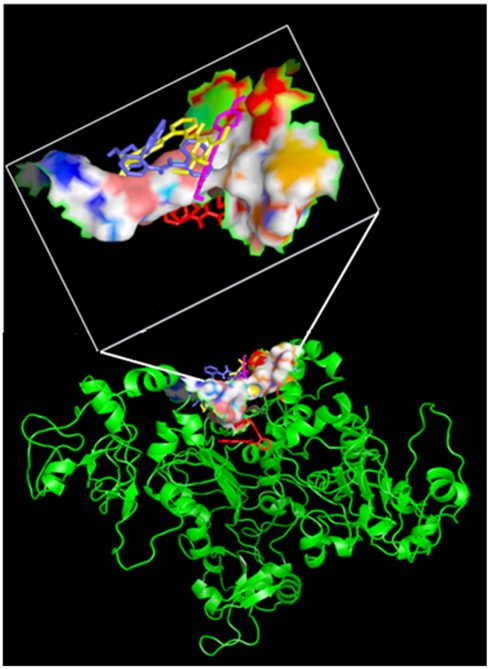
Figure 5. Side view of four activators docked on the active site. All the four activators are shown in “stick” confirmation with different colors: SRT1460 in yellow, SRT2183 in blue, resveratrol magenta and SRT1720 in red). The active site residues are shown in “surface” confirmation whereas Sirt-1 by cartoon. The active site region with the docked activators is highlighted in a white box.
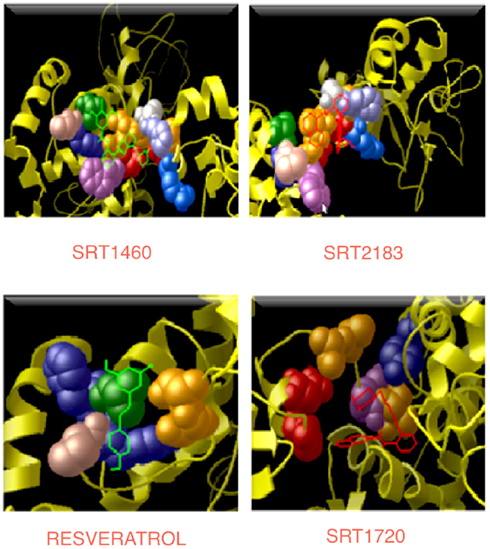
Figure 6. Interaction regions in four complexes. All the four activators are shown in the line/bond conformation with different colors and the residues (reported in Table 4), which are interacting with their respective activators, are shown in the CPK conformation.

Table 4. Interaction details of four complexes compared to experimental data reported in literature.
Discussion
Human Sirt-1 is an unordered protein (IDP) and may therefore adopt types of order (and conformations) that are not easily recognized by current secondary or tertiary structure prediction algorithm, which primarily recognize higher order assemblies stable in the time. Even the classic experimental structural techniques often fail in studying structural aspects of these proteins. Sirt-1 is a Hub protein because of its numerous partners and for its structural characteristic. Structural features that affect the ability of hubs in PPI networks to recognize and bind multiple partners are numerous. In this article we primarily focused on the role of intrinsic disorder in the Sirt-1 structure. However, a study in progress in our laboratory focuses on the charged residues on the surfaces of this protein and on the role of phosphorylations. Preliminary data support the idea that it has highly charged surfaces as compared to large, disorder containing hubs indicating its possible involvement in promiscuous binding (Patil and Nakamura, 2006).
Our interactomic analysis showed for the first time how much is vast the number of physiological partners of this hub protein. Sirt-1 is an interesting case because we are just beginning to understand some of the mechanisms that lead to multi-specificity in the binding of hub proteins. In particular, a huge number of articles have been published on the clinical, biological and, functional aspects of human sirtuins but we know only general details about their structures and molecular mechanisms which govern the functional behavior of these proteins.
The aim of this study was to evaluate and integrate functional and structural features by computational methods to predict the involvement of the human Sirt-1, the most studied of sirtuins, into the basic molecular mechanism describing the complex regulation of this protein. Since in vitro or in vivo experiments is time consuming and expensive; in silico prediction can provide functional candidates and help narrow down the experimental efforts. Moreover, we have also analyzed multiple large-scale experimental data sets describing the metabolic involvement of the Sirt-1 to understand the basic mechanism underlying the function of this hub protein. We have examined objective criteria that could infer organizations of the Sirt-1 network and the structural determinants featuring the interaction between Sirt-1 and some biological activators which are reported in the literature as potent modulators of the metabolic activities of sirtuin 1 (Milne et al., 2007; Dai et al., 2010). At the same time, we can make suggestions about the structural mechanisms underlying the interaction of small molecule activators on which there is currently much disagreement (Pacholec et al., 2010). This knowledge may also be used to direct the design of new and more specific sirtuin activators.
Sirt-1 interactomic study holds the key for understanding associations and interactions between various proteins to develop knowledgeable insights of highly diverse and complex biological systems, which are interwoven into each other. On the basis of the experimental data (see Materials and Methods) at disposal on various public databases, we have performed an interactomic analysis and found 136 direct partners interacting with Sirt-1 that are involved in the important pathways discussed above. Several proteins are biologically active in metabolic processes whereas several others proteins perform gene regulatory functions. Scale freeness of the Sirt-1 interaction map is exhibited by a trend shown by many proteins with logarithmically decreased connectivity and Sirt-1 interactome shows small-world property with smaller diameter and high connectivity (Figure 3). These properties make a network more robust to perturbations like mutations and viral infections. However, these parameters imply that the pleiotropicity nature or the complex associations of the proteins governing different biological processes are found implicated in many pathways. In particular, some proteins that connect more nodes in different pathways are, for example, the hub nodes like JUN, HDAC2, RELA, and SLC25A3. Promislow (2004) showed that the pleiotropicity is linked to higher connectivity of nodes, especially, in senescence. However, there is a significant amount of inferences on possible associations between Sirt-1 and caloric restriction and senescence. Probably, we could suggest that the pleiotropic nature of the proteins interacting with Sirt-1 may address the senescence through the involvement of multiple factors possibly related to stress and mitochondrial proteins or the processes associated with mitochondria. This derives from the fact that with aging there is the progression of many diseases like Parkinson, Huntington and Alzheimer that depend on mitochondrial dysfunction. In Sirt-1 interactome the mitochondrial sirtuins, i.e., Sirt-3, Sirt-4, and Sirt-5, interact with proteins implicated in different metabolic processes (Figure A8 in Appendix) and the deregulation of these proteins by any factor can lead to chronic metabolic disorders. Moreover, the direct interaction provides some insights about the involvement of Sirt-1 in cancer as this protein is also found to be acetylating TP53. In our studies of the Sirt-1 interactome, this protein and some hub proteins like JUN, RELA, and EP300 show to have interactions with many different proteins involved in some processes and in different cellular localizations. Further, Sirt-1 interaction map or other protein interaction networks often demonstrate static picture of bulk amount of complex dynamic interactions. To get perspective on modulation of Sirt-1, there will be necessary studies on dynamics interactions considering the interaction levels in strength, chronology in PPI maps and rate order reaction in case of metabolic processes. It would be very interesting to know the affinity values for the NAD moiety in Sirt family and PARP’s (second order interacting partner) of human Sirt-1 (Kolthur-Seetharam et al., 2006; Bai et al., 2011). Sirt-1 network analysis confined with GO studies showed agreement to the observations of Bai et al. (2011). Moreover, Sirtuin genes are found to be controlling the organism’s health in the times of adversity like in diseased conditions. CR is one of the phenomenon’s that switch on the Sirt-1 genes for regulatory functionality and controlling the metabolic pathways. Therefore, hyper activation of the sirtuin genes might be one of the possible contributory causes for healthier life.
Since Sirt-1 became an interesting and promising target for its importance in life span and for its role in various diseases (Camins et al., 2010), the exploration of its pharmacological aspects has been the topic of key research in last decade. In particular, the attention has been focused on the role of certain small activator molecules that affect the activity of Sirt-1. In literature there are some articles on the interaction between Sirt-1 and activators (Milne et al., 2007; Dai et al., 2010; Huber et al., 2010; Pacholec et al., 2010). In particular, Milne et al. (2007) showed that three synthetic activators, namely SRT1460, SRT1720, and SRT2183, are Sirt-1 activators better than the natural resveratrol because EC values of these three synthetic activators are lower than the natural ones. Moreover, these compounds were reported to bind the Sirt-1 enzyme – peptide substrate complex at an allosteric site. Therefore, these Authors suggested the possibility of developing a new therapeutic approach using both caloric restriction and the direct activation of Sirt-1 using these activators. In 2010, in contrast to Milne et al. (2007), other Authors (Huber et al., 2010; Pacholec et al., 2010) have evaluated the same Sirt-1 activators (SRT1460, SRT1720, SRT2183, and resveratrol) by employing biochemical assays containing native substrates such as the p53-derived peptide lacking the fluorophore as well as purified full-length protein p53 or acetyl-CoA synthetase 1. In these experiments the four activators did not lead to apparent activation of Sirt-1 with native peptide or full-length protein substrates, whereas they activated Sirt-1 with peptide substrate containing a covalently attached fluorophore. In particular, Huber et al. (2010) showed that SRT1720 and SRT2183 effectively decreased acetylated p53 in cells treated with DNA damaging agents but did so in cells that lack Sirt-1. Also Pacholec et al. (2010) evidenced that SRT1720, SRT2183, SRT1460, and resveratrol exhibited multiple off-target activities against receptors, enzymes, transporters, and ion channels. Therefore, they concluded that these four molecules were not direct activators of Sirt-1 and required a fluorophore (named TAMRA) for activating Sirt-1 (Pacholec et al., 2010). Recently, in contrast to Pacholec et al. (2010) and Huber et al. (2010) but in agreement with Milne et al. (2007), Dai et al. (2010) have demonstrated that there are many Sirtuin activating compounds (STACs) that produce biological effects consistent with direct Sirt-1 activation. In this study they evaluated again the three STACs (SRT1720, SRT2183, and SRT1460) and showed that they can accelerate the Sirt-1 catalyzed deacetylation of specific unlabeled peptides composed only of natural amino acids in contrast with those Authors which stated that fluorophores were required for Sirt-1 deacetylation. Therefore, they suggested that these three molecules interact directly with Sirt-1 and activate Sirt-1-catalyzed deacetylation through an allosteric mechanism demonstrating that the complex between STACs and specific fluorophores was not necessary for SIRT1 activation (Dai et al., 2010). As one can see the controversy essentially arises because of the lack of details on both structural and functional activity of Sirt-1. Moreover, in our opinion, authors do not take into account that disordered regions allow binding to multiple partners modulating their function. To achieve this capacity, these regions are able to interact with numerous and various enzymes that operate post-translational modifications of which the kinases are certainly the most studied. They phosphorylate sites that are found almost always in disordered zones modifying in this way both the ability to interact that the function. Therefore, the presence of specific kinases in the various cellular districts, where intrinsically disordered proteins have to be post-translationally modified, is fundamental for the activity of these proteins. In other words, if Sirt-1 with its long disordered terminal arms is controlled by its phosphorylation state (Autiero et al., 2009), its activity for the recognition of protein partners at any one time will be directly dependent on the activity of the kinases and phosphatases that act on it in a specific cellular district. In this regard, it is worth of note that we have found more than 90 putative sites on the human Sirt-1 arms specific for about 40 different human kinases (manuscript in preparation). All the above suggests that in vitro testing of one of these proteins should have in the assay also the kinase necessary for the specific recognition of partners or, at least, a sirtuin already post-translationally modified for the specific substrate. Only reasoning on this basis it will be possible to properly test and compare the functional activity of these proteins. However, often the experimentalists act with the traditional structure centric view characteristic of globular enzymes that cannot be applied to IDPs because their activity in respect of a substrate is strongly dependent on those post-translational modifications required to correctly recognize that substrate. It seems evident that a computational approach in these cases is useful for understanding and directing studies in solution. This has led to the lack of conclusive data particularly on small molecule activators due to the not easy comparability of the results of in vitro and in vivo experiments. We think that the field has been overfocused mainly with functional studies performed without taking account at structural level of the different structural behavior of the intrinsically disordered proteins and of the necessary recognition specificity determined by the presence of the numerous kinases. Moreover, conflicts at physiological level are probably due to animal models that are not genetically appropriate. The issue of longevity is extremely complicated because the aging involves many genes and the small molecules like polyphenols have gained attention because they can enter cellular machinery and exert epigenetic changes in hundreds of genes; therefore, higher standards for genetic analysis are required and it is important to assess if the longevity is due to a direct binding to Sirt-1 or to other physiological effects sirtuin independent. Therefore, in this work we have modeled by flexible docking studies the complexes between Sirt-1 and the four activators (SRT1460, SRT1720, SRT2183, and resveratrol) reported by Milne et al. (2007). Given that we recently modeled the interaction between AROS and the allosteric site of Sirt-1 (Autiero et al., 2009), Milne et al. (2007) and Dai et al. (2010) showed that these molecules can interact directly with Sirt-1 and activate it through an allosteric mechanism, therefore, we have decided to simulate these interactions. In particular, flexible docking study was chosen because of the highly flexible and unordered nature of Sirt-1 protein, that is composed of four different regions (Autiero et al., 2009), of which the two terminal domains are resulted highly unordered. In particular, the area selected for binding of these activators is a flexible loop joining N-terminal and allosteric site. In this particular scenario it is important to concern flexible binding area, as it will add more authenticity to the docking results. In fact, flexible docking environment can mimic the “in vivo” conditions of molecular interaction such as change in certain bond angles or bond lengths take place when two molecules tend to interact. In this work 13 residues present in the allosteric site were chosen to be flexible. In details, these 13 residues selected from the selective binding site area comprise four hydrophilic (SER172, SER173, SER174, TYR185), three hydrophobic (ALA171, VAL188, LEU192), one negatively charged (ASP175), two positively charged (HIS170, HIS191), and three aromatic residues. The significance of aromatic residues and charged residues in the area of active site is very important because they are involved in putative stacking and electrostatic interactions, respectively. Moreover our results evidence that aromatic residues form H-bonds that is important for the structural compactness and stability of the docked complexes. The comparison between flexible docking results and the experimental data indicates that the well known natural activator, resveratrol, does not show good binding affinity for Sirt-1 respect to other synthetic activators (SRT1460, SRT1720, SRT2183). In fact, resveratrol has lower affinity than its synthetic counterparts as shown from binding free energy values (expressed in Kcal/mol) and the lack of H-Bond formation with Sirt-1. Figure A9 in Appendix shows the correlation between the energy values found for the four tested small molecules and the values of EC, experimentally determined (see Table 4). As one can see, while for synthetic molecules there is a correlation coefficient of 0.97 which indicates a good agreement between our structural data of direct binding and physiological data, the resveratrol is the only molecule that does not correlate with the others due to its poor correlation coefficient. This suggests that the biological activity does not depend on a direct binding. Thus, our docking model resveratrol–sirtuin-1 clearly shows that resveratrol is a poor allosteric modulator. Its binding energy is lower than that of the other modulators (see Table 4).
On the basis of these results we can highlight that the use of a flexible docking in the case of intrinsically unordered and highly flexible proteins such as Sirt-1 is able to successfully simulate protein complexes since our docking data are in agreement with the functional data. This is the first example, to our knowledge, that a docking between a flexible and disordered protein and ligands is not only able to simulate the experimental data but also to clearly discriminate between different hypothesis. However recently it has also been reported that ligand-receptor docking studies of CXCR4 (Kufareva et al., 2011) failed to correctly predict the ligand binding sites despite the availability of template GPCR crystal structures. We observe that in the X-ray structure of CXCR4 (PDB: 3ODU) is missing the N-terminus of about 50 residues. This point as we will discuss later is important. Each chemokine receptor has an extracellular N-terminal region, seven helical transmembrane domains with three intracellular and three extracellular hydrophilic loops, and an intracellular C-terminal region. The first and second extracellular loops are linked together by disulfide bonding between two conserved cysteine residues. The N-terminal region of a chemokine receptor is structurally important because it is crucial for ligand specificity whereas the intracellular C-terminal region couples G-proteins and this mechanism is implicated for receptor signaling transduction. In a study in progress in our lab (manuscript in preparation) we have found diffuse presence of disorder in the family of the human chemokine membrane receptors. N and C terminal arms possess structural characteristics such that they can be considered intrinsically disordered with a high structural flexibility and the presence of numerous charged patches and phosphorylation sites. Without any consideration of these important structural aspects of CXCR4 (not resolved by X-ray), we think that dockings failed because evidently the structural features of N-terminus play a crucial role in the binding of those ligands and most of all the flexibility also plays a structural role which must carefully taken into account in docking as we have done with Sirt-1.
To better validate our docking results, we have compared the complexes between Sirt-1 and the four activators (SRT1460, SRT1720, SRT2183, and resveratrol) obtained by AutoDock4, a very useful tool for predicting the complexes conformation (Morris et al., 1998, 2009), with those performed by Glide, a program that uses a different protocol indicated as “flexible” (Halgren et al., 2004). The best complexes generated by this last program in terms of energetic values showed that (i) the four molecules bind the same allosteric site predicted for AROS with good affinity and use about 90% of interactions evidenced by AutoDock4 but with the same number and type of H-bonds and (ii) the correlation coefficient between energy score by AutoDock4 and Glide programs is 0.91 (Figure A9 in Appendix).
These results have evidenced the good accuracy of our complexes between Sirt-1 and four molecules even if the certainty of the result can be obtained only by experimental studies. Hence, further studies will be performed to validate experimentally our computational results by biochemistry assays.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The docking studies by Glide software were performed at Institute for Research in Biomedicine, Molecular Modelling and Bioinformatics group, Barcelona, Spain.
Abbreviations
ADP, adenine diphosphate; AR, androgen receptor; ARNTL, Aryl hydrocarbon receptor nuclear translocator-like; BRCA1, Breast cancer type 1 susceptibility protein; DLD, dihydrolipoamide dehydrogenase; DYNC1H1, dynein, cytoplasmic 1, heavy chain 1; EP300, E1A binding protein p300; FOXOs, forkhead box protein O; HIC1, hypermethylated in cancer 1; HDAC, histone deacetylase; KAT2, K (lysine) acetyltransferase 2; KRT1, keratin 1; MCF2L2, MCF.2 cell line derived transforming sequence-like 2; MYOD1, myogenic differentiation 1; NAD, nicotinamide adenine dinucleotide; NCOR1, nuclear receptor co-repressor 1; NEDD8, neural precursor cell expressed, developmentally down-regulated 8; NFkB, nuclear factor of kappa light polypeptide gene enhancer in B-cells; NUDC, nuclear distribution gene C homolog; PARP1, poly (ADP-ribose) polymerase 1; PPARGC1A, peroxisome proliferator-activated receptor gamma, coactivator 1 alpha; RELA, V-rel reticuloendotheliosis viral oncogene homolog A; RPS27L, ribosomal protein S27-like; RRP8, ribosomal RNA processing 8, methyltransferase, homolog; RTN4, reticulon 4; SLC25A3, solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3; SMAD4, SMAD family member 4; SYNCRIP, synaptotagmin binding, cytoplasmic RNA interacting protein; TP53, tumor protein 53; WRN, Werner syndrome, RecQ helicase-like.
References
Andrusier, N., Mashiach, E., Nussinov, R., and Wolfson, H. J. (2008). Principles of flexible protein-protein docking. Proteins 73, 271–289.
Appella, E., and Anderson, C. W. (2001). Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268, 2764–2772.
Assenov, Y., Ramirez, F., Schelhorn, S.-E., Lengauer, T., and Albrecht, M. (2008). Computing topological parameters of biological networks. Bioinformatics 24, 282–284.
Autiero, I., Costantini, S., and Colonna, G. (2009). Human sirt-1: molecular modeling and structure-function relationships of an unordered protein. PLoS ONE 4, 12. doi:10.1371/journal.pone.0007350
Bai, P., Cantó, C., Oudart, H., Brunyánszki, A., Cen, Y., Thomas, C., Yamamoto, H., Huber, A., Kiss, B., Houtkooper, R. H., Schoonjans, K., Schreiber, V., Sauve, A. A., Menissier-de Murcia, J., and Auwerx, J. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468.
Barabási, A. L., and Oltvai, Z. N. (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113.
Bemis, J. E., Vu, C. B., Xie, R., Nunes, J. J., and Ng, P. Y. (2009). Discovery of oxazolo4,5-b. Pyridines and related heterocyclic analogs as novel SIRT1 activators. Bioorg. Med. Chem. Lett. 19, 2350–2353.
Camins, A., Sureda, F. X., Junyent, F., Verdaguer, E., Folch, J., Pelegri, C., Vilaplana, J., Beas-Zarate, C., and Pallas, M. (2010). Sirtuin activators: designing molecules to extend life span. Biochim. Biophys. Acta 1799, 740–749.
Chatr-Aryamontri, A., Ceol, A., Palazzi, L. M., Nardelli, G., Schneider, M. V., Castagnoli, L., and Cesareni, G. (2007). MINT: the Molecular INTeraction database. Nucleic Acids Res. 35, D572–D574.
Chen, W. Y., Wang, D. H., Yen, R. C., Luo, J., Gu, W., and Baylin, S. B. (2005). Tumor suppressor HIC1 directly regulates SIRT-1 to modulate p53-dependent DNA damage responses. Cell 123, 437–448.
Dai, H., Kustigian, L., Carney, D., Case, A., Considine, T., Hubbard, B. P., Perni, R. B., Riera, T. V., Szczepankiewicz, B., Vlasuk, G. P., and Stein, R. L. (2010). SIRT1 activation by small molecules: kinetic and biophysical evidence for direct interaction of enzyme and activator. J. Biol. Chem. 285, 32695–32703.
Dartnell, L., Simeonidis, E., Hubank, M., Tsoka, S., Bogle, I. D. L., and Papageorgiou, L. G. (2005). Robustness of the p53 network and biological hackers. FEBS Lett. 579, 3037–3042.
Ford, J., Ahmed, S., Allison, S., Jiang, M., and Milner, J. (2008). JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle 19, 3091–3097.
Goh, K. I., Cusick, M. E., Valle, D., Childs, B., Vidal, M., and Barabasi, A. L. (2006). The human disease network. Proc. Natl. Acad. Sci. U.S.A. 104, 8685–8690.
Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., and Banks, J. L. (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759.
Halperin, I., Ma, B., Wolfson, H. J., and Nussinov, R. (2002). Principles of docking:an overview of search algorithms and a guide to scoring functions. Proteins 47, 409–442.
Han, J. D., Bertin, N., Hao, T., Goldberg, D. S., Berriz, G. F., Zhang, L. V., Dupuy, D., Walhout, A. J., Cusick, M. E., Roth, F. P., and Vidal, M. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93.
Harikumar, K. B., and Agarwal, B. B. (2008). Resveratrol: a multitargeted agent for age related chronic diseases. Cell Cycle 7, 1020–1035.
He, X., and Zhang, J. (2006). Why do hubs tend to be essential in protein networks? PLoS Genet. 2, e88. doi:10.1371/journal.pgen.0020088
Howitz, K. T., Bitterman, K. J., Cohen, H. Y., Lamming, D. W., and Lavu, S. (2003). Small molecule activators of sirtuins extend Sacchromyces cerevisae lifespan. Nature 425, 191–196.
Huber, J. L., McBurney, M. W., Distefano, P. S., and McDonagh, T. (2010). SIRT1-independent mechanisms of the putative sirtuin enzyme activators SRT1720 and SRT2183. Future Med. Chem. 2, 1751–1759.
Jeong, H., Mason, S. P., Barabasi, A. L., and Oltvai, Z. N. (2001). Lethality and centrality in protein networks. Nature 411, 41–42.
Joy, M. P., Brock, A., Ingber, D. E., and Huang, S. (2005). High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 96–103.
Keshava Prasad, T. S., Goel, R., Kandasamy, K., Keerthikumar, S., Kumar, S., Mathivanan, S., Telikicherla, D., Raju, R., Shafreen, B., Venugopal, A., Balakrishnan, L., Marimuthu, A., Banerjee, S., Somanathan, D. S., Sebastian, A., Rani, S., Ray, S., Harrys Kishore, C. J., Kanth, S., Ahmed, M., Kashyap, M. K., Mohmood, R., Ramachandra, Y. L., Krishna, V., Rahiman, B. A., Mohan, S., Ranganathan, P., Ramabadran, S., Chaerkady, R., and Pandey, A. (2009). Human Protein Reference Database – 2009 update. Nucleic Acids Res. 37, D767–D772.
Kohl, M., Wiese, S., and Warscheid, B. (2011). Cytoscape: software for visualization and analysis of biological networks. Methods Mol. Biol. 696, 291–303.
Kolthur-Seetharam, U., Dantzer, F., McBurney, M. W., de Murcia, G., and Sassone-Corsi, P. (2006). Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle 5, 873–877.
Kufareva, I., Rueda, M., Katritch, V., GPCR Dock 2010 Participants, Stevens, R. C., and Abagyan, R. (2011). Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 Assessment. Structure 19, 1108–1126.
Kyrylenko, S., Kyrylenko, O., Suuronen, T., and Salminen, A. (2003). Differential regulation of the Sir2 histone deacetylase gene family by inhibitors of class I and II histone deacetylases. Cell. Mol. Life Sci. 60, 1990–1997.
Lin, C.-Y., Chin, C.-H., Wu, H.-H, Chen, S. H., Ho, C. W., and Ko, M. T. (2008). Hubba: hub objects analyzer – a framework o interactome hubs identification for network biology. Nucleic Acids Res. 36, 1–6.
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449.
Marti-Renom, M. A., Stuart, A. C., Sanchez, R., Fiser, A., Melo, F., and Sali, A. (2000). Comparative protein structure modelling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325.
Mason, O., and Verwoerd, M. (2007). Graph theory and networks in biology. IET Syst. Biol. 1, 89–119.
Milne, J. C., Lambert, P. D., Schenck, S., Carney, D. P., and Smith, J. J. (2007). Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716.
Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., and Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an emperical binding free energy function. J. Comput. Chem. 19, 1639–1662.
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Goodsell, D. S., and Olson, A. J. (2009). AutoDock4 and AutoDockTools4: automated docking elective receptor flexibility. J. Comput. Chem. 30, 2785–2791.
Orallo, F. (2006). Comparative studies of antioxidant effects of cis- and trans resveratrol. Curr. Med. Chem. 13, 87–98.
Pacholec, M., Bleasdale, J. E., Chrunyk, B., Cunningham, D., Flynn, D., Garofalo, R. S., Griffith, D., Griffor, M., Laulakis, P., Pabst, B., Qiu, X., Stockman, B., Thanabal, V., Varghese, A., Ward, J., Withka, J., and Ahn, K. (2010). SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 285, 8340–8351.
Pallkes, M., Pizarro, J. G., Gutierrez-Cuesta, J., Crespo-Biel, N., Alvira, D., Tajes, M., Yeste-Velasco, M., Folch, J., Canudas, A. M., Sureda, F. X., Ferrer, I., and Camins, A. (2008). Modulation of SIRT-1 expression in different neurodegenerative models and human pathologies. Neuroscience 154, 1388–1397.
Patil, A., and Nakamura, H. (2006). Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 580, 2041–2045.
Promislow, D. E. (2004). Protein networks, pleiotropy and the evolution of senescence. Proc. Biol. Sci. 271, 1225–1234.
Przulj, N., Wigle, D. A., and Jurisica, I. (2004). Functional topology in a network of protein interactions. Bioinformatics 20, 340–348.
Ravasz, E., Somera, A. L., Mongru, D. A., Oltvai, Z. N., and Barabási, A. L. (2002). Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555.
Sasaki, T., Mayer, B., Koclega, K. D., Chruszcz, M., and Gluba, W. (2008). Phosphorylation regulates SIRT1 function. PLoS ONE 3, e4020. doi:10.1371/journal.pone.0004020
Scardoni, G., Petterlini, M., and Laudanna, C. (2009). Analyzing biological network parameters with CentiScaPe. Bioinformatics 25, 2857–2859.
Schaefer, C. F., Anthony, K., Krupa, S., Buchoff, J., Day, M., Hannay, T., and Buetow, K. H. (2009). PID: the Pathway Interaction Database. Nucleic Acids Res. 37, D674–D679.
Smith, J. J., Kenney, R. D., Gagne, D. J., Frushour, B. P., Ladd, W., Galonek, H. L., Israelian, K., Song, J., Razvadauskaite, G., Lynch, A. V., Carney, D. P., Johnson, R. J., Lavu, S., Iffland, A., Elliot, P. J., Lambert, P. D., Elliston, K. O., Jirousek, M. R., Milne, J. C., and Boss, O. (2009). Small molecule activator of SIRT1 replicate signaling pathway triggered by calorie restriction in vivo. BMC Syst. Biol. 3, 31. doi:10.1186/1752-0509-3-31
Soto, C. S., Fasnacht, M., Zhu, J., Forrest, L., and Honig, B. (2008). Loop Modeling: sampling, filtering and scoring. Proteins 70, 834–843.
Stark, C., Breitkreutz, B.-J., Reguly, T., Boucher, L., Breitkreutz, A., and Tyers, M. (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539.
Tanno, M., Sakamoto, J., Miura, T., Shimamoto, K., and Horio, Y. (2007). Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832.
Tompa, P., and Fuxreiter, M. (2008). Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8.
Tyler, A. L., Asselbergs, F. W., Williams, S. M., and Moore, J. H. (2009). Shadows of complexity: what biological networks reveal about epistasis and pleiotropy. Bioessays 31, 220–222.
Vu, C. B., Bemis, J. E., Disch, J. S., Ng, P. Y., Nunes, J. J., Milne, J. C., Carney, D. P., Lynch, A. V., Smith, J. J., Lavu, S. S., Lambert, P. D., Gagne, D. J., Jirousek, M. R., Schenk, S., Olefsky, J. M., and Perni, R. B. (2009). Discovery of imidazol[1,2-b] thiazole derivatives as novel SIRT1 activators. J. Med. Chem. 52, 1275–1283.
Watts, D. J., and Strogatz, S. (1998). Collective dynamics of “small-world” networks. Nature 393, 440–442.
Wodak, S. J., and Janin, J. (1978). Computer analysis of protein-protein interaction. J. Mol. Biol. 124, 323–342.
Yamazaki, Y., Usui, I., Kanatani, Y., Matsuya, Y., Tsuneyama, K., Fujisaka, S., Bukhari, A., Suzuki, H., Senda, S., Imanishi, S., Hirata, K., Ishiki, M., Hayashi, R., Urakaze, M., Nemoto, H., Kobayashi, M., and Tobe, K. (2009). Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am. J. Physiol. Endocrinol. Metab. 297, E1179–E1186.
Yang, H., Baur, J. A., Chen, A., Miller, C., Adams, J. K., Kisielewski, A., Howitz, K. T., Zipkin, R. E., and Sinclair, D. A. (2007). Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell 6, 35–43.
Yang, T., Fu, M., Pestell, R., and Sauve, A. A. (2006). SIRT1 and endocrine signaling. Trends Endocrinol. Metab. 17, 186–191.
Yu, H., Greenbaum, D., Xin Lu, H., Zhu, X., and Gerstein, M. (2004). Genomic analysis of essentiality within protein networks. Trends Genet. 206, 227–231.
Zhao, K., Harshaw, R., Chai, X., and Marmostein, R. (2004). Structural basis for nicotinamide cleavage and ADP ribose transfer by NAD(+)-dependent Sir2 histone/protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 101, 8563–8568.
Zhao, X., Sternsdorf, T., Bolger, T. A., Evans, R. M., and Yao, T.-P. (2005). Regulation of MEF2 by histone deacetylase 4- and SIRT-1 deacetylase-mediated lysine modifications. Mol. Cell. Biol. 25, 8456–8464.
Zhuang, H., Kim, Y. S., Koeheler, R. C., and Dore, S. (2003). Potential mechanism by which resveratrol, a red wine constituent, protects neurons. Ann. N. Y. Acad. Sci. 993, 276–286.
Appendix
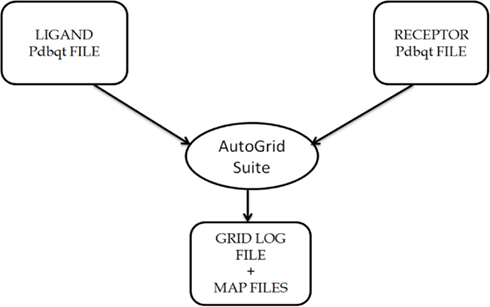
Figure A1. Description of the usage of AutoGrid Suite in the AutoDock4 software package, that generates map files specifying the area over the surface of protein for ligand binding on the basis of information provided in GPF files, i.e., Grid Parameter file.
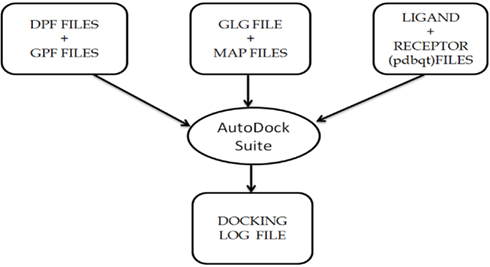
Figure A2. Description of the usage of AutoDock Suite in the AutoDock4 software package, that generates the final docking log file containing information about the final docked complex.
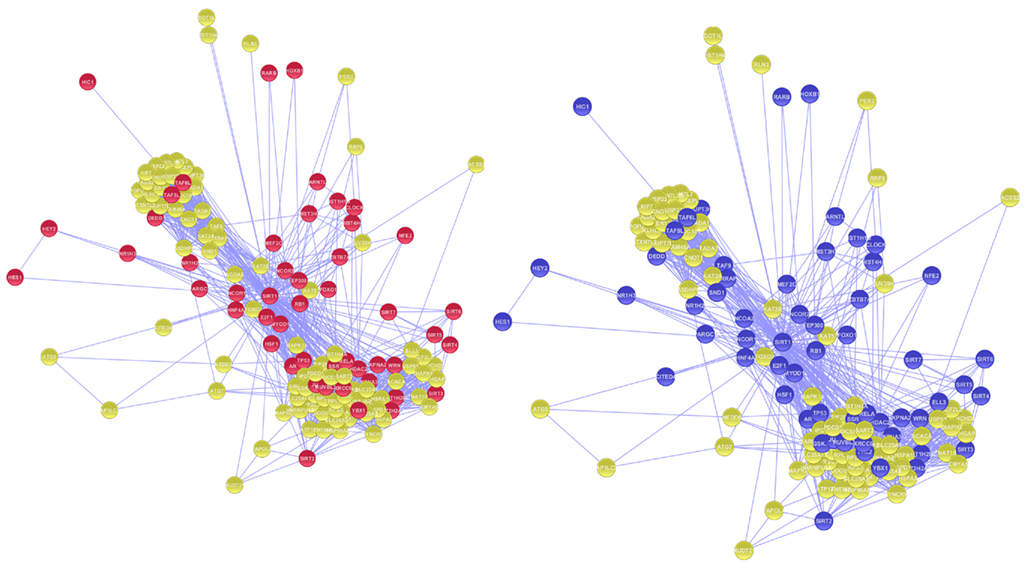
Figure A3. Nodes in magenta represents the proteins involved in DNA binding Activity whereas proteins involved in Transcription regulating activity in GO studies are represented in purple.
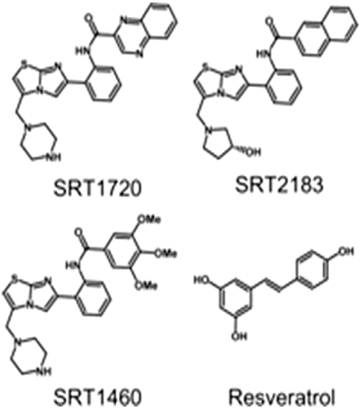
Figure A4. Chemical structures of four activators which are used in molecular docking studies against Sirt-1 (Pacholec et al., 2010).
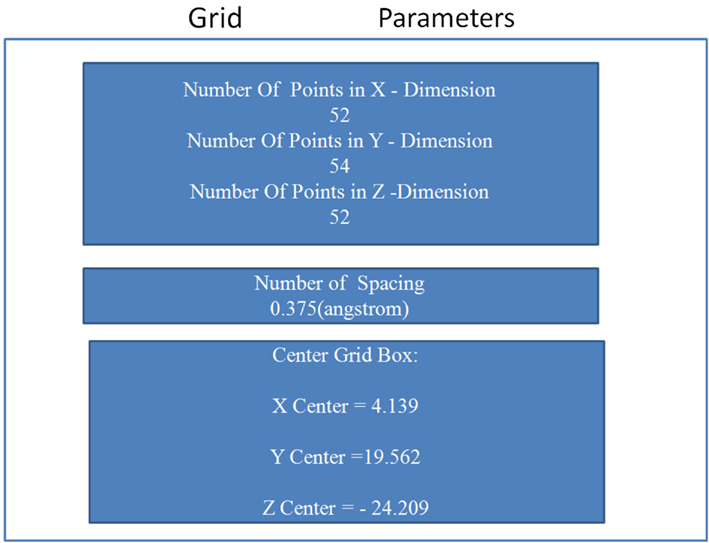
Figure A5. Details of the grid parameters such as “number of spacing,” “number of grid points,” and “center grid box,” in all three directions.
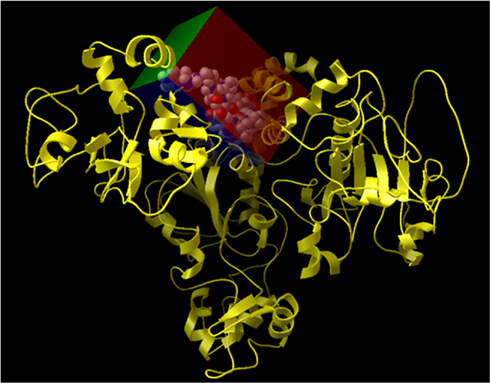
Figure A6. Representation of the grid box created on the surface of Sirt-1 around the allosteric site. In details, Sirt-1 is reported in yellow cartoon conformation and the area of active site in the cpk conformation.
Figure A7. Different view of active site (represented in the surface conformation by Pymol) of four directions. Concerning a clockwise direction, the first view shows the front view (A), the second shows the top view (B), the third shows the side view (C), and the fourth shows the rear side view of active site residues (D).
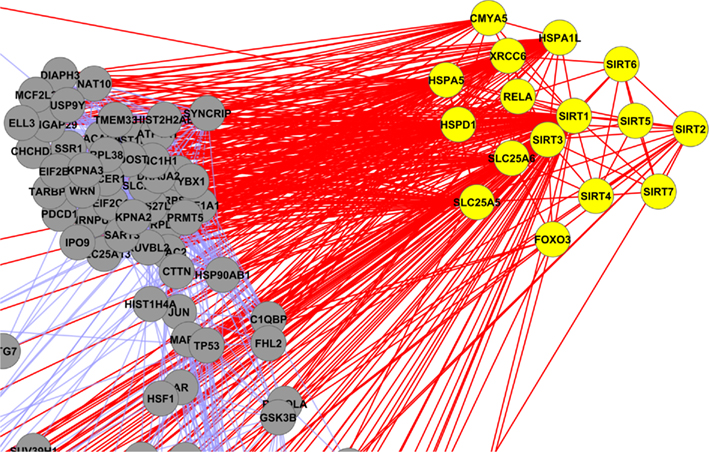
Figure A8. Yellow colored nodes are showing the interaction of mitochondrial sirtuins (Sirt-2, Sirt-3 and Sirt-4).
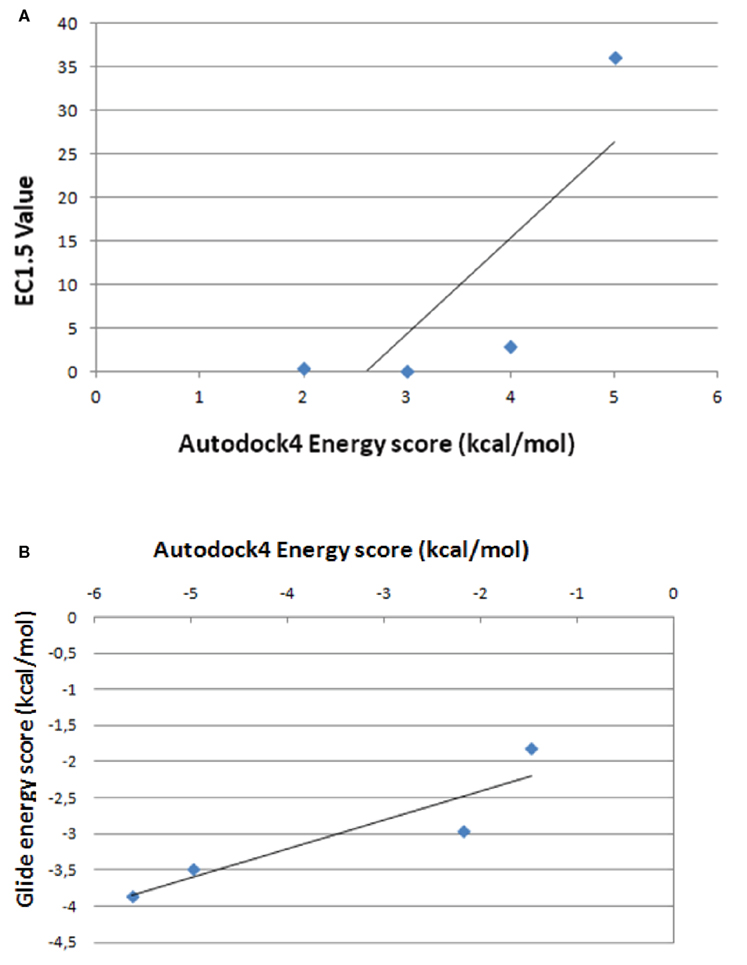
Figure A9. Correlation between energy scores by AutoDock4 and the values of EC1.5, experimentally determined (A) between energy scores by AutoDock4 and Glide programs (B).
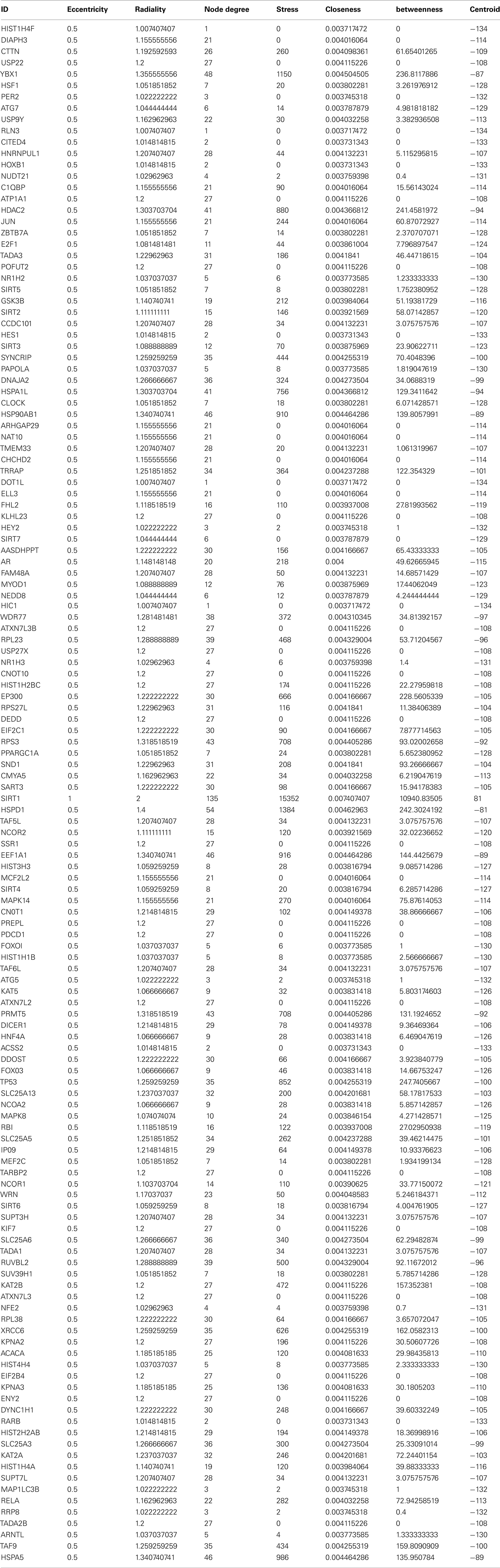
Table A1. The interacting partners and the centrality values associated with Sirt1 direct interacting partners.
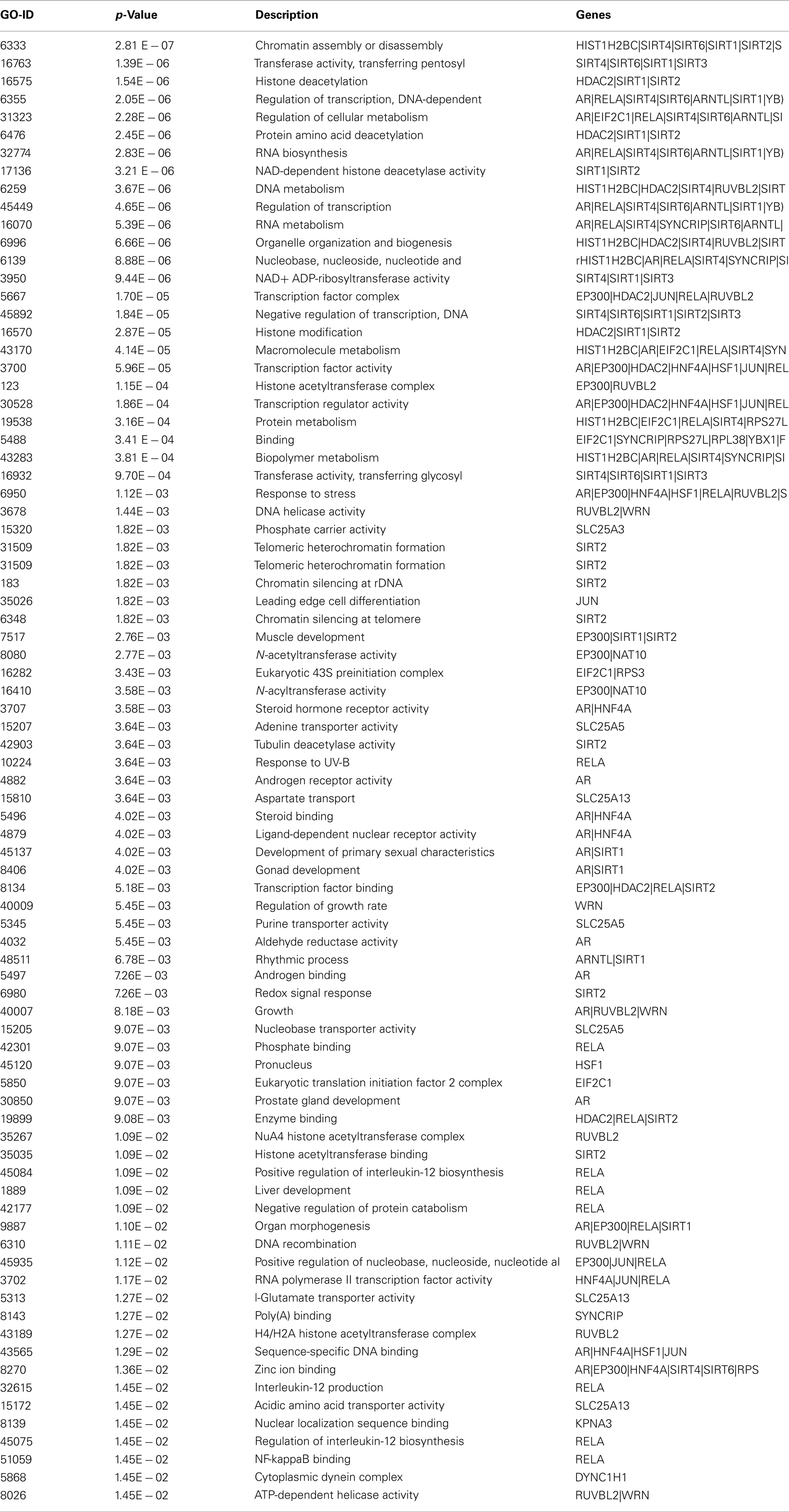
Table A2. Biological processes associated with interacting proteins in the Sirt 1 interaction maps with significant p-value.

Table A3. Interacting protein partner of Sirt-1 involved in various pathways.
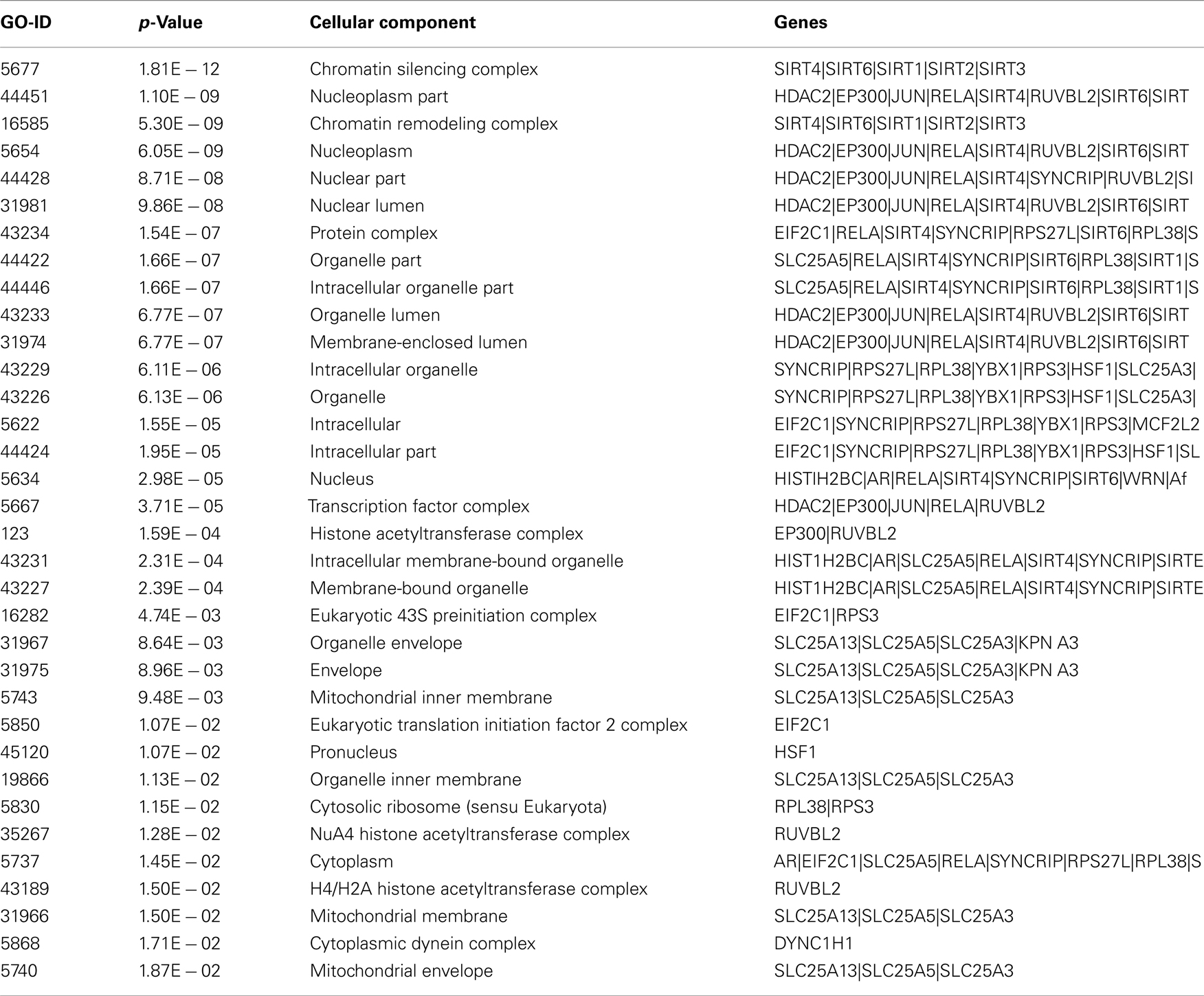
Table A4. Cellular localization of the Sirt-1 interacting proteins by GO studies.
Excel sheets with more the details can be found on these links.
• Complete details about the Biological processes of Sirt1 and its interacting partners as analyzed by GO studies: http://bit.ly/s0XBTz
• Details regarding HUB proteins, Average path length and biological processes and cellular localization associated with Sirtlhub nodes at bit.ly/hubproteinsofSIRT1Network
Keywords: Sirt-1, molecular docking, interactome, activators, interaction map
Citation: Sharma A, Gautam V, Costantini S, Paladino A and Colonna G (2012) Interactomic and pharmacological insights on human Sirt-1. Front. Pharmacol. 3:40. doi: 10.3389/fphar.2012.00040
Received: 25 November 2011; Accepted: 23 February 2012;
Published online: 23 March 2012.
Edited by:
Tiago F. Outeiro, University of Lisbon, PortugalReviewed by:
Roland Seifert, Medical School of Hannover, GermanyAleksey G. Kazantsev, Harvard Medical School and Massachusetts General Hospital, USA
Copyright: © 2012 Sharma, Gautam, Costantini, Paladino and Colonna. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Susan Costantini, INT “G. Pascale” – CROM “Fiorentino Lo Vuolo”, via Ammiraglio Bianco, 83013 Mercogliano Avellino, Italy. e-mail: susan.costantini@unina2.it; Giovanni Colonna, Dipartimento di Biochimica e Biofisica, CRISCEB, Seconda Università degli Studi di Napoli, Via Costantinopoli 16, 80138 Napoli, Italy. e-mail: giovanni.colonna@unina2.it