
- Department of Surgery, Transplant Institute, Beth Israel Deaconess Medical Center, Boston, MA, USA
Biliverdin (BV) has emerged as a cytoprotective and important anti-inflammatory molecule. Conversion of BV to bilirubin (BR) is catalyzed by biliverdin reductase (BVR) and is required for the downstream signaling and nuclear localization of BVR. Recent data by others and us make clear that BVR is a critical regulator of innate immune responses resulting from acute insult and injury and moreover, that a lack of BVR results in an enhanced proinflammatory phenotype. In macrophages, BVR is regulated by its substrate BV which leads to activation of the PI3K–Akt-IL-10 axis and inhibition of TLR4 expression via direct binding of BVR to the TLR4 promoter. In this review, we will summarize recent findings on the role of BVR and the bile pigments in inflammation in context with its activity as an enzyme, receptor, and transcriptional regulator.
History and Characterization of the Bile Pigments
Bilirubin has been accepted for centuries, if not longer, as a major clinical manifestation of jaundice, a yellow hue of the skin. In fact bile pigments can be observed in individuals everyday who have the misfortune of sustaining a contusion or bruise. Indeed the colorimetric nature of the pigments with black heme, green biliverdin, and yellow bilirubin allow one to observe in action the enzymes responsible for heme degradation. Hippocrates was believed to have crafted the notion that “the body of man has in itself blood, phlegm, yellow bile, and black bile and that these four humors make up the nature of his body, and through these man feels pain or enjoys health. The most perfect health is enjoyed when these elements are duly proportioned to one another in respect of compounding, power and bulk, and when they are perfectly mingled.” One might speculate whether Hippocrates was referring to heme or biliverdin as the black bile with BR as the yellow bile. BR is the most potent antioxidant in the serum of mammals, whereas BV is almost undetectable being readily converted to BR by biliverdin reductase (BVR) in most cells. In contrast, in lower vertebrates (avian or fish), BV is a critical pigment of the blood, bile, and egg shells of large bird species such as the Emu (Zhao et al., 2006). Cell energy expenditure for the reduction of BV to BR shows not only the importance for generation of a strong antioxidant, but also may underscore the mechanism for activation of critical BVR signaling cascades. Recent data describe kinase and transcriptional activities of BVR, which suggest important additional roles for BV as an intermediate in the catalysis of heme.
Physiology of Bile Pigments: Why is BV an Intermediate of Heme Catalysis? Is it Needed at All?
Biliverdin is formed in a single reaction during catalysis of heme by heme oxygenases (HO-1 and HO-2). Carbon monoxide and iron are also released at the same time. HO-1 is one of two HO isoforms and is inducible and designated as a protective gene mediating its effects through generation of one or more of its products (Otterbein et al., 2003). The rate of hemoglobin degradation in the reticulo-endothelial organs (spleen and liver) is slow, therefore all BV to BR is readily reduced particularly in the spleen, liver, kidney, and brain. Biliverdin, in contrast to BR, is soluble and readily excreted into bile. Bilirubin has to be glucuronidated to be excreted, which occurs in the liver. One of the major benefits in generating BR is its antioxidant power in the circulation. The normal concentrations of BR in humans varies between: 0.5 and 1 mg/dL (8.6–17.1 μM) which is sufficient to alleviate oxidative stress (McDonagh, 2001). In vitro, nanomolar concentrations of bilirubin inhibit ROS generation (Baranano et al., 2002). Bilirubin is toxic however, primarily in neonates in which the direct bilirubin may cause brain injury termed kernicterus if elevated prior to formation of the blood brain barrier. Bile pigments have a strong ability to absorb light and therefore phototherapy is used as a treatment option for newborns with hyperbilirubinemia. Under certain wavelengths of light, BR is converted to lumirubin and photobilirubin as isomers of BR which can readily be excreted in the urine (Pratesi et al., 1985). In contrast to the dangers of elevated BR in neonate, high normal concentrations of bilirubin (>1 mg/dL; >34.3 μM) in adults correlates with better cardiovascular function and less inflammation (Schwertner et al., 1994; Mayer, 2000; Novotnây and Vâitek, 2003).
Role of BR – Mild Hyperbilirubinemia
Approximately 5% of the human population carries a mutation in the glucuronyl transferase gene and therefore these individuals present with increased levels of direct bilirubin in the serum due to a low level of glucuronidation and secretion as bile. Gilbert’s syndrome is associated with high normal bilirubin levels (∼2 mg/dL; ∼34.3 μM), which correlates with decreased risk of coronary heart disease and arteriosclerosis (Bulmer et al., 2008) as well as systemic lupus erythematosus (Vitek et al., 2010). Gilbert’s syndrome is caused by a genomic homozygous polymorphism, A(TA)7TAA, in the promoter of the gene for UDP-glucuronosyltransferase 1A1 (UGT1A1), which leads to elevated serum levels of unconjugated bilirubin. Development and sustained presence of this mutation throughout evolution may indicate the importance of BR as a major cytoprotectant against ROS and RNS for mammals living in an oxidant environment. BR interacts with NO forming a N-nitroso derivative and therefore can be a scavenger of NO to counteract nitrosative stress (Minetti et al., 1998; Mancuso et al., 2003, 2006). BR is a more potent scavenger of superoxide radicals and peroxynitrite then BV, and therefore a lack of BVR may lead to accumulation of excessive oxidative stress in endothelial and other cells that leads to cell death and tissue injury (Jansen et al., 2010).
BVR-Identification and Characterization
Biliverdin reductase has been known for many decades as the second enzyme in the heme degradation pathway necessary for conversion of BV to BR (Frydman et al., 1987; Bell and Maines, 1988) which serves as both a radical scavenger (Stocker et al., 1987), but is also critical in lipid emulsion during digestion. There are two isoforms of BVR. BVR-A, which catalyzes conversion of BV-a specifically and is expressed in the majority of adult tissues and inducible with stress, and BVR-B which is present during embryogenesis and is an isoform specific for the BV-d and b isomers. Both enzymes catalyze reduction of a double-bond between the pyrrole ring into a single-bond using NADH or NADPH as electron donors dependent on a pH optima (6.75 and 8.7, respectively; McCoubrey et al., 1995). BR and BV, through BVR have been shown to prevent cellular senescence (Kim et al., 2011) and apoptosis (Jansen et al., 2010). Regulation of HO-1 and BVR expression and their enzymatic activity is critical for function of the heme degradation pathway. BVR expression is regulated negatively by NF-κB activation and positively by hypoxia-mediated HIF1α stabilization and specific HRE binding sites in the BVR promoter (Gibbs et al., 2010). BVR is strongly induced by its substrate, biliverdin as well as other agents that induce oxidative stress including LPS, heavy metals, and toxins (Maines et al., 2001; Wegiel et al., 2009). BVR has also been demonstrated to be the target for statins with this interaction leading to significant cognitive benefits in a preclinical model of Alzheimer’s disease (Barone et al., 2012). BVR is expressed ubiquitously in all tissues under basal conditions with high levels in the reticulo-macrophages in the spleen and liver. BVR can also be induced. We view this as placing BVR in the category of stress response genes, which is supported by the multiplicity of its functions within the cell, particularly in the context of inflammation. BVR is localized in different cellular compartments in response to stress. BVR has been detected in the membrane (Kim et al., 2004; Wegiel et al., 2009), cytoplasm/ER, mitochondria (Converso et al., 2006), and nucleus (Maines et al., 2001; Ahmad et al., 2002; Lerner-Marmarosh et al., 2008) and its translocation between different cellular compartments is regulated by nitrosylation, lipid modification, or phosphorylation (Wegiel et al., 2009, 2011). Importantly, BV induces NO generation allowing for the stabilization and translocation of BVR to the nucleus (Wegiel et al., 2011). We showed that BVR is strongly induced upon bacteria endotoxin treatment both in the nucleus and on the cell surface (BVRsurf). BVRsurf is newly characterized as an important isoform of BVR, which functions as a tyrosine kinase receptor-mediated signaling dimer in the membrane to bind and convert BV to BR. Simultaneously, with BV binding, BVR cross-phosphorylates Y 198MKM motifs allowing for interaction with PI3K p85α (Lerner-Marmarosh et al., 2005; Maines, 2007; Wegiel et al., 2009; Figure 1).
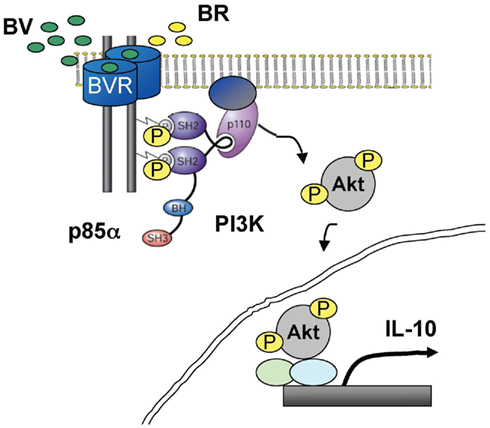
Figure 1. A role of BV in activation of PI3K–Akt through cell surface BVR to elicit anti-inflammatory effects. BVR is expressed on the cell surface of macrophages and mediates the anti-inflammatory and cytoprotective signaling of BV. BV is converted to BR by BVRsurf, which drives the recruitment and activation of PI3K–p85α/p110α to BVR to increase the IL-10 levels.
Conversion of BV by BVR has been shown to occur in any cellular compartment, however the majority of activity is detected in the ER and cell membrane. Modification of BVR by phosphorylation (Pachori et al., 2007) or S-nitrosylation (Wegiel et al., 2011) are important regulatory mechanisms for its activity as an enzyme. Phosphorylation of BVR on tyrosine occurs after ligand binding and is necessary for transmission of the signal from BVR to PI3K and downstream activation of Akt (Figure 1). Additionally, phosphorylation of serine/threonine as well as S-nitrosylation of BVR increases enzymatic activity and are important for bilirubin generation (Salim et al., 2001; Pachori et al., 2007). Post-translational modifications of BVR are evident in neurodegenerative models, where the presence of ROS and NO increases oxidative and nitrosative forms of BVR (Barone et al., 2011a,b). Nitrosylation of tyrosine residues in the brain of subjects with Alzheimer’s disease and those with mild cognitive impairment regulates BVR activity (Barone et al., 2011b).
Function and Regulation of BVR
Antioxidant Bilirubin Redox Cycle
The majority of the effects ascribed to BV are mimicked with BR, however the signaling pathways are vastly different including the effects on BVR. BR blocks iNOS expression as well as proinflammatory cytokine expression during endotoxemia (Wang et al., 2004; Lanone et al., 2005), improving the outcome in a lethal models of endotoxemia in rodents (Sarady-Andrews et al., 2005; Kadl et al., 2007; Wegiel et al., 2009). Some of the beneficial signaling and modification effects ascribed to BVR in response to BV, are also observed in response to BR (Baranano et al., 2002). In this cytoprotective amplification loop, biliverdin is able to be regenerated through oxidation of BR by ROS and then reduced back to BR by BVR (Baranano et al., 2002). Importantly, BR also interacts and neutralizes NO radicals forming NO-bilirubin (Barone et al., 2009). The cytoprotective function of BVR is therefore even more apparent with its dual ability to generate BV and BR interchangeably. However, this mechanism may not be fully functional as BR forms mainly non-specific oxidation products, which cannot be used by BVR in the amplification loop. Oxidation of BR by peroxyl radicals generates ∼30% BV as product, however when albumin-bound BR was used there was only a modest increase in BV (<10%; Maghzal et al., 2009). This amount however may be sufficient to drive activation of BVR and formation of additional BR in the amplification cycle described above due to exhaustion of the substrate. In this light, BVR can be labeled as a major protective enzyme. Sedlak et al. (2009) showed that knockdown of BVR in vitro leads to an increase in cell death in response to hydrogen peroxide. Similar antioxidant mechanisms of protection mediated by BVR have been reported in a model of experimental autoimmune encephalomyelitis (Liu et al., 2006).
Kinase Activity IGF/NF-κB
Biliverdin reductase exhibits theronine-serine and tyrosine kinase activity (Salim et al., 2001; Lerner-Marmarosh et al., 2005). While the enzymatic function of BVR was described in 1965, additional roles for BVR have just recently been described. BVR interacts and regulates the insulin receptor, extracellular regulated kinase (ERK), and phosphatidylinositol-3 kinase (PI3K), among other potential interacting proteins such as insulin receptor kinase-1 (IRK-1) or toll-like receptors (TLR). The unique Y198MKM motif of BVR allows BVR the ability to mimic the function of a receptor-like protein by interaction with upstream signaling proteins similar to growth factor receptors such as PDGFR or EGFR. We showed that BVR interacts with PI3K–p85α to drive Akt signaling and IL-10 production, while others demonstrate that BVR regulates the activity of PKC, ERK, and IRK by direct interaction and phosphorylation. BVR can also autophosphorylate and in a hyperphosphorylated form functions better as a catalyst for generating BR (Salim et al., 2001). One of the critical targets for BVR in insulin signaling is IRS-1, which increases phosphorylation of BVR by IRK (Lerner-Marmarosh et al., 2005) and is a key element to insulin resistance. Indeed knockdown of BVR leads to an increase in glucose uptake after insulin treatment in HEK 293 cells.
While biliverdin treatment inhibits NF-κB activation in response to TNFα (Gibbs and Maines, 2007) or LPS (Wegiel et al., 2009) as well as activity of PKC, the role of BVR remains controversial. On the one hand, BVR inhibits NF-κB activation in response to LPS in macrophages and the effects are amplified with BV (Wegiel et al., 2011). Similarly, BV inhibited transcriptional activity of NF-κB in HEK293A cells (Gibbs and Maines, 2007). However, on the other hand, in the same cells it has been shown that overexpression of hBVR enhanced both basal and TNF-α-mediated activation of NF-κB (Gibbs and Maines, 2007). The differences between macrophages and HEK293A cells may be due to the presence or lack of receptors (TLRs or TNFR), which mediate the upstream signaling through NF-κB.
Both PKC-β II and ERK MAPK are important signaling targets for BVR (Lerner-Marmarosh et al., 2005, 2008). Interaction of BVR with PKC-β II as well as BVR-mediated activation of PKC-β II through phosphorylation is critical for its translocation to the membrane. Both BVR and PKC-β II colocalize in the cell membrane and signal from this localization. Similarly, BVR is critical in assuring the localization of ERK proteins to the nucleus (Lerner-Marmarosh et al., 2008). BR induces phosphorylation of Erk in astrocytes and primary rat cerebellar granule neurons and therefore implicates BVR in MAPK activation (Fernandes et al., 2007; Mancuso et al., 2008). BVR forms a ternary complex with PKCδ–ERK2 that is essential for ERK2 signal transduction and activation of genes linked to cell proliferation and cancer (Gibbs et al., 2012). In addition to the protein–protein interactions with BVR, hematin is transported to the nucleus in a complex with BVR to regulate HO-1 expression (Ahmad et al., 2002; Kravets et al., 2004).
Surface BVR and PI3K–Akt Signaling
Biliverdin reductase is expressed in the membrane of endothelial cells as well as macrophages. It has not been formally evaluated in any other cell types. One of the reasons for the necessity of BVR to be located on the membrane of the cell is a need for rapid conversion of BV to BR as well as activation of signaling cascades initiated by BVR. Intravenous injection of BV leads to an immediate increase in serum BR and the process is completed 5–10 min after injection. BVR was first identified in caveolae together with HO-1 in EC (Kim et al., 2004). We further characterized expression on the external surface of the plasma membrane and, as such, colocalizes with surface markers such as TLR4 or F4.80 in macrophages (Wegiel et al., 2009). In this localization, BVR is a dimer and an active kinase, that upon encountering BV as its ligand, is cross-phosphorylated to transmit a signal to PI3K (Figure 1).
One of the major determinants of cellular fate is the Akt pathway. The broad number of targets of Akt kinase makes this cascade important not only for cell migration, survival, proliferation, growth, but also for metabolic control of the cell. BVR acts as a substrate for insulin receptor tyrosine kinase (IRK) as well as phosphorylation of IR substrate-1 (IRS-1) and therefore may regulate insulin signaling and glucose uptake (Lerner-Marmarosh et al., 2005). Tyrosine 198 in the YMKM motif is a substrate for insulin-activated IRK (Lerner-Marmarosh et al., 2005) as well as the formation of the BVR dimer (Wegiel et al., 2009) and when phosphorylated binds PI3K p85α to drive Akt phosphorylation. We have shown that BVR–Akt signaling is active in response to exogenous treatment with biliverdin and drives expression of the anti-inflammatory cytokine IL-10 production in macrophages (Figure 1).
Biliverdin reductase mediates hypoxia induced epithelial to mesenchymal transition via PI3K (Zeng et al., 2008). The crosstalk between BVR and cellular signaling is very pleiotropic and not limited to PI3K–Akt. Further the signaling control by BVR is strongly correlated with its nuclear function. We posit that BVR is the principal receptor for BV and one of the reasons for its immediate conversion to BR is control of BVR functionality as a signaling molecule.
BVR as a Transcriptional Modulator
It is well-established that BVR dimerizes both in the nucleus and acts as a leucine zipper–like transcription factor (Ahmad et al., 2002; Wegiel et al., 2009). Binding of BVR to Ap-1 sites either activates transcription (HO-1 promoter; Ahmad et al., 2002) or blocks the expression of the gene (TLR4; Wegiel et al., 2011). BVR as a dimer binds to a 100-mer DNA fragment of the mouse HO-1 promoter region as well as in the ATF2 promoter (−612 to +33) encompassing two activator protein (AP-1) sites (Ahmad et al., 2002). Mutation of Lys(143), Leu(150), or Leu(157) blocks the interaction between hBVR DNA complex formation. Further, BVR is essential as a transporter of heme to the nucleus to regulate HO-1 gene expression in the cell (Tudor et al., 2008). The feed-forward activation of HO-1 via BVR may therefore allow for additional production of BV as the substrate for BVR and amplification of signal under situations of persistent cellular stress.
We recently described the ability of BV to trigger Ca2+/CaMKK signaling that then leads to phosphorylation of eNOS and increased NO generation in macrophages with concomitant S-nitrosylation of BVR (Wegiel et al., 2011). This modification of BVR amplifies its enzymatic function and drives its nuclear translocation (Figure 3). In RAW264.7 macrophages, treatment with BV suppressed TLR4 expression and resulted in decreased proinflammatory cytokine release. Stable knockdown of BVR in macrophages resulted in elevated expression levels of TLR4 and TNFα. We further showed that inhibition of the TLR4 promoter occurs via a direct interaction of BVR with AP-1 sites on the TLR4 promoter. However due to the close proximity of the Ap-1 and GATA4 sites in the TLR4 promoter, BVR blocks TLR4 expression in contrast to the Ap-1-mediated effect on ATF2 where BVR activated the HO-1 promoter.
Knockdown of BVR results in a significantly heightened proinflammatory phenotype of macrophages with elevated levels of TNFα and activation of TLR4 expression basally reflecting the lack of BVR-regulated TLR4 expression. Further, we demonstrate that decreased NO bioavailability using an inhibitor of NOS in the liver prior to treatment with BV had similar effects as inhibition of BVR, suggesting that modification of BVR via S-nitrosylation is important for the protective effects of BV–BVR. Indeed, BV is required for the post-translational modifications of BVR that enable S-nitrosylation and phosphorylation (Figure 2). We speculate that inflammation or oxidative stress, which leads to induction of HO-1, generates BV to direct BVR to the nucleus to maintain the balance between the expression of cytoprotective genes (i.e., HO-1) and proinflammatory regulatory proteins (i.e., TLR4, TNFα) to prohibit unfettered inflammation.
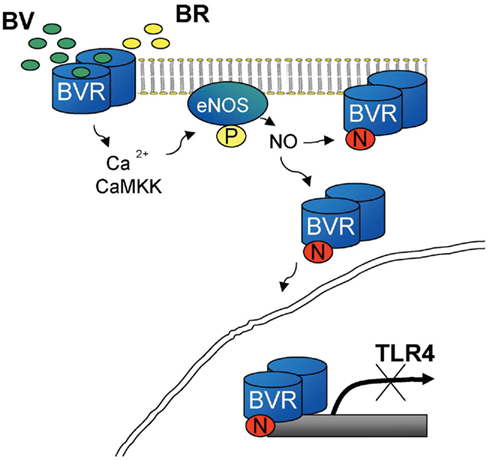
Figure 2. BV-mediated inhibition of TLR4 is dependent on NO-driven BVR translocation to the nucleus. BV drives Ca2+/CaMKK dependent activation of phosphorylation of eNOS and NO production. Presence of NO allows for S-nitrosylation of BVR and its translocation to the nucleus. Nuclear BVR as a transcriptional regulator binds to the TLR4 promoter to inhibit its expression. The mechanism is responsible for the preconditioning in the model of acute hepatitis.
Biliverdin/Bilirubin Effects in Disease Models
There are several preclinical models in which BV and BR have been shown to impart salutary effects in animals. Exogenous treatment with BV blocks inflammation including reducing infiltrating cells and proinflammatory cytokine expression in models of endotoxemia, ischemia reperfusion injury, or epithelial regeneration (Bellner et al., 2008, 2011). Further, biliverdin inhibits the complement cascade, especially at the C1 step in the classical pathway at low micromolar concentrations (Nakagami et al., 1993). A direct interaction between BV and C components of complement and its inhibition may be one of the cytoprotective mechanisms of BV during inflammation. Oral administration of BV before the anti-serum injection protects against anaphylaxic shock induced-death (Nakagami et al., 1993).
Salutary effects of bile pigments have also been described in models of vascular injury and transplantation as will be described in detail below (see also Figure 3). The general hypothesis for the similarity in the action of BV and BR in these models stands from the fast conversion of BV to BR. Amplification of the BVR signal may be due to BV, which is generated from the oxidized BR in the cytoprotective cycle (Baranano et al., 2002). However, not all of the effects of BV are interchangeable with BR, which may be due to requirement for BV to activate BVR. We would argue that generation of even low amounts of BV in the BR → BV → BR amplification loop may be sufficient to activate BVR. This signaling from the oxidized BR (BV) may be responsible for overlapping effects of both molecules, However, the signal may be too low in some instances. The evolutionary conserved generation of BV but not BR (in lower vertebrates) may be due to its additional anti-inflammatory function of BV in regulating BVR–Akt and BVR–TLR4 signaling. Reduction of BV to BR, even energy consuming, maybe a critical regulatory event in activation of signaling function of BVR.
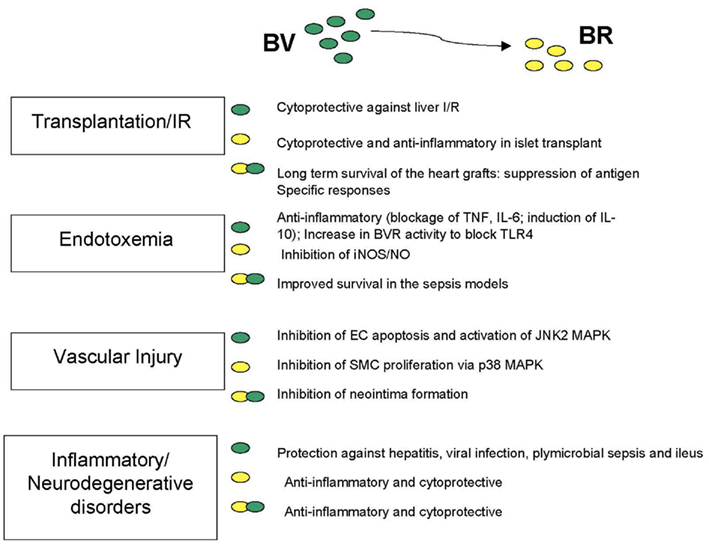
Figure 3. The cytoprotective and anti-inflammatory role of BV and BR in the disease models.
IRI/Transplantation
Biliverdin/BR treatment can mimic that observed with HO-1 induction, exemplified by imparting strong anti-inflammatory and immunosuppressive effect in macrophages (Sarady-Andrews et al., 2005) and T cells (Yamashita et al., 2004). In models of heart or islet transplantation, BV/BR suppresses antigen-specific responses and leads to long-term allograft tolerance (Yamashita et al., 2004; Lee et al., 2007). BV/BR inhibits proliferation of recipient splenocytes in the mixed leukocyte reaction interfering with IL-2 production (Yamashita et al., 2004) as well as MHCII expression (Wu et al., 2005). BR administration blocks inflammatory cell infiltration into the graft and promotes anti-apoptotic gene expression while inhibiting proinflammatory cytokine expression in transplanted islets (Lee et al., 2007). BV alone improved liver function and decreased ALT levels after IRI associated with liver transplantation. Strong antioxidant and anti-inflammatory effects were observed when BV was applied before surgery in a model of small bowel transplantation in rats that concomitantly ameliorated postoperative ileus (POI), a complication from excessive bowel manipulation (Nakao et al., 2004). POI is a major clinical problem for any abdominal surgical procedures and results from a profound inflammatory response in the gut that can lead to sepsis, shock and death from lack of bowel motility. Clinically, this syndrome is very common and leads to extended hospital stays because physicians will not discharge a patient without the presence of bowel sounds. The protective effects of BV in ameliorating POI was associated with inhibition of proinflammatory cytokines (IL-6, IL-1β), decreased infiltration of neutrophils and suppression of intestinal circular muscle contractility (Nakao et al., 2004). Whether the beneficial effects observed with BV in the IRI and POI models are driven by BVR directly, or involve only BR exerting global anti-inflammatory effects remains to be elucidated.
Endotoxic Shock and Hepatitis
Biliverdin has best been characterized as an anti-inflammatory molecule that can inhibit viral replication in vitro (Zhu et al., 2010) as well as protection against endotoxic shock (Sarady-Andrews et al., 2005; Wegiel et al., 2009) and microbial sepsis (Overhaus et al., 2006). Importantly, biliverdin has antiviral effects through inhibition of hepatitis C virus protease activity (Zhu et al., 2010). The effects of BV were mediated through BVR (Zhu et al., 2010). Further, BV ameliorates experimental dextran-sulfate-induced colitis (Berberat et al., 2005) inhibited IL-6 and MCP-1 while boosting IL-10 expression in a model of cecal ligation and puncture (CLP)-induced septic shock in rats. LPS induces BVR surface expression in macrophages. Expression of the anti-inflammatory cytokine IL-10 in response to BV–BVR activation is solely dependent on the Akt signaling pathway as BV-induced IL-10 is inhibited in the presence of dominant-negative Akt. Depletion of BVR in the liver using small interfering RNA (siRNA) against BVR with an adenoviral construct completely inhibited the protective effects of BV against acute hepatitis (Wegiel et al., 2009) which partially involves NO-dependent BVR-regulated TLR4 inhibition (Wegiel et al., 2011).
Vascular Injury
Biliverdin and BR have potent protective effects in models of vascular injury. Biliverdin prevents neointima formation in a model of vascular remodeling involving arterialization of vein grafts through inhibition of JNK1/2 MAPK in the graft as well as via inhibition of EC apoptosis (Nakao et al., 2005). Further, BV and BR blocked SMC proliferation through inhibition of p38 MAPK and blockade of cell cycle progression in G1-S phase, correlating with decreased hyperphosphorylation of Rb and YY1 expression (Ollinger et al., 2005). These in vitro studies were corroborated in vivo in rats in response to balloon trauma. Hyperbilirubinemic Gunn rats validated these observations as they were resistant to neointima formation in response to balloon trauma as compared to wild type controls (Ollinger et al., 2005). The role of BR remains to be determined. It has been well-described however with association studies that Gilbert’s individuals show very low incidence of atherosclerosis and heart disease which suggest an important role for BR (Novotnây and Vâitek, 2003; Bulmer et al., 2008).
Future Studies
BVR–fl/fl and Knockout Mice
Small interfering RNA is the only strategy to date to inhibit BVR expression in order to genetically assess the role of BVR. The field is lacking critical knockout and conditional knockout mice that allow detailed mechanistic testing of BVR in vitro and in vivo and its contribution to bile pigment turnover and immune regulation. We have generated conditional BVR–fl/fl mice (unpublished data) and have specifically deleted BVR in macrophages. Preliminary data show that a lack of BVR in macrophages mimics that observed with siRNA studies; both resulting in a proinflammatory phenotype of myeloid cells, their expansion in the lung and spleen, and subsequent alterations in response to organ injury (data not shown). Deletion of BVR in conjunction with HO-1 knockout mice allows the opportunity to study individually the role of each of the components of the heme degradation pathway in various tissues and under differing kinetics and stimuli. Unlike typical knockout strategies, the conditional approach to tissue specific deletion of BVR and/or HO-1 allows the study of these genes without any compensatory mechanisms in place. Indeed the HO-1 knockout shows <5% fecundity, which begs the question as to what is truly being studied and what conclusions can be drawn from these mice. Have these 5% somehow adapted or compensated? And as such, are they truly allowing appropriate conclusions to be drawn as to the role of HO-1 in naïve and disease scenarios? The conditional knockout mice will certainly shed important light on the role of HO-1 and BVR in basal physiology and importantly, their role in pathophysiology.
Summary and Conclusion – Potential Therapeutic Modalities and Applications
Biliverdin reductase, a pleiotropic signaling molecule and transcriptional regulator is a clear therapeutic target. The Asian community has been using bile salts and pigments for medicinal purposes for centuries if not millennia as a cure-all for everything from headaches to indigestion. In fact, it is firmly believed that bile from different species carries diverse biological effects. BV is an agonist of BVR and an activator of PI3K signaling, the activity of which is altered in various pathologies (insulin resistance, cancer). Recently BVR peptides have been designed to modulate insulin receptor signaling (Maines, 2010).
Importantly, a homozygous nonsense mutation in BVR (Cys214 → Ala) in humans has been described and results in accumulation of biliverdin during episodes of cholestasis (Nytofte et al., 2011). The functional consequences of this mutation and likely others are not yet known, however they may contribute to the development of proinflammatory syndromes, as we are starting to observe in the BVR-deficient mice. The complexity of BVR to both signal and modulate transcriptional activity in addition to enzymatic generation of BR, places it as a central control switch employed by the cell to respond appropriately to inflammation and stress. Exogenous delivery of BV is of potential therapeutic interest given its strong anti-inflammatory potential and defined mechanisms of action. That BV and BR are natural substances make clinical development perhaps more straightforward because the means by which the body metabolizes each pigment is known making pharmacokinetic and pharmacodynamic studies less complicated.
In conclusion, BVR is a truly remarkable molecule. It is triple-threat functionality places it in a category all its own with no other known molecule that has been defined that functions as an enzyme, a signaling kinase, and a regulator of transcription. Understanding what directs one activity over the other is the challenge for future research. Generating tools such as the cre–lox mice will no doubt open up knew avenues of research with exciting discoveries. Eventually, we envision BV and BR as powerful therapeutics that will prove useful in numerous disease pathologies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Julie Henry Fund at the Transplant Center of the BIDMC for their support. The work was supported by NIH 5R01GM088666 to Leo E. Otterbein and AHA 10SDG2640091 to Barbara Wegiel.
References
Ahmad, Z., Salim, M., and Maines, M. D. (2002). Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J. Biol. Chem. 277, 9226.
Baranano, D. E., Rao, M., Ferris, C. D., and Snyder, S. H. (2002). Biliverdin reductase: a major physiologic cytoprotectant. Proc. Natl. Acad. Sci. U.S.A. 99, 16093.
Barone, E., Di Domenico, F., Cenini, G., Sultana, R., Cini, C., Preziosi, P., Perluigi, M., Mancuso, C., and Butterfield, D. A. (2011a). Biliverdin reductase – a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 1812, 480.
Barone, E., Di Domenico, F., Cenini, G., Sultana, R., Coccia, R., Preziosi, P., Perluigi, M., Mancuso, C., and Butterfield, D. A. (2011b). Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 25, 623.
Barone, E., Mancuso, C., Di Domenico, F., Sultana, R., Murphy, M. P., Head, E., and Butterfield, D. A. (2012). Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 120, 135.
Barone, E., Trombino, S., Cassano, R., Sgambato, A., De Paola, B., Di Stasio, E., Picci, N., Preziosi, P., and Mancuso, C. (2009). Characterization of the S-denitrosylating activity of bilirubin. J. Cell. Mol. Med. 13, 2365.
Bell, J. E., and Maines, M. D. (1988). Kinetic properties and regulation of biliverdin reductase. Arch. Biochem. Biophys. 263, 1.
Bellner, L., Vitto, M., Patil, K. A., Dunn, M. W., Regan, R., and Laniado-Schwartzman, M. (2008). Exacerbated corneal inflammation and neovascularization in the HO-2 null mice is ameliorated by biliverdin. Exp. Eye Res. 87, 268.
Bellner, L., Wolstein, J., Patil, K. A., Dunn, M. W., and Laniado-Schwartzman, M. (2011). Biliverdin rescues the HO-2 null mouse phenotype of unresolved chronic inflammation following corneal epithelial injury. Invest. Ophthalmol. Vis. Sci. 52, 3246.
Berberat, P. O., A-Rahim, Y. I., Yamashita, K., Warny, M. M., Csizmadia, E., Robson, S. C., and Bach, F. H. (2005). Heme oxygenase-1-generated biliverdin ameliorates experimental murine colitis. Inflamm. Bowel Dis. 11, 350.
Bulmer, A. C., Blanchfield, J. T., Toth, I., Fassett, R. G., and Coombes, J. S. (2008). Improved resistance to serum oxidation in Gilbert’s syndrome: a mechanism for cardiovascular protection. Atherosclerosis 199, 390.
Converso, D. P., Taillé, C., Carreras, M. C., Jaitovich, A., Poderoso, J. J., and Boczkowski, J. (2006). HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 20, 1236.
Fernandes, A., Falcão, A. S., Silva, R. F., Brito, M. A., and Brites, D. (2007). MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur. J. Neurosci. 25, 1058.
Frydman, R. B., Tomaro, M. L., Rosenfeld, J., Awruch, J., Sambrotta, L., Valasinas, A., and Frydman, B. (1987). Biliverdin reductase: substrate specificity and kinetics. Biochim. Biophys. Acta 916, 500.
Gibbs, P. E., and Maines, M. D. (2007). Biliverdin inhibits activation of NF-kappaB: reversal of inhibition by human biliverdin reductase. Int. J. Cancer 121, 2567.
Gibbs, P. E., Miralem, T., Lerner-Marmarosh, N., Tudor, C., and Maines, M. D. (2012). Formation of ternary complex of human biliverdin reductase-protein kinase Cdelta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS). J. Biol. Chem. 287, 1066.
Gibbs, P. E., Miralem, T., and Maines, M. D. (2010). Characterization of the human biliverdin reductase gene structure and regulatory elements: promoter activity is enhanced by hypoxia and suppressed by TNF-{alpha}-activated NF-{kappa}B. FASEB J. 24, 3239–3254.
Jansen, T., Hortmann, M., Oelze, M., Opitz, B., Steven, S., Schell, R., Knorr, M., Karbach, S., Schuhmacher, S., Wenzel, P., Münzel, T., and Daiber, A. (2010). Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell. Cardiol. 49, 186–195.
Kadl, A., Pontiller, J., Exner, M., and Leitinger, N. (2007). Single bolus injection of bilirubin improves the clinical outcome in a mouse model of endotoxemia. Shock 28, 582.
Kim, H. P., Wang, X., Galbiati, F., Ryter, S. W., and Choi, A. M. (2004). Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 18, 1080.
Kim, S. Y., Kang, H. T., Choi, H. R., and Park, S. C. (2011). Biliverdin reductase A in the prevention of cellular senescence against oxidative stress. Exp. Mol. Med. 43, 15.
Kravets, A., Hu, Z., Miralem, T., Torno, M. D., and Maines, M. D. (2004). Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J. Biol. Chem. 279, 19916.
Lanone, S., Bloc, S., Foresti, R., Almolki, A., Taillé, C., Callebert, J., Conti, M., Goven, D., Aubier, M., Dureuil, B., El-Benna, J., Motterlini, R., and Boczkowski, J. (2005). Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J. 19, 1890.
Lee, S. S., Gao, W., Mazzola, S., Thomas, M. N., Csizmadia, E., Otterbein, L. E., Bach, F. H., and Wang, H. (2007). Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J. 21, 3450.
Lerner-Marmarosh, N., Miralem, T., Gibbs, P. E., and Maines, M. D. (2008). Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 6870.
Lerner-Marmarosh, N., Shen, J., Torno, M. D., Kravets, A., Hu, Z., and Maines, M. D. (2005). Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 7109.
Liu, Y., Liu, J., Tetzlaff, W., Paty, D. W., and Cynader, M. S. (2006). Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 40, 960.
Maghzal, G. J., Leck, M. C., Collinson, E., Li, C., and Stocker, R. (2009). Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J. Biol. Chem. 284, 29251.
Maines, M. D. (2007). Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid. Redox Signal. 9, 2187.
Maines, M. D. (2010). Potential application of biliverdin reductase and its fragments to modulate insulin/IGF-1/MAPK/PI3-K signaling pathways in therapeutic settings. Curr. Drug Targets 11, 1586–1594.
Maines, M. D., Ewing, J. F., Huang, T. J., and Panahian, N. (2001). Nuclear localization of biliverdin reductase in the rat kidney: response to nephrotoxins that induce heme oxygenase-1. J. Pharmacol. Exp. Ther. 296, 1091.
Mancuso, C., Bonsignore, A., Di Stasio, E., Mordente, A., and Motterlini, R. (2003). Bilirubin and S-nitrosothiols interaction: evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem. Pharmacol. 66, 2355.
Mancuso, C., Capone, C., Ranieri, S. C., Fusco, S., Calabrese, V., Eboli, M. L., Preziosi, P., Galeotti, T., and Pani, G. (2008). Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 86, 2235.
Mancuso, C., Pani, G., and Calabrese, V. (2006). Bilirubin: an endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep. 11, 207.
Mayer, M. (2000). Association of serum bilirubin concentration with risk of coronary artery disease. Clin. Chem. 46, 1723.
McCoubrey, W. K. Jr., Cooklis, M. A., and Maines, M. D. (1995). The structure, organization and differential expression of the rat gene encoding biliverdin reductase. Gene 160, 235.
Minetti, M., Mallozzi, C., Di Stasi, A. M., and Pietraforte, D. (1998). Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch. Biochem. Biophys. 352, 165.
Nakagami, T., Toyomura, K., Kinoshita, T., and Morisawa, S. (1993). A beneficial role of bile pigments as an endogenous tissue protector: anti-complement effects of biliverdin and conjugated bilirubin. Biochim. Biophys. Acta 1158, 189.
Nakao, A., Murase, N., Ho, C., Toyokawa, H., Billiar, T. R., and Kanno, S. (2005). Biliverdin administration prevents the formation of intimal hyperplasia induced by vascular injury. Circulation 112, 587.
Nakao, A., Otterbein, L. E., Overhaus, M., Sarady, J. K., Tsung, A., Kimizuka, K., Nalesnik, M. A., Kaizu, T., Uchiyama, T., Liu, F., Murase, N., Bauer, A. J., and Bach, F. H. (2004). Biliverdin protects the functional integrity of a transplanted syngeneic small bowel. Gastroenterology 127, 595.
Novotnây, L., and Vâitek, L. (2003). Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp. Biol. Med. (Maywood) 228, 568.
Nytofte, N. S., Serrano, M. A., Monte, M. J., Gonzalez-Sanchez, E., Tumer, Z., Ladefoged, K., Briz, O., and Marin, J. J. (2011). A homozygous nonsense mutation (c.214C->A) in the biliverdin reductase alpha gene (BLVRA) results in accumulation of biliverdin during episodes of cholestasis. J. Med. Genet. 48, 219.
Ollinger, R., Bilban, M., Erat, A., Froio, A., McDaid, J., Tyagi, S., Csizmadia, E., Graça-Souza, A. V., Liloia, A., Soares, M. P., Otterbein, L. E., Usheva, A., Yamashita, K., and Bach, F. H. (2005). Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation 112, 1030.
Otterbein, L. E., Soares, M. P., Yamashita, K., and Bach, F. H. (2003). Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 24, 449.
Overhaus, M., Moore, B. A., Barbato, J. E., Behrendt, F. F., Doering, J. G., and Bauer, A. J. (2006). Biliverdin protects against polymicrobial sepsis by modulating inflammatory mediators. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G695.
Pachori, A. S., Smith, A., McDonald, P., Zhang, L., Dzau, V. J., and Melo, L. G. (2007). Heme-oxygenase-1-induced protection against hypoxia/reoxygenation is dependent on biliverdin reductase and its interaction with PI3K/Akt pathway. J. Mol. Cell. Cardiol. 43, 580.
Pratesi, R., Agati, G., Fusi, F., Sbrana, G., Migliorini, M. G., Vecchi, C., and Donzelli, G. (1985). Laser investigation of bilirubin-photobilirubin photoconversion. Pediatr. Res. 19, 166.
Salim, M., Brown-Kipphut, B. A., and Maines, M. D. (2001). Human biliverdin reductase is autophosphorylated, and phosphorylation is required for bilirubin formation. J. Biol. Chem. 276, 10929.
Sarady-Andrews, J. K., Liu, F., Gallo, D., Nakao, A., Overhaus, M., Ollinger, R., Choi, A. M., and Otterbein, L. E. (2005). Biliverdin administration protects against endotoxin-induced acute lung injury in rats. Am. J. Physiol. Lung Cell Mol. Physiol. 289, L1131.
Schwertner, H. A., Jackson, W. G., and Tolan, G. (1994). Association of low serum concentration of bilirubin with increased risk of coronary artery disease. Clin. Chem. 40, 18.
Sedlak, T. W., Saleh, M., Higginson, D. S., Paul, B. D., Juluri, K. R., and Snyder, S. H. (2009). Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. U. S. A. 106, 5171–5176.
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043.
Tudor, C., Lerner-Marmarosh, N., Engelborghs, Y., Gibbs, P. E., and Maines, M. D. (2008). Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 413, 405.
Vitek, L., Muchová, L., Jancová, E., Pešicková, S., Tegzová, D., Peterová, V., Pavelka, K., Tesar, V., and Schwertner, H. (2010). Association of systemic lupus erythematosus with low serum bilirubin levels. Scand. J. Rheumatol. 39, 480.
Wang, W. W., Smith, D. L., and Zucker, S. D. (2004). Bilirubin inhibits iNOS expression and NO production in response to endotoxin in rats. Hepatology 40, 424.
Wegiel, B., Baty, C. J., Gallo, D., Csizmadia, E., Scott, J. R., Akhavan, A., Chin, B. Y., Kaczmarek, E., Alam, J., Bach, F. H., Zuckerbraun, B. S., and Otterbein, L. E. (2009). Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 284, 21369.
Wegiel, B., Gallo, D., Csizmadia, E., Roger, T., Kaczmarek, E., Harris, C., Zuckerbraun, B. S., and Otterbein, L. E. (2011). Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 18849–18854.
Wu, J., Ma, J., Fan, S. T., Schlitt, H. J., and Tsui, T. Y. (2005). Bilirubin derived from heme degradation suppresses MHC class II expression in endothelial cells. Biochem. Biophys. Res. Commun. 338, 890.
Yamashita, K., McDaid, J., Ollinger, R., Tsui, T. Y., Berberat, P. O., Usheva, A., Csizmadia, E., Smith, R. N., Soares, M. P., and Bach, F. H. (2004). Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. 18, 765.
Zeng, R., Yao, Y., Han, M., Zhao, X., Liu, X. C., Wei, J., Luo, Y., Zhang, J., Zhou, J., Wang, S., Ma, D., and Xu, G. (2008). Biliverdin reductase mediates hypoxia-induced EMT via PI3-kinase and Akt. J. Am. Soc. Nephrol. 19, 380.
Zhao, R., Xu, G. Y., Liu, Z. Z., Li, J. Y., and Yang, N. (2006). A study on eggshell pigmentation: biliverdin in blue-shelled chickens. Poult. Sci. 85, 546.
Keywords: heme oxygenase, oxidative stress, innate immunity, nitric oxide, toll-like receptor
Citation: Wegiel B and Otterbein LE (2012) Go green: the anti-inflammatory effects of biliverdin reductase. Front. Pharmacol. 3:47. doi: 10.3389/fphar.2012.00047
Received: 08 February 2012; Accepted: 29 February 2012;
Published online: 16 March 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Cesare Mancuso, Catholic University School of Medicine, ItalyAndreas Daiber, University Medical Center Mainz, Germany
Copyright: © 2012 Wegiel and Otterbein. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Barbara Wegiel, Department of Surgery, Harvard Medical School, Beth Israel Deaconess Medical Center, 3 Blackfan Circle EC/CLS 601, 02215 MA, Boston, USA. e-mail: bwegiel@bidmc.harvard.edu; Leo E. Otterbein, Department of Surgery, Harvard Medical School, Beth Israel Deaconess Medical Center, 3 Blackfan Circle EC/CLS 603, 02215 MA, Boston, USA. e-mail: lotterbe@bidmc.harvard.edu