
- 1 Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University, Nashville, TN, USA
- 2 Division of Cardiology, Department of Medicine and Cellular and Molecular Medicine, University of Ottawa, Ottawa, ON, Canada
Voltage-gated sodium channels (VGSC) are multi-molecular protein complexes expressed in both excitable and non-excitable cells. They are primarily formed by a pore-forming multi-spanning integral membrane glycoprotein (α-subunit) that can be associated with one or more regulatory β-subunits. The latter are single-span integral membrane proteins that modulate the sodium current (INa) and can also function as cell adhesion molecules. In vitro some of the cell-adhesive functions of the β-subunits may play important physiological roles independently of the α-subunits. Other endogenous regulatory proteins named “channel partners” or “channel interacting proteins” (ChiPs) like caveolin-3 and calmodulin/calmodulin kinase II (CaMKII) can also interact and modulate the expression and/or function of VGSC. In addition to their physiological roles in cell excitability and cell adhesion, VGSC are the site of action of toxins (like tetrodotoxin and saxitoxin), and pharmacologic agents (like antiarrhythmic drugs, local anesthetics, antiepileptic drugs, and newly developed analgesics). Mutations in genes that encode α- and/or β-subunits as well as the ChiPs can affect the structure and biophysical properties of VGSC, leading to the development of diseases termed sodium “channelopathies”. This review will outline the structure, function, and biophysical properties of VGSC as well as their pharmacology and associated channelopathies and highlight some of the recent advances in this field.
Introduction
In mammals, 11 genes (SCN1A–SCN11A) encode a family of nine functionally expressed voltage-gated sodium channels (VGSC; Nav1.1–Nav1.9) that share more than 50% amino acid sequence homology (Catterall et al., 2005). α-subunits encoded by these genes are organized into four homologous domains (DI–DIV), each one of which is composed of six transmembrane segments. Segments 1 through 4 of each domain form the voltage sensor, while segments 5 and 6 (and their connecting linker the P-loop) compose the pore region. In addition to the α-subunit, VGSC also include β-subunits (which are mainly regulatory molecules) as integral parts of the channel. VGSC can interact with other endogenous proteins called “channel partners” or “channel interactive proteins” (ChiPs) that modulate channel expression and/or function. The last group currently includes caveolin-3 (Lu et al., 1999; Yarbrough et al., 2002; Vatta et al., 2006; Cronk et al., 2007), calmodulin/calmodulin kinase II (CaMKII; Maier and Bers, 2002; Tan et al., 2002; Wagner et al., 2006; Pitt, 2007), connexin-43 (Sato et al., 2011; Chkourko et al., 2012), telethonin (Mazzone et al., 2008), plakophilin (Sato et al., 2009), ankyrins (Kordeli et al., 1995, 1998; Davis et al., 1996; Garrido et al., 2003; Mohler, 2006), neuronal precursor cell-expressed developmentally down regulated 4 (nedd4; Ingham et al., 2004; Rougier et al., 2005), fibroblast growth factor homologous factors (FHFs; Liu et al., 2001, 2003b; Laezza et al., 2007; Dover et al., 2010; Wang et al., 2011; Goldfarb, 2012), membrane-associated guanylate kinase synapse-associated proteins (SAPs; Petitprez et al., 2011; Milstein et al., 2012), and the syntrophin/dystrophin complex (Haenggi and Fritschy, 2006; Shao et al., 2009).
Mutations in the genes encoding the VGSC have been associated with a wide variety of diseases including Dravet syndrome and other types of epilepsy (Claes et al., 2001; Mantegazza et al., 2005; Mullen and Scheffer, 2009), pain-related syndromes [which includes congenital insensitivity to pain (CIP), primary erythromelalgia (PE), and paroxysmal extreme pain disorder (PEPD) (Dib-hajj et al., 2009; Lampert et al., 2010)], and cardiac arrhythmias [which includes congenital long QT syndrome (LQTS) type 3 (Wang et al., 1995), Brugada Syndrome (BS; Probst et al., 2003), progressive cardiac conduction defect (Scott et al., 1999), sick sinus syndrome (Benson et al., 2003), atrial fibrillation (AF; Olson et al., 2005; Darbar et al., 2008), slow ventricular conduction (Chambers et al., 2010; Sotoodehnia et al., 2010), and atrial stand still (Tan, 2006; Remme et al., 2008)]. Epigenetic up regulation of VGSC has recently been associated with aggressive metastatic carcinoma of prostate (Nav1.7) and breast (Nav1.5) (Onkal and Djamgoz, 2009). The up regulation of the VGSC seems to occur early in the dissemination of this type of cancer and ignite the metastatic status (the VGSC expression positively correlated in vivo with invasiveness and therefore metastatic spread) (Onkal and Djamgoz, 2009). Because of their central role in the pathophysiology of these diseases, VGSC are clear pharmacological targets as sites of action for antiepileptic drugs, newly developed analgesics and antiarrhythmic drugs, and potential disease markers in metastatic carcinomas (prostate, breast).
This review will focus on the structure, function, and biophysics of the VGSC, as well as their pharmacology, the sodium channel “partners” (or “ChiPs”) currently identified and the sodium “channelopathies”.
Structure of VGSC
Voltage-gated sodium channels are heteromeric integral membrane glycoproteins that can be differentiated by their primary structure, kinetics, and relative sensitivity to the neurotoxin tetrodotoxin (TTX). They are composed of an α-subunit of approximately 260 kDa (∼2000 amino acids), that is associated with one or more regulatory β-subunits (β1–β4) of approximately 35 kDa each (Catterall, 2000). We will describe in detail both subunits (α and β) that conform the VGSC.
α-Subunits
Ten different mammalian α-subunit isoforms (NaV1.1–NaV1.9 and NaX) have been characterized (Table 1) and at least seven of them are expressed in the nervous system. NaV1.1, NaV1.2, NaV1.3, and NaV1.6 isoforms are mainly expressed in the central nervous system (CNS). In contrast, NaV1.7, NaV1.8, and NaV1.9 isoforms are predominantly located in the peripheral nervous system (PNS; Ogata and Ohishi, 2002), are known to accumulate in the region of peripheral nerve injury and may be important in chronic, neuropathic pain (Devor, 2006; Table 1). In recent reports SCN10A/NaV1.8 has also been identified in human hearts (Facer et al., 2011; Yang et al., 2012) and in intracardiac neurons (Verkerk et al., 2012), where genetic variations in the SCN10A gene have been associated with alterations in the PR interval, QRS duration, and ventricular conduction (Chambers et al., 2010; Sotoodehnia et al., 2010). Because these isoforms (NaV1.1–1.3, NaV1.6–1.9) are mainly localized in nervous tissue they are generally referred as “brain type” or “neuronal-type” sodium channels. NaV1.4 isoform is mainly expressed in skeletal muscle, while NaV1.5 is the cardiac-specific isoform. The isoform referred to as “NaX channel” [also named NaG/SCL11 (rats), Nav2.3 (mice), and/or hNav2.1 (humans)] identifies a subfamily of sodium channel-like proteins (George et al., 1992). This channel has significant differences in the amino acid sequence in the voltage sensor, inactivation gate, and pore region when compared to the rest of VGSC (George et al., 1992; Goldin et al., 2000). NaX is normally expressed in a variety of organs including the heart, skeletal muscle, uterus, dorsal root ganglia (DRG), and brain [mainly in the circumventricular organs (CVOs)]. The difficulties in the characterization of the biophysical properties of this channel are mainly due to lack of success in expressing the functional protein in heterologous expression systems. Hiyama et al. (2002) generated a mouse model in which the NaX gene was knocked out. This group confirmed that Nax channel was expressed in neurons in the CVOs that play a fundamental role controlling body fluid and ionic balance. This group reported that under thirst conditions, mice lacking Nax showed hyperactivity of the neurons in these areas and ingested excessive salt, while wild-type mice did not. This led the investigators to propose that NaX was involved in the mechanism that senses sodium levels in the brain, where this protein might sense extracellular sodium concentration (Hiyama et al., 2002; Noda, 2006).
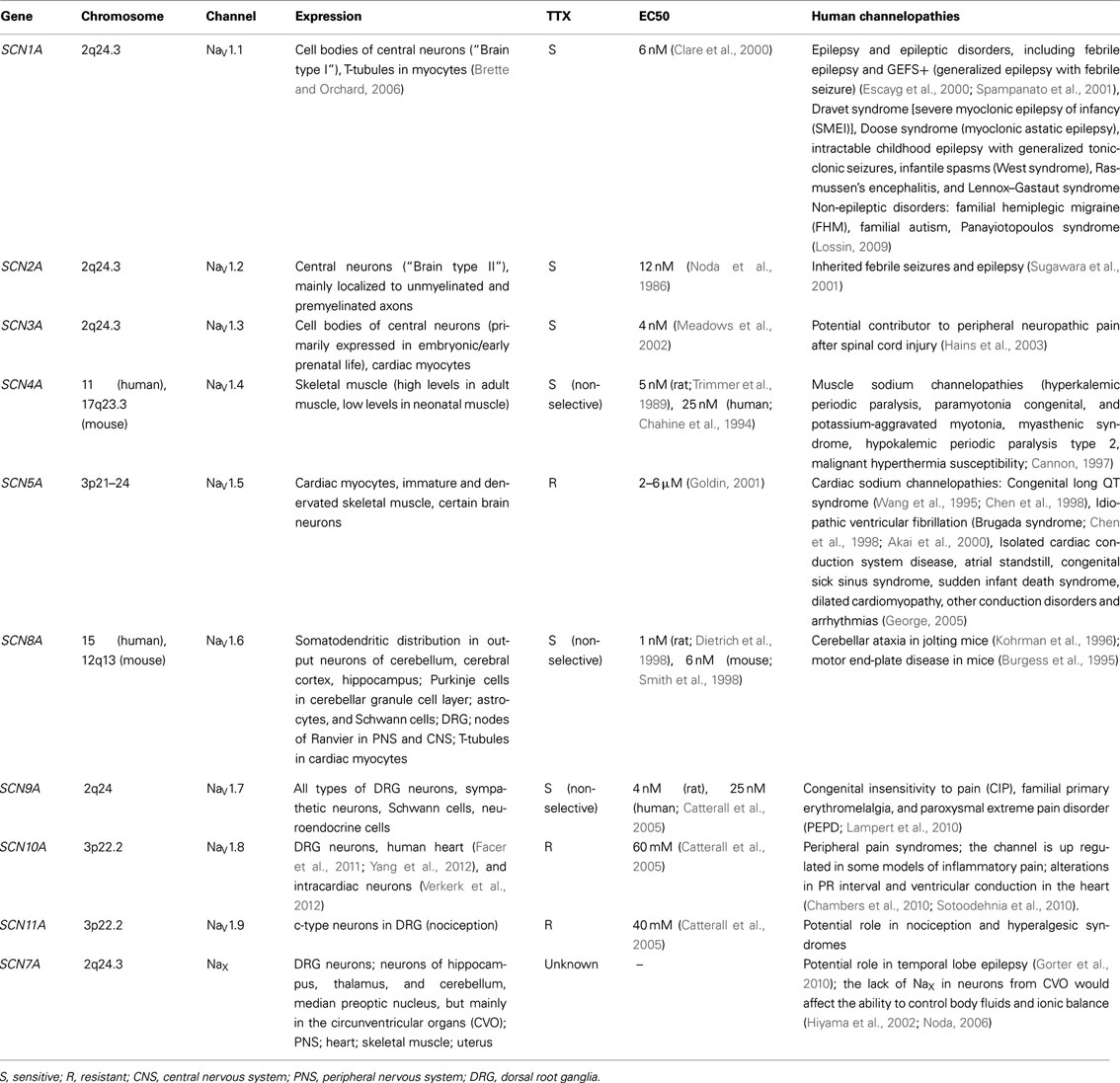
Table 1. Summary of the different types of VGSC, and the channelopathies associated to mutations in the genes encoding the α subunits.
Each α-subunit is arranged in four homologous domains (DI–DIV) that contain six transmembrane segments (S1–S6; Figure 1). Using cryo-electron microscopy Sato et al. (2001) showed that these four domains are arranged around the central pore of the channel. Segment 4 of each domain contains a high concentration of positive charges (mostly arginine) and functions as the core of the voltage sensor responsible for the voltage-dependent activation of the channels. Segment 6 from all four domains forms the inner surface of the pore. The hairpin-like loop between segments 5 and 6 [S5–S6 hairpin-like P(ore)-loop] is part of the pore of the channel and forms a narrow (ion-selective) filter that controls the ion selectivity and permeation at the extracellular side of the pore (Catterall, 2000; Yu and Catterall, 2003; George, 2005).
Payandeh et al. (2011) recently reported the crystal structure of NaVAb, a VGSC found in the bacterium Arcobacter butzleri. NaVAb is part of the NachBac channel family, which is a well-established model to study vertebrate NaV and CaV channels (Ren et al., 2001; Koishi et al., 2004; Payandeh et al., 2011). Payandeh et al. (2011) were able to capture this channel in the close configuration when the pore was closed with four activated voltage sensors at a resolution limit of 2.7 Å. Payandeh’s work provides the first insight into the structural basis for voltage-dependent gating ion selectivity and drug block in VGSC. The pore consists of an outer tubular vestibule, a selectivity filter, a central cavity (which can lodge partially hydrated sodium ions) and an intracellular activation gate. The helices that constitute the pore are positioned to stabilize cations in the central cavity through helical-dipole interactions (Doyle et al., 1998; Jogini and Roux, 2005). A second P2-helix forms an extracellular funnel and represents a highly conserved element in sodium channels (Payandeh et al., 2011).
Payandeh and coworkers proposed that in NaVAb the ion conduction pathway is electronegative and the selectivity filter (mainly composed of negatively charged glutamate (Glu) side chains) forms the narrowest constriction near the extracellular side of the membrane. There are 4 Glu 177 side chains that form a 6.5-Å × 6.5-Å scaffold with an orifice of approximately 4.6 Å wide. A profuse mesh of amino acid residue interactions, including hydrogen bonds between glutamine from the P-helix and the carbonyl of Glu, stabilizes the selectivity filter. The radius of the pore suggests that hydrated Na+ ions can conduct through the channel. Free diffusion then allows the hydrated Na+ to enter the central cavity and move through the open activation gate toward the cytoplasm (Payandeh et al., 2011). This permeation pathway contrasts with the selectivity filter in K+ channels, which is much narrower. In this case the smaller radius of the pore can only conduct dehydrated K+ ions through direct interactions with backbone carbonyls through a long, narrow pore (Morais-Cabral et al., 2001; Ye et al., 2010).
Identification of the primary structure of VGSC led to the development of the “sliding helix” (Catterall, 1986b) and the “helical screw” (Guy and Seetharamulu, 1986) models (validated by structure-function studies) to better understand how the voltage sensor operates. Both models suggest that positively charged residues in segment 4 within each domain serve as the gating charges moving outward across the membrane as a consequence of membrane depolarization, initiating the activation process (Catterall, 1986a,b; Guy and Seetharamulu, 1986; Catterall et al., 2010). Catterall and coworkers have extensively described these two models. Basically, four to seven residues positively charged within segment 4 would pair negatively charged residues in segments 1, 2, and/or 3. In this configuration, positively charged residues in segment 4 are pulled inward by the electric field of the resting membrane potential which is negative. As depolarization progresses, the change in the polarity of the membrane potential relieve the electrostatic force and the segments 4 move outward allowing each positive charged amino acid in the segment 4 pairs a negatively charged one. As described by Catterall (2010), this outward movement of the gating charges in segments 4 pulls the linker between segments 4 and 5, curves the segment 6 and initiates the opening of the central pore of the channel. The movement of charged particles to activate the sodium conductance (“gating charges” or “gating current”) was first predicted by Hodgkin and Huxley(Hodgkin and Huxley, 1952; Catterall, 2010), but Armstrong and Bezanilla (1973) were the first ones that measured it in 1973, combining the techniques of internal perfusion, voltage-clamp, and signal average. Using similar techniques, Keynes and Rojas (1973) confirmed the existence of the gating current the same year. Armstrong and Bezanilla (1974) reported additional properties of this current and strong evidence linking it to the gating of the sodium channels the following year.
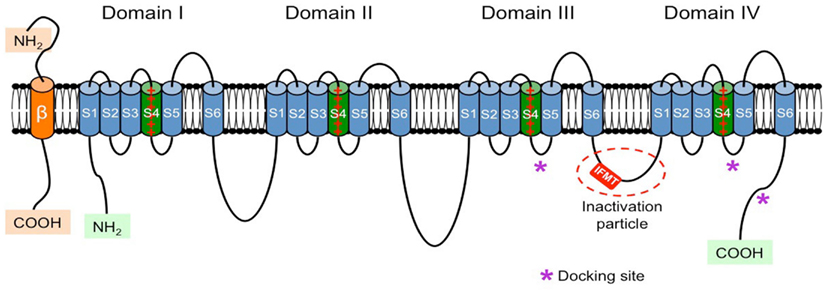
Figure 1. Schematic representation of the α- and β-subunits of the VGSC. The four homologous domains (I–IV) of the α-subunit are represented; S5 and S6 are the pore-lining segments and S4 is the core of the voltage sensor. In the cytoplasmic linker between domains III and IV the IFMT (isoleucine, phenylalanine, methionine, and threonine) region is indicated. This is a critical part of the “inactivation particle” (inactivation gate), and substitution of aminoacids in this region can disrupt the inactivation process of the channel. The “docking site” consists of multiple regions that include the cytoplasmic linker between S4–S5 in domains III and IV, and the cytoplasmic end of the S6 segment in domain IV (*). Depending on the subtype of β-subunit considered they could interact (covalently or non-covalently) with the α-subunit.
β-Subunits
These are integral proteins as well, composed of one extracellular domain (ECD, N-terminal domain), one transmembrane domain, and one intracellular domain (C-terminal domain). The β-subunits are expressed in excitable and non-excitable cells within the nervous system and the heart, and there is some evidence suggesting that these proteins can be expressed in the cells even in the absence of the α-subunit (Patino and Isom, 2010; Table 2). One or more regulatory β-subunits (β1–β4) can associate with one α-subunit. An individual α-subunit can be associated with one non-covalently (β1 or β3) and one covalently (β2 or β4) linked β-subunits (Yu and Catterall, 2003; Catterall et al., 2005; Patino and Isom, 2010). The role of β-subunits has been reviewed in detail by Patino and Isom (2010). The authors remark that β-subunits are regulatory proteins that can act both as cell adhesion molecules (CAMs) and modulate the cell surface expression of the VGSC, enhancing sodium channel density and cell excitability. The latter may be a very important mechanism that regulates nociceptor excitability in vivo (Lopez-Santiago et al., 2011). β1 association with contactin or neurofascin (NF)-186 also results in increased VGSC cell surface expression (Kazarinova-Noyes et al., 2001; McEwen and Isom, 2004). Furthermore, β1 and β2 are ankyrin-binding proteins. Mice lacking ankyrin exhibit reduced sodium current (INa) density and abnormal INa kinetics (Chauhan et al., 2000), suggesting that β-subunits play important roles in the VGSC–ankyrin complex (Patino and Isom, 2010). The interaction between α- and β-subunits may be particularly critical at the nodes of Ranvier of myelinated axons, since mice lacking β1-subunit have reduced numbers of nodes, alterations in the myelination process, and drastically altered contacts between neurons and glial cells (Chen et al., 2004). Even though proteins within nodal regions are localized normally in these mice, association between VGSC and contactin is disrupted. Loss of β1-subunit dependent protein–protein interactions can lead to changes in the structure of the Ranvier nodes and disrupted saltatory conduction (Chen et al., 2004; Davis et al., 2004). Similar to the β1- subunit, β2 can also modulate the expression of the channel at the cell surface and affect INa density (Isom et al., 1995). β2 (and β4) intracellular domains can translocate into the nucleus and enhance SCN1A expression, thus functioning as transcriptional regulators of the VGSC α-subunit.
β-subunits are also critical for cellular migration. β1 and β2 mediate migration of fibroblasts (Xiao et al., 1999) and cancer cells (Brackenbury and Isom, 2008), adhesion, and neurite outgrowth (β1 promotes and β2 inhibits this process, while β3 and 4 have no effect; Davis et al., 2004; McEwen et al., 2009). The effects of β-subunits on cell migration, adhesion, and neurite outgrowth also depends on intracellular transduction events like the activation of proto-oncogene tyrosine-protein kinase fyn by β1 to promote neurite (axon and/or dendrite) outgrowth (Brackenbury et al., 2008).
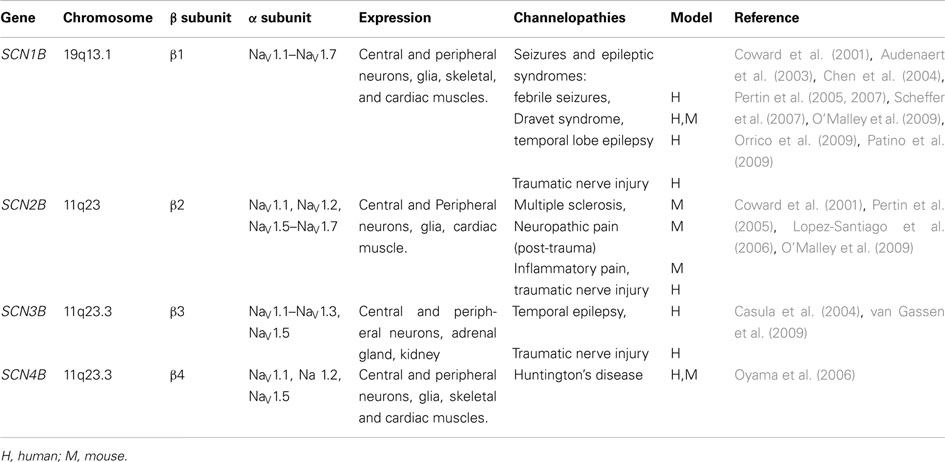
Table 2. Summary of the different types of β subunits associated with the different VGSC, and the related channelopathies associated with the mutations in the genes that encode them (modified from Patino and Isom, 2010).
Biophysical Properties of VGSC
When the cell is depolarized, the outward movement of all segments 4 generates a conformational change that opens the pore and thus activates the channel. This allows sodium to flow into the cell down its electrochemical gradient. INa reaches a maximum within milliseconds and then becomes smaller as the sodium channel stops conducting ions and starts closing. The closure of the channel during maintained depolarization is called inactivation. The activation and inactivation processes and the coupling between them (in particular, the outward movement of the voltage sensor) are voltage-dependent. All the downstream rearrangements are voltage-independent. The refractoriness of the cell is related to the inactivated sodium channels, which cannot reopen until they are completely recovered. This mechanism protects the cell and prevents firing during prolonged depolarization (Goldin, 2003).
The time that the channel remains in a specific state (open or close) as well as the rate at which it transitions from one state to the other affects its ionic conductance and thus the shape of the action potential (AP). During the last decade most of the advances in the biophysics of the ion channels has been obtained by expressing the channels in isolated systems (cell lines or lipid bilayers). But the interpretation of this data has been somehow challenging due to the lack of the correct physiological context. The importance of computational biology was first emphasized by Rudy and Silva (2006) by trying to understand the context in which ion channels operate. Still, such theoretical approach in electrophysiology was initiated by Hodgkin and Huxley (1952). Hodgkin and Huxley (1952) were the first that formulated a model of the AP based on the findings of their pioneering work with voltage-clamp techniques showing voltage-dependent changes in ionic conductance in squid axons. The data reported by Hodgkin and Huxley (1952) suggested that the inward flow of Na+ was responsible for the rapid initial positive upstroke of the membrane potential, whereas the outward flow of potassium determined the repolarization of the membrane back to resting levels. The mathematical model was designed to determine whether the sodium and potassium currents they identified could in fact generate an AP that was similar in morphology to their AP experimental recordings. The model proposed by Hodgkin and Huxley was reviewed in detail by Rudy and Silva (2006). Rudy and Silva explained that in this model the conductance of sodium and potassium currents was dependent upon the open probability of a series of activation gates. The activation gates in this model correspond to the α-subunits of the VGSC. Each gate can transition from a closed state (open probability = 0) to an open state (open probability = 1) that is independent of the state of the other gates. An ion can only pass through when all the gates (that is, the four α-subunits that constitute the tetramer VGSC) are open (open probability = 1). Rudy and Silva (2006) also pointed out that the original model has several limitations: the model assumes that intracellular concentrations of Na+ and K+ do not change during the AP, and it does not consider the inactivation of the channel. It also assumes that opening and closing transition rates are independent (Rudy and Silva, 2006). Rudy and Silva proposed that since the VGSC inactivation has a greater probability of occurring when the channel is open, then inactivation is highly dependent on activation. Thus the independent gating of the Hodgkin and Huxley model would not be valid. To improve the accuracy of the mathematical model, Rudy and Silva (2006) suggest considering the dependence of a given transition on the occupancy of different states of the channel. In the case of the VGSC, the model should consider the dependence of the inactivation transition on the probability that the channel occupies the open state. Finally, the authors report that the different states and the dependence of transitions (both voltage-dependent and independent) between states can then be more accurately predicted using Markov-type models (the importance of computational biology to study the integrated electrophysiology of ion channels has been extensively reviewed by Rudy and Silva (2006)).
Inactivation is the process by which an open-channel enters a stable non-conducting conformation when the cell membrane depolarizes. The inactivation process includes fast, slow and ultra-slow inactivation. In addition, long-term inactivation FHF- and β4-mediated are processes distinct from slow inactivation (Goldfarb, 2012). In general, while isoforms Nav1.1–Nav1.4, Nav1.6, and Nav1.7 have faster inactivation kinetics, Nav1.5, Nav1.8, and Nav1.9 have slower inactivation.
Fast inactivation mimics a “ball-and-chain” mechanism, where a cytoplasmic segment of the α-subunit of the VGSC (or inactivating particle) occludes the pore by binding to a docking site (Goldin, 2003). Rohl et al. (1999) were the group that first studied the inactivating particle. It consists of a portion of the cytoplasmic linker connecting domains III and IV, with the critical region centering on a 4-amino acid extent consisting of isoleucine (I), phenylalanine (F), methionine (M), and threonine (T) (IFMT; Goldin, 2003). Phenylalanine and threonine directly interact with the docking site and the distance between I- and T-residues correlates with the ability to inactivate the channel (Goldin, 2003). Amino acid substitution within this critical region can disrupt the inactivation of the channel (Kudora et al., 2000; Miyamoto et al., 2001a,b). The docking site includes the cytoplasmic linker connecting segments 4 and 5 in domains III and IV and the cytoplasmic end of segment 6 in domain IV. This “ball-and-chain” mechanism is equivalent to the N-type inactivation described for voltage-gated potassium channels (N-type fast inactivation), which involves occlusion of the intracellular mouth of the pore through binding of a short peptide segment from the N-terminal (Rasmusson et al., 1998). Fast inactivation is important during AP repolarization, and in some structures like mammalian nodes of Ranvier (which practically lack phasic potassium channels) it is the only repolarizing force besides the leakage current (Ulbricht, 2005).
Fast inactivation can be altered by the carboxyl (C)-terminus of the channel. This is due to electrostatic interactions involving the sixth helix in the C-terminus, which can modulate the interaction of the fast inactivating particle with its docking site. The different amino acid composition of the C-terminus explains the differences observed in fast inactivation between the VGSC isoforms (Mantegazza et al., 2001). Motoike et al. (2004) reported that the C-terminus is actually part of the inactivation gate, as it stabilizes the closed state minimizing the reopening of the channel. Mutations in the C-terminus disrupt fast inactivation and can lead to the LQTS type 3 (Goldin, 2003).
Fast inactivation can also be modulated by the interaction with β-subunits. The effect and mechanism is dependent on the specific α- and β-subunits involved and the heterologous expression system being used to express the channel. For example, the β1-subunit accelerates the recovery from inactivation of Nav1.5 (Zimmer and Benndorf, 2002) and Nav1.2 (Chen and Cannon, 1995; McCormick et al., 1998, 1999) and shifts the voltage-dependence of inactivation in the negative direction (Meadows et al., 2002). The β3-subunit has a similar effect on Nav1.5, but it increases persistent current through Nav1.2 in tsA-201 cells (cell line derived from human embryonic kidney cells; Goldin, 2003). β4-subunits disrupt VGSC inactivation in neurons, working as endogenous open-channel blockers. This subunit has a short cytoplasmic tail that basically blocks the channel in the open state but rapidly dissociates upon membrane repolarization to generate the resurgent current in some neurons like Purkinje cells (Grieco et al., 2005; Goldfarb, 2012).
Fast inactivation can be disrupted and transformed into long-term inactivation by the interaction of the α-subunit of the channel with a family of cytoplasmic proteins termed fibroblast growth factor homologous factors (FHFs) (Goldfarb, 2012). FHFs act as accessory channel subunits. Several FHFs delay fast inactivation by raising the voltage at which fast inactivation occurs. Dover et al. (2010) reported that all A-type FHFs (specially FHF2A and FHF4A) exert a rapid onset of a distinct mode of long-term inactivation of Nav1.6 and other VGSC including cardiac NaV1.5 (Dover et al., 2010; Goldfarb, 2012). A-type FHFs accomplish long-term inactivation by providing an independent cytoplasmic gating particle that competes with the channel’s intrinsic inactivating particle for blockade of the channel upon membrane depolarization. The authors reproduced this mechanism by injecting a synthetic peptide corresponding to the A-type FHF particle that mimics the long-term inactivation and opposes sustained firing of excitable cells (Dover et al., 2010). β4-mediated channel block and A-FHF-mediated long-term inactivation have a similar physical mechanism. Both processes are mediated by small cytoplasmic particles that interact with the channel after the depolarization has driven the channels into the open state. In both cases, the blocking particles dock at similar sites deep within the cytoplasmic opening of the channel pore. The main difference between the two processes is the rate of particle dissociation, where β4 particle dissociates rapidly and FHF dissociates far more slowly (Goldfarb, 2012).
Slow inactivation is a different process that involves conformational changes of the channel leading to rearrangement of the pore. The process also involves segment 4 of domain IV and segments 5 and 6 of domain II (Goldin, 2003). This mechanism is equivalent to the C-type inactivation mechanism described for potassium channels (Rasmusson et al., 1998). Slow inactivation may play a role in regulating excitability by, for example, modulating burst discharges. However, this modulation is complicated since slow inactivation depends on both resting membrane potential and the previous history of AP firing (Ulbricht, 2005).
In addition to fast and slow inactivation there is a third type of inactivation named ultra-slow inactivation. This process was described in Nav1.4 when the alanine in position 1529 (A1529) is replaced by aspartate (D) in the domain IV P-loop (Goldin, 2003). Binding of the fast inactivating particle inhibits this process. This result demonstrates that there are interactions (mostly, allosteric modulation) among the different inactivation events (Goldin, 2003).
“Sodium Channel Partners” or “Channel Interactive Proteins”
The current density and gating properties of VGSC can also be modulated by the differential expression of channel “partners” or ChiPs (Table 3). These terms designate molecules that affect the aggregation, density, function, and regulation of VGSC. Up-to-day, the main identified VGSC partners include caveolin-3 (and the membrane compartment “caveolae”), CaMKII, connexin-43, telethonin, plakophilin, ankyrins, fibroblast growth factor homologous factors (FHFs), nedd4, SAPs, and syntrophin/dystrophin complex.
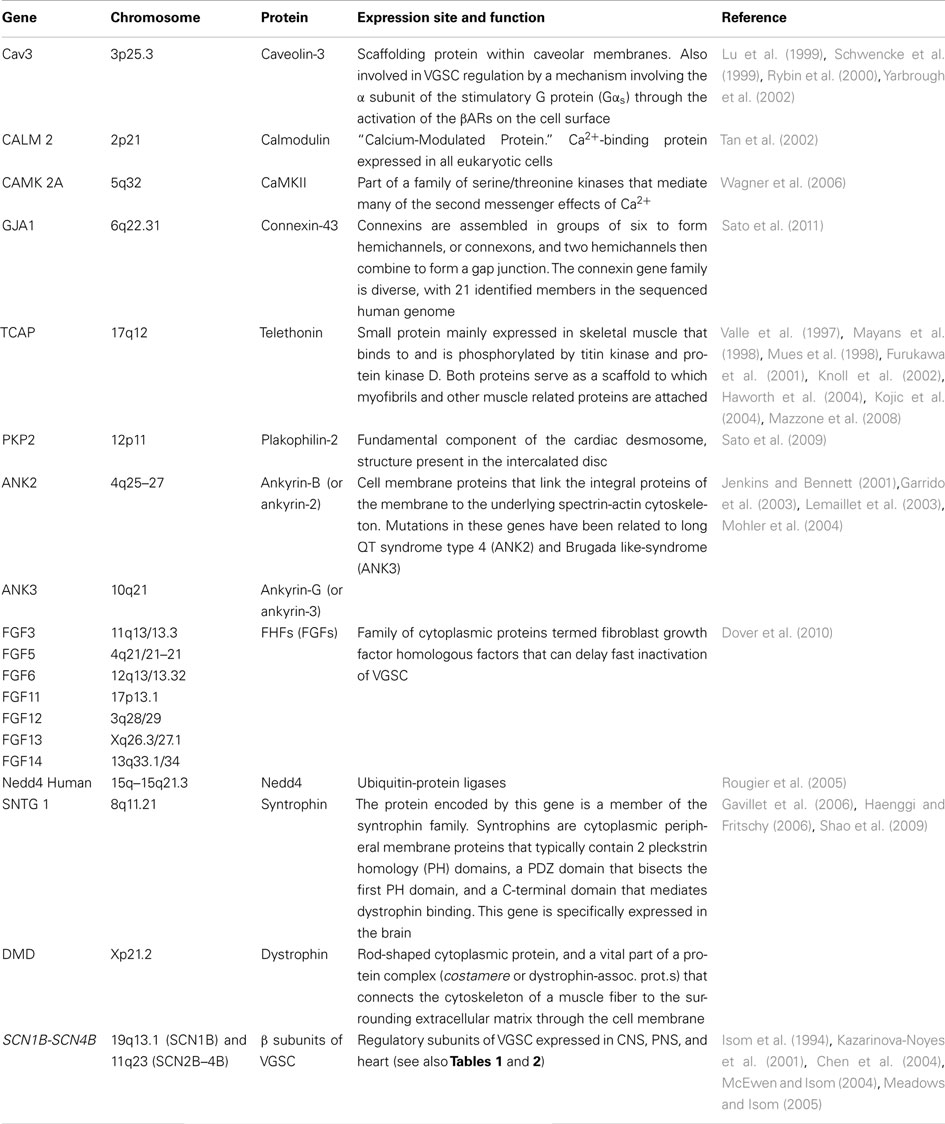
Table 3. VGSC protein partners.
Caveolae/Caveolin-3
Caveolae are sarcolemmal membrane invaginations that have been implicated in cellular trafficking cascades involving the β-adrenergic receptors (β-AR; Schwencke et al., 1999; Rybin et al., 2000). These membrane invaginations also contain scaffolding proteins named “caveolins”. Yarbrough et al. (2002) demonstrated both biochemically and functionally that caveolae are involved in VGSC regulation by a mechanism involving the α-subunit of the stimulatory G protein (Gαs) through the activation of the β-ARs on the cell surface. Because direct Gαs activation induces an increase in the number of functional channels at the sarcolemma (Lu et al., 1999), they hypothesized that functional channels were recruited from an intracellular store, allowing a faster presentation of channels to the cell surface after β-stimulation. The authors purified the caveolin-3 rich fraction using immunoprecipitation. VGSC and Gαs are colocalized in the Cav3(+)-fraction, suggesting a physical association of both proteins with the caveolar (Cav3-rich) membrane (Rook et al., 2012). They also reported that the increase in INa induced by isoproterenol stimulation (10 μM) in the presence of a protein kinase A (PKA) inhibitor (PKA-independent increase in INa) was abolished when an anti-Cav3 antibody was injected into the cytoplasm of the cell through the pipette. This suggests a direct action of the Gαs on the caveolae, resulting in the presentation of caveolar VGSC to the sarcolemma. Palygin et al. (2008) also demonstrated that the histidine residue at position 41 of Gαs (H41) is a critical residue for the functional increase of INa observed.
Calmodulin/Calmodulin Kinase II
Tan et al. (2002) demonstrated that calmodulin regulates sodium channel gating through binding to a region of 25 amino acids located at the C-terminus of the intracellular domain. Wagner et al. further studied the downstream signaling through Ca2+/CaM-dependent protein kinase II (CaMKIIδ) in heart cells from two heart failure animal models, where expression and activity of CaMKII are increased by twofold to threefold. They demonstrated that calmodulin regulates Na+ channel gating in part via CaMKII. Using two cell models of CaMKII overexpression, they concluded that both acute and chronic overexpression of CaMKIIδc significantly shifted voltage-dependence of Na+ channel availability by −6 mV, and the shift was Ca2+-dependent. CaMKII also enhanced the inactivation of the channel and slowed its recovery from inactivation. These effects were prevented using CaMKII inhibitors (KN93 or AIP). CaMKII over-expression also increased persistent (late) inward INa and the intracellular Na+ concentration (also prevented using CaMKII inhibitors). They reported that CaMKII coimmunoprecipitates with and phosphorylates sodium channels. In vivo data suggested that CaMKII overexpression mice were more prone to suffer ventricular arrhythmias, particularly monomorphic ventricular tachycardia. The data as a whole supports the hypothesis that CaMKII regulates sodium channel function in myocytes most likely by association with and phosphorylation of the channels (Wagner et al., 2006).
Telethonin
Telethonin is a small protein (19 kDa) that is mainly expressed in striated muscle (Valle et al., 1997). This protein binds to and is phosphorylated by titin kinase (Mayans et al., 1998) and protein kinase D (Haworth et al., 2004). One of its many functions includes acting as a stretch sensor in the heart (Knoll et al., 2002), linking sarcomeres to K+ channel subunits (Furukawa et al., 2001), and interacting with titin (Mues et al., 1998) and ankyrin-2 (also proposed to behave as stress sensors in muscle; Kojic et al., 2004). Mazzone et al. (2008) hypothesized that telethonin may be relevant in tissues different from striated muscle, where it might also behave as a ChiP. After screening 20 unrelated patients with primary intestinal pseudo-obstruction, the authors identified a patient with a heterozygous mutation, R76C, in the telethonin gene by direct DNA sequencing. The mutation is located in the region of telethonin where the protein has been shown to interact with sarcomeric proteins (muscle LIM protein and titin) in the heart. Using immunostaining and immunoprecipitation they demonstrated that telethonin and NaV1.5 were colocalized in mouse hearts. They studied the effects of the R67C mutation on the in vitro electrophysiology of SCN5A expressed in a Human embryonic kidney cell line (HEK)-293. The coexpression of R67C telethonin with SCN5A resulted in a leftward shift in the steady-state activation of the sodium channel, leading to increased Na+ entry at resting potential (depolarizing effect). The data supports the hypothesis that telethonin acts as a ChiP (Mazzone et al., 2008).
Plakophilin
Plakophilin-2 (PKP2) is a fundamental component of cardiac desmosomes. This structure is present in the intercalated disk, the site of end-to-end contact between cardiac myocytes, and provides mechanical integrity between adjacent cells. NaV1.5 is also highly localized at the intercalated disks. Combining immunochemistry and electrophysiological studies Sato et al. (2009) demonstrated that PKP2 associates with NaV1.5 in the same molecular complex, and that the knockout of PKP2 expression produced a decrease in peak current density, a shift in voltage-dependence inactivation, and a prolongation of time-dependence of recovery from inactivation.
Connexin-43
Connexin-43 peptides are localized at intercalated disks, where they form gap junctions for electrical coupling of adjacent cells. Sato et al. (2011) showed that AnkG, plakophilin, and connexin-43 are associated at the intercalated disks and that this macromolecular complex may interact with clusters of NaV1.5 also present in the disk. More recently, Chourko et al. characterized the remodeling of the gap junction (connexin-43) and VGSC in an ovine model of right ventricular pressure overload induced by pulmonary hypertension. The authors reported significant lateralization of connexin-43, which was colocalized with mechanical junction proteins and microtubule-associated proteins EB1 and Kifb5 (these proteins are responsible for the forward trafficking of connexin-43 to the intercalated disk). There was also a significant reduction in the peak INa and in V1/2 activation, a slower recovery from inactivation, with no lateralization of the VGSC (NaV1.5). The authors then speculate that the difference in the NaV1.5 remodeling respect to the connexin-43 could be explained due to the fact that trafficking of NaV1.5 might require molecules that cannot redirect the channel to the lateral membrane. In summary, the data reported support the idea of a partnership between these complexes, previously considered to be independent from each other (Chkourko et al., 2012).
Ankyrins
Ankyrins are a widely expressed family of “adaptor” proteins responsible for the localization of proteins at specialized membrane domains. From all the members that are included in the ankyrin family, ankyrin-G (“G” from “general”) was initially studied in the brain. In neurons, ankyrin-G colocalizes and copurifies with VGSC (Kordeli et al., 1995; Davis et al., 1996; Garrido et al., 2003; Mohler, 2006). Ankyrin-G is important for the clustering of NaV1.2 and 1.6 isoforms at the nodes of Ranvier (Jenkins and Bennett, 2001; Garrido et al., 2003) and also colocalizes with VGSC at the neuromuscular junction (Flucher and Daniels, 1989; Kordeli et al., 1998). A role for ankyrin-G for VGSC targeting in cardiac muscle was hypothesized based on the role of this protein in clustering neuronal VGSC (Mohler, 2006). Ankyrin-G binds to a nine residue domain in the DII–DIII loop in the α-subunit of VGSC (Lemaillet et al., 2003). This binding is required for Nav1.5 localization in heart cells (Garrido et al., 2003; Mohler et al., 2004). Since ankyrin-G is primarily expressed at the intercalated disk membrane and T-tubules, it colocalizes with Nav1.5 at these specific sites (Lemaillet et al., 2003; Mohler et al., 2004; Bennett and Healy, 2008; Lowe et al., 2008). Mutations in either the sodium channel domain at which ankyrin binds, or in ankyrin itself, can affect the channel expression (Mohler et al., 2004).
Fibroblast Growth Factor Homologous Factors
Fibroblast growth factor homologous factors (FHFs) is a family of cytoplasmic proteins that can interact with VGSC and delay fast inactivation by raising the voltage at which fast inactivation occurs. The role of FHFs as VGSC modulators was already discussed under “Biophysical properties of VGSC.”
Neuronal Precursor Cell-Expressed Developmentally Down Regulated 4
Neuronal precursor cell-expressed developmentally down regulated 4 (Nedd4) is the prototypical protein in a family of E3 ubiquitin. They select specific proteins for conjugation to ubiquitin, which acts as a marker for protein degradation but also in the sorting of proteins at different steps in biosynthetic and endocytic pathways. They are found in the nucleus and at the plasma membrane. Need4-2 refers to a subgroup of ubiquitin-protein ligases that binds the PY motif of Nav1.5 and reduces the sodium current (INa) in HEK293 cells by promoting its internalization (Rougier et al., 2005). For more details see the review written by Ingham et al. (2004).
Synapse-Associated Proteins
Synapse-associated proteins (also called MAGUK, membrane-associated guanylate kinase) are a family of proteins that include Dlg, SAP97/hDlg, SAP90/PSD-95, SAP102, and PSD-93/chapsyn110. They are composed of multiple sites of protein–protein interactions, like the PDZ domains. SAP are localized either to the pre- or postsynaptic sides of excitatory or inhibitory synapses and play a central role in the molecular organization of synapses, like PSD-95, SAP102, and distribution of the NMDA glutamate receptor at the postsynaptic level. One of the family members, SAP97, is also present in epithelial cells and localized at the lateral membrane between cells (Fujita and Kurachi, 2000). At the cardiomyocytes SAP97 colocalized with Nav1.5 at the intercalated disks, determining the existence of a second pool of sodium channels in addition to the channels targeted at lateral membranes by the syntrophin-dystrophin complex (Petitprez et al., 2011).
Syntrophin/Dystrophin Complex
Syntrophins (α, β, and Γ) bind and localize signaling proteins to the plasma membrane (Shao et al., 2009). Syntrophins can also interact with multiple proteins via two pleckstrin homology domains, a PDZ domain and a conserved syntrophin unique region. The PDZ domain can bind to the last three residues of the C-termini intracellular domain of NaV1.4 and 1.5 (Haenggi and Fritschy, 2006). The latter can also complex syntrophin and dystrophin (Gavillet et al., 2006). Syntrophin stabilizes the VGSC in the plasma membrane and reduce its internalization (Shao et al., 2009).
β-Subunits
In addition to the modulation of VGSC function (Johnson and Bennett, 2006), β-subunits play critical roles in the intracellular trafficking of α-subunits, regulating the channel expression levels at the plasma membrane and their role in cell adhesion (Isom et al., 1994). In vitro data suggests that β-subunits constitute communication links between adjacent cells, extracellular space (via their interaction with tenascin-C and R), cytoskeleton and intracellular signaling mechanisms, and other ion channels. In particular, the β1-subunit seems to be critical for the interaction of the VGSC with other CAMs and cytoskeletal proteins (Kazarinova-Noyes et al., 2001; Chen et al., 2004; McEwen and Isom, 2004). Due to their roles in the interactions with cytoskeletal proteins, CAMs, and other ion channels, Meadows and Isom (2005) proposed that β-subunits should also be considered as molecular scaffolds of the ion conducting pore (α-subunits), therefore critically affecting channel function, subcellular localization and cell surface expression in a cell-specific and subcellular domain-specific manner (see Table 2).
Pharmacology of VGSC
Voltage-gated sodium channels are the site of action of many toxins and drugs. At least six sites of action for neurotoxins (sites 1–6) and one receptor site for class I antiarrhythmic drugs, local anesthetics and related antiepileptic drugs are known to exist on the VGSC (Cestele and Catterall, 2000). All of them are located on the α-subunit of the channel. Receptor site 1 binds TTX and saxitoxin (Noda et al., 1989; Hille, 2001). This receptor site is formed by amino acid residues in the pore loops and on the extracellular side of them at the outer end of the pore. The sensitivity of the VGSC to TTX segregates them into two groups (Table 1):
1. TTX-sensitive channels (TTX-S; blocked with TTX in the nanomolar concentration range). This group includes Nav1.1, NaV1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7 isoforms.
2. TTX-resistant channels (TTX-R; blocked with TTX in the micromolar-millimolar concentration range). This group includes Nav1.5, Nav1.8, and Nav1.9 isoforms.
Biophysical and pharmacological properties of TTX-S and TTX-R Na+ channels are different. TTX-R Na+ channels can be blocked by inorganic (Co2+, Mn2+, Ni2+, Cd2+, Zn2+, La3+) and organic Ca2+ channel blockers. Typically, TTX-R Na+ channels show smaller single-channel conductance, slower kinetics, and a more positive current-voltage relation than TTX-sensitive ones. Li and Zhu (2011) recently reported two chimeric peptides of drosotoxin that can block the activity of both TTX-R and TTX-S channels. The authors proposed that this approach of understanding the molecular determinants of toxins affecting VGSC would allow a more rational design of subtype-specific sodium channel blockers.
The overlapping sites of action of antiarrhythmic drugs, local anesthetics and related antiepileptic drugs are located in the inner cavity of the pore of the channel, and they are formed by amino acid residues located in S6 in domains I, III, and IV (Ragsdale et al., 1994, 1996; Hockerman et al., 1997; Catterall, 2000; Yarov-Yarovoy et al., 2001, 2002; Liu et al., 2003a). Drug affinity can be reduced by mutations in critical residues in the pore. Fundamentally, these drugs bind to their corresponding site of action to change the function of the channel (decrease the sodium current density). They can also change the affinity with which the channel binds the drug depending on the functional conformation or state in which the channel is found (rest, active, inactive; Bruton et al., 2011). Most sodium channel-blocking agents block the channel when it is open or inactivated, and have very little or no effect at all while the channel is in the resting state. Thus, with each AP, the drug binds to the VGSC and blocks them, and then dissociates during repolarization, with the consequent loss of blockage.
The dissociation rate is a key determinant of steady-state block of sodium channels. AP frequency and duration, membrane potential level, and the physicochemical properties of the drug will determine the rate of recovery from blockage. When depolarization frequency increases, the rest interval decreases, and so does the time available for drug dissociation as the drug remains attached to the channel for a longer time and consequently the steady-state channel block increases. The increase in depolarization frequency also represents repetitive openings of the pore that increase the access of drugs to the intracellular site of action (use-dependent block; Hille, 1977, 2001). The rate of recovery from blockage also slows as cells are depolarized, as occurs during ischemia. Increased AP duration results in a relative increase in the time the channel remains in the inactive state and this can also increase the block by drugs that mainly bind to sodium channels in the inactivated state such as lidocaine.
Current treatment of neuropathic pain includes tricyclic antidepressants (amitriptyline, nortriptyline), local anesthetics (lidocaine, mexiletine), and antiepileptic drugs (phenytoin, carbamazepine, lamotrigine). These drugs however have low efficacy in terms of pain control and are associated with adverse effects involving the heart and CNS.
Diseases Associated with VGSC Mutations (“Channelopathies”)
Ten genes (SCN1A–SCN11A; Table 1) encoding the α-subunit isoforms of the VGSC and four genes encoding the β-subunits (SCN1B–SCN4B; Table 2) have been identified in the human genome. Mutations in any of these genes can affect the structure of the channel and, thus, its biophysical properties leading to the development of “channelopathies” (Tables 1 and 2). All these conditions are associated with autosomal dominant inheritance and de novo mutations have been identified. These channelopathies can be divided in four disease groups depending on the predominant organ involved (George, 2005):
1. Brain sodium channelopathies, which include mutations in SCN1A, SCN2A, SCN3A, and some mutations in SCN8A observed in cases of familial human ataxia and in mice models of ataxia and end-plate diseases (these genes encode the channels NaV1.1, NaV1.2, β1-subunit, and NaV1.6 respectively). SCN1A, SCN2A, and SCN3A gene mutations may give rise to epilepsy and epileptic/convulsive disorders.
2. Skeletal muscle sodium channelopathies. This group involves mutations in the SCN4A, the gene that encodes the NaV1.4 isoform (skeletal muscle specific isoform). SCN4A gene mutations are associated with myotonia, myasthenia syndromes, and paralysis.
3. Cardiac sodium channelopathies, which involve mutations in SCN5A (the gene that encodes NaV1.5, which is predominantly found in cardiac muscle) and SCN10A (the gene that encodes NaV1.8, which has been recently identified in the heart (Facer et al., 2011; Verkerk et al., 2012; Yang et al., 2012) and has been associated in genome-wide association studies (GWAS) with alterations in the ventricular conduction (Chambers et al., 2010; Sotoodehnia et al., 2010).
4. Peripheral nerve sodium channelopathies, which include mutations in SCN9A (NaV1.7), SCN10A (NaV1.8), and SCN11A (NaV1.9). Mutations in these genes have been associated with peripheral pain syndromes (hyperalgesic syndrome) including neuropathic and inflammatory pain.
Brain Sodium Channelopathies
The most commonly affected gene is SCN1A1 , 2. Functional studies of SCN1A missense epileptogenic mutations in vitro have been controversial but several results are consistent with loss of function (hypoexcitability) mutations (Ragsdale, 2008; Mantegazza et al., 2010) and data obtained with animal models have confirmed this (Tang et al., 2009; Martin et al., 2010). Data obtained with a mouse model of Dravet syndrome expressing a truncated Nav1.1 showed that loss of function of this VGSC causes reduced sodium current and excitability in GABAergic neurons, consistent with reduced GABAergic inhibition (Yu et al., 2006). Nav1.1 missense mutations can induce loss of function because of folding defects and these mutants can be rescued by molecular interactions with co-expressed proteins and drugs; this may be one of the causes of the phenotypic variability in GEFS+ and may be exploited for therapeutic potential (Escayg et al., 2000; Meisler and Kearney, 2005; Rusconi et al., 2007, 2009; Catterall et al., 2008). Epilepsy has also been related with SCN1A mutations that alter channel inactivation, resulting in persistent inward sodium current [gain-of-function (hyperexcitability) mutations; Lossin et al., 2002]. The above paragraph describes functional studies with Nav1.1 mutants that yield a wide range of biophysical phenotypes from loss of function to gain-of-function. At first sight this seems to be contradictory. Therefore, questions arise as to how mutations with such diverse functional effects can be associated with the same epileptic syndrome or disease. To better understand this, it is critical to remember that SCN1A is widely expressed in most neurons in the brain. It has also been previously reported that a single sodium channel mutation can produce hyper- or hypoexcitability in different types of neurons (Rush et al., 2006). Therefore, the net effect of the SCN1A mutations on the brain excitability will not only depend on the type of neuron where the mutant channel is expressed but also on the electrical balance between all the ionic currents that contribute to the neuronal AP and the mutant currents. There also might be several additional pathogenic mechanisms involved in the production of epilepsy that are not completely understood yet but still make a significant contribution to the production of the disease.
SMEI is a rare disorder characterized by generalized tonic, clonic, or tonic-clonic seizures that are initially induced by fever and begin during the first year of life. Typically, children with Dravet syndrome [or myoclonic epilepsy of infancy (SMEI); Claes et al., 2001] carry de novo mutations not inherited from their parents. Later, patients also manifest other seizure types, including absence, myoclonic, and simple and complex partial seizures. Psychomotor development delay is observed around the second year of life. SMEI is considered to be the most severe phenotype within the spectrum of generalized epilepsies with febrile seizures-plus. Because of this, genetic screening for SCN1A has become the diagnostic tool for children with early-onset seizures. More than half of the SMEI mutations cause loss of function as a result of stop codons or deletions, leading to decreased levels of functional sodium channels.
SCN1A mutations have also been associated with three other epileptic disorders: intractable childhood epilepsy with generalized tonic-clonic (ICEGTC) seizures, familial febrile convulsions type 3A (FEB3A), and familial hemiplegic migraine type 3 (FHM3). ICEGTC has been included in the Dravet syndrome (Mullen and Scheffer, 2009). A mutation causing simple familial febrile convulsions has been studied by Mantegazza et al. (2005). Familial hemiplegic migraine type 3 is a distinct disease caused by missense mutations of Nav1. Here again the functional effects are controversial, but gain-of-function effects have been observed and this is consistent with the pathogenic mechanism of migraines with aura (Cestele et al., 2008; Kahlig et al., 2008).
Missense mutations of SCN2A were also identified in a small percentage of GEFS+ patients and mainly in patients with benign familial neonatal-infantile seizures (BFNIS), a syndrome of mild seizures that remit during the first year of life without neurologic sequelae. BFNIS mutations produced abnormalities in the sodium channels that led to a reduced channel activity (loss of function; Misra et al., 2008). Other groups have reported mutations in SCN2A that result in a gain-of-function, consistently with the role of Nav1.2 in excitatory cortical neurons (Scalmani et al., 2006; Liao et al., 2010).
The first SCN3A mutation (K353Q) was identified in a patient with partial epilepsy resistant to antiepileptic drugs (Holland et al., 2008). Even though the missense mutation described caused an increase in late current, the pathogenic role of mutated Nav1.3 is still debated. In mouse models, mutations in SCN8A lead to ataxia and end-plate disease. These conditions can be reproduced by conditional knockout of SCN8A in cerebellar Purkinje and granule cell neurons (Levin et al., 2006). In rare cases of human familial ataxia, one frame-shift mutation has been identified in SCN8A that truncates the protein at the DIV and lead to loss of channel function (Vicart et al., 2005; Trudeau et al., 2006) (For more detailed information about mutations of brain VGSC see Catterall, 2010; Mantegazza et al., 2010).
Skeletal Muscle Sodium Channelopathies
The second group of channelopathies includes mutations in the SCN4A gene, which is expressed in skeletal muscle. These skeletal muscle channelopathies (sodium channel myotonic disorders) are part of a group of diseases called non-dystrophic myotonias (Matheus et al., 2010). The clinical disorders can be split between two groups based on the presence or absence of episodic weakness: paramyotonia congenita (characterized by a marked worsening of myotonia by cold and by the presence of clear episodes of weakness), and sodium channel myotonia (notable for the absence of episodic weakness but still have cold sensitivity). The latter group includes all the pure myotonic phenotypes, including the potassium-aggravated myotonias (Fournier et al., 2004).
Causative mutations in the SCN4A gene result in a gain of sodium channel function that may show marked temperature dependence. Almost all mutations (over 40) that have been described are missense mutations with an exception of a three base pair deletion (Michel et al., 2007). Exons 22 and 24 are the main exons involved in paramyotonia congenita, including mutations T1313M, V1589M, and mutations at the R1448 and G1306 position (Vicart et al., 2005; Matheus et al., 2008). Lerche et al. (1993) reported a group of heterozygous mutations at the G1306 position of the SCN4A gene. Electrophysiological studies on patient muscle samples showed slower sodium fast channel inactivation and an increase in late channel opening resulting in a steady-state inward current, sustained muscle depolarization, and muscle fiber hyperexcitability. These findings suggest that SCN4A residue 1306 is important for sodium channel inactivation (Lerche et al., 1993).
Finally, numerous mutations in SCN4A gene have also been related to hypokalemic periodic paralysis. This is a muscle disease characterized by episodes of extreme muscle weakness, and it usually begins in infancy or early childhood. Most often, these episodes involve a temporary inability to move muscles in the arms and legs. Sokolov et al. (2007) reported three mutations in gating-charge-carrying arginine residues in an S4 segment that cause hypokalemic periodic paralysis. The mutations induce a hyperpolarization-activated cationic leak through the voltage sensor of the skeletal muscle NaV1.4 channel, consistent with a gain-of-function. This “gating pore current” is active at the resting membrane potential and closed by depolarizations that activate the voltage sensor. The results reported by these authors showed a clear correlation between mutations that cause gating pore current and hypokalemic periodic paralysis.
Cardiac Sodium Channelopathies
Nav1.5, encoded by SCN5A, conducts the inward sodium current (INa) that initiates the cardiac AP. SCN5A-mediated late sodium current also influences repolarization and refractoriness. Mutations in the SCN5A gene result in alterations in the function of the α-subunit of the cardiac isoform Nav1.5 channel that have been associated with several inherited arrhythmia syndromes. The main entities related to SCN5A mutations include an autosomal dominant form of the LQTS (LQT3; Wang et al., 1995), BS (Probst et al., 2003), progressive cardiac conduction disease (CCD; Scott et al., 1999), sinus node dysfunction (SND; Benson et al., 2003), AF (Olson et al., 2005; Darbar et al., 2008), atrial standstill (Tan, 2006; Remme et al., 2008), and dilated cardiomyopathy (DCM; McNair et al., 2004). Most of these diseases are associated with an increased risk of sudden cardiac death (SCD). The malfunction of the β-subunits (β1 through β4) as well as some of the protein partners that interact with Nav1.5 α-subunit (like caveolin-3 and α-1 syntrophin) have been recently associated with diseases that resemble these arrhythmia phenotypes (Vatta et al., 2006; Cronk et al., 2007; Wu et al., 2008; Watanabe et al., 2009).
Long QT syndrome is characterized by a cardiac repolarization abnormality, with a prolonged QT interval duration observed on 12-lead ECG and vulnerability to a polymorphic ventricular tachycardia called Torsade de Pointes. About 5–10% of LQTS cases are related to mutations in SCN5A (LQT3) or the genes that encode the ChiPs. Mutations in SCN5A compromise the II–IV linker and disrupt fast inactivation, allowing repeated reopening of the channel during sustained depolarization. As a consequence, a small persistent sodium current is evoked during the AP plateau. This excessive inward current (gain-of-function) delays the repolarization of the cell, prolonging AP duration, and increasing the risk for ventricular arrhythmias.
Brugada Syndrome is a genetic disease that has been associated with ventricular fibrillation and SCD in young people. Approximately 20% of these patients have mutations in the SCN5A gene. More than 200 mutations have been associated with this disease (Kapplinger et al., 2010). In contrast to LQTS3 mutations, SCN5A mutations related to BS result in a loss of function of the channel. This can be produced by a confluence of different mechanisms, such as trafficking defects, generation of defective or truncated proteins, faster channel inactivation, shift of voltage-dependence inactivation toward a more depolarized membrane potential, or even slow recovery from inactivation. The electrical consequence of this is the presence of a slower conduction substrate. BS has also been associated with mutations in the genes that encode β1- (SCN1B, BS type 5) and β3-subunits (SCN3B, BS type 7; Abriel, 2010) of the cardiac sodium channel.
Genetic mutations in SCN5A specific only to AF have recently been described. Recently, Li et al. (2009) identified a novel coding variant, K1493R, which altered a highly conserved residue in the DIII–IV linker and was located six amino acids downstream from the fast inactivation motif of sodium channels. Biophysical studies of K1493R in tsA-201 cells demonstrated a significant positive shift in voltage-dependence of inactivation and a large ramp current near resting membrane potential, indicating a gain-of-function. Enhanced cellular excitability was observed in transfected HL-1 atrial cardiomyocytes, including spontaneous AP depolarizations and a lower threshold for AP firing. These novel biophysical observations provide molecular evidence linking cellular “hyperexcitability” as a mechanism inducing vulnerability to AF.
Other pathologies related to mutations in SCN5A include progressive familial heart block type 1A (PFHB1A), sick sinus syndrome type 1 (SSS1), sudden infant death syndrome (SIDS), familial atrial standstill, and DCM. For a more detailed review on SCN5A channelopathies see Zimmer and Surber (2008), and Wilde and Brugada (2011).
In addition to SCN5A mutations, variants in SCN10A (the gene that encodes NaV1.8) can also lead to alterations in the cardiac rhythm. NaV1.8 has only recently been identified in the heart (Facer et al., 2011; Verkerk et al., 2012) and GWAS have identified common genetic variants in this gene that modulate ventricular conduction (Chambers et al., 2010; Sotoodehnia et al., 2010).
Peripheral Nerve Sodium Channelopathies
Lampert et al. and Theile and Cummins, recently published extensive reviews on the role of sodium channels in chronic and neuropathic pain syndromes (Lampert et al., 2010; Theile and Cummins, 2011). Neuropathic pain is defined as “pain caused by a lesion or disease of the somatosensory nervous system,” and can be divided into central and peripheral neuropathic pain. Typical examples of neuropathic pain include post-herpetic neuralgia, painful diabetic neuropathy, phantom limb pain, and spinal cord injury pain. The fundamental mechanism involved in the production of neuropathic pain is an increase in nerve excitability (and thus changes in VGSC properties), generally manifested in impulses generated ectopically or with minimal stimulation. Nerve injury (classically associated with neuropathic pain) can result in changes in sodium channel trafficking, gene expression, and/or channel kinetics, all of which contribute to neuronal membrane remodeling and hyperexcitability associated with neuropathic pain (Devor, 2006). VGSC Nav1.7, Nav1.8, and Nav1.9 have been particularly identified in the PNS (peripheral neurons and DRG neurons) and seem to have a central role in the genesis of neuropathic pain. Thus, these channels are the new targets for analgesia in peripheral neuropathy pain syndromes. In particular, Nav1.7 is considered to be one of the main mediators of peripheral pain. It has been recently reported that Nav1.8 sodium channel is part of the molecular machinery involved in mechanotransduction of joint pain and other pain syndromes (Schuelert and McDougall, 2012). On the other hand, the role of Nav1.3 in diseased states is still controversial.
Recent human association studies have directly linked SCN9A, the gene that encodes Nav1.7, to three human pain disorders: dominantly inherited gain-of-function mutations in inherited erythromelalgia (IEM; nine mutations), paroxysmal extreme pain disorder (PEPD; eight mutations), and recessively inherited loss-of-function mutations in Nav1.7-related congenital insensitivity to pain (CIP; fourteen mutations) (Dib-hajj et al., 2009).
Inherited erythromelalgia (IEM) is a chronic neuropathic pain syndrome that is characterized by excruciating painful attacks in the extremities that begin in childhood and progress over life. A shift to voltage-dependent activation toward more negative potentials seems to be a common factor in all the mutations of SCN9A that lead to this disease. This leftward shift of activation can lead to a hyperexcitability state (gain-of-function mutations). Many mutations also delay inactivation, and therefore, larger currents result from slow depolarizing stimuli (“ramp currents”).
Paroxysmal extreme pain disorder (PEPD), previously referred to as familial rectal pain (Fertleman et al., 2006), is characterized by severe pain accompanied by flushing which are induced by bowel movements or probing of the perianal areas, and are sometimes accompanied by tonic non-epileptic seizures, syncope, bradycardia, and occasionally asystole.
Congenital insensitivity to pain (CIP) is characterized by complete absence of pain perception in patients with non-functional Nav1.7. These patients also exhibit partial anosmia. In this case, the mutations in SCN9A identified introduce a stop codon leading to the production of truncated proteins that are non-functional. For further details on the mutations related to each of these diseases see Lampert et al. (2010).
Summary
Voltage-gated sodium channels are widely distributed in excitable and non-excitable cells, and play a critical role in electrical activation in the body. VGSC constitute macromolecular complexes, in which their function relies on both the specific structure of the channel protein (α- and β-subunits) as well as their protein partners (ChiPs). Since VGSC occur predominantly in the central and PNS, and striated (skeletal and cardiac) muscles, mutations in genes encoding VGSC and ChiPs will culminate in diseases named “channelopathies” that can be grouped into four main categories: epileptic syndromes, skeletal myopathies, cardiac arrhythmias, and neuropathies (with pain-related syndromes). Pathologic conditions can also arise from the up regulation of the VGSC, as for example in highly aggressive prostate (Nav1.7) and breast (Nav1.5) metastatic carcinomas, An improved understanding of the critical role of the molecular composition of ion channel complexes, the influence of protein partners, and the specific cellular domains underlying protein interactions, are essential for the development of new therapies to treat channelopathies associated with VGSC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by U19 HL65962, HL092217, AHA Established Investigator Award (0940116N), and the Heart and Stroke Foundation of Ontario Clinician-Scientist Award.
Footnotes
References
Abriel, H. (2010). Cardiac sodium channel Nav1.5 and interacting proteins: physiology and pathophysiology. J. Mol. Cell. Cardiol. 48, 2–11.
Akai, J., Makita, N., Sakurada, H., Shirai, N., Ueda, K., Kitabatake, A., Nakazawa, K., Kimura, A., and Hiraoka, M. (2000). A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 479, 29–34.
Armstrong, C. M., and Bezanilla, F. (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242, 459–461.
Armstrong, C. M., and Bezanilla, F. (1974). Charge movement associated with the opening and closing of the activation gates of the Na+ channels. J. Gen. Physiol. 63, 533–552.
Audenaert, D., Claes, L., Ceulemans, B., Lofgren, A., Van Broeckhoven, C., and De Jonghe, P. (2003). A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 61, 854–856.
Bennett, V., and Healy, J. (2008). Being there: cellular targeting of voltage-gated sodium channels in the heart. J. Cell Biol. 180, 13–15.
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., Rhodes, T. H., and George, A. L. Jr. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028.
Brackenbury, W. J., Davis, T. H., Chen, C., Slat, E. A., Detrow, M. J., Dickendesher, T. L., Ranscht, B., and Isom, L. L. (2008). Voltage-gated Na+ channel beta1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J. Neurosci. 28, 3246–3256.
Brackenbury, W. J., and Isom, L. L. (2008). Voltage-gated Na+ channels: potential for beta subunits as therapeutic targets. Expert Opin. Ther. Targets 12, 1191–1203.
Brette, F., and Orchard, C. H. (2006). Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem. Biophys. Res. Commun. 348, 1163–1166.
Bruton, L. L., Chabner, B. A., and Knollmann, B. C. (2011). Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edn. New York: Mc Graw-Hill Medical Books.
Burgess, D. L., Kohrman, D. C., Galt, J., Plummer, N. W., Jones, J. M., Spears, B., and Meisler, M. H. (1995). Mutation of a new sodium channel gene, Scn8a, in the mouse mutant motor endplate disease. Nat. Genet. 10, 461–465.
Cannon, S. C. (1997). From mutation to myotonia in sodium channel disorders. Neuromuscul. Disord. 7, 241–249.
Casula, M. A., Facer, P., Powell, A. J., Kinghorn, I. J., Plumpton, C., Tate, S. N., Bountra, C., Birch, R., and Anand, P. (2004). Expression of the sodium channel beta3 subunit in injured human sensory neurons. Neuroreport 15, 1629–1632.
Catterall, W. A. (2010). Ion channel voltage sensor: structure, function, and pathophysiology. Neuron 67, 915–928.
Catterall, W. A. (1986a). Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 55, 953–985.
Catterall, W. A. (1986b). Voltage-dependent gating of sodium channels: correlating structure and function. Trends Neurosci. 9, 7–10.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Catterall, W. A., Dib-Hajj, S., Meisler, M. H., and Pietrobon, D. (2008). Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J. Neurosci. 28, 11768–11777.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). Nav1.1 channels and epilepsy. J. Physiol. (Lond.) 588.11, 1849–1859.
Cestele, S., and Catterall, W. A. (2000). Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochemistry 82, 883–892.
Cestele, S., Scalmani, P., Rusconi, R., Terragni, B., Franceschetti, S., and Mantegazza, M. (2008). Self-limited hyperexcitability: functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na channel. J. Neurosci. 28, 7273–7283.
Chahine, M., Bennett, P. B., George, A. L. Jr., and Horn, R. (1994). Functional expression and properties of the human skeletal muscle sodium channel. Pflugers Arch. Eur. J. Physiol. 427, 136–142.
Chambers, J. C., Zhao, J., Terracciano, C. M. N., Bezzina, C. R., Zhang, W., Kaba, R., Navaratnarajah, M., Lotlikar, A., Sehmi, J. S., Kooner, M. K., Deng, G., Siedlecka, U., Parasramka, S., El-Hamamsy, I., Wass, M. N., Dekker, L. R., de Jong, J. S., Sternberg, M. J., McKenna, W., Severs, N. J., de Silva, R., Wilde, A. A., Anand, P., Yacoub, M., Scott, J., Elliott, P., Wood, J. N., and Kooner, J. S. (2010). Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 42, 149–152.
Chauhan, V. S., Tuvia, S., Buhusi, M., Bennett, V., and Grant, A. O. (2000). Abnormal cardiac Na+ channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ. Res. 86, 441–447.
Chen, C., and Cannon, S. C. (1995). Modulation of Na+ channel inactivation by the beta1 subunit: a deletion analysis. Pflugers Arch. 431, 186–195.
Chen, C., Westenbroek, R. E., Xu, X., Edwards, C. A., Sorenson, D. R., Chen, Y., McEwen, D. P., O’Malley, H. A., Bharucha, V., Meadows, L. S., Knudsen, G. A., Vilaythong, A., Noebels, J. L., Saunders, T. L., Scheuer, T., Shrager, P., Catterall, W. A., and Isom, L. L. (2004). Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J. Neurosci. 24, 4030–4042.
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., Potenza, D., Moya, A., Borggrefe, M., Breithardt, G., Ortiz-Lopez, R., Wang, Z., Antzelevitch, C., O’Brien, R. E., Schulze-Bahr, E., Keating, M. T., Towbin, J. A., and Wang, Q. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296.
Chkourko, H. S., Guerrero-Serna, G., Lin, X., Darwish, N., Pohlmann, J. R., Cook, K. E., Martens, J. R., Rothenberg, E., Musa, H., and Delmar, M. (2012). Remodeling of mechanical junctions and of microtubule-associated proteins accompanies cardiac connexin 43 lateralization. Heart Rhythm 9, 1133–1140.
Claes, L., Del-Favero, J., Ceulemans, B., Lagae, L., Van Broeckhoven, C., and De Jonghe, P. (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 68, 1327–1332.
Clare, J. J., Tate, S. N., Nobbs, M., and Romanos, M. A. (2000). Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today 5, 506–520.
Coward, K., Jowett, A., Plumpton, C., Powell, A., Birch, R., Tate, S., Bountra, C., and Anand, P. (2001). Sodium channel β1 and β2 subunits parallel SNS/PN3 α-subunit changes in injured human sensory neurons. Neuroreport 12, 483–488.
Cronk, L. B., Ye, B., Kaku, T., Tester, D. J., Vatta, M., Makielski, J. C., and Ackerman, M. J. (2007). Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm 4, 161–166.
Darbar, D., Kannankeril, P. J., Donahue, B. S., Kucera, G., Stubblefield, T., Haines, J. L., George, A. Jr., and Roden, D. M. (2008). Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 117, 1927–1935.
Davis, J. Q., Lambert, S., and Bennett, V. (1996). Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J. Cell Biol. 135, 1355–1367.
Davis, T. H., Chen, C., and Isom, L. L. (2004). Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J. Biol. Chem. 279, 51424–51432.
Dib-hajj, S. D., Binshtok, A. M., Cummins, T. R., Jarvis, M. F., Samad, T., and Zimmermann, K. (2009). Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res. Rev. 60, 65–83.
Dietrich, P. S., McGivern, J. G., Delgado, S. G., Koch, B. D., Eglen, R. M., Hunter, J. C., and Sangameswaran, L. (1998). Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglion. J. Neurochem. 70, 2262–2272.
Dover, K., Solinas, S., D’Angelo, E., and Goldfarb, M. (2010). Long-term inactivation particle for voltage-gated sodium channels. J. Physiol. (Lond.) 588, 3695–3711.
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T., and MacKinnon, R. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77.
Escayg, A., MacDonald, B. T., Meisler, M. H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., Brice, A., LeGuern, E., Moulard, B., Chaigne, D., Buresi, C., and Malafosse, A. (2000). Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24, 343–345.
Facer, P., Phil, M., Punjabi, P. P., Abrari, A., Kaba, R. A., Severs, N. J., Chambers, J., Kooner, J. S., and Anand, P. (2011). Localisation of SCN10A gene product Nav1.8 and novel pain-related ion channels in human heart. Int. Heart J. 52, 146–152.
Fertleman, C. R., Baker, M. D., Parker, K. A., Moffatt, S., Elmsile, F. V., Abrahamsen, B., Ostman, J., Klugbauer, N., Wood, J. N., Gardiner, R. M., and Rees, M. (2006). SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774.
Flucher, B. E., and Daniels, M. P. (1989). Distribution of Na+ channels and ankyrin in neuromuscular junctions is complementary to that of acetylcholine receptors and the 43 kd protein. Neuron 3, 163–175.
Fournier, E., Arzel, M., Sternberg, D., Vicart, S., Laforet, P., Eymard, B., Willer, J. C., Tabti, N., and Fontaine, B. (2004). Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann. Neurol. 56, 650–661.
Furukawa, T., Ono, Y., Tsuchiya, H., Katayama, Y., Bang, M.-L., Labeit, D., Labeit, S., Inagaki, N., and Gregorio, C. C. (2001). Specific interaction of the potassium channel β-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J. Mol. Biol. 313, 775–784.
Garrido, J. J., Fernandes, F., Moussif, A., Fache, M. P., Giraud, P., and Dargent, B. (2003). Dynamic compartmentalization of the voltage-gated sodium channels in axons. Biol. Cell 95, 437–445.
Gavillet, B., Rougier, J. S., Domenighetti, A. A., Behar, R., Boixel, C., Ruchat, P., Lehr, H. A., Pedrazzini, T., and Abriel, H. (2006). Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 99, 407–414.
George, A. L. J. (2005). Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115, 1990–1999.
George, A. L. J., Knittle, T. J., and Tamkun, M. M. (1992). Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc. Natl. Acad. Sci. U.S.A. 89, 4893–4897.
Goldfarb, M. (2012). Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell. Mol. Life Sci. 69, 1067–1076.
Goldin, A. L. (2003). Mechanisms of sodium channel inactivation. Curr. Opin. Neurobiol. 13, 284–290.
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., Kallen, R. G., Mandel, G., Meisler, M. H., Netter, Y. B., Noda, M., Tamkun, M. M., Waxman, S. G., Wood, J. N., and Catterall, W. A. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368.
Gorter, J. A., Zurolo, E., Lyer, A., Fluiter, K., Van Vliet, E. A., Baayen, J. C., and Aronica, E. (2010). Induction of sodium channel Nax (SCN7A) expression in rat and human hippocampus in temporal lobe epilepsy. Epilepsia 51, 1791–1800.
Grieco, T. M., Malhotra, J. D., Chen, C., Isom, L. L., and Raman, I. M. (2005). Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 45, 233–244.
Guy, H. R., and Seetharamulu, P. (1986). Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. U.S.A. 508, 508–512.
Haenggi, T., and Fritschy, J. M. (2006). Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell. Mol. Life Sci. 63, 1614–1631.
Hains, B. C., Klein, J. P., Saab, C. Y., Craner, M. J., Black, J. A., and Waxman, S. G. (2003). Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 23, 8881–8892.
Haworth, R. S., Cuello, F., Herron, T. J., Franzen, G., Kentish, J. C., Gautel, M., and Avkiran, M. (2004). Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ. Res. 95, 1091–1099.
Hille, B. (1977). Local anesthetics: hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69, 497–515.
Hiyama, T. Y., Watanabe, E., Ono, K., Inenaga, K., Tamkun, M. M., Yoshida, S., and Noda, M. (2002). Nax channel involved in CNS sodium-level sensing. Nat. Neurosci. 5, 511–512.
Hockerman, G. H., Peterson, B. Z., Johnson, B. D., and Catterall, W. A. (1997). Molecular determinants of drug binding and action on L-type calcium channels. Annu. Rev. Pharmacol. Toxicol. 37, 361–396.
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544.
Holland, K. D., Kearney, J. A., Glauser, T. A., Buck, G., Keddache, M., Blankston, J. R., Glaaser, I. W., Kass, R. S., and Meisler, M. H. (2008). Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci. Lett. 433, 65–70.
Ingham, R. J., Gish, G., and Pawson, T. (2004). The Nedd4 of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23, 1972–1984.
Isom, L. L., De Jongh, K. S., and Catterall, W. A. (1994). Auxiliary subunits of voltage-gated ion channels. Neuron 12, 1183–1194.
Isom, L. L., Scheuer, T., Brownstein, A. B., Ragsdale, D. S., Murphy, B. J., and Catterall, W. A. (1995). Functional co-expression of the beta1 and type IIA alpha subunits of sodium channels in a mamalian cell line. J. Biol. Chem. 270, 3306–3312.
Jenkins, S. M., and Bennett, V. (2001). Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J. Cell Biol. 155, 739–746.
Jogini, V., and Roux, B. (2005). Electrostatics of the intracellular vestibule of K+ channels. J. Mol. Biol. 354, 272–288.
Johnson, D., and Bennett, E. S. (2006). Isoform-specific effects of the beta 2 subunit on voltage-gated sodium channel gating. J. Biol. Chem. 281, 25875–25881.
Kahlig, K. M., Rhodes, T. H., Pusch, M., Freilinger, T., Pereira-Monteiro, J. M., Ferrari, M. D., van den Maagdenberg, A. M. J. M., Dichgans, M., and George, A. L. Jr. (2008). Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. U.S.A. 105, 9799–9804.
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., Brugada, P., Fressart, V., Guerchicoff, A., Harris-Kerr, C., Kamakura, S., Kyndt, F., Koopmann, T. T., Miyamoto, Y., Pfeiffer, R., Pollevick, G. D., Probst, V., Zumhagen, S., Vatta, M., Towbin, J. A., Shimizu, W., Schulze-Bahr, E., Antzelevitch, C., Salisbury, B. A., Guicheney, P., Wilde, A. A., Brugada, R., Schott, J. J., and Ackerman, M. J. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46.
Kazarinova-Noyes, K., Malhotra, J. D., McEwen, D. P., Mattei, L. N., Berglund, E. O., Ranscht, B., Levinson, S. R., Schachner, M., Shrager, P., Isom, L. L., and Xiao, Z. C. (2001). Contactin associates with Na+ channels and increases their functional expression. J. Neurosci. 21, 7517–7525.
Keynes, R. D., and Rojas, E. (1973). Characteristics of the sodium gating current in squid giant axons. J. Physiol. 233, 28P.
Knoll, R., Hoshijima, M., Hoffman, M. H., Person, V., Lorenzen-Schmidt, I., Bang, M.-L., Hayashi, T., Shiga, N., Yasukawa, H., Schaper, W., McKenna, W., Yokoyama, M., Schork, J. N., Omens, H. J., McCulloch, D. A., Kimura, A., Gregorio, C. C., Poller, W., Schaper, J., Schultheiss, H. P., and Chien, K. R. (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955.
Kohrman, D. C., Smith, M. R., Goldin, A. L., Harris, J., and Meisler, M. H. (1996). A missense mutation in the sodium channel SCN8A is responsible for cerebellar ataxia in the mouse mutant jolting. J. Neurosci. 16, 5993–5999.
Koishi, R., Xu, H., Ren, D., Navarro, B., Spiller, B. W., Shi, Q., and Clapham, D. E. (2004). A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem. 279, 9532–9538.
Kojic, S., Medeot, E., Guccione, E., Krmac, H., Zara, I., Martinelli, V., Valle, G., and Faulkner, G. (2004). The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J. Mol. Biol. 339, 313–325.
Kordeli, E., Lambert, S., and Bennett, V. (1995). AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J. Biol. Chem. 270, 2352–2359.
Kordeli, E., Ludosky, M. A., Deprette, C., Frappier, T., and Cartaud, J. (1998). AnkyrinG is associated with the postsynaptic membrane and the sarcoplasmic reticulum in the skeletal muscle fiber. J. Cell Sci. 111, 2197–2207.
Kudora, Y., Miyamoto, K., Matsumoto, M., Maeda, Y., Kanaori, K., Otaka, A., Fujii, N., and Nakagawa, T. (2000). Structural study of the sodium channel inactivation gate peptide including an isoleucine-phenylalanine-methionine motif and its analogous peptide (phenylalanine/glutamine) in trifluoroethanol solutions and SDS micelles. J. Pept. Res. 56, 172–184.
Laezza, F., Gerber, B. R., Lou, J.-Y., Kozel, M. A., Hartman, H., Craig, A. M., Ornitz, D. M., and Nerbonne, J. M. (2007). The FGF14F145S mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J. Neurosci. 27, 12033–12044.
Lampert, A., O’Reilly, A. O., Reeh, P., and Leffler, A. (2010). Sodium channelopathies and pain. Pflugers Arch. 460, 249–263.
Lemaillet, G., Walker, B., and Lambert, S. (2003). Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 278, 27333–27339.
Lerche, H., Heine, R., Pika, U., George, A. L. Jr., Mitrovic, N., Browatzki, M., Weiss, T., Rivet-Bastide, M., Franke, C., and Lomonaco, M. (1993). Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J. Physiol. 470, 13–22.
Levin, S. I., Khaliq, Z. M., Aman, T. K., Grieco, T. M., Kearney, J. A., Raman, I. M., and Meisler, M. H. (2006). Impaired motor function and learning in mice with conditional knockout of the Na channel SCN8A (Nav1.6) in cerebellar Purkinje neurons and granule cells. J. Neurophysiol. 96, 785–793.
Li, P., and Zhu, S. (2011). Molecular design of new sodium channel blockers. Biochem. Biophys. Res. Commun. 414, 321–325.
Li, Q., Huang, H., Liu, G., Lam, K., Rutberg, J., Green, M. S., Birnie, D. H., Lemery, R., Chahine, M., and Gollob, M. H. (2009). Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem. Biophys. Res. Commun. 380, 132–137.
Liao, Y., Deprez, L., Maljevic, S., Pitsch, J., Claes, L., Hristova, D., Jordanova, A., Ala-Mello, S., Bellan-Koch, A., Blazevic, D., Schubert, S., Thomas, E. A., Petrou, S., Becker, A. J., De Jonghe, P., and Lerche, H. (2010). Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 133, 1403–1414.
Liu, C.-J., Dib-Hajj, S. D., and Waxman, S. G. (2001). Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN). J. Biol. Chem. 276, 18925–18933.
Liu, G., Yarov-Yarovoy, V., Nobbs, M., Clare, J. J., Scheuer, T., and Catterall, W. A. (2003a). Differential interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage-gated sodium channels. Neuropharmacology 44, 413–422.
Liu, C.-J., Dib-Hajj, S. D., Renganathan, M., Cummins, T. R., and Waxman, S. G. (2003b). Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 278, 1029–1036.
Lopez-Santiago, L. F., Brackenbury, W. J., Chen, C., and Isom, L. L. (2011). Na channel Scn1b gene regulates dorsal root ganglion nociceptor excitability in vivo. J. Biol. Chem. 286, 22913–22923.
Lopez-Santiago, L. F., Pertin, M., Morisod, X., Chen, C., Hong, S., Wiley, J., Decosterd, I., and Isom, L. L. (2006). Sodium channel beta2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J. Neurosci. 26, 7984–7994.
Lossin, C. (2009). Compilation of Genetic Variations in the SCN1A Gene that Alter the Expression or Function of Nav1.1. “SCN1A Infobase.” Available at: http://www.scn1a.info/
Lossin, C., Wang, D. W., Rhodes, T. H., Vanoye, C. G., and George, A. L. (2002). Molecular basis of an inherited epilepsy. Neuron 34, 877–884.
Lowe, J. S., Palygin, O., Bhasin, N., Hund, T. J., Boyden, P. A., Shibata, E., Anderson, M. E., and Mohler, P. J. (2008). Nav channel targeting in the heart requires an ankyrin-G-dependent cellular pathway. J. Cell Biol. 180, 173–186.
Lu, T., Lee, H. C., Kabat, J. A., and Shibata, E. F. (1999). Modulation of rat cardiac sodium channel by the stimulatory G protein α subunit. J. Physiol. 518, 371–384.
Maier, L. S., and Bers, D. M. (2002). Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J. Mol. Cell. Cardiol. 34, 919–939.
Mantegazza, M., Gambardella, A., Rusconi, R., Schiavon, E., Annesi, F., Cassulini, R. R., Labate, A., Carrideo, S., Chifari, R., Canevini, M. P., Canger, R., Franceschetti, S., Annesi, G., Wanke, E., and Quattrone, A. (2005). Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. U.S.A. 102, 18177–18182.
Mantegazza, M., Rusconi, R., Scalmani, P., Avanzini, G., and Franceschetti, S. (2010). Epileptogenic ion channel mutations: from bedside to bench and, hopefully, back again. Epilepsy Res. 92, 1–29.
Mantegazza, M., Yu, F. H., Catterall, W. A., and Scheuer, T. (2001). Role of the C-terminal domain in inactivation of brain and cardiac sodium channels. Proc. Natl. Acad. Sci. U.S.A. 98, 15348–15353.
Martin, M. S., Dutt, K., Papale, L. A., Dube, C. M., Dutton, S. B., de Haan, G., Shankar, A., Tufik, S., Meisler, M. H., Baram, T. Z., Goldin, A. L., and Escayg, A. (2010). Altered function of the SCN1A voltage-gated sodium channel leads to -aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 285, 9823–9834.
Matheus, E., Fialho, D., Tan, S. V., Venance, S. L., Cannon, S. C., Sternberg, D., Fontaine, B., Amato, A. A., Barohn, R. J., Griggs, R. C., Hanna, M. G., and the CINCH Investigators. (2010). The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 133, 9–22.
Matheus, E., Tan, S. V., Fialho, D., Sweeney, M. G., Sud, M. G., Haworth, A., Stanley, E., Cea, G., Davis, M. B., and Hanna, M. G. (2008). What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology 70, 50–53.
Mayans, O., van der Ven, P. F. M., Wilm, M., Mues, A., Young, P., Furst, D. O., Wilmanns, M., and Gautel, M. (1998). Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature 395, 863–869.
Mazzone, A., Strege, P. R., Tester, D. J., Bernard, C. E., Faulkner, G., De Giorgio, R., Makielski, J. C., Stanghellini, V., Gibbons, S. J., Ackerman, M. J., and Farrugia, G. (2008). A mutation in telethonin alters Nav1.5 function. J. Biol. Chem. 283, 16537–16544.
McCormick, K. A., Isom, L. L., Ragsdale, D., Smith, D., Scheuer, T., and Catterall, W. A. (1998). Molecular determinants of Na+ channel function in the extracellular domain of the β1 subunit. J. Biol. Chem. 273, 3954–3962.
McCormick, K. A., Srinivasan, J., White, K., Scheuer, T., and Catterall, W. A. (1999). The extracellular domain of the β1 subunit is both necessary and sufficient for β1-like modulation of sodium channel gating. J. Biol. Chem. 274, 32638–32646.
McEwen, D. P., Chen, C., Meadows, L. S., Lopez-Santiago, L., and Isom, L. L. (2009). The voltage-gated Na+ channel β3 subunit does not mediate trans homophilic cell adhesion or associate with the cell adhesion molecule contactin. Neurosci. Lett. 462, 272–275.
McEwen, D. P., and Isom, L. L. (2004). Heterophilic interactions of sodium channel β1 subunits with axonal and glial cell adhesion molecules. J. Biol. Chem. 279, 52744–52752.
McNair, W. P., Ku, L., Taylor, M. R., Fain, P. R., Dao, D., Wolfel, E., and Mestroni, L. (2004). SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 110, 2163–2167.
Meadows, L. S., Chen, Y. H., Powell, A. J., Clare, J. J., and Ragsdale, D. S. (2002). Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary β1, β2 and β3 subunits. Neuroscience 114, 745–753.
Meadows, L. S., and Isom, L. L. (2005). Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc. Res. 67, 448–458.
Meisler, M. H., and Kearney, J. A. (2005). Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest. 115, 2010–2017.
Michel, P. D. S., Jeannet, P. Y., Dunand, M., Thonney, F., Kress, W., Fontaine, B., Fournier, E., and Kuntzer, T. (2007). Comparative efficacy of repetitive nerve stimulation, exercise, and cold in differentiating myotonic disorders. Muscle Nerve 36, 643–650.
Milstein, M. L., Musa, H., Ponce Balbuena, D., Anumonwo, J. M., Auerbach, D. S., Furspan, P. B., Hou, L., Hu, B., Schumacher, S. M., Vaidyanathan, R., Martens, J. R., and Jalife, J. (2012). Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 1–10.
Misra, S. N., Kahlig, K. M., and George, A. L. (2008). Impaired Nav1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 49, 1535–1545.
Miyamoto, K., Nakagawa, T., and Kuroda, Y. (2001a). Solution structure of the cytoplasmic linker between domain III-S6 and domain IV-S1 (III-IV linker) of the rat brain sodium channel in SDS micelles. Biopolymers 59, 380–393.
Miyamoto, K., Kanaori, K., Nakagawa, T., and Kuroda, Y. (2001b). Solution structures of the inactivation gate particle peptides of rat brain type IIA and human heart sodium channels in SDS micelles. J. Pept. Res. 57, 203–214.
Mohler, P. J. (2006). Ankyrins and human disease: what the electrophysiologist should know. J. Cardiovasc. Electrophysiol. 17, 1153–1159.
Mohler, P. J., Rivolta, I., Napolitano, C., LeMaillet, G., Lambert, S., Priori, S. G., and Bennett, V. (2004). Nav1.5 E 1053K mutation causing Brugada Syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 101, 17533–17538.
Morais-Cabral, J. H., Zhou, Y., and MacKinnon, R. (2001). Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature 414, 37–42.
Motoike, H. K., Liu, H., Glaaser, I. W., Yang, A., Tateyama, M., and Kass, R. S. (2004). The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J. Gen. Physiol. 123, 155–165.
Mues, A., van der Ven, P. F. M., Young, P., Fürst, D. O., and Gautel, M. (1998). Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett. 428, 111–114.
Mullen, S. A., and Scheffer, I. E. (2009). Translational research in epilepsy genetics. Arch. Neurol. 66, 21–26.
Noda, M. (2006). The subfornical organ, a specialized sodium channel, and the sensing of sodium levels in the brain. Neuroscientist 12, 80–91.
Noda, M., Ikeda, T., Kayano, T., Suzuki, H., Takeshima, H., Kurasaki, M., Takahashi, H., and Numa, S. (1986). Existence of distinct sodium channel messenger RNAs in rat brain. Nature 320, 188–192.
Noda, M., Suzuki, H., Numa, S., and Stuhmer, W. A. (1989). A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 259, 213–216.
Ogata, N., and Ohishi, Y. (2002). Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 88, 365–377.
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., Horton, S. C., Rodeheffer, R. J., and Anderson, J. L. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454.
O’Malley, H. A., Shreiner, A. B., Chen, G. H., Huffnagle, G. B., and Isom, L. L. (2009). Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Mol. Cell. Neurosci. 40, 143–155.
Onkal, L., and Djamgoz, M. B. A. (2009). Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav1.5 in breast cancer. Eur. J. Pharmacol. 625, 206–219.
Orrico, A., Galli, L., Grosso, S., Buoni, S., Pianigiani, R., Balestri, P., and Sorrentino, V. (2009). Mutational analysis of the SCN1A, SCN1B and GABRG2 genes in 150 Italian patients with idiopathic childhood epilepsies. Clin. Genet. 75, 579–581.
Oyama, F., Miyazaki, H., Sakamoto, N., Becquet, C., Machida, Y., Kaneko, K., Uchikawa, C., Suzuki, T., Kurosawa, M., Ikeda, T., Tamaoka, A., Sakurai, T., and Nukina, N. (2006). Sodium channel beta4 subunit: down-regulation and possible involvement in neuritic degeneration in Huntington’s disease transgenic mice. J. Neurochem. 98, 518–529.
Palygin, O. A., Pettus, J. M., and Shibata, E. F. (2008). Regulation of caveolar cardiac sodium current by a single Gs alpha histidine residue. Am. J. Physiol. Heart Circ. Physiol. 294, H1693–H1699.
Patino, G. A., Claes, L. R., Lopez-Santiago, L. F., Slat, E. A., Dondeti, R. S., Chen, C., O’Malley, H. A., Gray, C. B., Miyazaki, H., Nukina, N., Oyama, F., De Jonghe, P., and Isom, L. L. (2009). A functional null mutation of SCN1B in a patient with Dravet syndrome. J. Neurosci. 29, 10764–10778.
Patino, G. A., and Isom, L. L. (2010). Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci. Lett. 486, 53–59.
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358.
Pertin, M., Allchorne, A. J., Beggah, A. T., Woolf, C. J., and Decosterd, I. (2007). Delayed sympathetic dependence in the spared nerve injury (SNI) model of neuropathic pain. Mol. Pain 3, 21.
Pertin, M., Ji, R. R., Berta, T., Powell, A. J., Karchewski, L., Tate, S. N., Isom, L. L., Woolf, C. J., Gilliard, N., Spahn, D. R., and Decosterd, I. (2005). Upregulation of the voltage-gated sodium channel beta2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J. Neurosci. 25, 10970–10980.
Petitprez, S., Zmoos, A. F., Ogrodnik, J., Balse, E., Raad, N., El-Haou, S., Albesa, M., Bittihn, P., Luther, S., Lehnart, S. E., Hatem, S. N., Coulombe, A., and Abriel, H. (2011). SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ. Res. 108, 294–304.
Pitt, G. S. (2007). Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc. Res. 73, 641–647.
Probst, V., Kyndt, F., Potet, F., Trochu, J. N., Mialet, G., Demolombe, S., Schott, J. J., Baro, I., Escande, D., and Le, M. H. (2003). Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenegre disease. J. Am. Coll. Cardiol. 41, 643–652.
Ragsdale, D. S. (2008). How do mutant Nav1.1 sodium channels cause epilepsy? Brain Res. Rev. 58, 149–159.
Ragsdale, D. S., McPhee, J. C., Scheuer, T., and Catterall, W. A. (1994). Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265, 1724–1728.
Ragsdale, D. S., McPhee, J. C., Scheuer, T., and Catterall, W. A. (1996). Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U.S.A. 93, 9270.
Rasmusson, R. L., Morales, M. J., Wang, S., Liu, S., Campbell, D. L., Brahmajothi, M. V., and Strauss, H. C. (1998). Inactivation of voltage-gated cardiac K+ channels. Circ. Res. 82, 739–750.
Remme, A. C., Wilde, A. A., and Bezzina, C. R. (2008). Cardiac sodium channel overlap syndromes: different faces for SCN5A mutations. Trends Cardiovasc. Med. 18, 78–87.
Ren, D., Navarro, B., Xu, H., Yue, L., Shi, Q., and Clapham, D. E. (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2371–2375.
Rohl, C. A., Boeckman, F. A., Baker, C., Scheuer, T., Catterall, W. A., and Klevit, R. E. (1999). Solution structure of the sodium channel inactivation gate. Biochemistry 38, 855–861.
Rook, M. B., Evers, M. M., Vos, M. A., and Bierhuizen, M. F. A. (2012). Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc. Res. 93, 12–23.
Rougier, J. S., van Bemmelen, M. X., Bruce, M. C., Jespersen, T., Gavillet, B., Apotheloz, F., Cordonier, S., Staub, O., Rotin, D., and Abriel, H. (2005). Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am. J. Physiol. Cell Physiol. 288, C692–C701.
Rudy, Y., and Silva, J. R. (2006). Computational biology in the study of cardiac ion channels and cell electrophysiology. Q. Rev. Biophys. 39, 57–116.
Rusconi, R., Combi, R., Cestele, S., Grioni, D., Franceschetti, S., Dalpra, L., and Mantegazza, M. (2009). A rescuable folding defective Nav1.1 (SCN1A) sodium channel mutant causes GEFS+: common mechanism in Nav1.1 related epilepsies? Hum. Mutat. 30, E747–E760.
Rusconi, R., Scalmani, P., Cassulini, R. R., Giunti, G., Gambardella, A., Franceschetti, S., Annesi, G., Wanke, E., and Mantegazza, M. (2007). Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na channel mutant. J. Neurosci. 27, 11037–11046.
Rush, A. M., Dib-Hajj, S. D., Liu, S., Cummins, T. R., Black, J. A., and Waxman, S. G. (2006). A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 8245–8250.
Rybin, V. O., Xiaohong, X., Lisanti, M. P., and Steinberg, S. F. (2000). Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. J. Biol. Chem. 275, 41447–41457.
Sato, C., Ueno, Y., Asai, K., Takahashi, K., Sato, M., Engel, A., and Fujiyoshi, Y. (2001). The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature 409, 1047–1051.
Sato, P. Y., Coombs, W., Lin, X., Nekrasova, O., Green, K. J., Isom, L. L., Taffet, S. M., and Delmar, M. (2011). Interactions between ankyrin-G, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201.
Sato, P. Y., Musa, H., Coombs, W., Guerrero-Serna, G., Patinio, G. A., Taffet, S. M., Isom, L. L., and Delmar, M. (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 105, 523–526.
Scalmani, P., Rusconi, R., Armatura, E., Zara, F., Avanzini, G., Franceschetti, S., and Mantegazza, M. (2006). Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J. Neurosci. 26, 10100–10109.
Scheffer, I. E., Harkin, L. A., Grinton, B. E., Dibbens, L. M., Turner, S. J., Zielinski, M. A., Xu, R., Jackson, G., Adams, J., Connellan, M., Petrou, S., Wellard, R. M., Briellmann, R. S., Wallace, R. H., Mulley, J. C., and Berkovic, S. F. (2007). Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130, 100–109.
Schuelert, N., and McDougall, J. J. (2012). Involvement of NaV 1.8 sodium ion channels in the transduction of mechanical pain in a rodent model of osteoarthritis. Arthritis Res. Ther. 14, 1–9.
Schwencke, C., Okumura, S., Yamamoto, M., Geng, Y. J., and Ishikawa, Y. (1999). Colocalization of β-adrenergic receptors and caveolin within the plasma membrane. J. Cell. Biochem. 75, 64–72.
Scott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M., Wilde, A. A., Escande, D., Mannens, M. M., and Le, M. H. (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21.
Shao, D., Okuse, K., and Djamgoz, M. B. A. (2009). Protein–protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. Int. J. Biochem. Cell Biol. 41, 1471–1481.
Smith, M. R., Smith, R. D., Plummer, N. W., Meisler, M. H., and Goldin, A. L. (1998). Functional analysis of the mouse SCN8A sodium channel. J. Neurosci. 18, 6093–6102.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78.
Sotoodehnia, N., Isaacs, A., de Bakker, P. I. W., Dörr, M., Newton-Cheh, C., Nolte, I. M., van der Harst, P., Müller, M., Eijgelsheim, M., Alonso, A., Hicks, A. A., Padmanabhan, S., Hayward, C., Smith, A. V., Polasek, O., Giovannone, S., Fu, J., Magnani, J. W., Marciante, K. D., Pfeufer, A., Gharib, S. A., Teumer, A., Li, M., Bis, J. C., Rivadeneira, F., Aspelund, T., Köttgen, A., Johnson, T., Rice, K., Sie, M. P., Wang, Y. A., Klopp, N., Fuchsberger, C., Wild, S. H., Mateo Leach, I., Estrada, K., Völker, U., Wright, A. F., Asselbergs, F. W., Qu, J., Chakravarti, A., Sinner, M. F., Kors, J. A., Petersmann, A., Harris, T. B., Soliman, E. Z., Munroe, P. B., Psaty, B. M., Oostra, B. A., Cupples, L. A., Perz, S., de Boer, R. A., Uitterlinden, A. G., Völzke, H., Spector, T. D., Liu, F. Y., Boerwinkle, E., Dominiczak, A. F., Rotter, J. I., van Herpen, G., Levy, D., Wichmann, H. E., van Gilst, W. H., Witteman, J. C., Kroemer, H. K., Kao, W. H., Heckbert, S. R., Meitinger, T., Hofman, A., Campbell, H., Folsom, A. R., van Veldhuisen, D. J., Schwienbacher, C., O’Donnell, C. J., Volpato, C. B., Caulfield, M. J., Connell, J. M., Launer, L., Lu, X., Franke, L., Fehrmann, R. S., te Meerman, G., Groen, H. J., Weersma, R. K., van den Berg, L. H., Wijmenga, C., Ophoff, R. A., Navis, G., Rudan, I., Snieder, H., Wilson, J. F., Pramstaller, P. P., Siscovick, D. S., Wang, T. J., Gudnason, V., van Duijn, C. M., Felix, S. B., Fishman, G. I., Jamshidi, Y., Stricker, B. H., Samani, N. J., Kääb, S., and Arking, D. E. (2010). Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 42, 1068–1076.
Spampanato, J., Escayg, A., Meisler, M. H., and Goldin, A. L. (2001). Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J. Neurosci. 21, 7481–7490.
Sugawara, T., Tsurubuchi, Y., Agarwala, K. L., Ito, M., Fukuma, G., Mazaki-Miyazaki, E., Nagafuji, H., Noda, M., Imoto, K., Wada, K., Mitsudome, A., Kaneko, S., Montal, M., Nagata, K., Hirose, S., and Yamakawa, K. (2001). A missense mutation of the Na+ channel alpha II subunit gene NaV1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc. Natl. Acad. Sci. U.S.A. 98, 6384–6389.
Tan, H. L. (2006). Sodium channel variants in heart disease: expanding horizons. J. Cardiovasc. Electrophysiol. 17(Suppl. 1), S151–S157.
Tan, H. L., Kupershmidt, S., Zhang, R., Stepanovic, S., and Roden, D. M. (2002). A calcium sensor in the sodium channel modulates cardiac excitability. Nature 415, 442–447.
Tang, B., Dutt, K., Papale, L., Rusconi, R., Shankar, A., Hunter, J., Tufik, S., Yu, F. H., Catterall, W. A., Mantegazza, M., Goldin, A. L., and Escayg, A. (2009). A BAC transgenic mouse model reveals neuron subtype-specific effects of a generalized epilepsy with febrile seizures plus (GEFS+) mutation. Neurobiol. Dis. 35, 91–102.
Theile, J. W., and Cummins, T. R. (2011). Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front. Pharmacol. 2:54. doi:10.3389/fphar.2011.00054
Trimmer, J. S., Cooperman, S. S., Tomiko, S. A., Zhou, J., Crean, S. M., Boyle, M. B., Kallen, R. G., Sheng, Z., Barchi, R. L., Sigworth, F. J., Goodman, R. H., Agnew, W. S., and Mandel, G. (1989). Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 3, 33–49.
Trudeau, M. M., Dalton, J. C., Day, J. W., Ranum, L. P., and Meisler, M. H. (2006). Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia and mental retardation. J. Med. Genet. 43, 527–530.
Ulbricht, W. (2005). Sodium channel inactivation: molecular determinants and modulation. Physiol. Rev. 85, 1271–1301.
Valle, G., Faulkner, G., De Antoni, A., Pacchioni, B., Pallavicini, A., Pandolfo, D., Tiso, N., Toppo, S., Trevisan, S., and Lanfranchi, G. (1997). Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett. 415, 163–168.
van Gassen, K. L., de Wit, M., van Kempen, M., van der Hel, W. S., van Rijen, P. C., Jackson, A. P., Lindhout, D., and de Graan, P. N. (2009). Hippocampal Na beta3 expression in patients with temporal lobe epilepsy. Epilepsia 50, 957–962.
Vatta, M., Ackerman, M. J., Ye, B., Makielski, J. C., Ughanze, E. E., Taylor, E. W., Tester, D. J., Balijepalli, R. C., Foell, J. D., Li, Z., Kamp, T. J., and Towbin, J. A. (2006). Mutant caveolin-3 induces persistent late sodium current and is associated with long QT-syndrome. Circulation 114, 2104–2112.
Verkerk, A. O., Remme, C. A., Schumacher, C. A., Scicluna, B. P., Wolswinkel, R., de Jonge, B., Bezzina, C. R., and Veldkamp, M. W. (2012). Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ. Res. PMID: 22723301. [Epub ahead of print].
Vicart, S., Sternberg, D., Fontaine, B., and Meola, G. (2005). Human skeletal muscle sodium channelopathies. Neurol. Sci. 26, 194–202.
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., Maier, S. K. G., Zhang, T., Hasenfuss, G., Brown, J. H., Bers, D. M., and Maier, L. S. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138.
Wang, C., Wang, C., Hoch, E. G., and Pitt, G. S. (2011). Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J. Biol. Chem. 286, 24253–24263.
Wang, Q., Shen, J., Splawski, I., Atkinson, D., Li, Z., Robinson, J. L., Moss, A. J., Towbin, J. A., and Keating, M. T. (1995). SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–811.
Watanabe, H., Darbar, D., Kaiser, D. W., Jiramongkolchai, K., Chopra, S., Donahue, B. S., Kannankeril, P. J., and Roden, D. M. (2009). Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275.
Wilde, A. A. M., and Brugada, R. (2011). Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res. 108, 884–897.
Wu, G., Ai, T., Kim, J. J., Mohapatra, B., Xi, Y., Li, Z., Abbasi, S., Purevjav, E., Samani, K., Ackerman, M. J., Qi, M., Moss, A. J., Shimizu, W., Towbin, J. A., Cheng, J., and Vatta, M. (2008). Alpha-1-syntrophin mutation and the long QT syndrome: a disease of sodium channel disruption. Circ. Arrhythm. Electrophysiol. 1, 193–201.
Xiao, Z. C., Ragsdale, D. S., Malhotra, J. D., Mattei, L. N., Braun, P. E., Schachner, M., and Isom, L. L. (1999). Tenascin-R is a functional modulator of sodium channel beta subunits. J. Biol. Chem. 274, 26511–26517.
Yang, T., Atack, T. C., Stroud, D. M., Zhang, W., Hall, L., and Roden, D. M. (2012). Blocking SCN10A channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. PMID: 22723299. [Epub ahead of print].
Yarbrough, T. L., Lu, T., Lee, H.-C., and Shibata, E. F. (2002). Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ. Res. 90, 443–449.
Yarov-Yarovoy, V., Brown, J., Sharp, E. M., Clare, J. J., Scheuer, T., and Catterall, W. A. (2001). Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na channel alpha subunit. J. Biol. Chem. 276, 20–27.
Yarov-Yarovoy, V., McPhee, J. C., Idsvoog, D., Pate, C., Scheuer, T., and Catterall, W. A. (2002). Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277, 35393–35401.
Ye, S., Li, Y., and Jiang, Y. (2010). Novel insights into K+ selectivity from high-resolution structures of an open K+ channel pore. Nat. Struct. Mol. Biol. 17, 1019–1023.
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4, 207.
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., Spain, W. J., McKnight, G. S., Scheuer, T., and Catterall, W. A. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149.
Zimmer, T., and Benndorf, K. (2002). The human heart and rat brain IIA Na+ channels interact with different molecular regions of the beta1 subunit. J. Gen. Physiol. 120, 887–895.
Keywords: voltage-gated sodium channels, channelopathies, electrophysiology, sodium channels, pharmacology, biophysics
Citation: Savio-Galimberti E, Gollob MH and Darbar D (2012) Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front. Pharmacol. 3:124. doi: 10.3389/fphar.2012.00124
Received: 15 November 2011; Accepted: 11 June 2012;
Published online: 11 July 2012.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Massimo Mantegazza, University of Nice Sophia Antipolis, FrancePeter Enyedi, Semmelweis University, Hungary
Copyright: © 2012 Savio-Galimberti, Gollob and Darbar. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Dawood Darbar, Division of Cardiovascular Medicine, Vanderbilt University School of Medicine, 2215B Garland Avenue, Room 1285A MRB IV, Nashville,TN 37323-6602, USA. e-mail: dawood.darbar@vanderbilt.edu