
- CNRS, UMR6293/INSERM U1103, Génétique, Reproduction et Développement, Clermont Université, Aubière, France
The aldose reductase (AR; human AKR1B1/mouse Akr1b3) has been the focus of many research because of its role in diabetic complications. The starting point of these alterations is the massive entry of glucose in polyol pathway where it is converted into sorbitol by this enzyme. However, the issue of AR function in non-diabetic condition remains unresolved. AR-like enzymes (AKR1B10, Akr1b7, and Akr1b8) are highly related isoforms often co-expressed with bona fide AR, making functional analysis of one or the other isoform a challenging task. AKR1B/Akr1b members share at least 65% protein identity and the general ability to reduce many redundant substrates such as aldehydes provided from lipid peroxidation, steroids and their by-products, and xenobiotics in vitro. Based on these properties, AKR1B/Akr1b are generally considered as detoxifying enzymes. Considering that divergences should be more informative than similarities to help understanding their physiological functions, we chose to review specific hallmarks of each human/mouse isoforms by focusing on tissue distribution and specific mechanisms of gene regulation. Indeed, although the AR shows ubiquitous expression, AR-like proteins exhibit tissue-specific patterns of expression. We focused on three organs where certain isoforms are enriched, the adrenal gland, enterohepatic, and adipose tissues and tried to connect recent enzymatic and regulation data with endocrine and metabolic functions of these organs. We presented recent mouse models showing unsuspected physiological functions in the regulation of glucido-lipidic metabolism and adipose tissue homeostasis. Beyond the widely accepted idea that AKR1B/Akr1b are detoxification enzymes, these recent reports provide growing evidences that they are able to modify or generate signal molecules. This conceptually shifts this class of enzymes from unenviable status of scavenger to upper class of messengers.
Introduction
Aldose reductases (AR) are cytosolic monomeric enzymes, belonging to the aldo-keto reductase (AKR) superfamily. This superfamily encompasses more than 150 NAD(P)(H)-dependent oxidoreductases distributed in all prokaryotic and eukaryotic kingdoms including yeast, plant, invertebrates, and vertebrates. They catalyze the reduction of carbonyl groups from a wide variety of natural or synthetic substrates such as aliphatic and aromatic aldehydes, ketones, keto prostaglandins, ketosteroids, and xenobiotics. Because of overlapping substrates and coenzyme specificities that could lead to confusion between these closely conserved proteins, a nomenclature system for the AKR superfamily has been established based on their structural and genetic properties and is available at www.med.upenn.edu/akr/. Indeed, based on sequence identity, these proteins are divided in 15 families termed AKR1–AKR15, each family having less than 40% amino acid sequence identity with the others. Some families are further subdivided into subfamilies containing proteins with more than 60% sequence identity (Jez et al., 1997; Hyndman et al., 2003). To date, the AKR1 family is the major group encompassing 50 variants of the referring founder protein AKR1A1.
Among the AKR1 family, the AR subgroup (AKR1B) is one of the most characterized because of its involvement in human diseases such as diabetic complications resulting from the ability of AKR1B1 to reduce glucose into sorbitol in a NADPH + H+dependent manner. In addition to glucose conversion, AKR1B proteins display multiple other activities including reduction of aldehydes generated by lipid peroxidation, steroids and their derivatives or by-products, retinoids, xenobiotics, and prostaglandins.
The AKR1B subfamily contains several proteins with very high structural similarity to the human former AKR1B1. The AKR1B proteins share more than 65% sequence identity (Table 1). According to their phylogenetic features and their ability to reduce glucose, they can be classified into two subgroups, i.e., AR (AKR1B1–6) and aldose reductase-like proteins (ARLP; Akr1b7–16), respectively.
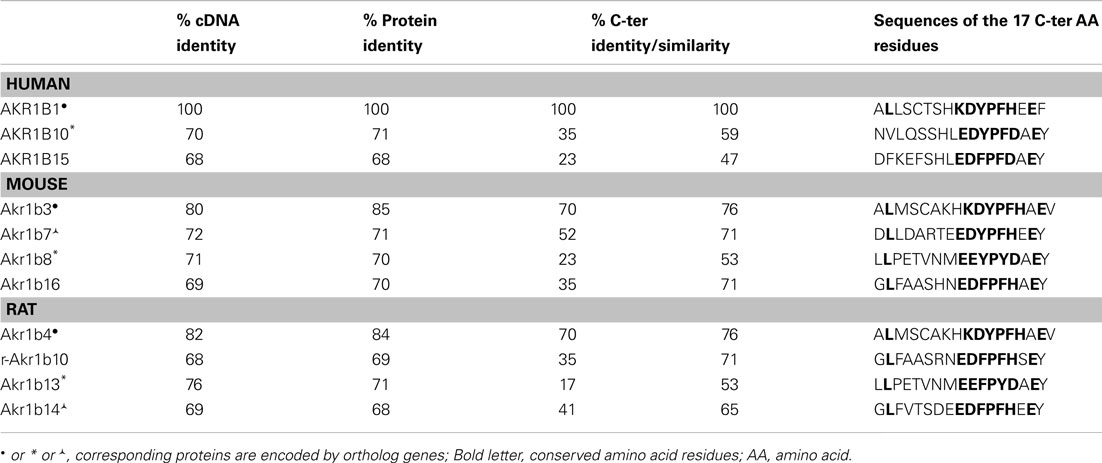
Table 1. Comparison of protein sequence of AKR1B.
To date, three human AKR1B have been characterized: AKR1B1 (human AR; Bohren et al., 1989), AKR1B10 [also designated as human small intestine (HSI) reductase; Cao et al., 1998; Hyndman and Flynn, 1998], and AKR1B15 (Barski et al., 2008; Salabei et al., 2011), which are encoded by genes tandemly arrayed on chromosome 7q33–35. AKR1B1 seems to be ubiquitously expressed whereas AKR1B10 expression was only reported in small intestine, colon, liver, and thymus (Cao et al., 1998). A genetic study recently identified a new gene named AKR1B15 closely related to the AKR1B1 and AKR1B10 cluster on chromosome 7, encoding a putative protein sharing 68 and 91% sequence identity with AKR1B1 and AKR1B10, respectively. AKR1B15 tissue expression has not been explored so far (Barski et al., 2008; Salabei et al., 2011).
Four murine Akr1b have been described: Akr1b3 (murine AR; Gui et al., 1995), Akr1b7 [previously named Mouse vas deferens protein (MVDP); Pailhoux et al., 1990], Akr1b8 [previously named fibroblast growth factor-related protein (FR-1); Donohue et al., 1994], and Akr1b16 (Salabei et al., 2011). Murine AR genes are located on chromosome 6 (locus 6 B1) and their tandem arrangement suggests (as for the three human AKR1B) that these four genes arise from an ancestral gene duplication event (Ho et al., 1999; Ruiz et al., 2011). Like AKR1B1, Akr1b3 and Akr1b16 seem to be ubiquitously expressed (Joshi et al., 2010; Salabei et al., 2011) whereas Akr1b7 and Akr1b8 display more restricted tissue distribution: Akr1b7 is expressed in vas deferens, adrenal glands, gonads, intestine, white adipose tissue, eye, liver, and kidney (Pailhoux et al., 1990; Lau et al., 1995; Tirard et al., 2007; Brunskill et al., 2011; Schmidt et al., 2011). Akr1b8 expression is detected in testis, heart, adrenal glands, intestine, and liver (Donohue et al., 1994; Lau et al., 1995; Joshi et al., 2010).
Among rat AR, Akr1b4 seems to be ubiquitously expressed whereas the other related proteins have a more restricted expression pattern (MacLeod et al., 2010). Transcripts for Akr1b14 have been detected in liver, kidney, and adrenals. Those of r-Akr1b10 share this tissue expression pattern but are also found in brain, heart, and lungs (Endo et al., 2010b). Akr1b13 expression has been observed at mRNA level in almost all organs, except brain and liver but the protein remains undetectable in small intestine and colon (Endo et al., 2011).
Several studies allowed identification of Akr1b4, Akr1b13, and Akr1b14 as the orthologs of mouse Akr1b3, Akr1b8, and Akr1b7, respectively (Barski et al., 2008; Endo et al., 2011). This phylogenetic analysis between rat and mouse AR has some limits: Akr1b14 and Akr1b7 do not exactly display the same expression pattern. Indeed, vas deferens is the major site of Akr1b7 expression whereas Akr1b14 is barely detectable in this tissue. Despite high sequence identity, the two promoters differ by a short 77-bp region absent in rat sequence which confers vas deferens targeted expression and androgen responsiveness to the Akr1b7 gene (Val et al., 2002).
Identification of the mechanisms regulating expression of AKR1B/Akr1b is a necessary step to understand their physiological functions. Nevertheless, the precise identification and determination of spatial distribution of AKR1B/Akr1b in healthy tissues remains a difficult task, as it requires powerful and specific immunological or nucleic probes to allow discrimination between these closely related isoforms. For that reason, we designed immunological probes targeting the most divergent domain among these enzymes that encompass the 17 C-terminal amino acid residues (Table 1). Indeed, using this C-terminal region critical to substrate specificity (Bohren et al., 1992), we developed immunological tools discriminating Akr1b7, Akr1b3, and Akr1b8 isoforms (Lefrançois-Martinez et al., 2004). Similarly, the most discriminating nucleotide probes or primers should be carefully designed from 5′- to 3′-untranslated regions while open reading frame should be avoided whenever possible. Knowing that in mammals, AR genes (i.e., human AKR1B1 gene, murine Akr1b3, and rat Akr1b4) display a broad tissue expression pattern, it can be assumed that in normal conditions, most of their activities are involved in maintaining general cellular homeostasis through osmotic regulation or detoxification processes.
This review will focus on the most characterized AKR1B proteins: AKR1B1, Akr1b3, Akr1b7, Akr1b8, and AKR1B10. To date many in vitro and ex vivo studies have examined and carefully described their enzymatic activities and defined their substrate specificities. These studies have enlightened some redundancy in substrate specificities that can be confusing when considering that various isoforms may coexist in the same tissue. In light of these enzymatic data, one of the most challenging issue regarding AKR1B enzymes would now be to explore their distinct biological functions in specific physiological or pathological processes. The focus of this review is to integrate most recent data on specific regulations of AR genes with enzymatic and functional data, in selected organs involved in endocrine and metabolic function, i.e., the adrenal gland, enterohepatic tissue, and white adipose tissue.
AKR1B and Adrenal Endocrine Function
The adrenal gland has two anatomically and functionally different components: the outer cortex which provides mineralocorticoids from the zona glomerulosa, glucocorticoids from the zona fasciculate, and the inner medulla in which chromaffin cells produce adrenal catecholamines, i.e., epinephrine and norepinephrine and various neuropeptides.
Acute and chronic adrenal cortex steroidogenesis is regulated mainly through activation of the cAMP-dependent protein kinase (PKA) signaling pathway mediated by the pituitary adrenocorticotropin hormone (ACTH).
Excess levels of glucocorticoid in the plasma in turn induce a negative feedback on ACTH production resulting in blunted ACTH-dependent steroidogenesis in the adrenal gland. This blockade of the hypothalamic-pituitary-adrenal axis can be experimentally recapitulated by dexamethasone treatment (a synthetic glucocorticoid). In adrenals, cAMP-induced PKA activation results at least in the phosphorylation of transcription factors such as steroidogenic factor 1 (SF-1), CCAAT Enhancer Binding Protein (C/EBP), and cAMP response element-binding protein. These, in turn stimulate transcription of genes encoding steroidogenic enzymes and proteins responsible for cholesterol metabolism, mobilization and transport. Therefore, adrenal steroidogenesis is strongly associated with production of endogenous harmful lipid aldehyde by-products including isocaproaldehyde (4-methylpentanal) derived from cholesterol side chain cleavage (the first step of steroid synthesis) and 4-hydroxynonenal (4-HNE). Interestingly, previous studies have established that the adrenal gland is one of the major sites for human and murine AR expression (Lau et al., 1995; Hyndman and Flynn, 1998).
Akr1b8/AKR1B10: Expression Profile, Detoxification Function (Tables 2 and 3)
Akr1b8 messenger was observed by in situ hybridization analysis in fetal and adult murine adrenal cortex and remained undetected in the medulla (Lau et al., 1995). Nevertheless, dexamethasone-induced ACTH suppression had no effect on Akr1b8 protein levels (Lambert-Langlais et al., 2009) suggesting that its biological function would not be associated to the ACTH-dependent steroidogenic activity. Indeed, isocaproaldehyde reductase activity was abolished in adrenocortical Y1 cells lacking Akr1b7 (due to stable antisense expression) in the presence of unaltered Akr1b8 protein levels (Lefrancois-Martinez et al., 1999). Accordingly, isocaproaldehyde accumulated and resulted in cellular toxicity despite the presence of Akr1b8. On the basis of these functional studies, of its enzymatic constants and constitutive expression, Akr1b8 does not appear to be the major isocaproaldehyde reductase in the adrenal cortex (Martinez et al., 2001). Although all murine AR exhibit 4-HNE reductase activity, Akr1b8 seems to be the most efficient HNE reductase in mouse tissues (Srivastava et al., 1998; Martinez et al., 2001). This suggests that adrenocortical expression of Akr1b8 could be dedicated to detoxification of aldehyde lipids that are present in large amounts in the cortex (Burczynski et al., 1999). Nevertheless, functional demonstration remains to be established.
AKR1B10 mRNA has been detected in adrenal glands using a human RNA Master Blot, but to date, there is no information available on its in situ localization and transcriptional control in this organ (Hyndman and Flynn, 1998). Although both AKR1B1 and AKR1B10 human AR display 4-HNE reductase activity, AKR1B10 exhibits a higher activity and product turn over than AKR1B1 (Shen et al., 2011). Both AKR1B1 and AKR1B10 isoforms are expressed in the human adrenal gland and ex vivo studies revealed that they also share the ability to reduce isocaproaldehyde (Hyndman and Flynn, 1998). Nevertheless, in a comparative enzymatic study, Hara and colleagues showed that AKR1B1 had a more effective isocaproaldehyde reductase activity than AKR1B10, suggesting that the latter was unlikely to play a major role in the detoxification of steroidogenic by-products (Endo et al., 2009b).
Since its identification in human hepatocellular carcinoma, AKR1B10 was shown to be differentially expressed in other tumor and normal tissues (Cao et al., 1998; Fukumoto et al., 2005; Yoshitake et al., 2007; Rajkumar et al., 2010). AKR1B10 was suggested to be a biomarker of smoker’s non-small cell lung carcinomas (Fukumoto et al., 2005) and to be involved in drug resistance (Matsunaga et al., 2012).
Human adrenocortical carcinoma (ACC) are rare malignant tumors generally associated with a poor prognosis (Libè et al., 2007). In order to identify molecular predictors of malignancy and of survival, 153 unilateral adrenocortical tumors were studied by microarray (http://www.ebi.ac.uk/arrayexpress, experiment E-TABM-311). Unsupervised clustering analysis was performed which allowed robust discrimination of malignant and benign tumors. On the basis of this analysis, AKR1B10 was not found to be associated with the ACC group (de Reyniès et al., 2009).
Akr1b7: Expression Profile, Detoxification Function, and Paracrine Action (Tables 2 and 3)
High levels of Akr1b7 transcripts were initially observed by in situ hybridization in fetal and adult murine adrenal cortex but were undetectable in the medulla (Lau et al., 1995). We confirmed these results by immunohistochemistry experiments which allowed us to further restrict Akr1b7 expression to the zona fasciculata (Aigueperse et al., 1999). In vivo, ACTH suppression with dexamethasone treatment resulted in a marked decrease of Akr1b7 mRNA levels that were restored when the treated mice were injected with exogenous ACTH. This ACTH or cAMP-induced Akr1b7 transcription was blocked by a PKA inhibitor (H89) in the murine adrenocortical ATC and Y1 cell lines, respectively (Aigueperse et al., 1999; Ragazzon et al., 2006).
Adrenal expression and ACTH regulation of Akr1b7 are supported by three SF-1 binding sites and other cis-elements located in the 5′-flanking regulatory region of the gene. Using transgenic mice and cell transfection experiments, we delimited a cryptic SF-1 response element (SFRE) 102 bp upstream of the transcription start site. This SFRE supported basal adrenal promoter activity. Two other SFREs were identified further upstream. The site at −458 was a bona fide SFRE playing an essential role for both basal promoter activity and cAMP responsiveness whereas the site at −503 conferred intrinsic cAMP-sensing ability (Martinez et al., 2003; Val et al., 2004). Two other binding sites for the trans-acting factors Sp1 and C/EBPβ at position −52 and −61, respectively, also contributed to the transcriptional cAMP responsiveness (Aigueperse et al., 2001; Table 2 and Figure 1).
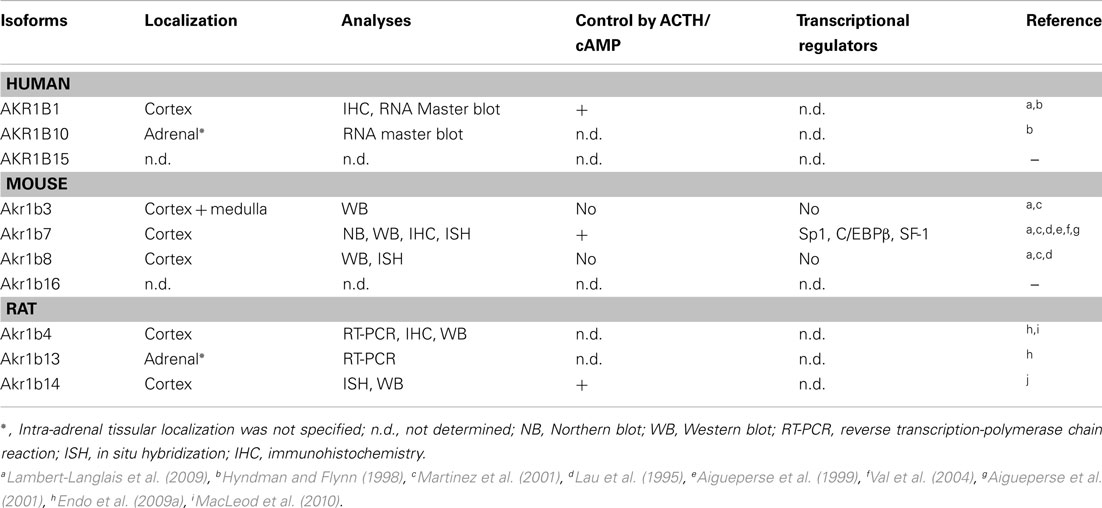
Table 2. Localization and regulation of AKR1B expression in adrenal gland.
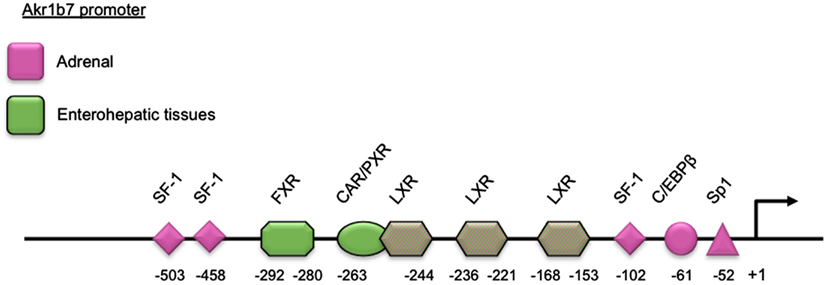
Figure 1. Schematic representation of the Akr1b7 promoter. The DNA binding site for transcription factors and nuclear receptors required for the Akr1b7 specific expression in adrenal gland (pink boxes) and in enterohepatic tissue (green boxes) are shown. LXR binding sites are involved in both adrenal and intestine Akr1b7 expression.
Aldose reductases are able to reduce isocaproaldehyde that is produced in large amount in the adrenal cortex during steroidogenesis. Indeed, the first step of steroidogenesis is the removal of the cholesterol side chain by the P450scc enzyme, resulting in the formation of pregnenolone and isocaproaldehyde. Furthermore, isocaproaldehyde is a cytotoxic aldehyde whose accumulation in Y1 cells decreased their viability (Lefrancois-Martinez et al., 1999). In vitro studies revealed that Akr1b3, Akr1b7, and Akr1b8 all had the ability to reduce isocaproaldehyde. Their kinetic constants suggested that isocaproaldehyde was a major substrate for Akr1b3 and Akr1b7, and a poor substrate for Akr1b8 (Martinez et al., 2001; Table 3). Akr1b7 silencing in Y1 adrenocortical cells disrupted cAMP-induced isocaproaldehyde reductase activity. This strongly suggested that Akr1b7, rather than Akr1b3, was the main isocaproaldehyde reductase in the adrenal gland (Lefrancois-Martinez et al., 1999). These observations further indicate that ACTH not only coordinates expression of enzymes responsible for the biosynthesis of steroids, but also of non-steroidogenic enzymes involved in the detoxification of reactive aldehydes generated during steroidogenesis.
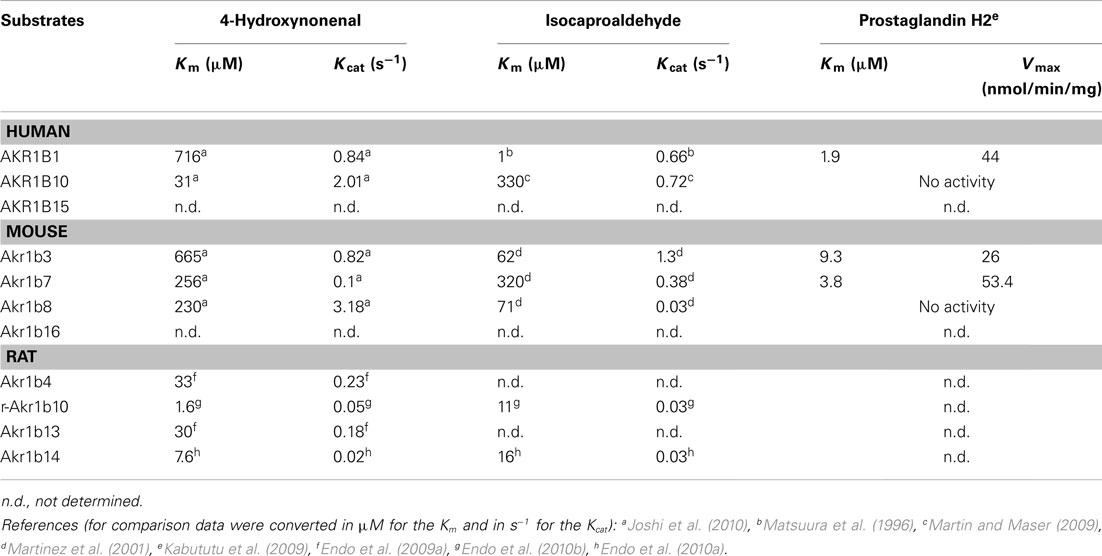
Table 3. Kinetic constants of AKR1B toward 4-hydroxynonenal, isocaproaldehyde, and prostaglandin H2.
In a previous study, Madore and colleagues proposed that AKR1B5, initially characterized as the bovine 20α-hydroxysteroid dehydrogenase, ensured prostaglandin F2α synthase (PGFS) activity in the endometrium (Madore et al., 2003). Thereafter, we have established by in vitro studies, that this property could be extended to other but not all AKR1B enzymes. Indeed, AKR1B1, Akr1b3, and Akr1b7 were shown to catalyze the reduction of prostaglandin H2 (PGH2) into PGF2α (Table 3). In contrast, Akr1b8 and AKR1B10 recombinant proteins were devoid of this PGH2 9-,11-endoperoxide reductase activity. This activity has not been investigated for Akr1b16, AKR1B15, and rat AR so far. Importantly, based on their kinetic parameters, recombinant AKR1B1, Akr1b3, and Akr1b7 displayed better PGF synthase activities than the previously characterized PGF synthases in mammals (Kabututu et al., 2009).
Prostaglandins are paracrine/autocrine cell mediators sharing a common precursor, PGH2, which is synthesized from free arachidonic acid by the cyclooxygenases type 1 (COX-1) or type 2 (COX-2). COX-1 is regarded as a constitutively expressed enzyme. COX-2, on the other hand, is undetectable in most tissues in basal conditions but can be induced by various mitogenic agents and inflammatory stimuli (Ramsay et al., 2003). Following these observations, we carefully examined the PGF2α biosynthetic pathway in the adrenal gland (Lambert-Langlais et al., 2009).
PGF2α was secreted by both cortical (steroidogenic cells) and medullary (chromaffin cells) compartments of the adrenal gland. In primary adrenocortical cell culture, PGF2α release was induced 2.5-fold by ACTH exposure which was correlated with ACTH-responsiveness of both COX-2 and Akr1b7. Using over- and down-expression strategies in cell lines, we demonstrated the pivotal role of Akr1b7 in ACTH-induced PGF2α release and its functional coupling with COX-2. In the adrenal medulla, PGF2α production seemed to be resulting from the coordinated activities of Akr1b3 and COX-1. In the adrenal, expression of the PGF2α specific receptor (FP) was restricted to chromaffin cells, suggesting that both autocrine (within the medulla) and paracrine (between steroidogenic and chromaffin cells) mechanisms were relaying PGF2α action. Indeed in the chromaffin cell line MPC862L, PGF2α was able to repress both basal and glucocorticoid-induced dopamine release. By comparing PGF2α-responsiveness of isolated cells and whole adrenal cultures, we demonstrated that PGF2α repressed glucocorticoid secretion by an indirect mechanism involving a decrease in catecholamine release, which in turn decreased adrenal steroidogenesis.
These results allowed us to propose a new mechanism for an intra-adrenal feedback loop, in which AR play a pivotal role in the regulation of adrenal endocrine functions (Figure 2). The mechanism that we proposed was the following: (1) In basal conditions, PGF2α is constitutively secreted by chromaffin cells (by the coupling of COX-1 and Akr1b3), thus regulating catecholamines production and also limiting their paracrine action on steroidogenesis. (2) During a stress situation, ACTH transiently induces COX-2 and Akr1b7 expression, which results in PGF2α production inside the cortex. PGF2α produced in the cortex then represses catecholamines release by the medulla, via a paracrine action on its FP receptor. Decreased catecholamines release in turn reduces the effect of ACTH on glucocorticoids production (Lambert-Langlais et al., 2009).
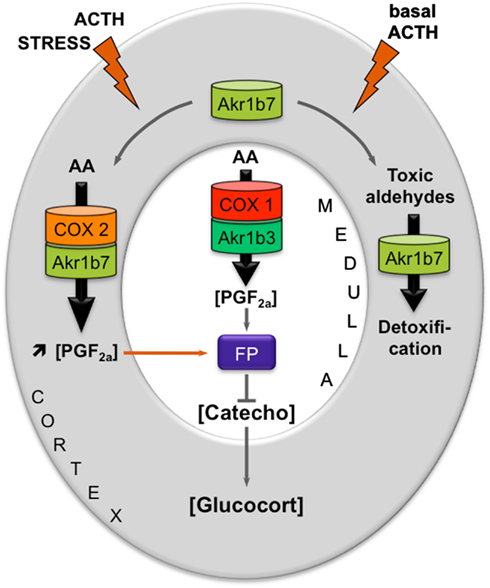
Figure 2. Proposed model integrating dual functions of aldose reductases in the regulation of adrenal endocrine functions. Free arachidonic acid (AA) is metabolized into PGH2 by COX enzymes and then converted into PGF2α by PGFS of the AKR1B family. FP receptor expression is restricted to the medullary zone. PGF2α synthesized in both the cortex and medulla thus signals in an autocrine/paracrine manner on chromaffin cells. This inhibits catecholamines production. Catecholamines produced in the medulla normally stimulate glucocorticoids release by the cortex. Decreased catecholamines production in response to PGF2α stimulation thus results in a decrease in glucocorticoids production. The differential expression and regulation of both COX and AKR1B enzymes within the adrenal zones could allow the adjustment of PGF2α production to limit stress response or control basal steroidogenesis by finely tuning glucocorticoids secretion. In basal conditions, chromaffin cells of the medullary zone constitutively secrete PGF2α, through the functional coupling between COX-1 and possibly the PGFS Akr1b3. Under stress conditions, the resulting ACTH surge induces COX-2 expression and sustains Akr1b7 levels in the cortex. The functional coupling between COX-2 and Akr1b7 triggers a PGF2α surge that could act as a local paracrine feedback to limit catecholamine-mediated glucocorticoid release. After the stress response has ended, COX-2 returns to undetectable levels. The coupling between Akr1b7 and COX-2 does not take place. Akr1b7 then functions only as a detoxifying enzyme of the harmful aldehydes produced under chronic/basal stimulation of steroidogenesis. Catecho, catecholamines; Gluco, glucocorticoids.
Akr1b3/AKR1B1 Expression Profile, Detoxification Function, and Paracrine Action
By using different adrenal cell lines, we managed to detect Ak1b3 protein in the adrenocortical Y1 cells, and in the chromaffin MPC862L cells. These results confirmed that, unlike other murine AR, Akr1b3 is constitutively expressed in the whole adrenal gland (Lambert-Langlais et al., 2009). In Y1 adrenocortical cell line and during in vivo hormonal manipulations, cAMP appeared to have no significant effect on Akr1b3 expression suggesting that its expression was insensitive to ACTH (Martinez et al., 2001; Table 2).
Akr1b3 is also involved in the detoxification of toxic carbonyls. Even if Akr1b7 is the main isocaproaldehyde reductase and Akr1b8 the principal 4-HNE reductase, Akr1b3 is also able to reduce these toxic compounds. This suggests that Akr1b3 can take part in the elimination of these compounds in basal physiological conditions (Lefrancois-Martinez et al., 1999; Martinez et al., 2001). Moreover, Akr1b3 is constitutively expressed in cortical and medullary cells, where it could be coupled to COX-1 to synthesize PGF2α (see above paragraph).
Despite its expected participation in the elimination of toxic compounds and in the production of signal molecules (PGF2α), there is no report of adrenal dysfunction in mice lacking Akr1b3 (Akr1b3−/−; Aida et al., 2000; Ho et al., 2000). This absence of adrenal phenotype may result from functional redundancy between the different family members present in the gland (Akr1b7 and Akr1b8).
AKR1B1 expression has initially been detected using a RNA master blot in human adrenal gland (Hyndman and Flynn, 1998). More recently, immunohistochemistry experiments allowed us to assign AKR1B1 to the cortical compartment of the gland (Lambert-Langlais et al., 2009). In NC1-H295, a human adrenocortical tumor cell line, AKR1B1 mRNA levels were induced by forskolin (adenylyl cyclase inducer) treatment (Lefrançois-Martinez et al., 2004). This suggested that similarly to Akr1b7, AKR1B1 gene expression could be sensitive to ACTH control. However, the molecular mechanisms and cis-acting elements ensuring ACTH/cAMP responsiveness of AKR1B1 gene have remained unexplored.
For years, AKR1B1 had been considered as the major isocaproaldehyde reductase in the adrenal gland (Matsuura et al., 1996). The NADPH-dependent isocaproaldehyde reductase activity harbored by AKR1B1 was inhibited by tolrestat while the murine isocaproaldehyde reductase Akr1b7 was insensitive to this pharmacological inhibitor (Matsuura et al., 1996; Lefrancois-Martinez et al., 1999). We demonstrated that AKR1B1 was also endowed with 9-,11-endoperoxide reductase activity (Kabututu et al., 2009). The conversion of PGH2 into PGF2α, catalyzed by AKR1B1, strictly dependent on the presence of NADPH, was inhibited by tolrestat whereas Akr1b7 PGFS activity was insensitive to AR inhibitors.
We showed that in the normal human adrenal gland, AKR1B1 and COX-2 were co-localized in steroidogenic cortical cells (Lambert-Langlais et al., 2009). Therefore, the human adrenal cortex could also have the potential to produce PGF2α in response to ACTH. With respect to their hormonal regulation in the adrenal cortex and their reductase activity toward common substrates, we have postulated that AKR1B1 could be considered as a functional ortholog of Akr1b7 in the human adrenal cortex (Lefrançois-Martinez et al., 2004). The possibility that the PGFS activity of AKR1B1 could be involved in an intra-adrenal feedback loop between endocrine activities of cortical and medullary compartments in human adrenal gland remains to be explored.
Given the high expression of AKR1B1 in the adrenal cortex we evaluated alterations in its expression in association with human adrenal disorders. The relative abundance of AKR1B1 mRNA was decreased in ACCs when compared to benign tumors, Cushing’s hyperplasia, or normal adrenals (Lefrançois-Martinez et al., 2004). These data provided evidence that expression of AKR1B1 was decreased in adrenocortical cancer. This was further confirmed by the unsupervised clustering analysis of the human adrenal tumors transcriptome performed by de Reyniès et al. (2009), indicating that decreased expression of AKR1B1 correlated with malignancy for the molecular diagnosis of adrenal tumors.
AKR1B and Glucido-Lipidic Homeostasis
AKR1B in Enterohepatic Tissues
Excessive nutrient intake is detrimental for cells and tissues. In mammals, the liver converts excess dietary carbohydrates into triglycerides through de novo lipogenesis. Two transcription factors, carbohydrate-responsive element-binding protein (ChREBP) and sterol responsive element-binding protein-1c (SREBP-1c) emerged as major mediators of glucose and insulin action in the control of both glycolysis and lipogenesis in the liver. The liver X receptors (LXR) are oxysterol activated transcription factors acting as important metabolic regulators of the lipogenic pathway. Indeed, LXRs ensure the transcriptional control of SREBP-1c in response to insulin and of ChREBP. Moreover, direct targets of LXRs include other lipogenic genes such as fatty acid synthase (Fas) and stearoyl-CoA desaturase 1 (SCD1; Chen et al., 2004; Postic and Girard, 2008; Kim et al., 2009). The farnesoid X receptor (FXR)/bile acid receptor is another nuclear receptor that plays an important role in maintaining bile acid, lipid, and glucose homeostasis since its activation has been shown to lower blood triglyceride and cholesterol levels and to improve insulin sensitivity in diabetic mouse models (Zhang and Edwards, 2008). In enterocytes, FXR was shown to protect against the cytotoxic effects of bile acids by increasing expression of binding proteins and transporters (Schmidt and Mangelsdorf, 2008).
Akr1b8/AKR1B10: From cell detoxification to lipid synthesis
Previous analyses performed by RNAse protection assays reported very weak levels of Akr1b8 transcript (Lau et al., 1995) in the mouse intestine and absence of expression in the adult liver. These observations were partially revised by Joshi et al. (2010) who showed high Akr1b8 expression all along the adult intestinal tract and moderate expression in the liver. Akr1b8 expression was initially described to be up-regulated during the early phase of the cell cycle and induced by growth promoting agents [fibroblast growth factor (FGF) and epidermal growth factor (EGF)], as well as by hypertonic stress (Donohue et al., 1994; Hsu et al., 1997). Although Akr1b8 displayed a strong expression in mouse colon cancer cells (Joshi et al., 2010) and was suggested to be a target gene for NF-E2 related factor 2 (Nrf2) transcription factor that controls intestinal detoxification response (Varady et al., 2011), its expression and regulation in healthy liver and intestine remained elusive. A recent study investigating bile acids impacts on Akr1b7 expression showed that enterohepatic expression of Akr1b8 was insensitive to bile acids (Schmidt et al., 2011). Akr1b8 enzymatic activities were extensively studied regarding its ability to reduce the highly reactive 4-HNE produced during peroxidation of polyunsaturated fatty acids (Srivastava et al., 1998). In vitro enzymatic studies reported that multiple members of the AKR1B group, including Akr1b8, were endowed with the ability to reduce phospholipid aldehydes derived from lipid peroxidation. Compared with AKR1B1 or Akr1b7, Akr1b8 activity was more efficient with the reduction of short chain aldehydes such as 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine and 1-pamitoyl-2-arachidonoyl-sn-glycero-3-phosphoglycerol (Spite et al., 2007). In line with the high lipid metabolic activities in Akr1b8 producing sites, it was proposed that Akr1b8 was acting physiologically as a scavenger of toxic aldehydes derived from peroxidation of endogenous or dietary lipids.
Beside its aldehyde reductase activity, Akr1b8 was shown to associate with the lipogenic acetyl-CoA carboxylase α (ACCA) in murine colon cancer cells (Joshi et al., 2010). ACCA is a rate-limiting enzyme of de novo synthesis of fatty acids, catalyzing the formation of malonyl-coA by ATP-dependent carboxylation of acetyl-coA. This interaction protects ACCA from proteasomal degradation and consequently allows increased fatty acid synthesis that leads to production of lipid second messengers that promote cell proliferation (Chajès et al., 2006). Whether Akr1b8 is also involved in the stability of ACCA in healthy enterocytes has not yet been determined. In order to associate gene signature with metabolic network, a genetic study integrating quantitative trait locus (QTL) mapping and network modeling led to knock-out (KO) the three corresponding identified genes, among which Akr1b8 (Derry et al., 2010). Akr1b8−/− mice phenotype was very briefly described in this study suggesting a more specific perturbation in fat tissue homeostasis rather than in the liver or intestine (see below).
The AKR1B10 gene has recently been determined as the human ortholog of Akr1b8 (Joshi et al., 2010). Its expression was primarily detected in healthy colon, small intestine, and liver (Cao et al., 1998) and was found up-regulated with their corresponding tumorigenic transformation (Cao et al., 1998; Yan et al., 2007; Heringlake et al., 2010; Liu et al., 2012). This was also observed in lung and breast cancers suggesting that AKR1B10 overexpression could be associated with a broader tumor phenotype. In human hepatocarcinoma, insulin or EGF enhanced AKR1B10 expression through the activator protein-1 (AP1) mitogenic signaling. This enhanced tumor development and progression through elimination of cytotoxic carbonyls and promotion of lipogenesis (Liu et al., 2012). The treatment of human colon cancer SW-480 and HT-29 cell lines with proteasome inhibitors known to increase the expression of Nrf2-regulated genes induced AKR1B10 expression suggesting that AKR1B10 could be a target of the Nrf2 transcription factor (Ebert et al., 2011). Nrf2 responsiveness of AKR1B10 gene was further demonstrated by co-transfection experiments of AKR1B10 promoter luciferase reporter constructs in human lung adenocarcinoma cell lines (Nishinaka et al., 2011; Figure 3).
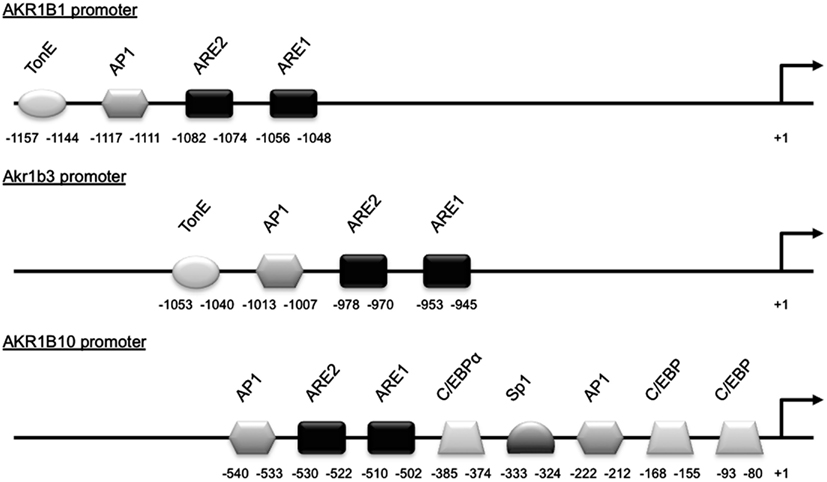
Figure 3. Schematic representation of the AKR1B1, Akr1b3, and AKR1B10 promoters. Cis- and trans-acting factors shown to be involved in stress responsiveness are indicated. TonE, tonicity response element; AP1, activator protein-1 binding site; ARE, antioxidant response element; C/EBP, CCAAT enhancer binding protein binding site; Sp1, selective promoter factor 1 binding site.
AKR1B10 was the first AKR1B protein which was demonstrated to enhance cell proliferation and promote cell survival through its ability to interact with and stabilize ACCA in breast cancer cells (Ma et al., 2008) and in colon cancer cells (Wang et al., 2009). AKR1B10 mediates ACCA stability through physical association. This was shown to affect fatty acid/lipid synthesis, mitochondrial function, and oxidative status. Identification of Akr1b8/AKR1B10 protein domain interacting with ACCA would be helpful to predict this characteristic for other AKR1B proteins and to develop targeted therapeutic strategies for the modulation of de novo fatty acid synthesis in cancer cells.
We are exposed daily to α,β-unsaturated aldehyde and trans-2-hexenal through food and drink consumption (Stout et al., 2008). This persistent exposure to electrophilic carbonyls requires an effective defense system to protect intestinal cells from irreversible damage. Similarly to Akr1b8, in vitro enzymatic studies on AKR1B10 demonstrated a highly specific ability to reduce physiological levels of 4-HNE and other alpha and beta-unsaturated carbonyls in both their free or glutathione-conjugated form (Martin and Maser, 2009; Zhong et al., 2009). Expression of AKR1B10 may reflect the requirement for intestinal cells to protect against the dietary, lumen microbial and lipid-derived carbonyls.
Although physiological integration of all these data remains difficult, this suggests that the beneficial role of Akr1b8/AKR1B10 in the protection of healthy cells against lipid peroxidation could switch, in the case of cancer, into a deleterious role promoting cell proliferation through stabilization of ACCA (Figure 4).
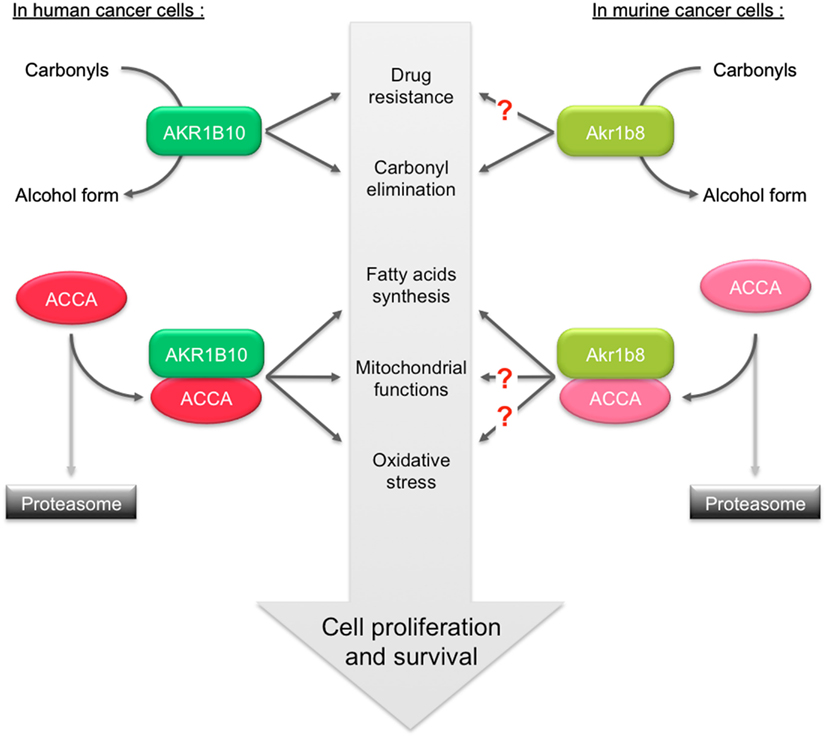
Figure 4. Role of AKR1B10 and Akr1b8 in the regulation of lipid synthesis and contribution in promoting cancer. In human cancer cells, ACCA is protected against ubiquitination-proteasome dependent degradation through its association with AKR1B10. Stabilization of ACCA increases fatty acids synthesis. This protects cell from apoptosis by preventing depletion of phospholipids, mitochondrial membrane lesions, and oxidative stress. The dual capacity of AKR1B10 to stimulate lipogenesis and to detoxify carbonyls and drugs thus contributes to enhance cell proliferation/survival. In murine cancer cells, Akr1b8 was also shown to stabilize ACCA thereby modulating fatty acid synthesis. The impact of Akr1b8-mediated ACCA stabilization on mitochondrial function and oxidative stress is still unknown.
Akr1b7: Improving (liver) metabolic capacity
A target for metabolic nuclear receptors (figure 1 and table 4). Several studies reported specific Akr1b7 expression in the small intestine (Volle et al., 2004; Ge et al., 2011; Schmidt et al., 2011) within the epithelial cells of the villi (Lau et al., 1995) with a decreasing gradient from the duodenum to the ileum. This expression has been shown to be under the control of oxysterol activated LXR/retinoic X receptor (RXR) heterodimers acting through three LXR response elements (LXRE) located in the Akr1b7 proximal promoter (LXRE3: −259 to −244, LXRE1: −236 to −221, LXRE2: −153 to −168 from the transcription start site). Two of these cis-acting elements were specific for regulation by the alpha LXR isoform (Volle et al., 2004).
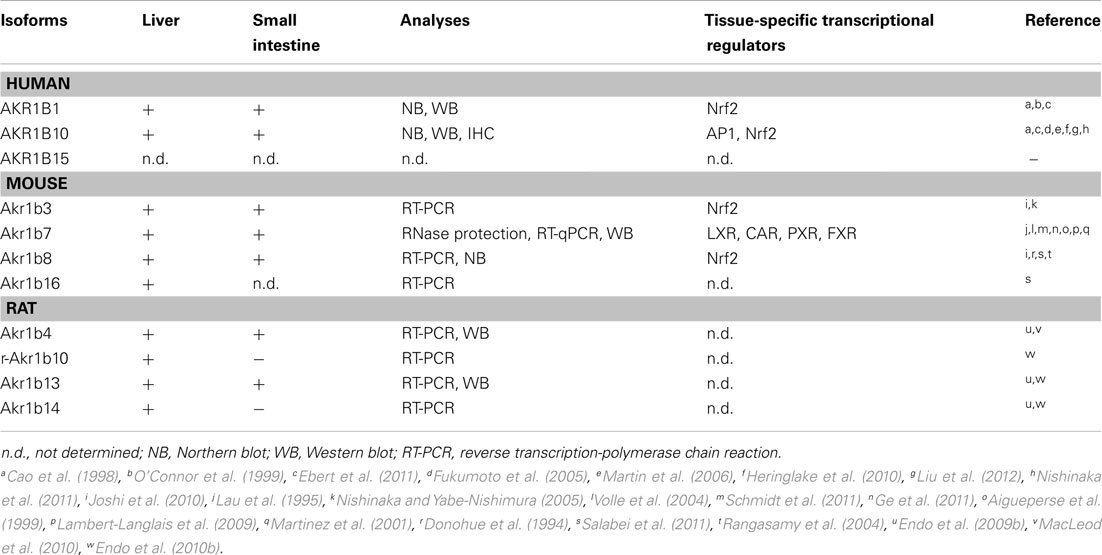
Table 4. Expression of aldose reductases in liver and small intestine.
Two other recent reports showed that Akr1b7 gene transcription in the small intestine, colon and liver was also controlled by bile acids through a FXR response elements (FXRE) located at position −292 to −280 on the promoter (Ge et al., 2011; Schmidt et al., 2011).
Several nuclear receptors, including FXR, pregnane X receptor (PXR) and constitutive androstane receptor (CAR), have been shown to protect against the cytotoxic effects of bile acids by increasing expression of binding proteins, transporters, and enzymes that detoxify bile acids (Zollner et al., 2006; Schmidt and Mangelsdorf, 2008). The combined use of KO mouse models for PXR/CAR receptors and pharmacological activators allowed identification of Akr1b7 as one of their target genes in the liver (Liu et al., 2009; Table 4 and Figure 1).
Although molecular mechanisms mediating metabolic regulation of Akr1b7 expression in the liver and intestine were profusely examined using genetic models or forced expression systems, there is no direct evidence for Akr1b7 expression in liver sections or in isolated hepatocytes, to date.
Detoxifying lipid aldehydes. Wild-type mice treated with PCN (pregnenolone-16α-carbonitrile; a PXR agonist) exhibited a decrease in malondialdehyde (MDA) levels (another by-product of polyunsaturated fatty acid peroxidation) compared with their vehicle-treated counterparts. The effect of PCN on MDA was abolished in Pxr−/− mice. The authors suggested that PXR played a role in alleviating lipid peroxidation in small intestine. Since the induction of Akr1b7 expression by PCN was also abolished by PXR KO, the effects of PXR on lipid peroxidation could be mediated, at least in part, by modulation of Akr1b7 expression (Liu et al., 2009; Ge et al., 2011; Schmidt et al., 2011).
In light of the ability of Akr1b7 to reduce 4-HNE, Volle et al. have compared the status of lipid peroxidation in the small intestine of wild-type and Lxrα−/− mice by measuring MDA levels. In the duodenum, a significant decrease in MDA concentrations was seen when wild-type mice were treated with T091317 (a synthetic agonist of LXR). This effect was not observed in Lxrα−/− mice, where T091317 was unable to induce Akr1b7. The authors suggested that increased levels of LXRα in the small intestine and its activation by oxysterols could in turn, up-regulate the expression of detoxifying genes which could ultimately reduce oxidative stress (Volle et al., 2004). However, although the decrease in MDA contents is clearly a LXR dependent mechanism, the direct involvement of Akr1b7 seems unlikely. Indeed overexpression of Akr1b7 in vivo does not alter MDA levels (Ge et al., 2011).
Detoxifying bile acids or lipid peroxidation induced by bile acids excess (figure 5). Recently three independent studies proposed that Akr1b7 or its rat ortholog Akr1b14 could be involved in bile acid metabolism and/or signaling (Endo et al., 2011; Ge et al., 2011; Schmidt et al., 2011).
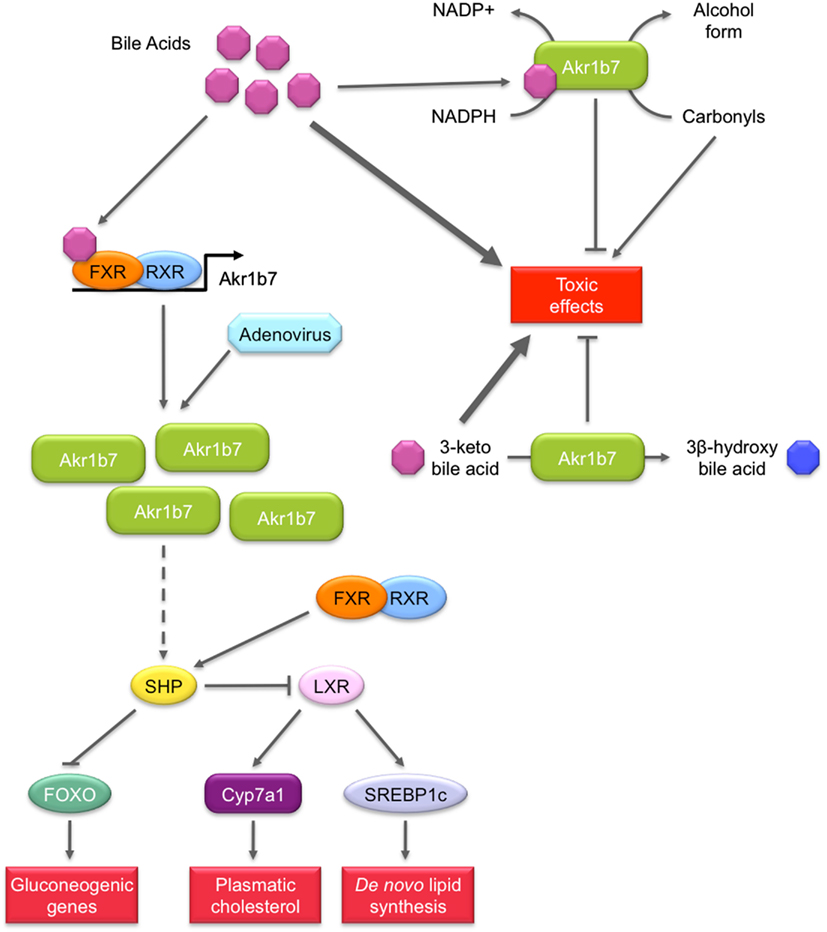
Figure 5. Role of Akr1b7 in bile acids metabolism and signaling pathway. Akr1b7 has the ability to reduce 3-keto bile acids to their less toxic 3β-hydroxy derivatives. In turn, bile acids can stimulate detoxification activity of Akr1b7, enhancing its capacity to reduce carbonyl and 3-keto bile acids. In addition to bile acids detoxification, Akr1b7 is also one of their target genes. In liver and intestine, bile acids induce the FXR-dependent Akr1b7 transcription. The forced expression of Akr1b7 in mouse liver by the mean of recombinant adenovirus results in the down-regulation of gluconeogenesis and lipid metabolism. Mechanisms involved are not unravel yet but could rely on the up-regulation of SHP expression. Increased SHP levels could in turn repress both FOXO and LXR, leading to the inhibition of gluconeogenic genes and lipogenic genes, respectively. The mechanism by which Akr1b7 accumulation modulates the SHP gene expression appears independent from FXR and remains to be discovered (dashed lines).
Bile acids are cholesterol-derived molecules produced for its solubilization in the gallbladder and intestine by forming mixed micelles with cholesterol and phospholipids. They are required for the activation of some pancreatic enzymes and for the absorption of cholesterol, lipid soluble vitamins and to a lesser extent, triglycerides and fatty acids from the intestine (Hylemon et al., 2009). They also act as signaling molecules by acting as ligands for several nuclear receptors including FXR and PXR or the membrane Gαs protein-coupled receptors TGR5 as well as Gαi protein-coupled receptors. Hence, bile acids contribute to the regulation of their own synthesis, fatty acid, lipid, and lipoprotein synthesis as well as glucose metabolism in the liver.
An in vitro enzymatic study using recombinant Akr1b14 (rat) and Akr1b7 (mouse) proteins showed that conjugated and unconjugated bile acids (chenodeoxycholic acid (CDCA)/glyco-CDCA and hyodeoxycolic acid (HDCA)/glyco-HDCA) specifically, quickly, and significantly activated the NADPH-linked reductase activity of Akr1b14 and to a lesser extent Akr1b7 activity (Endo et al., 2011). More accurately, higher specific activation of Akr1b14 was mediated by an interaction of bile acids with the His269 facilitating the release of NADP+. In Akr1b7, the histidine residue found at position 269 in the Akr1b14 protein is replaced by an arginine residue. This may account for weaker activation by bile acids. The authors suggested that bile acids could activate Akr1b14/Akr1b7 when their blood concentrations were elevated. As high concentrations of hydrophobic bile acids could induce cell injury through several pathways, e.g., lipid peroxidation, activation of Akr1b14 by bile acids may increase detoxification of the harmful lipid peroxidation by-products, and contribute to the attenuation of toxic effects of bile acids.
Because members of the AKR family (i.e., AKR1C4 and AKR1D1) were shown to be involved in the bile acid synthesis, Mangelsdorf’s group hypothesized that FXR-inducible Akr1b7 could be involved in bile acid metabolism in the intestine (Schmidt et al., 2011). They showed that Akr1b7 overexpression in heterologous HEK293 cells was associated with a reduction activity of 3-keto bile acids to their 3β-hydroxy derivatives, which are less toxic than their 3α-hydroxy epimers. Altogether, these results suggest a novel function for Akr1b7 in the detoxification of bile acids.
Possible role in metabolic action of bile acids signaling (figure 5). In the liver, bile acids are able to influence glucose metabolism by at least two mechanisms. First, conjugated bile acids rapidly activate the insulin signaling pathway via Gαi protein-coupled receptors or superoxide ions. In this aspect, they function much like insulin through the AKT pathway to activate glycogen synthase and repress neoglucogenic genes (Hylemon et al., 2009). Importantly, bile acids also mediate another control on liver metabolism through the transcriptional activation of the gene encoding the small heterodimer partner (SHP) via a functional FXRE in its promoter. The orphan receptor SHP can exert inhibitory interactions with forkhead box O1 (FOXO1), C/EBPα, and hepatocyte nuclear factor 4α transcription factors, known to activate neoglucogenic genes (Yamagata et al., 2004). The inhibitory control of bile acids on gluconeogenesis has also been demonstrated in FXR null mice or with the treatment of diabetic animals with the FXR agonist GW4064 (Ma et al., 2006; Zhang et al., 2006). SHP is also able to interact with LXR to down-regulate the gene encoding SREBP-1c which is the predominant transactivator for genes encoding enzymes involved in fatty acid, triglyceride, and VLDL biosynthesis (Horton et al., 2002).
These observations led Ge et al. (2011) to investigate the implication of FXR target gene Akr1b7 in liver glucose and lipid metabolism using adenovirus-mediated Akr1b7 overexpression. Overexpression of hepatic Akr1b7 significantly reduced plasma glucose levels. This was associated with reduced hepatic mRNA levels of gluconeogenic genes encoding phosphoenolpyruvate carboxykinase and glucose 6-phosphatase and of peroxisome proliferator-activated receptor γ co-activator 1α, as well as markedly increased hepatic SHP mRNA levels. Hepatic lipogenesis was also affected: mRNA levels of SREBP-1c and several lipogenic genes including Fas, diacylglycerol acyltransferase 2, cholesterol 7α-hydroxylase, and the ATP binding cassette G5 were significantly reduced. Nevertheless other FXR target genes such as the bile salt export protein or the multi drug resistance 2 were not affected. This excluded the hypothesis that FXR was activated through overexpression of Akr1b7. Forced hepatic expression of Akr1b7 also significantly lowered plasma glucose and hepatic triglyceride and cholesterol levels in db/db mice. These results show that overexpression of Akr1b7 in the liver selectively down-regulates gluconeogenic and lipogenic gene expression, even though the molecular mechanisms linking Akr1b7 to these metabolic changes are not elucidated. Yet, we can speculate that up-regulation of the receptor SHP upon Akr1b7 overexpression has a central role in the coordinated repression of both gluconeogenic and lipogenic metabolisms.
Unlike overexpression of Akr1b8/AKR1B10 that leads to increased lipogenesis, overexpression of Akr1b7 in the liver reduced hepatic lipid accumulation suggesting that in vivo these closely related AKR1B proteins display non-overlapping functions. The possibility that other AR (including AKR1B10) endowed with 4-HNE reductase activity could modulate bile acid metabolism under the control of FXR, LXR, or PXR, remains to be determined. Transcripts for a novel murine ARLP (Akr1b16) were recently described in the liver but expression of Akr1b16 has not been investigated in the small intestine (Salabei et al., 2011). Moreover, mechanisms controlling their expression in these tissues have not yet been elucidated.
Akr1b3/AKR1B1: Altering (liver) metabolic capacity
Both Akr1b3 and AKR1B1 are ubiquitously expressed. Akr1b3 transcripts were detected in the liver and small intestine at similar levels (Joshi et al., 2010). AKR1B1 messenger RNAs are found in the liver and small intestine (Cao et al., 1998). Western blot analyses confirmed AKR1B1 expression in the small intestine although it was undetectable in the normal adult liver. However AKR1B1 expression could be induced by alcoholic cirrhosis (O’Connor et al., 1999).
In diabetic mice, Akr1b3 is involved in glucose metabolism in some tissues through its participation in the polyol pathway. In response to chronic hyperglycemia, Akr1b3 catalyzes the rate-limiting reduction of glucose into sorbitol which is in turn converted into fructose by the sorbitol dehydrogenase (SDH). Activation of this pathway is involved in the development and progression of diabetic complications (Brownlee, 2001). Some evidences suggest that it could also be implicated in lipid metabolism. First, diabetic patients exhibit elevated blood triglycerides and non-esterified fatty acids and second, fructose-fed rats display an elevation of blood triglycerides and a reduction of peroxisome proliferator-activated receptor alpha (PPARα) activation (Roglans et al., 2007). These data led Qiu et al. to investigate the effects of hepatic activation of the Akr1b3/polyol pathway on lipid metabolism (Figure 6). In a murine hepatocyte cell line, overexpression of Akr1b3 was able to suppress PPARα transcriptional activity through extracellular signal-regulated kinase (ERK1/2)-mediated inactivating phosphorylations on Ser-12 and Ser-21. This resulted in decreased expression of genes involved in peroxisomal and mitochondrial β-oxidation pathway. In addition, Akr1b3 is up-regulated under high glucose concentrations in mouse hepatocyte AML12 cells and in vivo in diabetic mice. The genetic ablation or pharmacological inhibition of Akr1b3 in diabetic mice decreased ERK1/2-dependent phosphorylation of PPARα and blood triglycerides levels. In diabetic animals, SDH KO was accompanied by a reduction of blood triglycerides. This study clearly indicated that Akr1b3 and the polyol pathway sensed intracellular levels of glucose and adjusted PPARα activity through its phosphorylation/dephosphorylation, which in turn affected cellular lipid homeostasis (Qiu et al., 2008). In db/db mice, inhibition and knock-down of Akr1b3 induced the decrease of both plasma and liver triglycerides levels. Inhibition of Akr1b3 in db/db mice led to an improvement of hepatosteatosis due to the up-regulation of acetyl-coA oxidase and apolipoprotein A-V, two PPARα target genes involved in lipid catabolism (Qiu et al., 2012). Mechanisms by which Akr1b3 influences ERK-dependent inactivation of PPARα remain to be established.
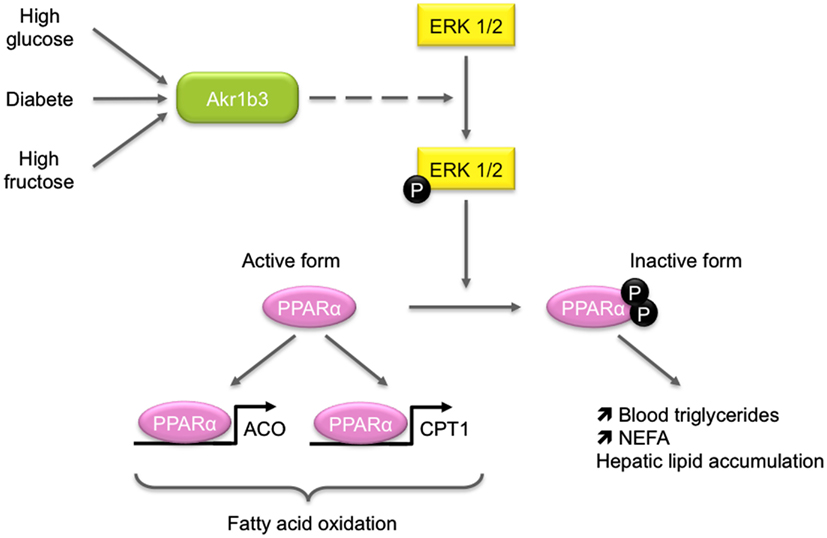
Figure 6. Role of Akr1b3 in hepatic lipid homeostasis. Diabetic conditions (high glucose or high fructose) lead to Akr1b3 up-regulation and Akr1b3-dependent ERK1/2 activation and are often associated to dyslipidemia. The ERK1/2 mediated phosphorylation of PPARγ inhibits its transcriptional activity resulting in down-regulation of β-oxidation master genes. Impaired β-oxidation favors hepatic lipid accumulation and increases blood triglycerides and non-esterified fatty acids (NEFA). The mechanism by which Akr1b3 induces ERK1/2 phosphorylation is still unknown (dashed lines).
AKR1B1 is also involved in the polyol pathway. Therefore, the relevance of these results in human remains to be determined. However, studies using Akr1b3−/− mice or transgenic mice overexpressing AKR1B1 revealed no abnormalities in either the liver or small intestine (Yamaoka et al., 1995; Aida et al., 2000).
In addition to this possible implication in metabolism, AKR1B1 could be an actor of small intestine protection against electrophilic carbonyls. Its expression is also induced in human colon cancer cell lines SW-480 and HT-29, by proteasome inhibitors known to induce Nrf2 expression (Ebert et al., 2011). Characterization of Akr1b3 promoter has shown that Nrf2 could regulate its activity through an antioxidant response element 1 (−953 to −945) and an AP1 site (Nishinaka and Yabe-Nishimura, 2005). The arrangement of these elements in the stress response region was conserved between Akr1b3 and AKR1B1 promoters (Figure 3).
Moreover, in a similar way as AKR1B10, AKR1B1 displayed enzymatic properties allowing protection of intestinal cells against dietary electrophilic carbonyls. Indeed, AKR1B1 kinetic parameters are compatible with reduction of glutathione-conjugated carbonyl compounds (Shen et al., 2011). Studies in healthy tissues could allow a better understanding of AKR1B1 implication in cytoprotection against oxidative damage.
AKR1B in White Adipose Tissue
For decades, adipose tissue was considered an inert mass of energy storage in which adipocytes were unique in the quantity of lipid that they can store during period of energy excess and mobilize as free fatty acids when required. In fat pads, adipocytes are intermingled with other cells including blood cells, endothelial cells, adipose precursors of varying degree of differentiation, and fibroblasts (Ailhaud et al., 1992). Adipose tissue plays a crucial role in the regulation of energy homeostasis, insulin sensitivity, and lipid/carbohydrate metabolism. These actions are mediated by both non-secreted proteins and hormones produced by adipocytes. These adipokines have wide-ranging effect on energy intake, energy expenditure, carbohydrate and lipid metabolism, including nutrient partitioning, and fuel selection (Trayhurn and Beattie, 2001; Fonseca-Alaniz et al., 2007; Vázquez-Vela et al., 2008). White adipose tissue expansion takes place rapidly after birth and it retains some plasticity throughout life (Frühbeck, 2008). Adipose precursors are able to differentiate in mature adipocytes and acquire features of fully differentiated cells during adipogenesis. Acquisition of the adipocyte phenotype is characterized by chronological changes in the expression of numerous genes. Studies on 3T3-L1 cells, a line of murine preadipocytes that have the ability to accumulate lipids during their hormonal-induced differentiation into adipocytes, have allowed identification of C/EBPs and PPARγ as master regulators of adipogenesis (Tontonoz et al., 1994; Wang et al., 1995; Tanaka et al., 1997; Rosen et al., 1999). The earliest event of adipogenesis is the transient induction of C/EBPβ and C/EBPδ expression, which in turn induces PPARγ and C/EBPα expression (Tang et al., 2004). This is followed by the synergic action of PPARγ and C/EBPα that enhance the expression of several genes characterizing the adipocyte phenotype along with massive triglyceride accumulation (Wu et al., 1999). SCD1, phosphoenolpyruvate carboxykinase, Fas, ACCA, malic enzyme, insulin receptor, adipocyte specific fatty acid binding protein, leptin (Rosen and Spiegelman, 2000) are some of the essential genes induced during terminal differentiation of adipocytes. Adipogenesis is tightly regulated by a set of pro- and anti-adipogenic factors including insulin, FGF, EGF, and prostaglandins (Gregoire et al., 1998). Adipose tissue expansion is resulting from adipocyte hyperplasia (generated by an increased number of adipocytes) and/or hypertrophy (caused by an enlargement of adipocyte resulting from increased lipid accumulation). Disruption of the mechanisms that control adipose tissue homeostasis can result in massive expansion of the tissue as exemplified during development of obesity (Henry et al., 2012). One of the most exciting challenges in the field of adipose tissue homeostasis is to identify the key regulators of adipose tissue expansion, which would allow a better understanding of the mechanisms involved in the pathogenesis of adipose tissue disorders. Recent reports based on functional studies and identification of novel enzymatic properties suggest that some AKR1B enzymes can regulate adipose tissue homeostasis.
Akr1b7: A negative regulator of adipose expansion
Until 2003, there was no mention of AR expression in white adipose tissue. In a transcriptomic study, Moraes et al. reported for the first time the detection of Akr1b7 mRNA in mouse white adipose tissue. In this study, the authors highlighted a decrease in Akr1b7 expression in abdominal white fat tissue from diet-induced obese mice when compared with mice fed a standard diet (Moraes et al., 2003). This decrease reflected either the down-regulation of Akr1b7 gene expression level per cell or the lower proportion of cells expressing the gene. Indeed, Akr1b7 expression appeared to vary depending on the location of white adipose depots and was found enriched in the stromal vascular fraction (containing adipocyte progenitors) whereas it was virtually absent from mature adipocytes (Tirard et al., 2007). Accordingly, Akr1b7 expression decreased during adipogenic differentiation of primary preadipocytes from the stromal vascular fraction and was detectable in the 3T3-L1 preadipocyte cell line.
The transcription factor SF-1, an essential regulator of Akr1b7 expression in the adrenal cortex is absent from adipose tissue. Instead, adipocyte precursors express liver receptor homolog-1 (LRH-1; Clyne et al., 2002), a nuclear receptor that also binds to SF-1 regulatory elements and can substitute for SF-1 in tissues where it is not expressed (Sirianni et al., 2002). Consistent with decreased expression of Akr1b7 during adipogenesis, LRH-1 expression is rapidly down-regulated during adipocyte differentiation (Clyne et al., 2002). We thus evaluated the possibility that LRH-1 stimulated Akr1b7 promoter activity in preadipocytes. Indeed, in co-transfection experiments in 3T3-L1 preadipocytes, LRH-1 induced Akr1b7 promoter through the SF-1 binding site at −458 (unpublished observations). Therefore, the dynamic expression profile of LRH-1 may account for expression of Akr1b7 in the preadipocytes-enriched stromal fraction (stromal vascular fraction) of various fat depots and its transitory expression in the early differentiation steps of 3T3-L1 cells.
Overexpression of Akr1b7 in 3T3-L1 preadipocytes prevented lipid droplets accumulation during adipogenic differentiation. In contrast, knock-down of Akr1b7 accelerated differentiation and lipid accumulation in 3T3-L1 cells. These results allowed us to demonstrate that Akr1b7 is a negative regulator of adipogenesis in vitro, which prevents differentiation by reducing lipid storage (Tirard et al., 2007). The mechanisms through which Akr1b7 inhibits adipocyte differentiation in vivo were recently unraveled by our group using Akr1b7−/− mice. These will be discussed below.
Akr1b7 has two different enzymatic activities that could potentially be involved in the regulation of white adipose tissue homeostasis. First, just like Akr1b3, Akr1b7 is endowed with PGFS activity allowing synthesis of PGF2α, a potent inhibitor of adipogenesis (Serrero et al., 1992; Kabututu et al., 2009). Indeed, PGF2α is able to inhibit differentiation of 3T3-L1 into adipocytes, through binding to the FP receptor. Activation of FP receptor in turn blocks PPARγ and C/EBPα expression through a Gαq-calcium-calcineurin-dependent signaling pathway (Liu and Clipstone, 2007). PGF2α was also shown to block adipogenesis through activation of mitogen-activated protein kinase, resulting in inhibitory phosphorylation of PPARγ (Reginato et al., 1998). Second, Akr1b7 reduces 4-HNE, the accumulation of which is known to induce an excessive development of adipose tissue through adipocytes hypertrophy. Detailed analysis of Akr1b7−/− mice allowed us to show that Akr1b7 behaved as an anti-adipogenic factor limiting white adipose tissue expansion, essentially through the regulation of PGF2α levels in vivo (Volat et al., 2012; Figure 7).
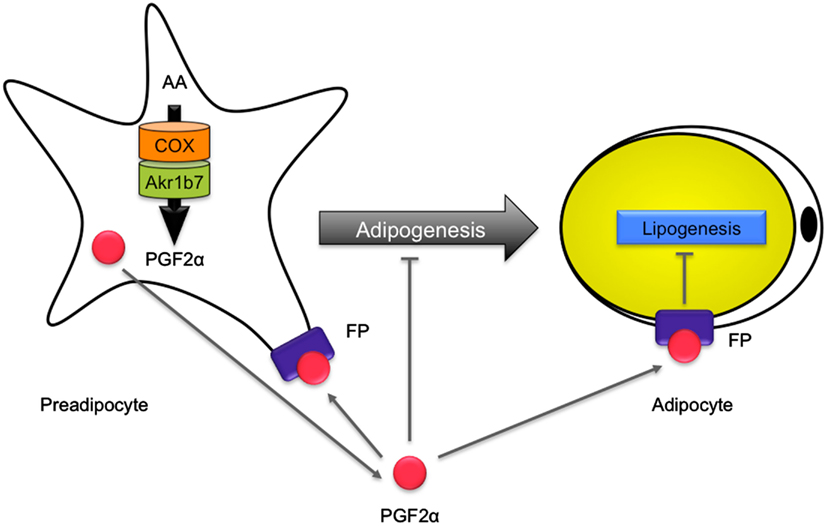
Figure 7. Role of Akr1b7 in adipose tissue homeostasis. In preadipocytes, arachidonic acid (AA) is metabolized into PGH2 by COX and is further converted into PGF2α by the PGFS activity of Akr1b7. PGF2α is supposed to mediate an autocrine activation of FP receptors on preadipocytes that inhibits their differentiation into adipocytes. In addition, PGF2α inhibits lipogenesis in mature adipocytes. Accordingly, altered production of PGF2α in Akr1b7−/− mice leads to an expansion of adipose tissue due to both hyperplasia and hypertrophy of adipocytes.
Akr1b7−/− mice displayed excessive basal adiposity resulting from both adipocyte hyperplasia and hypertrophy. They further exhibited increased sensitivity to diet-induced obesity. Following adipose enlargement and irrespective of the diet, they developed liver steatosis and progressive insulin-resistance. Akr1b7 loss was associated with decreased PGF2α white adipose tissue contents. Cloprostenol (a PGF2α agonist) administration in Akr1b7−/− mice normalized white adipose tissue expansion by altering both de novo adipocyte differentiation and size. Treatment of 3T3-L1 adipocytes and Akr1b7−/− mice with cloprostenol suggested that decreased adipocyte size resulted from inhibition of lipogenic gene expression. Hence, Akr1b7 is a major regulator of white adipose tissue development through at least two PGF2α-dependent mechanisms: inhibition of adipogenesis and lipogenesis (Volat et al., 2012).
Akr1b8/AKR1B10
Although we showed that Akr1b8 is devoid of PGH2 9-,11-endoperoxide reductase activity (Kabututu et al., 2009), QTL mapping allowed identification of Akr1b8 as a possible regulator of adiposity in mouse (Derry et al., 2010). Akr1b8−/− male mice had a tendency to be fatter than their wild-type littermates, under both standard and high fat diets. This was particularly obvious for the gonadal fat pad. Under high fat diet, Akr1b8−/− mice also displayed higher serum cholesterol than their wild-type littermates. The mechanisms involving Akr1b8 in the regulation of white adipose tissue homeostasis remain completely unknown and are not necessarily resulting from expression of the gene in the adipose tissue. Indeed, we were unable to detect significant Akr1b8 protein expression in various adipose depots in mouse (Volat et al., 2012). Although we cannot exclude that Akr1b8 could be expressed in variable amounts depending on the mouse genetic background and the location of fat pads, it should be considered that the effect of Akr1b8 ablation could be indirect. Thus the involvement of Akr1b8 in adipose tissue homeostasis should be carefully examined with respect to its expression sites and enzymatic activities.
AKR1B10 expression in the adipose tissue has not yet been determined. Because AKR1B10 and Akr1b8 are not only detoxifying enzymes but also affect de novo fatty acids synthesis in cancer cells, it would be interesting to determine if and which of the enzymatic activities of AKR1B10 could be involved in adipose tissue homeostasis (Ma et al., 2008; Wang et al., 2009; Joshi et al., 2010).
Akr1b3/AKR1B1
Akr1b3 is expressed in undifferentiated 3T3-L1 preadipocytes and during the early phase of their differentiation. Akr1b3 can synthesize PGF2α in 3T3-L1 cells, which inhibits their differentiation through stimulation of the FP receptor (Fujimori et al., 2010).
In addition to their PGF2α synthase activity, Akr1b3 and AKR1B1 also catalyze the isomerization of PGH2 to PGD2 (Nagata et al., 2011). The impact of PGD2 on white adipose tissue homeostasis is still very disputed. Indeed, lipocalin-type prostaglandin D synthase (L-PGDS−/−) mice show hypertrophy of adipocytes (Ragolia et al., 2005). In contrast, the knock-down of L-PGDS decreases lipid accumulation in 3T3-L1 cells (Fujimori et al., 2007). In agreement with these observations, transgenic mice overexpressing (human hematopoietic-type prostaglandin D synthase (H-PGDS) overproduce PGD2 and show signs of obesity and pronounced adipogenesis on a high fat diet (Fujitani et al., 2010). These investigations suggest that PGD2 promotes adipogenesis in vivo.
Because of its involvement in the synthesis of PGF2α and PGD2, the involvement of Akr1b3 in white adipose tissue physiology may result from a balance between both enzymatic activities. However the relevance of these data has not yet been studied in vivo. Indeed, no defect in adipose tissue homeostasis have been reported in Akr1b3−/− mice and the prostaglandin contents of their fat depots have not been analyzed (Aida et al., 2000; Ho et al., 2000).
Although the murine AR Akr1b3 could potentially participate in the homeostatic maintenance of adipose tissue, there is no available information about its human ortholog AKR1B1. Previous studies using transgenic mice with constitutive overexpression of AKR1B1 did not report any effect on adipose tissue (Yamaoka et al., 1995). On the basis of its enzymatic properties (PGF2α and PGD2 synthesis and reduction of 4-HNE), AKR1B1 could be considered as a potential actor of adipose tissue physiology (Kabututu et al., 2009; Nagata et al., 2011; Shen et al., 2011). However its expression pattern in human fat depots remains to be determined.
Conclusive Remarks
Understanding physiological functions of AKR1B/Akr1b enzymes is a really challenging task. In that regard, review of the literature highlights the marked contrast between the abundance of enzymatic data and the small number of reports dedicated to functional studies in a physiological context. The main reason for this discrepancy is likely to be rooted in the functional redundancy resulting from the obvious structure conservation of the members of this enzyme family. Moreover, Akr1b genes being tandemly arranged on a same chromosome, classical genetic approaches to study multigene families that consist in combining KO models for each isoforms to obtain double or triple mutant mice are almost impossible to setup. Thus, alternative approaches to genetically disrupt Akr1b in various combinations and locations are mandatory to advance our understanding of their functions. Nuclease targeted invalidation or RNA interference approaches combined to existing KO models could present as valuable options.
Gain/loss-of-function models available for the Akr1b7 isoform revealed its implication in metabolic function and adipose tissue homeostasis. Beside the widely accepted idea that Akr1b are detoxification enzymes, several teams have now provided functional demonstration that some isoforms are endowed with the capacity to produce directly (prostaglandins) or indirectly (fatty acids) signal molecules. We think that further exploration of the functions of AKR1B/Akr1b enzymes must now take this dual potentiality into account.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funds from the Center National de la Recherche Scientifique, Université Blaise Pascal, Université d’Auvergne, Institut National de la Santé et de la Recherche Médicale, and grant from the Région Auvergne. We are grateful to Dr Pierre Val (CNRS UMR6293/INSERM U1103-GReD, Clermont Université, Aubière, France) for critical reading of the manuscript.
References
Aida, K., Ikegishi, Y., Chen, J., Tawata, M., Ito, S., Maeda, S., and Onaya, T. (2000). Disruption of aldose reductase gene (Akr1b1) causes defect in urinary concentrating ability and divalent cation homeostasis. Biochem. Biophys. Res. Commun. 277, 281–286.
Aigueperse, C., Martinez, A., Lefrancois-Martinez, A. M., Veyssière, G., and Jean, C. I. (1999). Cyclic AMP regulates expression of the gene coding for a mouse vas deferens protein related to the aldo-keto reductase superfamily in human and murine adrenocortical cells. J. Endocrinol. 160, 147–154.
Aigueperse, C., Val, P., Pacot, C., Darne, C., Lalli, E., Sassone-Corsi, P., Veyssière, G., Jean, C., and Martinez, A. (2001). SF-1 (steroidogenic factor-1), C/EBPbeta (CCAAT/enhancer binding protein), and ubiquitous transcription factors NF1 (nuclear factor 1) and Sp1 (selective promoter factor 1) are required for regulation of the mouse aldose reductase-like gene (AKR1B7) expression in adrenocortical cells. Mol. Endocrinol. 15, 93–111.
Ailhaud, G., Grimaldi, P., and Négrel, R. (1992). Cellular and molecular aspects of adipose tissue development. Annu. Rev. Nutr. 12, 207–233.
Barski, O. A., Tipparaju, S. M., and Bhatnagar, A. (2008). The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 40, 553–624.
Bohren, K. M., Bullock, B., Wermuth, B., and Gabbay, K. H. (1989). The aldo-keto reductase superfamily. cDNAs and deduced amino acid sequences of human aldehyde and aldose reductases. J. Biol. Chem. 264, 9547–9551.
Bohren, K. M., Grimshaw, C. E., and Gabbay, K. H. (1992). Catalytic effectiveness of human aldose reductase. Critical role of C-terminal domain. J. Biol. Chem. 267, 20965–20970.
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820.
Brunskill, E. W., Sequeira-Lopez, M. L. S., Pentz, E. S., Lin, E., Yu, J., Aronow, B. J., Potter, S. S., and Gomez, R. A. (2011). Genes that confer the identity of the renin cell. J. Am. Soc. Nephrol. 22, 2213–2225.
Burczynski, J. M., Hayes, J. R., Longhurst, P. A., and Colby, H. D. (1999). Species differences in adrenal lipid peroxidation: role of alpha-tocopherol. Free Radic. Biol. Med. 26, 987–991.
Cao, D., Fan, S. T., and Chung, S. S. (1998). Identification and characterization of a novel human aldose reductase-like gene. J. Biol. Chem. 273, 11429–11435.
Chajès, V., Cambot, M., Moreau, K., Lenoir, G. M., and Joulin, V. (2006). Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 66, 5287–5294.
Chen, G., Liang, G., Ou, J., Goldstein, J. L., and Brown, M. S. (2004). Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. U.S.A. 101, 11245–11250.
Clyne, C. D., Speed, C. J., Zhou, J., and Simpson, E. R. (2002). Liver receptor homologue-1 (LRH-1) regulates expression of aromatase in preadipocytes. J. Biol. Chem. 277, 20591–20597.
de Reyniès, A., Assié, G., Rickman, D. S., Tissier, F., Groussin, L., René-Corail, F., Dousset, B., Bertagna, X., Clauser, E., and Bertherat, J. (2009). Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J. Clin. Oncol. 27, 1108–1115.
Derry, J. M. J., Zhong, H., Molony, C., MacNeil, D., Guhathakurta, D., Zhang, B., Mudgett, J., Small, K., El Fertak, L., Guimond, A., Selloum, M., Zhao, W., Champy, M. F., Monassier, L., Vogt, T., Cully, D., Kasarskis, A., and Schadt, E. E. (2010). Identification of genes and networks driving cardiovascular and metabolic phenotypes in a mouse F2 intercross. PLoS ONE 5, e14319. doi:10.1371/journal.pone.0014319
Donohue, P. J., Alberts, G. F., Hampton, B. S., and Winkles, J. A. (1994). A delayed-early gene activated by fibroblast growth factor-1 encodes a protein related to aldose reductase. J. Biol. Chem. 269, 8604–8609.
Ebert, B., Kisiela, M., Wsól, V., and Maser, E. (2011). Proteasome inhibitors MG-132 and bortezomib induce AKR1C1, AKR1C3, AKR1B1, and AKR1B10 in human colon cancer cell lines SW-480 and HT-29. Chem. Biol. Interact. 191, 239–249.
Endo, S., Matsunaga, T., Fujita, A., Kuragano, T., Soda, M., Sundaram, K., Dhagat, U., Tajima, K., El-Kabbani, O., and Hara, A. (2011). Activation of aldo-keto reductase family member 1B14 (AKR1B14) by bile acids: activation mechanism and bile acid-binding site. Biochimie 93, 1476–1486.
Endo, S., Matsunaga, T., Fujita, A., Tajima, K., El-Kabbani, O., and Hara, A. (2010a). Rat aldose reductase-like protein (AKR1B14) efficiently reduces the lipid peroxidation product 4-oxo-2-nonenal. Biol. Pharm. Bull. 33, 1886–1890.
Endo, S., Matsunaga, T., Kuragano, T., Ohno, S., Kitade, Y., Tajima, K., El-Kabbani, O., and Hara, A. (2010b). Properties and tissue distribution of a novel aldo-keto reductase encoding in a rat gene (Akr1b10). Arch. Biochem. Biophys. 503, 230–237.
Endo, S., Matsunaga, T., Mamiya, H., Hara, A., Kitade, Y., Tajima, K., and El-Kabbani, O. (2009a). Characterization of a rat NADPH-dependent aldo-keto reductase (AKR1B13) induced by oxidative stress. Chem. Biol. Interact. 178, 151–157.
Endo, S., Matsunaga, T., Mamiya, H., Ohta, C., Soda, M., Kitade, Y., Tajima, K., Zhao, H.-T., El-Kabbani, O., and Hara, A. (2009b). Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch. Biochem. Biophys. 487, 1–9.
Fonseca-Alaniz, M. H., Takada, J., Alonso-Vale, M. I. C., and Lima, F. B. (2007). Adipose tissue as an endocrine organ: from theory to practice. J. Pediatr. (Rio. J.) 83, S192–S203.
Frühbeck, G. (2008). Overview of adipose tissue and its role in obesity and metabolic disorders. Methods Mol. Biol. 456, 1–22.
Fujimori, K., Aritake, K., and Urade, Y. (2007). A novel pathway to enhance adipocyte differentiation of 3T3-L1 cells by up-regulation of lipocalin-type prostaglandin D synthase mediated by liver X receptor-activated sterol regulatory element-binding protein-1c. J. Biol. Chem. 282, 18458–18466.
Fujimori, K., Ueno, T., Nagata, N., Kashiwagi, K., Aritake, K., Amano, F., and Urade, Y. (2010). Suppression of adipocyte differentiation by aldo-keto reductase 1B3 acting as prostaglandin F2alpha synthase. J. Biol. Chem. 285, 8880–8886.
Fujitani, Y., Aritake, K., Kanaoka, Y., Goto, T., Takahashi, N., Fujimori, K., and Kawada, T. (2010). Pronounced adipogenesis and increased insulin sensitivity caused by overproduction of prostaglandin D2 in vivo. FEBS J. 277, 1410–1419.
Fukumoto, S.-I., Yamauchi, N., Moriguchi, H., Hippo, Y., Watanabe, A., Shibahara, J., Taniguchi, H., Ishikawa, S., Ito, H., Yamamoto, S., Iwanari, H., Hironaka, M., Ishikawa, Y., Niki, T., Sohara, Y., Kodama, T., Nishimura, M., Fukayama, M., Dosaka-Akita, H., and Aburatani, H. (2005). Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin. Cancer Res. 11, 1776–1785.
Ge, X., Yin, L., Ma, H., Li, T., Chiang, J. Y. L., and Zhang, Y. (2011). Aldo-keto reductase 1B7 is a target gene of FXR and regulates lipid and glucose homeostasis. J. Lipid Res. 52, 1561–1568.
Gregoire, F. M., Smas, C. M., and Sul, H. S. (1998). Understanding adipocyte differentiation. Physiol. Rev. 78, 783–809.
Gui, T., Tanimoto, T., Kokai, Y., and Nishimura, C. (1995). Presence of a closely related subgroup in the aldo-keto reductase family of the mouse. Eur. J. Biochem. 227, 448–453.
Henry, S. L., Bensley, J. G., Wood-Bradley, R. J., Cullen-McEwen, L. A., Bertram, J. F., and Armitage, J. A. (2012). White adipocytes: more than just fat depots. Int. J. Biochem. Cell Biol. 44, 435–440.
Heringlake, S., Hofdmann, M., Fiebeler, A., Manns, M. P., Schmiegel, W., and Tannapfel, A. (2010). Identification and expression analysis of the aldo–ketoreductase1-B10 gene in primary malignant liver tumours. J. Hepatol. 52, 220–227.
Ho, H. T., Chung, S. K., Law, J. W., Ko, B. C., Tam, S. C., Brooks, H. L., Knepper, M. A., and Chung, S. S. (2000). Aldose reductase-deficient mice develop nephrogenic diabetes insipidus. Mol. Cell. Biol. 20, 5840–5846.
Ho, H. T., Jenkins, N. A., Copeland, N. G., Gilbert, D. J., Winkles, J. A., Louie, H. W., Lee, F. K., Chung, S. S., and Chung, S. K. (1999). Comparisons of genomic structures and chromosomal locations of the mouse aldose reductase and aldose reductase-like genes. Eur. J. Biochem. 259, 726–730.
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131.
Hsu, D. K., Guo, Y., Peifley, K. A., and Winkles, J. A. (1997). Differential control of murine aldose reductase and fibroblast growth factor (FGF)-regulated-1 gene expression in NIH 3T3 cells by FGF-1 treatment and hyperosmotic stress. Biochem. J. 328(Pt 2), 593–598.
Hylemon, P. B., Zhou, H., Pandak, W. M., Ren, S., Gil, G., and Dent, P. (2009). Bile acids as regulatory molecules. J. Lipid Res. 50, 1509–1520.
Hyndman, D., Bauman, D. R., Heredia, V. V., and Penning, T. M. (2003). The aldo-keto reductase superfamily homepage. Chem. Biol. Interact. 143–144, 621–631.
Hyndman, D. J., and Flynn, T. G. (1998). Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochim. Biophys. Acta 1399, 198–202.
Jez, J. M., Flynn, T. G., and Penning, T. M. (1997). A new nomenclature for the aldo-keto reductase superfamily. Biochem. Pharmacol. 54, 639–647.
Joshi, A., Rajput, S., Wang, C., Ma, J., and Cao, D. (2010). Murine aldo-keto reductase family 1 subfamily B: identification of AKR1B8 as an ortholog of human AKR1B10. Biol. Chem. 391, 1371–1378.
Kabututu, Z., Manin, M., Pointud, J.-C., Maruyama, T., Nagata, N., Lambert, S., Lefrançois-Martinez, A.-M., Martinez, A., and Urade, Y. (2009). Prostaglandin F2alpha synthase activities of aldo-keto reductase 1B1, 1B3, and 1B7. J. Biochem. 145, 161–168.
Kim, T.-H., Kim, H., Park, J.-M., Im, S.-S., Bae, J.-S., Kim, M.-Y., Yoon, H.-G., Cha, J.-Y., Kim, K.-S., and Ahn, Y.-H. (2009). Interrelationship between liver X receptor alpha, sterol regulatory element-binding protein-1c, peroxisome proliferator-activated receptor gamma, and small heterodimer partner in the transcriptional regulation of glucokinase gene expression in liver. J. Biol. Chem. 284, 15071–15083.
Lambert-Langlais, S., Pointud, J.-C., Lefrançois-Martinez, A.-M., Volat, F., Manin, M., Coudoré, F., Val, P., Sahut-Barnola, I., Ragazzon, B., Louiset, E., Delarue, C., Lefebvre, H., Urade, Y., and Martinez, A. (2009). Aldo keto reductase 1B7 and prostaglandin F2α are regulators of adrenal endocrine functions. PLoS ONE 4, e7309. doi:10.1371/journal.pone.0007309
Lau, E. T., Cao, D., Lin, C., Chung, S. K., and Chung, S. S. (1995). Tissue-specific expression of two aldose reductase-like genes in mice: abundant expression of mouse vas deferens protein and fibroblast growth factor-regulated protein in the adrenal gland. Biochem. J. 312(Pt 2), 609–615.
Lefrançois-Martinez, A.-M., Bertherat, J., Val, P., Tournaire, C., Gallo-Payet, N., Hyndman, D., Veyssière, G., Bertagna, X., Jean, C., and Martinez, A. (2004). Decreased expression of cyclic adenosine monophosphate-regulated aldose reductase (AKR1B1) is associated with malignancy in human sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 89, 3010–3019.
Lefrancois-Martinez, A. M., Tournaire, C., Martinez, A., Berger, M., Daoudal, S., Tritsch, D., Veyssière, G., and Jean, C. (1999). Product of side-chain cleavage of cholesterol, isocaproaldehyde, is an endogenous specific substrate of mouse vas deferens protein, an aldose reductase-like protein in adrenocortical cells. J. Biol. Chem. 274, 32875–32880.
Libè, R., Fratticci, A., and Bertherat, J. (2007). Adrenocortical cancer: pathophysiology and clinical management. Endocr. Relat. Cancer 14, 13–28.
Liu, L., and Clipstone, N. A. (2007). Prostaglandin F2alpha inhibits adipocyte differentiation via a G alpha q-calcium-calcineurin-dependent signaling pathway. J. Cell. Biochem. 100, 161–173.
Liu, M.-J., Takahashi, Y., Wada, T., He, J., Gao, J., Tian, Y., Li, S., and Xie, W. (2009). The aldo-keto reductase Akr1b7 gene is a common transcriptional target of xenobiotic receptors pregnane X receptor and constitutive androstane receptor. Mol. Pharmacol. 76, 604–611.
Liu, Z., Yan, R., Al Salman, A., Shen, Y., Bu, Y., Ma, J., Luo, D. X., Huang, C., Jiang, Y., Wilber, A., Mo, Y. Y., Huang, M. C., Zhao, Y., and Cao, D. (2012). Epidermal growth factor induces tumour marker AKR1B10 expression through activator protein-1 signalling in hepatocellular carcinoma cells. Biochem. J. 442, 273–282.
Ma, J., Yan, R., Zu, X., Cheng, J.-M., Rao, K., Liao, D.-F., and Cao, D. (2008). Aldo-keto reductase family 1 B10 affects fatty acid synthesis by regulating the stability of acetyl-CoA carboxylase-alpha in breast cancer cells. J. Biol. Chem. 283, 3418–3423.
Ma, K., Saha, P. K., Chan, L., and Moore, D. D. (2006). Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Invest. 116, 1102–1109.
MacLeod, A. K., Kelly, V. P., Higgins, L. G., Kelleher, M. O., Price, S. A., Bigley, A. L., Betton, G. R., and Hayes, J. D. (2010). Expression and localization of rat aldo-keto reductases and induction of the 1B13 and 1D2 isoforms by phenolic antioxidants. Drug Metab. Dispos. 38, 341–346.
Madore, E., Harvey, N., Parent, J., Chapdelaine, P., Arosh, J. A., and Fortier, M. A. (2003). An aldose reductase with 20 alpha-hydroxysteroid dehydrogenase activity is most likely the enzyme responsible for the production of prostaglandin f2 alpha in the bovine endometrium. J. Biol. Chem. 278, 11205–11212.
Martin, H.-J., Breyer-Pfaff, U., Wsól, V., Venz, S., Block, S., and Maser, E. (2006). Purification and characterization of akr1b10 from human liver: role in carbonyl reduction of xenobiotics. Drug Metab. Dispos. 34, 464–470.
Martin, H.-J., and Maser, E. (2009). Role of human aldo-keto-reductase AKR1B10 in the protection against toxic aldehydes. Chem. Biol. Interact. 178, 145–150.
Martinez, A., Aigueperse, C., Val, P., Dussault, M., Tournaire, C., Berger, M., Veyssière, G., Jean, C., and Lefrançois Martinez, A. (2001). Physiological functions and hormonal regulation of mouse vas deferens protein (AKR1B7) in steroidogenic tissues. Chem. Biol. Interact. 130–132, 903–917.
Martinez, A., Val, P., Sahut-Barnola, I., Aigueperse, C., Veyssière, G., and Lefrançois-Martinez, A.-M. (2003). Steroidogenic factor-1 controls the aldose reductase akr1b7 gene promoter in transgenic mice through an atypical binding site. Endocrinology 144, 2111–2120.
Matsunaga, T., Wada, Y., Endo, S., Soda, M., El-Kabbani, O., and Hara, A. (2012). Aldo-keto reductase 1B10 and its role in proliferation capacity of drug-resistant cancers. Front. Pharmacol. 3:5. doi:10.3389/fphar.2012.00005
Matsuura, K., Deyashiki, Y., Bunai, Y., Ohya, I., and Hara, A. (1996). Aldose reductase is a major reductase for isocaproaldehyde, a product of side-chain cleavage of cholesterol, in human and animal adrenal glands. Arch. Biochem. Biophys. 328, 265–271.
Moraes, R. C., Blondet, A., Birkenkamp-Demtroeder, K., Tirard, J., Orntoft, T. F., Gertler, A., Durand, P., Naville, D., and Begeot, M. (2003). Study of the alteration of gene expression in adipose tissue of diet-induced obese mice by microarray and reverse transcription-polymerase chain reaction analyses. Endocrinology 144, 4773–4782.
Nagata, N., Kusakari, Y., Fukunishi, Y., Inoue, T., and Urade, Y. (2011). Catalytic mechanism of the primary human prostaglandin F2α synthase, aldo-keto reductase 1B1 – prostaglandin D2 synthase activity in the absence of NADP(H). FEBS J. 278, 1288–1298.
Nishinaka, T., Miura, T., Okumura, M., Nakao, F., Nakamura, H., and Terada, T. (2011). Regulation of aldo-keto reductase AKR1B10 gene expression: involvement of transcription factor Nrf2. Chem. Biol. Interact. 191, 185–191.
Nishinaka, T., and Yabe-Nishimura, C. (2005). Transcription factor Nrf2 regulates promoter activity of mouse aldose reductase (AKR1B3) gene. J. Pharmacol. Sci. 97, 43–51.
O’Connor, T., Ireland, L. S., Harrison, D. J., and Hayes, J. D. (1999). Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem. J. 343(Pt 2), 487–504.
Pailhoux, E. A., Martinez, A., Veyssiere, G. M., and Jean, C. G. (1990). Androgen-dependent protein from mouse vas deferens. cDNA cloning and protein homology with the aldo-keto reductase superfamily. J. Biol. Chem. 265, 19932–19936.
Postic, C., and Girard, J. (2008). Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118, 829–838.
Qiu, L., Lin, J., Xu, F., Gao, Y., Zhang, C., Liu, Y., Luo, Y., and Yang, J. Y. (2012). Inhibition of aldose reductase activates hepatic peroxisome proliferator-activated receptor-α and ameliorates hepatosteatosis in diabetic db/db mice. Exp. Diabetes Res. 2012, 789730.
Qiu, L., Wu, X., Chau, J. F. L., Szeto, I. Y. Y., Tam, W. Y., Guo, Z., Chung, S. K., Oates, P. J., Chung, S. S. M., and Yang, J. Y. (2008). Aldose reductase regulates hepatic peroxisome proliferator-activated receptor alpha phosphorylation and activity to impact lipid homeostasis. J. Biol. Chem. 283, 17175–17183.
Ragazzon, B., Lefrançois-Martinez, A.-M., Val, P., Sahut-Barnola, I., Tournaire, C., Chambon, C., Gachancard-Bouya, J.-L., Begue, R.-J., Veyssière, G., and Martinez, A. (2006). Adrenocorticotropin-dependent changes in SF-1/DAX-1 ratio influence steroidogenic genes expression in a novel model of glucocorticoid-producing adrenocortical cell lines derived from targeted tumorigenesis. Endocrinology 147, 1805–1818.
Ragolia, L., Palaia, T., Hall, C. E., Maesaka, J. K., Eguchi, N., and Urade, Y. (2005). Accelerated glucose intolerance, nephropathy, and atherosclerosis in prostaglandin D2 synthase knock-out mice. J. Biol. Chem. 280, 29946–29955.
Rajkumar, T., Vijayalakshmi, N., Gopal, G., Sabitha, K., Shirley, S., Raja, U. M., and Ramakrishnan, S. A. (2010). Identification and validation of genes involved in gastric tumorigenesis. Cancer Cell Int. 10, 45.
Ramsay, R. G., Ciznadija, D., Vanevski, M., and Mantamadiotis, T. (2003). Transcriptional regulation of cyclo-oxygenase expression: three pillars of control. Int. J. Immunopathol. Pharmacol. 16, 59–67.
Rangasamy, T., Cho, C. Y., Thimmulappa, R. K., Zhen, L., Srisuma, S. S., Kensler, T. W., Yamamoto, M., Petrache, I., Tuder, R. M., and Biswal, S. (2004). Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 114, 1248–1259.
Reginato, M. J., Krakow, S. L., Bailey, S. T., and Lazar, M. A. (1998). Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 273, 1855–1858.
Roglans, N., Vilà, L., Farré, M., Alegret, M., Sánchez, R. M., Vázquez-Carrera, M., and Laguna, J. C. (2007). Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology 45, 778–788.
Rosen, E. D., Sarraf, P., Troy, A. E., Bradwin, G., Moore, K., Milstone, D. S., Spiegelman, B. M., and Mortensen, R. M. (1999). PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617.
Rosen, E. D., and Spiegelman, B. M. (2000). Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 16, 145–171.
Ruiz, F. X., Moro, A., Gallego, O., Ardèvol, A., Rovira, C., Petrash, J. M., Parés, X., and Farrés, J. (2011). Human and rodent aldo-keto reductases from the AKR1B subfamily and their specificity with retinaldehyde. Chem. Biol. Interact. 191, 199–205.
Salabei, J. K., Li, X.-P., Petrash, J. M., Bhatnagar, A., and Barski, O. A. (2011). Functional expression of novel human and murine AKR1B genes. Chem. Biol. Interact. 191, 177–184.
Schmidt, D. R., and Mangelsdorf, D. J. (2008). Nuclear receptors of the enteric tract: guarding the frontier. Nutr. Rev. 66, S88–S97.
Schmidt, D. R., Schmidt, S., Holmstrom, S. R., Makishima, M., Yu, R. T., Cummins, C. L., Mangelsdorf, D. J., and Kliewer, S. A. (2011). AKR1B7 is induced by the farnesoid X receptor and metabolizes bile acids. J. Biol. Chem. 286, 2425–2432.
Serrero, G., Lepak, N. M., and Goodrich, S. P. (1992). Paracrine regulation of adipose differentiation by arachidonate metabolites: prostaglandin F2 alpha inhibits early and late markers of differentiation in the adipogenic cell line 1246. Endocrinology 131, 2545–2551.
Shen, Y., Zhong, L., Johnson, S., and Cao, D. (2011). Human aldo-keto reductases 1B1 and 1B10: a comparative study on their enzyme activity toward electrophilic carbonyl compounds. Chem. Biol. Interact. 191, 192–198.
Sirianni, R., Seely, J. B., Attia, G., Stocco, D. M., Carr, B. R., Pezzi, V., and Rainey, W. E. (2002). Liver receptor homologue-1 is expressed in human steroidogenic tissues and activates transcription of genes encoding steroidogenic enzymes. J. Endocrinol. 174, R13–R17.
Spite, M., Baba, S. P., Ahmed, Y., Barski, O. A., Nijhawan, K., Petrash, J. M., Bhatnagar, A., and Srivastava, S. (2007). Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem. J. 405, 95–105.
Srivastava, S., Harter, T. M., Chandra, A., Bhatnagar, A., Srivastava, S. K., and Petrash, J. M. (1998). Kinetic studies of FR-1, a growth factor-inducible aldo-keto reductase. Biochemistry 37, 12909–12917.
Stout, M. D., Bodes, E., Schoonhoven, R., Upton, P. B., Travlos, G. S., and Swenberg, J. A. (2008). Toxicity, DNA binding, and cell proliferation in male F344 rats following short-term gavage exposures to trans-2-hexenal. Toxicol. Pathol. 36, 232–246.
Tanaka, T., Yoshida, N., Kishimoto, T., and Akira, S. (1997). Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 16, 7432–7443.
Tang, Q.-Q., Zhang, J.-W., and Daniel Lane, M. (2004). Sequential gene promoter interactions by C/EBPbeta, C/EBPalpha, and PPARgamma during adipogenesis. Biochem. Biophys. Res. Commun. 318, 213–218.
Tirard, J., Gout, J., Lefrançois-Martinez, A.-M., Martinez, A., Begeot, M., and Naville, D. (2007). A novel inhibitory protein in adipose tissue, the aldo-keto reductase AKR1B7: its role in adipogenesis. Endocrinology 148, 1996–2005.
Tontonoz, P., Hu, E., and Spiegelman, B. M. (1994). Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79, 1147–1156.
Trayhurn, P., and Beattie, J. H. (2001). Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 60, 329–339.
Val, P., Aigueperse, C., Ragazzon, B., Veyssière, G., Lefrançois-Martinez, A.-M., and Martinez, A. (2004). Adrenocorticotropin/3′,5′-cyclic AMP-mediated transcription of the scavenger akr1-b7 gene in adrenocortical cells is dependent on three functionally distinct steroidogenic factor-1-responsive elements. Endocrinology 145, 508–518.
Val, P., Martinez, A., Sahut-Barnola, I., Jean, C., Veyssière, G., and Lefrançois-Martinez, A.-M. (2002). A 77-base pair LINE-like sequence elicits androgen-dependent mvdp/akr1-b7 expression in mouse vas deferens, but is dispensable for adrenal expression in rats. Endocrinology 143, 3435–3448.
Varady, J., Eder, K., and Ringseis, R. (2011). Dietary oxidized fat activates the oxidative stress-responsive transcription factors NF-κB and Nrf2 in intestinal mucosa of mice. Eur. J. Nutr. 50, 601–609.
Vázquez-Vela, M. E. F., Torres, N., and Tovar, A. R. (2008). White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 39, 715–728.
Volat, F. E., Pointud, J. C., Pastel, E., Morio, B., Sion, B., Hamard, G., Guichardant, M., Colas, R., Lefrançois, A. M., and Martinez, A. (2012). Depressed levels of PGF2α in mice lacking Akr1b7 increase basal adiposity and predispose to diet-induced obesity. Diabetes 61 (in press).
Volle, D. H., Repa, J. J., Mazur, A., Cummins, C. L., Val, P., Henry-Berger, J., Caira, F., Veyssière, G., Mangelsdorf, D. J., and Lobaccaro, J.-M. A. (2004). Regulation of the aldo-keto reductase gene akr1b7 by the nuclear oxysterol receptor LXRalpha (liver X receptor-alpha) in the mouse intestine: putative role of LXRs in lipid detoxification processes. Mol. Endocrinol. 18, 888–898.
Wang, C., Yan, R., Luo, D., Watabe, K., Liao, D.-F., and Cao, D. (2009). Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J. Biol. Chem. 284, 26742–26748.
Wang, N. D., Finegold, M. J., Bradley, A., Ou, C. N., Abdelsayed, S. V., Wilde, M. D., Taylor, L. R., Wilson, D. R., and Darlington, G. J. (1995). Impaired energy homeostasis in C/EBP alpha knockout mice. Science 269, 1108–1112.
Wu, Z., Rosen, E. D., Brun, R., Hauser, S., Adelmant, G., Troy, A. E., McKeon, C., Darlington, G. J., and Spiegelman, B. M. (1999). Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 3, 151–158.
Yamagata, K., Daitoku, H., Shimamoto, Y., Matsuzaki, H., Hirota, K., Ishida, J., and Fukamizu, A. (2004). Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 279, 23158–23165.
Yamaoka, T., Nishimura, C., Yamashita, K., Itakura, M., Yamada, T., Fujimoto, J., and Kokai, Y. (1995). Acute onset of diabetic pathological changes in transgenic mice with human aldose reductase cDNA. Diabetologia 38, 255–261.
Yan, R., Zu, X., Ma, J., Liu, Z., Adeyanju, M., and Cao, D. (2007). Aldo-keto reductase family 1 B10 gene silencing results in growth inhibition of colorectal cancer cells: implication for cancer intervention. Int. J. Cancer 121, 2301–2306.
Yoshitake, H., Takahashi, M., Ishikawa, H., Nojima, M., Iwanari, H., Watanabe, A., Aburatani, H., Yoshida, K., Ishi, K., Takamori, K., Ogawa, H., Hamakubo, T., Kodama, T., and Araki, Y. (2007). Aldo-keto reductase family 1, member B10 in uterine carcinomas: a potential risk factor of recurrence after surgical therapy in cervical cancer. Int. J. Gynecol. Cancer 17, 1300–1306.
Zhang, Y., Lee, F. Y., Barrera, G., Lee, H., Vales, C., Gonzalez, F. J., Willson, T. M., and Edwards, P. A. (2006). Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 103, 1006–1011.
Zhong, L., Liu, Z., Yan, R., Johnson, S., Zhao, Y., Fang, X., and Cao, D. (2009). Aldo–keto reductase family 1 B10 protein detoxifies dietary and lipid-derived alpha, beta-unsaturated carbonyls at physiological levels. Biochem. Biophys. Res. Commun. 387, 245–250.
Keywords: aldose reductases, adipose tissue, prostaglandins, enterohepatic tissue, metabolism
Citation: Pastel E, Pointud J-C, Volat F, Martinez A and Lefrançois-Martinez A-M (2012) Aldo-keto reductases 1B in endocrinology and metabolism. Front. Pharmacol. 3:148. doi: 10.3389/fphar.2012.00148
Received: 15 June 2012; Accepted: 11 July 2012;
Published online: 02 August 2012.
Edited by:
Yi Jin, University of Pennsylvania, USAReviewed by:
Toshiyuki Matsunaga, Gifu Pharmaceutical University, JapanMark Petrash, University of Colorado SOM, USA
Copyright: © 2012 Pastel, Pointud, Volat, Martinez and Lefrançois-Martinez. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Antoine Martinez, CNRS, UMR6293/INSERM U1103, Génétique, Reproduction et Développement, Clermont Université, BP80026 24, Avenue des Landais, 63171 Aubière Cedex, France. e-mail: antoine.martinez@univ-bpclermont.fr