
- Institute of Pharmacology and Clinical Pharmacy, University of Marburg, Marburg, Germany
New concepts on potassium channel function in neuroinflammation suggest that they regulate mechanisms of microglial activation, including intracellular calcium homeostasis, morphological alterations, pro-inflammatory cytokine release, antigen presentation, and phagocytosis. Although little is known about voltage independent potassium channels in microglia, special attention emerges on small (SK/KCNN1-3/KCa2) and intermediate (IK/KCNN4/KCa3.1)-conductance calcium-activated potassium channels as regulators of microglial activation in the field of research on neuroinflammation and neurodegeneration. In particular, recent findings suggested that SK/KCa2 channels, by regulating calcium homeostasis, may elicit a dual mechanism of action with protective properties in neurons and inhibition of inflammatory responses in microglia. Thus, modulating SK/KCa2 channels and calcium signaling may provide novel therapeutic strategies in neurological disorders, where neuronal cell death and inflammatory responses concomitantly contribute to disease progression. Here, we review the particular role of SK/KCa2 channels for [Ca2+]i regulation in microglia and neurons, and we discuss the potential impact for further experimental approaches addressing novel therapeutic strategies in neurological diseases, where neuronal cell death and neuroinflammatory processes are prominent.
Introduction
An increasing number of recent reports on potassium SK/KCa2 and IK/KCa3.1 channels elucidate their multifaceted functions in cell motility, neuronal excitability, neuroprotection, and neuroinflammation (Stocker, 2004; Schlichter et al., 2010; Allen et al., 2011; Dolga et al., 2011). In fact, based on the plethora of functions exerted by SK/KCa2 channels, modulators of these potassium channels have been proposed for therapeutic applications in a wide variety of pathological conditions, including brain tumors, cerebral ischemia, schizophrenia, Parkinson’s and Alzheimer’s diseases, and other neurological diseases where neuroinflammatory mechanisms are considered as major hallmarks (see Table 1; Rupalla et al., 1998; Wyss-Coray and Mucke, 2000; Liégeois et al., 2003; Yan et al., 2003; Stocker, 2004; Judge et al., 2007; Hirsch and Hunot, 2009; Schlichter et al., 2010; Allen et al., 2011; Dolga et al., 2011, 2012; Varas-Lorenzo et al., 2011; Herrik et al., 2012; Kuiper et al., 2012). For example, riluzole, a drug used in patients with amyotrophic lateral sclerosis (ALS) and hereditary cerebellar ataxia, is a potent activator of SK/KCa2 channels (Grunnet et al., 2001; Cao et al., 2002) that reduces neuroinflammation and neuronal excitability (Judge et al., 2007; Waubant, 2007; Ristori et al., 2010; Liu et al., 2012; see Table 1). Recently, derivatives of 2-(phenylamino)benzimidazole and 2-amino benzimidazole were patented as novel SK/KCa2 channel modulators for therapeutic applications in Alzheimer’s disease, anxiety, and ataxia (Sørensen et al., 2006a,b).

Table 1. Targeting KCa2.1/KCa2.2/KCa2.3 channels in CNS disorders – patents and clinical trials.
Potassium Channels in Microglia
Patch-clamp studies of microglial cells showed that these cells express a wide variety of potassium channels. These include inward rectifier K+ (Kir) channels (described in rat, murine, bovine, and human microglia), delayed outwardly rectifying K+ (Kdr) channels (described in rat, mouse, and human microglia), human ether-a-go-go-related gene (HERG) K+ channels (described in rat microglia), G protein-activated K+ channels, ATP-sensitive potassium (KATP) channels, and voltage independent calcium-activated potassium (SK/KCa2) channels (described in murine, bovine, and human microglia; Eder et al., 1997; Eder, 1998). Several studies demonstrated that microglial potassium channel activity promotes neuronal survival and reduces microglial activation and related neuroinflammation (Polazzi and Monti, 2010; Kettenmann et al., 2011). For example, KATP channel activation using diazoxide exerted anti-inflammatory effects in LPS and interferon gamma (IFNγ)-activated mouse primary microglial cells in vitro (Virgili et al., 2011). Diazoxide, a classical KATP channel activator, prevented rotenone-induced rat microglial activation (Zhou et al., 2008), inhibited NO release, tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6) production, and inducible nitric oxide synthase (iNOS) expression in a mouse model for multiple sclerosis in an induced experimental autoimmune encephalomyelitis (Virgili et al., 2011).
KCNN3/SK3/Kca2.3 Channel Function
The expression of all subtypes KCNN1-3/SK1-3/KCa2.1–3 channels have been described in microglial cells (Kaushal et al., 2007; Schlichter et al., 2010; Hayashi et al., 2011). However, among all SK1-3/KCa2.1–3 channel subtypes, the SK3/KCa2.3 channel subtype plays the most critical role in microglial activation. The mRNA of SK3/KCa2.3 channels was detected in mouse and rat microglial cells (Khanna et al., 2001; Dolga et al., 2012) and was found significantly up-regulated after LPS stimulation in rat primary microglial cells (Schlichter et al., 2010). In recent studies, contributions of SK3/KCa2.3 channels to microglial activation processes were elucidated using a pharmacological approach (Schlichter et al., 2010) based on affinities of apamin and tamapin for cloned SK/KCa2 channels (Pedarzani and Stocker, 2008). In these experiments, 100 nM apamin or 5 nM tamapin inhibited the function of SK2/KCa2.2 and SK3/KCa2.3 channels, while the role for SK3/KCa2.3 channels was deduced when the cellular microglial functions were compared with tamapin at concentrations of 250 pM that should not inhibit SK3/KCa2.3 channel function (Schlichter et al., 2010). Using this pharmacological approach, the authors found that inhibition of SK3/KCa2.3 channels reduced the neurotoxic effects of activated rat primary microglia.
In the same study, apamin or tamapin reduced LPS-induced NO release, and decreased the activity of p38-mitogen-activated protein kinase (MAPK) in microglia cells and attenuated tyrosine-nitrated proteins in the neurons exposed to activated microglia. Interestingly, LPS-mediated nuclear factor-kappa B (NF-κB) activation was not affected by low doses of either apamin or tamapin (Schlichter et al., 2010).
Furthermore, it has not yet been determined how SK3/KCa2.3 channel modulation regulates microglial activation pathways; including cytokine release and calcium deregulation and if there are any species-dependent differences. Since the previous studies using small molecules or toxins seem to reveal critical roles of SK3/KCa2.3 channels in microglial activation, studies using SK/KCa2 channel siRNA or knockout animals are required to resolve the limitations of these pharmacological approaches.
Potassium KCNN/SK/Kca2 Channels in Neurons
All subtypes of small-conductance calcium-activated potassium KCNN1-3/SK1-3/KCa2.1–3 channels have been identified in neuronal cells of different species, including mouse, rat, and human neurons (Köhler et al., 1996; Sailer et al., 2004; Dolga et al., 2011). In neuronal cells, SK/KCa2 channels are activated by submicromolar calcium concentrations (KD ∼ 0.5 μM) due to the calcium sensing binding site, a calmodulin (CaM)-binding domain located at the C-terminus of β-subunits (Adelman et al., 2012).
Functional SK/KCa2 channels consist of four subunits, each with six transmembrane segments and cytosolic N and C termini (Stocker, 2004). Upon increasing intracellular calcium concentrations, CaM interaction to CaM-binding domain (CaMBD) alters the geometry of the C-lobe E-F hands from one subunit of SK/KCa2 channels by binding to a neighboring subunit and forming a dimer that promotes a conformational rotation of CaMBD, allowing potassium flow through the channel pore (Xia et al., 1998; Adelman et al., 2012). It is well established that SK/KCa2 channels mediate afterhyperpolarization (AHP) in response to an action potential in excitable cells. Opening of SK/KCa2 channels promotes a negative resting membrane potential or even hyperpolarizing, preventing the onset of a second action potential. Thus, SK/KCa2 channels act as fine tuning regulators of action potential frequencies, neuronal excitability, and [Ca2+]i (Stocker, 2004).
SK/KCa2 channels are responsible for the medium AHP (mAHP), which decays in 200 ms (Bond et al., 2004), while BK channels are responsible for the fast AHP component, which decays in 50 ms (Faber and Sah, 2003). SK/KCa2 channels possess a small unitary conductance (∼10 pS in symmetrical potassium; Köhler et al., 1996) and exhibit an inward rectification. The general knowledge of the inward rectification of SK/KCa2 channels is attributed to the block of outward current by divalent ions, such as Ca2+ and Mg2+ (Lu, 2004), as it is the case for inward rectifier K+ channels (Kir, IRK). However, it was recently shown by inside-out patch-clamp recordings of rat SK2/KCa2.2 channel activity that three negatively charged residues in the sixth transmembrane domain could also be responsible for the SK/KCa2 channel inward rectification (Li and Aldrich, 2011). Importantly, these negatively charged residues affect the Ca2+ affinity for SK/KCa2 channel gating, and the open probability (Po) of the channel in the absence of Ca2+ and this intrinsic inward rectification is largely attributed to a voltage-dependent reduction in outward single-channel conductance (Li and Aldrich, 2011). However, in specific neurons, such as in the rat nucleus basalis, in the rat ventral midbrain, or in preoptic rat hypothalamus, SK/KCa2 channels are responsible for slow “spontaneous miniature” outward currents (Arima et al., 2001; Cui et al., 2004; Klement et al., 2010). The precise function of these outward currents has not yet been elucidated, however, it has been suggested that they may also contribute to spontaneous firing, hyperpolarization, and regulation of burst firing (Adelman et al., 2012).
SK2/Kca2.2 Channel Function
In hippocampal and cortical mouse neurons, SK2/KCa2.2 channel subtypes are localized at dendritic spines, where they are closely associated with NMDA receptors and contribute to AHP, regulate calcium homeostasis and reduce the amplitude of evoked synaptic potential in an NMDA receptor-dependent manner, thereby attenuating neuronal excitability (Faber et al., 2005; Ngo-Anh et al., 2005). Very little has been reported on the regulation of SK2/KCa2.2 channel expression under physiological conditions and in disease. The physiological regulatory functions of SK2/KCa2.2 receptors on NMDAR function and calcium homeostasis become even more relevant under conditions of excitotoxic stress. For example, pharmacological activation of SK2/KCa2.2 receptors attenuates NMDA-mediated intracellular [Ca2+] increases and excitotoxicity in rat primary cortical neurons (Dolga et al., 2011). In contrast, NMDAR-mediated excitotoxicity and cell death is associated with rapid SK2/KCa2.2 down-regulation in both rat and mouse primary neurons (Allen et al., 2011; Dolga et al., 2011), and such reduced SK2/KCa2.2 protein levels resulted in a failure to regulate neuronal excitability. Overall these data point at a pivotal regulative function of SK2/KCa2.2 channels on NMDAR-dependent calcium homeostasis under physiological conditions and suggest a therapeutic potential in excitotoxic conditions.
Interestingly, two responsive elements of NF-κB were found in the intronic promoter region of SK2/KCa2.2 channels, suggesting that NF-κB regulates SK2/KCa2.2 channel gene transcription in rat adrenal medulla PC12 cells (Kye et al., 2007). In activated microglia, NF-κB signaling and the p38 MAPK pathway are strongly activated (Koistinaho and Koistinaho, 2002; Mattson, 2005). In addition, TNF-α production is also augmented following microglial activation (Kettenmann et al., 2011). In mouse primary cortical neurons, TNF-α facilitates activation of NF-κB signaling, which in turn may up-regulate SK2/KCa2.2 channel gene transcription in neurons (Dolga et al., 2008). For example, it is not yet known whether NF-κB activity also modulates SK2/KCa2.2 channel gene transcription in microglial cells. However, in activated rat primary microglial cells, inhibition of SK/KCa2 channels by apamin further decreased inhibitory factor κB (IκB-α) levels (Schlichter et al., 2010). These data suggested that inhibition of SK/KCa2 channels does not affect LPS-induced NF-κB activation in activated rat primary microglial cells. It is not yet resolved whether SK2/KCa2.2 channel function is different in various mammalian species or whether in activated microglia, the persistent increase in [Ca2+]i and TNF-α production is associated with altered SK2/KCa2.2 channel expression and activity.
Neuronal and Microglial Pathways of SK1-3/Kca2.1–3 Channel Activation
Neuronal Pathways
In excitable cells, SK/KCa2 channels are activated by increases in intracellular calcium after release from several different sources: Ca2+ influx from the extracellular space via ionotropic receptors, including NMDARs and nicotinic acetylcholine receptors (nAChRs), Ca2+ influx via voltage-dependent calcium channels (VDCC, Cav), and Ca2+ release from the intracellular calcium stores, such as the endoplasmic reticulum (ER; Oliver et al., 2000; Ngo-Anh et al., 2005). For example, at the dendritic spines of mouse hippocampal neurons, NMDARs are closely associated with SK2/KCa2.2 channels (Ngo-Anh et al., 2005). NMDARs-dependent Ca2+ transients induce excitatory postsynaptic currents that are followed by SK/KCa2 channel activity in the rat brain (Faber et al., 2005). While the slow kinetics of the NMDARs does not provide the major Ca2+ source for SK/KCa2 channel activation in rat hippocampal brain slices (Sabatini et al., 2002), Ca2+ influx through α9-nAChRs is direct and more effective in activating SK/KCa2 channels in the rat basolateral amygdala (Power and Sah, 2008). In the inner and outer hair cells of the rat cochlea, SK/KCa2 channels and nAChRs are functionally coupled and are situated ∼10–20 nm apart (Oliver et al., 2000). SK/KCa2 channels translate Ca2+ influx through nAChRs inducing an inhibitory postsynaptic current that hyperpolarizes the plasma membrane and reduces action potential frequencies (Oliver et al., 2000). Action potentials induce Ca2+ influx via VDCC generating fast, large (larger than micromolar concentrations), and local Ca2+ increases. Activation of SK/KCa2 channels through VDCC channels is largely attributed to their proximity to these channels (Fakler and Adelman, 2008).
Further, inositol 1,4,5-triphosphate (IP3) generation via metabotropic receptor activation is another neuronal Ca2+ source for SK/KCa2 channel activation. IP3 generation in the presence of an action potential further promote Ca2+ release from the intracellular stores, such as ER, that in turn activates SK/KCa2 channels (Power and Sah, 2008). Using whole-cell patch-clamp recordings and high-speed fluorescence imaging it was shown that muscarinic acetylcholine receptors (mAChR) agonists promote large rises in cytosolic calcium via IP3 receptors in the soma and proximal dendrites of neurons in the rat basolateral amygdala. This calcium release was associated with activation of an outward current and hyperpolarization, which resulted from the activation of SK/KCa2 channel (Power and Sah, 2008). The functional relevance of this SK/KCa2 channel activation was demonstrated in rat midbrain dopaminergic neurons, in rat cortical neurons, and rat amygdala, where glutamate and acetylcholine release in response to an action potential mediated a slow inhibitory postsynaptic potential, largely attributed to SK/KCa2 channel activation via calcium release from the IP3 intracellular stores (Fiorillo and Williams, 1998; Morikawa et al., 2000; Seutin et al., 2000; Faber et al., 2005; Gulledge and Stuart, 2005).
Another calcium source for SK/KCa2 channel activation is promoted by P/Q-type Ca2+ channel activity and ryanodine receptor (RyR)-induced Ca2+ release (Adelman et al., 2012). Mutations in P/Q-type Ca2+ channels are associated with human episodic ataxia type 2, irregular pacemaking, and reduced firing precision in mouse Purkinje cells. Reduced activity of P/Q-type Ca2+ channels is attributed to a decreased mAHP as a consequence of reduced activity of postsynaptic P/Q-coupled SK/KCa2 channels. Indeed, in vivo application of the SK/KCa2 channel opener 1-EBIO into the cerebellum significantly improved the motor coordination deficits, dyskinesia, and ataxia in the P/Q-type Ca2+ channel mutant mice (Walter et al., 2006). This particular cross-talk between SK/KCa2 channels and P/Q-type Ca2+ channels is of critical importance in the cerebellum where together with RyR they cooperate to generate and maintain AHP (Kakizawa et al., 2007).
Microglial Pathways
In microglial cells, intracellular calcium signals are modulated by calcium diffusion through membrane ion channels and by active and passive transport through calcium pumps and co-transporters (Kettenmann et al., 2011; Figure 1). Like in all non-excitable cells, Ca2+ signals in microglia are regulated by Ca2+ release mechanisms from the intracellular stores and by extracellular Ca2+ entry into the cytosol through membrane-located store-operated Ca2+ (SOC) channels and ligand-gated channels (Kettenmann et al., 2011). Release of free Ca2+ into the cytosol is mainly attributed to the dynamic release from intracellular stores, such as ER and mitochondria. In the ER, sarcoendoplasmic reticulum Ca2+-ATPases (SERCA) transfer Ca2+ to the lumen of the ER, while the Ca2+ release from ER into the cytoplasm is accomplished by RyRs and IP3-gated calcium channels (Verkhratsky and Kettenmann, 1996; Burdakov et al., 2005; Klegeris et al., 2007).
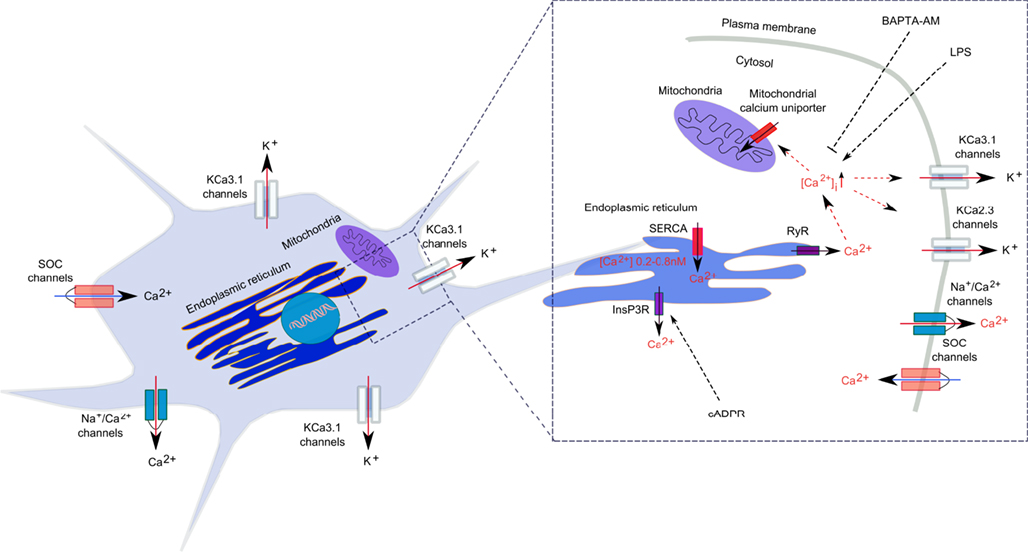
Figure 1. Calcium regulation in microglia. Calcium signal generation is achieved by a well-regulated relationship between Ca2+ release from the intracellular stores and the Ca2+ entry through plasmalemma. In the ER, sarcoendoplasmic reticulum Ca2+-ATPases (SERCA) transfer Ca2+ to the lumen of the ER, while the Ca2+ release from ER into the cytoplasm is accomplished by ryanodine receptors (RyRs) and inositol 1,4,5-triphosphate (IP3)-gated calcium channels. Ca2+ also accumulates in mitochondria through a Ca2+-selective uniporter. Ca2+ extrusion from the cytosol is achieved by a Na+/Ca2+ exchanger.
Although there is no evidence so far for an association between SK/IK/KCa2/KCa3 channels and RyR receptors in microglia, it was demonstrated in neurons of the rat and mouse substantia nigra pars reticulata and the rat medial preoptic nucleus, that RyR-mediated Ca2+ release from intracellular stores activated SK2/KCa2.2 and SK3/KCa2.3 channels, respectively (Yanovsky et al., 2005; Klement et al., 2010). Further, combined electrophysiological, immunohistochemical, and two-photon Ca2+ imaging techniques applied to the rat nucleus reticularis thalami indicated that calcium-induced calcium release (CICR) via RyRs activated plasma membrane SK2/KCa2.2 channels, which together with SERCA pumps and low-voltage-activated Ca2+ channels, shaped rhythmic [Ca2+]i oscillations (Coulon et al., 2009). In rat smooth muscle cells, CICR have a critical implication in the regeneration of the contractile cycle, since Ca2+ release via RyRs facilitates the activation of IK/KCa3.1 channels, which, in turn, mediates smooth muscle cell hyperpolarization and relaxation (Haddock and Hill, 2002). Since both RyR receptors and SK/IK/KCa2/KCa3.1 channels are expressed and functional in microglial cells, research on expression, and function of KCa channels in the ER requires further in depth investigation in order to demonstrate their functional interconnectivity, potential role in the regulation of intracellular calcium homeostasis, and influence on inflammatory responses in activated microglia.
Several studies have reported that inflammatory activation promoted dysbalanced calcium homeostasis in microglia (Hoffmann et al., 2003; Beck et al., 2008; Kettenmann et al., 2011). For example, LPS triggered mouse primary microglial activation, NO, and cytokine release, an increase in [Ca2+]i, and a decrease of calcium signals in response to UTP and complement factor 5a (Hoffmann et al., 2003). The critical role of [Ca2+]i in microglial activation was demonstrated by the intracellular calcium chelator BAPTA-AM that reverted LPS-induced microglial activation and reduced the associated NO and cytokine production in both mouse and rat primary microglia (Hoffmann et al., 2003; Nagano et al., 2006). Extracellular Ca2+ is likely of major importance for microglial activation, since depletion of extracellular Ca2+ or EDTA diminished LPS-induced microglial activation and proliferation in vitro in mouse primary microglia (Dolga et al., 2012). Interestingly, an increase in [Ca2+]i is more a facilitator than a trigger of microglial activation, since, for example, ionomycin increased [Ca2+]i but it did not induce cytokine or NO releases from microglia (Hoffmann et al., 2003). Studies addressing the influence of calcium homeostasis on cell survival pathways demonstrated that extracellular calcium chelation did not trigger microglial cell death, whereas increasing [Ca2+]i with ionomycin or thapsigargin induced apoptotic cell death (Hoffmann et al., 2003; Nagano et al., 2006). Furthermore, in LPS-stimulated microglia, thapsigargin and ionomycin induced necrotic cell death, and these effects were attenuated by lowering [Ca2+]i with BAPTA-AM (Nagano et al., 2006). These data suggest that deregulated [Ca2+]i concentration in activated microglia is critical for cell survival and shifts the mode of cell death from apoptosis to necrosis (Hoffmann et al., 2003; Nagano et al., 2006). Better understanding of the consequences of deregulated intracellular Ca2+ concentration in microglial cells warrants comprehensive investigation for establishing potential therapeutic approaches for inflammation-related CNS disorders.
Protective Role of SK/Kca2 Channels in Neurons and Microglia
Several acute and chronic pathologies, including Alzheimer and Parkinson’s disease, multiple sclerosis, ALS, cerebral ischemia, and cardiovascular disease are endowed with an inflammatory component. The function of SK/KCa2 channel in stroke and Alzheimer’s disease was recently reviewed by Kuiper et al. (2012) and on atherosclerosis and cardiovascular diseases by Wulff and colleagues (Wulff et al., 2007; Köhler et al., 2010). Neurodegenerative diseases triggered by neuronal hyperexcitability, progressive dysregulated Ca2+ homeostasis, and excitotoxic neuronal death could benefit from small molecules that enhance SK/KCa2 channel activity (Kuiper et al., 2012). Other reviews present data on opening of endothelial KCa3.1/KCa2.3 channels, which stimulate endothelium-derived-hyperpolarizing-factor (EDHF)-mediated arteriolar dilation (Brähler et al., 2009) and lower blood pressure (Wulff et al., 2007; Köhler et al., 2010). On the other hand, inhibition of smooth muscle IK/KCa3.1 channels is considered for the treatment of pathological vascular remodeling and sickle cell anemia, since IK/KCa3.1 channel inhibition has particular beneficial effects in restenosis disease, atherosclerosis, and autoimmune encephalomyelitis (Wulff et al., 2007; Köhler et al., 2010).
Further, potassium IK1/SK4/KCa3.1 channels are also under intense investigation because of their high potential in therapeutic drug development for various pathophysiological conditions, including sickle cell anemia, pancreatic cancer, arterial restenosis, immune diseases, and CNS inflammation (Jensen et al., 2002; Köhler et al., 2003; Wulff et al., 2003; Jager et al., 2004). In particular, IK/KCa3.1 channels are highly expressed in murine microglial cells (Khanna et al., 2001) and are implicated in the production of reactive oxygen species, nitric oxide as well as peroxynitrite and protein tyrosine nitration (Skaper, 2011). Increasing evidence for beneficial effects of IK/KCa3.1 channel inhibition is mainly derived from studies using the selective antagonist triarylmethane-34 (TRAM-34), which does not exert any action on KCNN1-3/SK1-3/KCa2.1-KCa2.3 channels (Wulff et al., 2000). Interestingly, TRAM-34 reduces the neurotoxicity mediated by LPS-activated rat primary microglia, and this effect is associated with diminished iNOS expression and reduced caspase 3 activation (Kaushal et al., 2007). TRAM-34 also reduced the degeneration of retinal ganglion cells after optic nerve transection in adult female Sprague Dawley rats (Kaushal et al., 2007). It improved locomotor function, reduced the secretion of TNF-α and interleukin-1β, and diminished the expression of iNOS in a rodent model of spinal cord injury (Bouhy et al., 2011). In a rat model of ischemia/reperfusion stroke, TRAM-34 reduced infarct area by ≈50% as determined by hematoxylin and eosin staining and improved neurological deficits related to the secondary inflammatory damage (Chen et al., 2011).
Neuroinflammation and dysfunction of dopaminergic midbrain neurons have been implicated in the etiology of Parkinson’s disease (Qian et al., 2010; Whitton, 2010). Low-level activity of dopaminergic neurons and alterations of dopamine release have been associated with impairment in voluntary movements, working memory, and reward-based learning (Dunnett and Bjorklund, 1999; Spanagel and Weiss, 1999; Svensson, 2000). Therefore, it is of paramount importance to identify molecular mechanisms underlying dopaminergic activity and neurotransmitter release. During early postnatal development, dopaminergic neurons display anomalous firing patterns and exhibit spontaneous miniature hyperpolarizations, which are not present in adults (Cui et al., 2004). These spontaneous hyperpolarizations exhibit outward currents and depend on SK/KCa2 channel regulation, T-type Ca2+ channel activation, and RyR-dependent Ca2+-induced Ca2+ release in mouse and rat midbrain areas (Wolfart et al., 2001; Cui et al., 2004). In vivo recordings demonstrated that adult rat dopaminergic neurons exhibit either a single-spike pacemaker in a burst firing pattern or show irregular firing patterns (Wilson et al., 1977; Grace and Bunney, 1984), while in vitro recordings in rat dopamine neurons demonstrated a regular, low-frequency pacemaker activity (Grace and Onn, 1989). These discrepancies between in vitro and in vivo recordings reside in a particular type of synaptic activity able to switch dopaminergic neurons into burst firing (Cui et al., 2004).
SK/KCa2 channels are emerging candidates to control NMDAR activation (Ping and Shepard, 1996), GABA receptor-mediating inhibitory activities (Yanovsky et al., 2005) and postsynaptic conductances in dopaminergic neurons. In fact, a subtype of SK/KCa2 channels, SK3/KCa2.3 subtype was shown to be expressed in mouse, rat, and guinea pig dopaminergic neurons (Wolfart et al., 2001; Bosch et al., 2002; Sarpal et al., 2004; Benítez et al., 2011). The function of SK3/KCa2.3 channels was addressed in mouse dopaminergic neurons of the substantia nigra, where they regulate the frequency and precision of pacemaker spiking (Wolfart et al., 2001). However, SK3/KCa2.3 channels were not found in a subpopulation of mouse dopaminergic neurons in the ventral tegmental area (Wolfart et al., 2001), suggesting that these significant differences in the density of apamin-binding sites in dopaminergic neurons might be responsible for the functional differences between ventral tegmental area (A10) and substantia nigra (A9; Grenhoff et al., 1988). Recently, immune-electron microscopy established also the presence of SK2/KCa2.2 channel subtype in mouse dopaminergic neurons (Deignan et al., 2012). SK2/KCa2.2 channels are expressed exclusively in the dendrites of dopaminergic neurons, while SK3/KCa2.3 channels are expressed in the soma and, to a lesser extent, in the dendritic neuronal network. To gain further knowledge of the precise function of SK/KCa2 channels in dopaminergic neurons, experiments performed in subtype specific null mice have demonstrated that SK2/KCa2.2-containing channels are responsible for the precision of action potential timing, while SK3/KCa2.3-containing channels contribute to action potential frequencies (Deignan et al., 2012). Indeed, modulation of SK/KCa2 channels was shown to regulate the excitability of rat midbrain neurons (Ji et al., 2009). Positive SK/KCa2 channel modulation using a non-specific SK/KCa2 channel subtype compound, NS309 (Strøbaek et al., 2004) decreased the responsiveness of rat dopaminergic neurons to depolarizing currents, enhanced spike frequency adaptation, and slowed spontaneous firing, due to an increase in the amplitude and duration of AHP (Ji et al., 2009).
Further, CyPPA, a positive modulator of SK2/KCa2.2 and SK3/KCa2.3 channel subtypes (Hougaard et al., 2007), decreases spontaneous firing rate and increases the duration of the apamin-sensitive AHP, causing an activity-dependent inhibition of current-evoked action potentials in both mouse and rat midbrain slices via SK3/KCa2.3 channel activation (Herrik et al., 2012). Although CyPPA repressed dopamine release in a concentration-dependent manner in vivo systemic administration of CyPPA attenuated methylphenidate-induced hyperactivity and stereotypic behaviors in mice (Herrik et al., 2012). In vitro studies further demonstrated that CyPPA prevented neuronal loss in an AMPA-dependent toxicity model in rat dopaminergic neurons (Benítez et al., 2011). In contrast, negative pharmacological modulation by NS8593 (Strøbaek et al., 2006), a non-specific SK/KCa2 channel subtype compound increased rat dopaminergic excitability, induced an irregular pacemaker and bursting discharge (Ji et al., 2009) and reduced the number of rat dopaminergic neurons in a dose-dependent manner (Benítez et al., 2011).
Taken together, modulation of SK/KCa2 channels in dopaminergic neurons regulates neuronal excitability, survival, and neurotransmitter release, making them suitable candidates for therapeutic intervention in pathological conditions related to dopaminergic dysfunction, such as Parkinson’s disease.
Disturbed Ca2+ homeostasis is one of the major causes of delayed cell death and infarct development after acute brain damage, e.g., after cerebral ischemia. Consequently, reducing intracellular [Ca2+]i levels by blocking NMDA receptors has been widely investigated in experimental and clinical stroke studies. However, inhibitors of NMDA receptors largely failed in clinical studies and, therefore, novel strategies controlling [Ca2+]i homeostasis are warranted for the development of effective therapies. An alternative approach avoiding direct NMDA receptor targeting would be to modulate [Ca2+]i homeostasis indirectly via [Ca2+]i sensors, such as KCa channels. Earlier studies showed that pyramidal cells from the CA3 area of the rat hippocampus respond to chemical-induced ischemia with an initial transient hyperpolarization that is responsible for delayed neuroprotective effects (Tanabe et al., 1999). A potential neuroprotective role for SK/KCa2 channels was identified when apamin-sensitive SK/KCa2 conductance channels were activated and generated a pronounced outward current in response to brief ischemia in rat hippocampal organotypic slice cultures (Tanabe et al., 1999). Indeed, activation of SK2/KCa2.2 channels by NS309 or 1-EBIO positive modulators decreased brain damage area (Allen et al., 2011; Dolga et al., 2011) and also regulated calcium homeostasis (Dolga et al., 2011). In addition, SK2/KCa2.2 channel activation is required for neuroprotection in neurons against glutamate-induced excitotoxicity and also in vivo in a mouse model of middle cerebral artery occlusion (MCAo; Allen et al., 2011; Dolga et al., 2011).
Summary and Perspectives
Neuroinflammation is a critical component for the initiation and progression of several neurodegenerative pathologies, such as cerebral ischemia, schizophrenia, Parkinson’s, and Alzheimer’s diseases. Potential therapeutic approaches directed against both inflammation and neuronal degeneration might benefit from modulation of SK/IK/KCa channels since SK/KCa2 channel activity elicits a dual mechanism of action with direct protective effects in neuronal cells and inhibition of inflammatory activities in microglial cells. However, the role of SK/IK/KCa channels in microglial activity and maintenance needs further in depth investigation in order to reveal the culpable molecular mechanisms. It is of interest to determine whether SK/IK/KCa channels are endowed with similar characteristics among different species, whether their activity alters during aging or during the progression of neurodegeneration, and whether modulating SK/IK/KCa channel activity can limit neurodegeneration and related neuroinflammation. Altogether, the hitherto accumulated evidence exposes modulation of SK/IK/KCa channel functions as potential target in neurodegenerative diseases where inflammatory processes significantly contribute to the progress of neuronal dysfunction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Emma Jane Esser and Goutham K Ganjam for editing of the manuscript. This work was partly supported by a grant of the Alzheimer Forschung Initiative e.V.
References
Adelman, J. P., Maylie, J., and Sah, P. (2012). Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol. 74, 245–269.
Allen, D., Nakayama, S., Kuroiwa, M., Nakano, T., Palmateer, J., Kosaka, Y., et al. (2011). SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J. Cereb. Blood Flow Metab. 31, 2302–2312.
Arima, J., Matsumoto, N., Kishimoto, K., and Akaike, N. (2001). Spontaneous miniature outward currents in mechanically dissociated rat Meynert neurons. J. Physiol. 534, 99–107.
Beck, A., Penner, R., and Fleig, A. (2008). Lipopolysaccharide-induced down-regulation of Ca2+ release-activated Ca2+ currents (I CRAC) but not Ca2+-activated TRPM4-like currents (I CAN) in cultured mouse microglial cells. J. Physiol. 586, 427–439.
Benítez, B. A., Belálcazar, H. M., Anastasía, A., Mamah, D. T., Zorumski, C. F., Mascó, D. H., et al. (2011). Functional reduction of SK3-mediated currents precedes AMPA-receptor-mediated excitotoxicity in dopaminergic neurons. Neuropharmacology 60, 1176–1186.
Bond, C. T., Herson, P. S., Strassmaier, T., Hammond, R., Stackman, R., Maylie, J., et al. (2004). Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J. Neurosci. 24, 5301–5306.
Bosch, M. A., Kelly, M. J., and Rønnekleiv, O. K. (2002). Distribution, neuronal colocalization, and 17beta-E2 modulation of small conductance calcium-activated K(+) channel (SK3) mRNA in the guinea pig brain. Endocrinology 143, 1097–1007.
Bouhy, D., Ghasemlou, N., Lively, S., Redensek, A., Rathore, K. I., Schlichter, L. C., et al. (2011). Inhibition of the Ca2+-dependent K+ channel, KCNN4/KCa3.1, improves tissue protection and locomotor recovery after spinal cord injury. J Neurosci. 31, 16298–16308.
Brähler, S., Kaistha, A., Schmidt, V. J., Wölfle, S. E., Busch, C., Kaistha, B. P., et al. (2009). Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 119, 2323–2332.
Burdakov, D., Petersen, O. H., and Verkhratsky, A. (2005). Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 38, 303–310.
Cao, Y. J., Dreixler, J. C., Couey, J. J., and Houamed, K. M. (2002). Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur. J. Pharmacol. 449, 47–54.
Chen, Y. J., Raman, G., Bodendiek, S., O’Donnell, M. E., and Wulff, H. (2011). The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 31, 2363–2374.
Coulon, P., Herr, D., Kanyshkova, T., Meuth, P., Budde, T., and Pape, H. C. (2009). Burst discharges in neurons of the thalamic reticular nucleus are shaped by calcium-induced calcium release. Cell Calcium 46, 333–346.
Cui, G., Okamoto, T., and Morikawa, H. (2004). Spontaneous opening of T-type Ca2+ channels contributes to the irregular firing of dopamine neurons in neonatal rats. J. Neurosci. 24, 11079–11087.
Deignan, J., Luján, R., Bond, C., Riegel, A., Watanabe, M., Williams, J.T., et al. (2012). SK2 and SK3 expression differentially affect firing frequency and precision in dopamine neurons. Neuroscience 217, 67–76.
Dolga, A. M., Granic, I., Blank, T., Knaus, H. G., Spiess, J., Luiten, P. G., et al. (2008). TNF-alpha-mediates neuroprotection against glutamate-induced excitotoxicity via NF-kappaB-dependent up-regulation of K2.2 channels. J. Neurochem. 107, 1158–1167.
Dolga, A. M., Letsche, T., Gold, M., Doti, N., Bacher, M., Chiamvimonvat, N., et al. (2012). Activation of KCNN3/SK3/K(Ca)2.3 channels attenuates enhanced calcium influx and inflammatory cytokine production in activated microglia. Glia 60, 2050–2064.
Dolga, A. M., Terpolilli, N., Kepura, F., Nijholt, I. M., Knaus, H. G., D’Orsi, B., et al. (2011). KCa2 channels activation prevents [Ca2+]i deregulation and reduces neuronal death following glutamate toxicity and cerebral ischemia. Cell Death Dis. 2, e147.
Dunnett, S. B., and Bjorklund, A. (1999). Prospects for new restorative and neuroprotective treatments in Parkinson’s disease. Nature 399, A32–A39.
Eder, C., Klee, R., and Heinemann, U. (1997). Pharmacological properties of Ca2+-activated K+ currents of ramified murine brain macrophages. Naunyn Schmiedebergs Arch. Pharmacol. 356, 233–239.
Eriksen, B. L., Teuber, L., Hougaard, C., and Sørenson, U. S. (2006). Pyrazolyl-pyrimidines as potassium channel modulating agents and their medical use. Patent WO/2006/100212.
Faber, E. S., Delaney, A. J., and Sah, P. (2005). SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat. Neurosci. 8, 635–644.
Faber, E. S., and Sah, P. (2003). Ca2+-activated K+ (BK) channel inactivation contributes to spike broadening during repetitive firing in the rat lateral amygdala. J. Physiol. 552, 483–497.
Fakler, B., and Adelman, J. P. (2008). Control of KCa channels by calcium nano/microdomains. Neuron 59, 873–881.
Fiorillo, C. D., and Williams, J. T. (1998). Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature 394, 78–82.
Grace, A. A., and Bunney, B. S. (1984). The control of firing pattern in nigral dopamine neurons: burst firing. J. Neurosci. 4, 2877–2890.
Grace, A. A., and Onn, S. P. (1989). Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J. Neurosci. 9, 3463–3481.
Grenhoff, J., Ugedo, L., and Svensson, T. H. (1988). Firing patterns of midbrain dopamine neurons: differences between A9 and A10 cells. Acta Physiol. Scand. 134, 127–132.
Grunnet, M., Jespersen, T., Angelo, K., Frøkjaer-Jensen, C., Klaerke, D. A., Olesen, S. P., et al. (2001). Pharmacological modulation of SK3 channels. Neuropharmacology 40, 879–887.
Gulledge, A. T., and Stuart, G. J. (2005). Cholinergic inhibition of neocortical pyramidal neurons. J. Neurosci. 25, 10308–10320.
Haddock, R. E., and Hill, C. E. (2002). Differential activation of ion channels by inositol 1,4,5-trisphosphate (IP3)- and ryanodine-sensitive calcium stores in rat basilar artery vasomotion. J. Physiol. (Lond.) 545, 615–627.
Hayashi, Y., Kawaji, K., Sun, L., Zhang, X., Koyano, K., Yokoyama, T., et al. (2011). Microglial Ca(2+)-activated K(+) channels are possible molecular targets for the analgesic effects of S-ketamine on neuropathic pain. J. Neurosci. 31, 17370–17382.
Herrik, K. F., Redrobe, J. P., Holst, D., Hougaard, C., Sandager-Nielsen, K., Nielsen, A. N., et al. (2012). CyPPA, a positive SK3/SK2 modulator, reduces activity of dopaminergic neurons, inhibits dopamine release, and counteracts hyperdopaminergic behaviors induced by methylphenidate. Front. Pharmacol. 3:11. doi:10.3389/fphar.2012.00011
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397.
Hoffmann, A., Kann, O., Ohlemeyer, C., Hanisch, U. K., and Kettenmann, H. (2003). Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 23, 4410–4419.
Hougaard, C., Eriksen, B. L., Jørgensen, S., Johansen, T. H., Dyhring, T., Madsen, L. S., et al. (2007). Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br. J. Pharmacol. 151, 655–665.
Jager, H., Dreker, T., Buck, A., Giehl, K., Gress, T., and Grissmer, S. (2004). Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol. Pharmacol. 65, 630–638.
Jensen, B. S., Hertz, M., Christophersen, P., and Madsen, L. S. (2002). The Ca2+-activated K+ channel of intermediate conductance: a possible target for immune suppression. Expert Opin. Ther. Targets 6, 623–636.
Ji, H., Hougaard, C., Herrik, K. F., Strøbaek, D., Christophersen, P., and Shepard, P. D. (2009). Tuning the excitability of midbrain dopamine neurons by modulating the Ca2+ sensitivity of SK channels. Eur. J. Neurosci. 29, 1883–1895.
Judge, S. I., Smith, P. J., Stewart, P. E., and Bever, C. T. Jr. (2007). Potassium channel blockers and openers as CNS neurologic therapeutic agents. Recent Pat. CNS Drug Discov. 2, 200–228.
Kakizawa, S., Kishimoto, Y., Hashimoto, K., Miyazaki, T., Furutani, K., Shimizu, H., et al. (2007). Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 26, 1924–1933.
Kaushal, V., Koeberla, P. D., Wang, Y., and Schlichter, L. C. (2007). The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J. Neurosci. 27, 234–244.
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553.
Khanna, R., Roy, L., Zhu, X., and Schlichter, L. C. (2001). K+ channels and the microglial respiratory burst. Am. J. Physiol. Cell Physiol. 280, C796–C806.
Klegeris, A., Choi, H. B., McLarnon, J. G., and McGeer, P. L. (2007). Functional ryanodine receptors are expressed by human microglia and THP-1 cells: their possible involvement in modulation of neurotoxicity. J. Neurosci. Res. 85, 2207–2215.
Klement, G., Druzin, M., Haage, D., Malinina, E., Arhem, P., and Johansson, S. (2010). Spontaneous ryanodine-receptor-dependent Ca2+-activated K+ currents and hyperpolarizations in rat medial preoptic neurons. J. Neurophysiol. 103, 2900–2911.
Köhler, M., Hirschberg, B., Bond, C. T., Kinzie, J. M., Marrion, N. V., Maylie, J., et al. (1996). Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273, 1709–1714.
Köhler, R., Kaistha, B. P., and Wulff, H. (2010). Vascular KCa-channels as therapeutic targets in hypertension and restenosis disease. Expert Opin. Ther. Targets 14, 143–155.
Köhler, R., Wulff, H., Eichler, I., Kneifel, M., Neumann, D., Knorr, A., et al. (2003). Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation 108, 1119–1125.
Koistinaho, M., and Koistinaho, J. (2002). Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia 40, 175–183.
Kuiper, E. F., Nelemans, A., Luiten, P., Nijholt, I., Dolga, A., and Eisel, U. (2012). K(Ca)2 and K(Ca)3 channels in learning and memory processes, and neurodegeneration. Front. Pharmacol. 3, 107.
Kye, M. J., Spiess, J., and Blank, T. (2007). Transcriptional regulation of intronic calcium-activated potassium channel SK2 promoters by nuclear factor-kappa B and glucocorticoids. Mol. Cell. Biochem. 300, 9–17.
Li, W., and Aldrich, R. W. (2011). Electrostatic influences of charged inner pore residues on the conductance and gating of small conductance Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. U.S.A. 108, 5946–5953.
Liégeois, J. F., Mercier, F., Graulich, A., Graulich-Lorge, F., Scuvée-Moreau, J., and Seutin, V. (2003). Modulation of small conductance calcium-activated potassium (SK) channels: a new challenge in medicinal chemistry. Curr. Med. Chem. 10, 625–647.
Liu, B. S., Ferreira, R., Lively, S., and Schlichter, L. C. (2012). Microglial SK3 and SK4 currents and activation state are modulated by the neuroprotective drug, riluzole. J. Neuroimmune Pharmacol. [Epub ahead of print].
Lu, Z. (2004). Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 66, 103–129.
Mattson, M. P. (2005). NF-kappaB in the survival and plasticity of neurons. Neurochem. Res. 30, 883–893.
Morikawa, H., Imani, F., Khodakhah, K., and Williams, J. T. (2000). Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J. Neurosci. 20, RC103.
Nagano, T., Kimura, S. H., Takai, E., Matsuda, T., and Takemura, M. (2006). Lipopolysaccharide sensitizes microglia toward Ca(2+)-induced cell death: mode of cell death shifts from apoptosis to necrosis. Glia 53, 67–73.
Ngo-Anh, T. J., Bloodgood, B. L., Lin, M., Sabatini, B. L., Maylie, J., and Adelman, J. P. (2005). SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 8, 642–649.
Oliver, D., Klocker, N., Schuck, J., Baukrowitz, T., Ruppersberg, J. P., and Fakler, B. (2000). Gating of Ca2+-activated K+ channels controls fast inhibitory synaptic transmission at auditory outer hair cells. Neuron 26, 595–601.
Pedarzani, P., and Stocker, M. (2008). Molecular and cellular basis of small- and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell. Mol. Life Sci. 65, 3196–3217.
Ping, H. X., and Shepard, P. D. (1996). Apamin-sensitive Ca2+-activated K+ channels regulate pacemaker activity in nigral dopamine neurons. Neuroreport 7, 809–814.
Polazzi, E., and Monti, B. (2010). Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog. Neurobiol. 92, 293–315.
Power, J. M., and Sah, P. (2008). Competition between calcium-activated K+ channels determines cholinergic action on firing properties of basolateral amygdala projection neurons. J. Neurosci. 28, 3209–3220.
Qian, L., Flood, P. M., and Hong, J. S. (2010). Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J. Neural Transm. 117, 971–979.
Ristori, G., Romano, S., Visconti, A., Cannoni, S., Spadaro, M., Frontali, M., et al. (2010). Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology 74, 839–845.
Rupalla, K., Allegrini, P. R., Sauer, D., and Wiessner, C. (1998). Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 96, 172–178.
Sabatini, B. L., Oertner, T. G., and Svoboda, K. (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33, 439–452.
Sailer, C. A., Kaufmann, W. A., Marksteiner, J., and Knaus, H. G. (2004). Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol. Cell. Neurosci. 26, 458–469.
Sarpal, D., Koenig, J. I., Adelman, J. P., Brady, D., Prendeville, L. C., and Shepard, P. D. (2004). Regional distribution of SK3 mRNA-containing neurons in the adult and adolescent rat ventral midbrain and their relationship to dopamine-containing cells. Synapse 53, 104–113.
Schlichter, L. C., Kaushal, V., Moxon-Emre, I., Sivagnanam, V., and Vincent, C. (2010). The Ca2+ activated SK3 channel is expressed in microglia in the rat striatum and contributes to microglia-mediated neurotoxicity in vitro. J. Neuroinflammation 7, 4.
Seutin, V., Mkahli, F., Massotte, L., and Dresse, A. (2000). Calcium release from internal stores is required for the generation of spontaneous hyperpolarizations in dopaminergic neurons of neonatal rats. J. Neurophysiol. 83, 192–197.
Skaper, S. D. (2011). Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol. Disord. Drug Targets 10, 44–56.
Sørensen, U. S., Teuber, L., Peters, D., Stroebaek, D., Johansen, T. H., Nielsen, K. S., et al. (2006a). Novel 2-amino benzimidalzole derivatives and their use as modulators of small conductance calcium-activated potassium channels. Patent WO/2006/074991.
Sørensen, U. S., Teuber, L., Peters, D., Stroebaek, D., Johansen, T. H., Nielsen, K. S., et al. (2006b). 2-(phenylamino)benzimidalzole derivatives and their use as modulators of small conductance calcium-activated potassium channels. Patent WO/2006/136580.
Spanagel, R., and Weiss, F. (1999). The dopamine hypothesis of reward: past and current status. Trends Neurosci. 22, 521–527.
Stocker, M. (2004). Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5, 758–770.
Strøbaek, D., Hougaard, C., Johansen, T. H., Sørensen, U. S., Nielsen, E. Ø., Nielsen, K. S., et al. (2006). Inhibitory gating modulation of small conductance Ca2+-activated K+ channels by the synthetic compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyper-polarizing current in hippocampal CA1 neurons. Mol. Pharmacol. 70, 1771–1782.
Strøbaek, D., Teuber, L., Jørgensen, T. D., Ahring, P. K., Kjaer, K., Hansen, R. S., et al. (2004). Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim. Biophys. Acta 1665, 1–5.
Svensson, T. H. (2000). Dysfunctional brain dopamine systems induced by psychotomimetic NMDA-receptor antagonists and the effects of antipsychotic drugs. Brain Res. Brain Res. Rev. 31, 320–329.
Tanabe, M., Mori, M., Gähwiler, B. H., and Gerber, U. (1999). Apamin-sensitive conductance mediates the K(+) current response during chemical ischemia in CA3 pyramidal cells. J. Neurophysiol. 82, 2876–2882.
Varas-Lorenzo, C., Riera-Guardia, N., Calingaert, B., Castellsague, J., Pariente, A., Scotti, L., et al. (2011). Stroke risk and NSAIDs: a systematic review of observational studies. Pharmacoepidemiol. Drug Saf. 20, 1225–1236.
Verkhratsky, A., and Kettenmann, H. (1996). Calcium signalling in glial cells. Trends Neurosci. 19, 346–352.
Virgili, N., Espinosa-Parrilla, J. F., Mancera, P., Pastén-Zamorano, A., Gimeno-Bayon, J., Rodríguez, M. J., et al. (2011). Oral administration of the KATP channel opener diazoxide ameliorates disease progression in a murine model of multiple sclerosis. J. Neuroinflammation 8, 149.
Walter, J. T., Alvina, K., Womack, M. D., Chevez, C., and Khodakhah, K. (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 9, 389–397.
Wang, X., Fulp, A. B., van Rhee, A. M., and Spear, K. L. (2003). Bis-benzimidazoles and related compounds as potassium channel modulators. Patent WO/2003/094861.
Waubant, E. (2007). Neuroprotection with riluzole in patients with early multiple sclerosis. Patent NCT00501943.
Whitton, P. S. (2010). Neuroinflammation and the prospects for anti-inflammatory treatment of Parkinson’s disease. Curr. Opin. Investig. Drugs 11, 788–794.
Wilson, C. J., Young, S. J., and Groves, P. M. (1977). Statistical properties of neuronal spike trains in the substantia nigra: cell types and their interactions. Brain Res. 136, 243–260.
Wolfart, J., Neuhoff, H., Franz, O., and Roeper, J. (2001). Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J. Neurosci. 21, 3443–3456.
Wulff, H., Calabresi, P. A., Allie, R., Yun, S., Pennington, M., Beeton, C., et al. (2003). The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J. Clin. Invest. 111, 1703–1713.
Wulff, H., Kolski-Andreaco, A., Sankaranarayanan, A., Sabatier, J. M., and Shakkottai, V. (2007). Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem. 14, 1437–1457.
Wulff, H., Miller, M. J., Hansel, W., Grissmer, S., Cahalan, M. D., and Chandy, K. G. (2000). Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. U.S.A. 97, 8151–8156.
Wyss-Coray, T., and Mucke, L. (2000). Ibuprofen, inflammation and Alzheimer’s disease. Nat. Med. 6, 973–974.
Xia, X. M., Fakler, B., Rivard, A., Wayman, G., Johnson-Pais, T., Keen, J. E., et al. (1998). Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507.
Yan, Q., Zhang, J., Liu, H., Babu-Khan, S., Vassar, R., Biere, A. L., et al. (2003). Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J. Neurosci. 23, 7504–7509.
Yanovsky, Y., Zhang, W., and Misgeld, U. (2005). Two pathways for the activation of small-conductance potassium channels in neurons of substantia nigra pars reticulata. Neuroscience 136, 1027–1036.
Keywords: calcium regulation, KCa2/SK channels, KCa3/IK channels, microglia, neuroprotection
Citation: Dolga AM and Culmsee C (2012) Protective roles for potassium SK/KCa2 channels in microglia and neurons. Front. Pharmacol. 3:196. doi: 10.3389/fphar.2012.00196
Received: 09 March 2012; Accepted: 01 November 2012;
Published online: 26 November 2012.
Edited by:
Nick Andrews, Harvard Medical School, USAReviewed by:
Marianthi Papakosta, Grunenthal GmbH, GermanyChristian Von Hehn, Children’s Hospital Boston, USA
Copyright: © 2012 Dolga and Culmsee. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Amalia M. Dolga, Fachbereich Pharmazie, Institut für Pharmakologie und Klinische Pharmazie, Philipps-Universität Marburg, Karl-von-Frisch-Straße 1, 35032 Marburg, Germany. e-mail: dolga@staff.uni-marburg.de