 S. Sokolov1
S. Sokolov1- 1Molecular Cardiac Physiology Group, Department of Biomedical Physiology and Kinesiology, Simon Fraser University, Burnaby, BC, Canada
- 2Department of Biology, Cardiovascular Therapeutic Area, Gilead Sciences, Inc., Fremont, CA, USA
Ranolazine is clinically approved for treatment of angina pectoris and is a potential candidate for antiarrhythmic, antiepileptic, and analgesic applications. These therapeutic effects of ranolazine hinge on its ability to inhibit persistent or late Na+ currents in a variety of voltage-gated sodium channels. Extracellular acidosis, typical of ischemic events, may alter the efficiency of drug/channel interactions. In this study, we examined pH modulation of ranolazine's interaction with the cardiac sodium channel, Nav1.5. We performed whole-cell path clamp experiments at extracellular pH 7.4 and 6.0 on Nav1.5 transiently expressed in HEK293 cell line. Consistent with previous studies, we found that ranolazine induced a stable conformational state in the cardiac sodium channel with onset/recovery kinetics and voltage-dependence resembling intrinsic slow inactivation. This interaction diminished the availability of the channels in a voltage- and use-dependent manner. Low extracellular pH impaired inactivation states leading to an increase in late Na+ currents. Ranolazine interaction with the channel was also slowed 4–5 fold. However, ranolazine restored the voltage-dependent steady-state availability profile, thereby reducing window/persistent currents at pH 6.0 in a manner comparable to pH 7.4. These results suggest that ranolazine is effective at therapeutically relevant concentrations (10 μM), in acidic extracellular pH, where it compensates for impaired native slow inactivation.
Introduction
Ranolazine is a piperazine derivative that was clinically approved by the FDA in 2006 for treatment of angina pectoris. It decreases late sodium currents (INa) through cardiac sodium channel, Nav1.5, and thereby reduces calcium influx through the sodium-calcium exchanger NCX during reverse mode activity (Sossalla et al., 2008). Recently, more potential applications for ranolazine have been explored and reported. Ranolazine demonstrated antiarrhythmic properties in both atria and ventricles (Wu et al., 2004; Undrovinas et al., 2006; Burashnikov et al., 2007; Dobrev and Nattel, 2010). The proposed basic mechanism underlying ranolazine's antiarrythmic action in ventricles is inhibition of late INa (Wasserstrom et al., 2009; Undrovinas et al., 2010). In the atria, use-dependent inhibition of peak INa as well as IKr is thought to play an important role in treatment of atrial fibrillation, in addition to suppression of late INa (Burashnikov et al., 2007; Sossalla et al., 2010). Additionally, ranolazine displayed cardioprotection during ischemia (Hale et al., 2006, 2008; Stone et al., 2010), and effectively diminished late INa in Long QT syndrome type 3 mutations (Fredj et al., 2006; Moss et al., 2008; Kahlig et al., 2010; Huang et al., 2011).
Ranolazine may also be effective in non-cardiac tissue. It blocks the skeletal muscle sodium channel, Nav1.4, (Wang et al., 2008) and shows increased potency for paramyotonia congenita mutants (El-Bizri et al., 2011). Ranolazine inhibits neuronal channels, Nav1.7 (Rajamani et al., 2008; Wang et al., 2008) and Nav1.1, (Kahlig et al., 2010) and shows potent reduction of persistent currents in GEFS+, SMEI, and FHM3 mutants (Kahlig et al., 2010). Ranolazine also decreases cell excitability of dorsal root ganglion neurons (Estacion et al., 2010; Hirakawa et al., 2012), thus demonstrating an analgesic utility in the treatment of neuropathic pain (Gould et al., 2009). We previously reported the effects of ranolazine on the brain isoform, Nav1.2 at normal and acidic extracellular pH (Peters et al., 2013).
Similar to local anesthetic drugs, ranolazine's action on voltage-gated ion channels (including some Ca2+ and K+ channels, see Antzelevitch et al., 2011, for review) involves use-dependent as well as tonic block. However, therapeutic benefits of ranolazine for cardioprotection in ischemic conditions are attributed to its distinctive ability to diminish late sodium current in a variety of channels (Belardinelli et al., 2006). Ranolazine's IC50 for peak INa in Nav1.5 is reported to be several hundred μM, while late Na+ current is inhibited in the 7–10 μM range (Rajamani et al., 2009; Antzelevitch et al., 2011), close to therapeutically achieved plasma concentrations (2 ÷ 6 μM, Chaitman, 2006; 5.8 μM, Antzelevitch et al., 2011). Thus, while not exhibiting prominent isoform specificity, ranolazine possesses an intriguing type of functional specificity.
In the present study we examined effects of ranolazine on cardiac sodium channel Nav1.5 transiently expressed in HEK293 cells as well as the effects of extracellular acidification to pH 6.0, typically exhibited during myocardial ischemia (Maruki et al., 1993), on drug channel interaction. We suggest the capacity of ranolazine to inhibit late INa at therapeutically relevant concentrations (10 μM) may be related to its ability to potentiate slow inactivation. Ranolazine shifts the midpoint of the steady-state availability curve to more hyperpolarized voltages, accelerates entry into, and slows recovery from, a conformational state that shares kinetic- and voltage-dependent profiles of Nav channel slow inactivation. Extracellular acidification impairs inactivation states of Nav1.5, including slow inactivation, leading to an increase in persistent current. Ranolazine remedies this deficiency, restoring inactivation over a time course of seconds to tens of seconds.
Methods
Expression of hNav1.5 in HEK293 Cells
The human variant of the cardiac sodium channel, hNav1.5, was in the pRC-CMV vector (A.L. George, Vanderbilt University, Nashville, TN). Rat β1 subunit was in the pBK/CMV vector. HEK293 cells were cultured using media comprising DMEM (Gibco), FBS 20% (Gibco), and 10,000 U penicillin/streptomycin solution (Sigma). Cells were transiently transfected (Polyfect, Qiagen) with the hNav1.5 α-subunit, β1 subunit, and enhanced Green Fluorescent Protein (pEGFP, graciously provided by Brett Adams, Utah State University, Logan, UT) to identify channel expression. Fluorescing cells were used for recording 24–36 h after transfection.
Materials
Ranolazine was obtained from Gilead Sciences (Foster City, CA) in powder form, diluted to 100 mM stock in 0.1 M HCl, aliquoted at 10 mM and stored at −20°C. Working concentrations of 10 or 100 μM were freshly prepared in bath solution. pH was readjusted before performing electrophysiological experiments.
Electrophysiology
Ionic currents were measured with whole-cell patch clamp using an EPC-9 amplifier, an ITC-16 interface, and an iMac running Patchmaster (HEKA, Lambrecht, Germany). Data were sampled at 50 kHz and low pass filtered at 10 kHz. Pipettes were made from borosilicate glass (Sutter Instruments, Novato, CA), coated with dental wax, and fire polished to a resistance of 1–1.5 MΩ. Series resistance, Rs, was typically 3 MΩ or less, Rs greater than 3 MΩ was compensated by 60–75%. Only cells with a seal resistance of 1 GΩ or greater were used. All measurements were conducted at room temperature (22°C).
The pipette solution contained (in mM): 130 CsF, 10 NaCl, 10 EGTA, and 10 HEPES adjusted to pH 7.4 with CsOH. The extracellular saline contained (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4) or MES (pH 6.0). pH was adjusted with CsOH.
A −130 mV holding potential was used for most voltage protocols except 10 and 25 Hz use-dependent trains, where an intermittent −100 mV resting potential was used between test pulses. We recognize −130 mV is more negative than the physiological resting potential in cardiac myocytes of −80 mV and the pathological resting potential of −65 mV during ischemia and we thus may miss some physiologically relevant information. However, the nature of our measurements require the channels to be in a “ground” state in which they are all closed and fully recovered from inactivation. All test pulses were to −10 mV. Cells were perfused at the holding potential for 5 min after whole-cell configuration was achieved before recordings started to allow dialysis of internal solution and stabilization of current amplitude. In matched pair experiments (Figure 7: ramps; Figure 9: tonic block and steady-state slow inactivation) control data were obtained first, followed by perfusion of cells for 5 min with either 10 or 100 μM ranolazine while holding at −130 mV. Voltage protocols were then repeated in the presence of ranolazine. This procedure minimized run down and gradual deterioration of the seal during prolonged recordings. A −P/4 leak subtraction was used for most protocols except use-dependent trains, ramps, and slow inactivation kinetics protocols. Details of voltage pulse protocols are given in the figure legends. Fit parameters are reported in Tables A1–A9 (Appendix).
Student's t-tests with two-tailed p-values were used for statistical analysis using the Instat software package (GraphPad Software, San Diego, CA). Results are presented as means ± SEM unless otherwise stated. Statistical significance is assumed to be p < 0.05 if not specifically given.
Results
Activation and Fast Inactivation
We examined the effects of extracellularly applied ranolazine on Nav1.5, transiently co-expressed in HEK293 cells along with the β1 subunit and eGFP marker. We studied ranolazine action in two different extracellular pH conditions: pH 7.4 (Figures 1A,B) corresponding to normal physiological conditions and pH 6.0 (Figures 1C,D) that approximates extracellular pH during ischemic cardiac events.
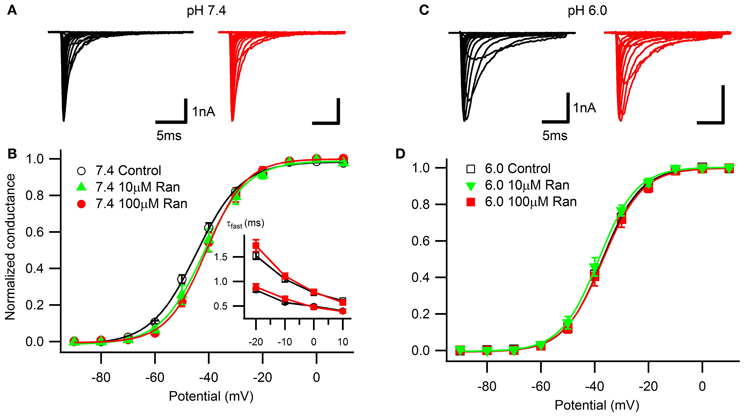
Figure 1. Voltage-dependence of activation and open-state inactivation time constants. Representative currents in response to 20 ms depolarizations in 10 mV steps from −130 mV at extracellular pH 7.4 (A) and 6.0 (C) in drug-free conditions (black) and with 100 μM ranolazine (red). (B) Conductance-voltage relationships at pH 7.4 in control (open black circles), 10 μM ranolazine (solid green triangles), and 100 μM ranolazine (solid red circles). (D) Conductance-voltage relationships at pH 6.0 in control (open black squares), 10 μM ranolazine (solid green triangles), and 100 μM ranolazine (solid red squares). Fit parameters are summarized in Table A1. (B, inset) Time constants of open-state inactivation were obtained by fitting current decay with a single exponential function in control (open black symbols) and with 100 μM ranolazine (solid red symbols) at pH 7.4 (circles) and 6.0 (squares). Time constants are reported in Table A2.
Consistent with our previous studies (Jones et al., 2011; Vilin et al., 2012) and others (Murphy et al., 2011), we observed a ~7 mV depolarizing shift in the voltage-dependence of activation at pH 6.0 (Figure 1; Table A1). At pH 7.4, the presence of 10 or 100 μM ranolazine in the extracellular solution caused a small but statistically significant depolarizing shift of the activation curves. Ranolazine did not significantly affect the V1/2 of activation at pH 6.0 compared to Ranolazine at pH 7.4.
Open-state fast inactivation kinetics were quantified by fitting current decay with an exponential function from test pulses between −20 and +10 mV (Figure 1B, inset; Table A2). Acidic pH significantly slowed channel fast inactivation. 100 μM ranolazine did not significantly affect fast inactivation time constants at either pH.
Steady-state fast inactivation was assessed after 500 ms conditioning pulses to voltages from −130 to +10 mV (Figure 2). Acidic pH caused a ~10 mV depolarizing shift of the V1/2 with no effect on the apparent valence (Table A3). 100 μM ranolazine did not significantly shift the V1/2 or apparent valence at either pH.
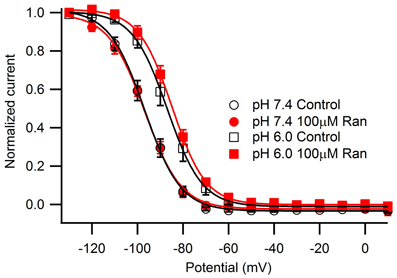
Figure 2. Steady-state fast inactivation induced by 500 ms conditioning pulses ranging from −130 mV to +10 mV in 10 mV intervals and measured by a test pulse to −10 mV. Channels were held at −130 mV for 10 s between sweeps. pH 7.4 control (open black circles) differed significantly from pH 6.0 control (open black squares). 100 μM ranolazine had no effect on steady-state fast inactivation (pH 7.4, solid red circles; pH 6.0, solid red squares). Fit parameters are summarized in Table A3.
We next studied the kinetics of fast inactivation recovery after a 500 ms conditioning pulse to −10 mV measured by 20 ms test pulses to −10 mV applied at various intervals (Figure 3). Consistent with previous studies (Jones et al., 2011; Vilin et al., 2012), we detected accelerated fast inactivation recovery kinetics at pH 6.0 (Table A4). Interestingly, 100 μM ranolazine induced a profound slow component of recovery at pH 7.4, but not at pH 6.0 (Figures 3A,B).
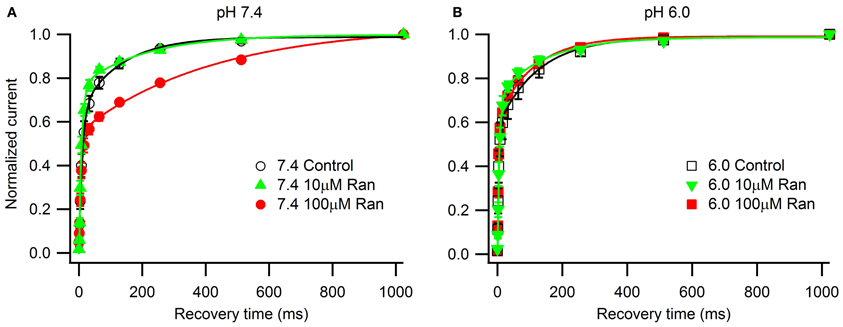
Figure 3. Fast inactivation recovery at −130 mV. Fast inactivation was induced by a 500 ms conditioning pulse to −10 mV. Cells were then hyperpolarized to −130 mV for 0–1000 ms and current measured with a −10 mV test pulse was plotted as a function of recovery pulse duration and fitted with a double exponential function. (A) pH 7.4 in control (open black circles), 10 μM ranolazine (solid green triangles), and 100 μM ranolazine (solid red circles). (B) pH 6.0 in control (open black squares), 10 μM ranolazine (solid green triangles), and 100 μM ranolazine (solid red squares). Fit parameters are in Table A4.
Use-Dependent Inhibition by Ranolazine
We next examined the extracellular pH effects on use-dependent channel inactivation and block by ranolazine. At first, we used high frequency depolarization trains (10 and 25 Hz, Figure 4) that many studies employ to assess use-dependent block by ranolazine, local anesthetics, and other compounds with state-dependent action (Fredj et al., 2006; Kahlig et al., 2010; El-Bizri et al., 2011; Huang et al., 2011; Hirakawa et al., 2012).
Figure 4. Use-dependent inactivation induced by one hundred 5 ms pulses to −10 mV applied at 10 Hz (A,C) or 25 Hz (B,D) from holding potential −100 mV at extracellular pH 7.4 (A,B) and 6.0 (C,D). Error bars removed for clarity. Use-dependence in presence of 100 μM ranolazine (red symbols) was fit with single exponential function (solid lines). Fit parameters are reported in Table A5.
In the presence of 100 μM ranolazine at pH 7.4, a series of 100 pulses produced 67 ± 1% peak current inhibition at 10 Hz and 74 ± 3% at 25 Hz (Table A5). At pH 6.0 the degree of use-dependent inhibition by ranolazine was significantly reduced at both pulsing frequencies. This result correlates with our observation of drug-induced slow recovery component at pH 7.4, but not at pH 6.0 (Figure 3). Use-dependent inhibition developed 4–5 fold slower at pH 6.0 than at pH 7.4, and subsequently did not reach steady-state equilibrium within 100 pulses.
To assess the pH-dependence of ranolazine effect at more physiologically relevant conditions, we applied trains of 500 pulses 300 ms in length at 1 Hz, roughly corresponding to cardiac action potential frequency and duration (Figure 5). pH 6.0 significantly slowed inhibition kinetics by 100 μM ranolazine (Figure 5, Table A6). Interestingly, use-dependent block by 100 μM ranolazine reached similar magnitudes at pH 7.4 and 6.0 by the 500th pulse (pH 7.4: 51 ± 4%; pH 6.0: 50 ± 4%).
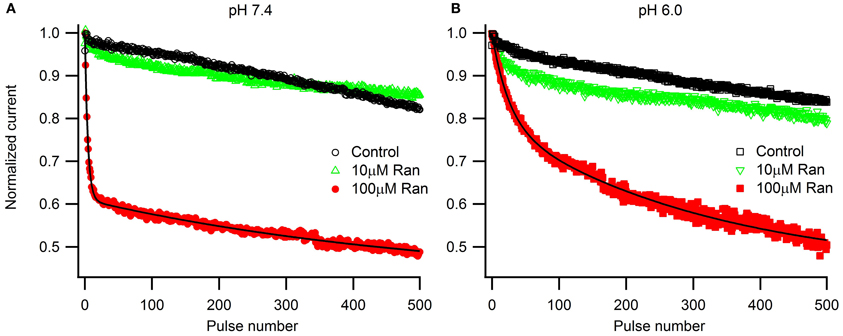
Figure 5. Use-dependent inactivation by trains of five hundred 300 ms pulses to −10 mV applied at 1 Hz from holding potential −130 mV at extracellular pH 7.4 (A) and 6.0 (B). Error bars removed for clarity. Data in 100 μM ranolazine (red symbols) was fit with double exponential functions (solid lines). Fit parameters are summarized in Table A6.
Ranolazine Effects on Late Sodium Current and Window Current
The clinical benefits of ranolazine are predominantly ascribed to its ability to block late or window Na+ currents (Reddy et al., 2010; Antzelevitch et al., 2011). We measured late sodium current (late INa) with 50 ms depolarizations to −10 mV from a holding potential of −130 mV (Figure 6). Fifty sweeps were averaged to improve the signal to noise ratio. In line with previous studies (Magyar et al., 2004; Huang et al., 2011) we found that Nav1.5 channels exhibit a very small late INa at pH 7.4 (0.94 ± 0.34% of peak current amplitude). In the presence of 100 μM ranolazine, we observed a small reduction of late INa (0.73 ± 0.17%). This effect was, however, not statistically significant (p > 0.05).
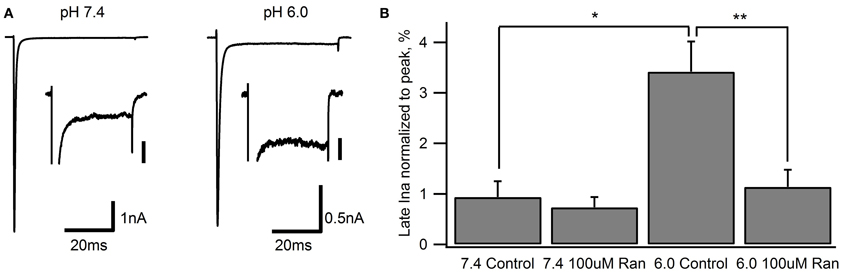
Figure 6. Late Na+ current during 50 ms depolarization to −10 mV from a −130 mV holding potential. Representative traces from recordings at extracellular pH 7.4 and 6.0 are shown in (A) Inset scale bars are 40 pA. (B) Late INa measured at 45–50 ms normalized to peak current in drug-free conditions (pH 7.4: 0.94 ± 0.34%, n = 7; pH 6.0: 3.4 ± 0.6%, n = 7) and with 100 μM ranolazine (pH 7.4: 0.73 ± 0.17%, n = 10; pH 6.0: 1.1 ± 0.4%, n = 8). Statistically significant differences are indicated with asterisks.
Extracellular pH 6.0 substantially reduces Nav channel peak current amplitude (Jones et al., 2011; Murphy et al., 2011; Vilin et al., 2012). When normalized to peak current, late INa was significantly increased to 3.4 ± 0.6% at pH 6.0 (Figure 6). Addition of 100 μM ranolazine to the extracellular solution significantly reduced the relative late INa to 1.1 ± 0.4% (p < 0.01).
Window currents arise due to partial activation and incomplete inactivation of voltage-gated ion channels at intermediate membrane potentials. To examine the effects of ranolazine on window currents, we applied 0.3 mV/ms ramps before and after perfusion of 100 μM of the drug (Figure 7). Fifty sweeps were averaged to improve the signal to noise ratio (Figure 7A). In these matched recordings, 100 μM ranolazine significantly reduced inward sodium influx (pH 7.4: 44 ± 7%; pH 6.0: 31 ± 6%). The difference between reduction of window currents at pH 7.4 and 6.0 was not statistically significant. The effect of 10 μM ranolazine was significant at pH 7.4 (23 ± 3%, p = 0.02), but not at pH 6.0 (10 ± 4%, p > 0.05).
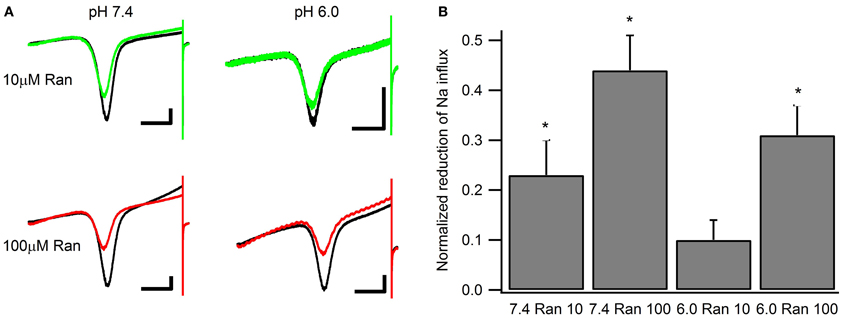
Figure 7. Window currents measured with 500 ms ramps (0.3 mV/ms) from −130 to +20 mV. (A) Representative matched pairs before and after perfusion of either 10 μM ranolazine (top) or 100 μM ranolazine (bottom). Scale bars are 100 ms and 50 pA. (B) Total charge flow was calculated by integrating current traces after subtracting the linear leak component. Charge for every cell was then normalized to that in the absence of ranolazine and plotted as a function of drug and pH. Statistically significant reductions in charge influx are indicated with asterisks.
Effects of Ranolazine on Slow Inactivation
We studied the effects of acidic pH on ranolazine modulation of slow inactivation. The kinetics of onset and recovery from slow inactivation states and ranolazine block were assessed using a triple pulse protocol. Cells were given conditioning pulses to −10 mV varying in length from 500 ms to 64 s, followed by a −130 mV recovery pulse. Available current was measured with 5 ms test pulses to −10 mV applied at 20, 100, 500 ms, 2, 5, 15, 30 and 60 s after the end of the conditioning pulse (Figure 8). Here we report data from the 100 ms and 2 s recovery intervals (Figures 8A–D). These recovery periods allowed us to concurrently assess the slow inactivation onset and the onset of ranolazine block.
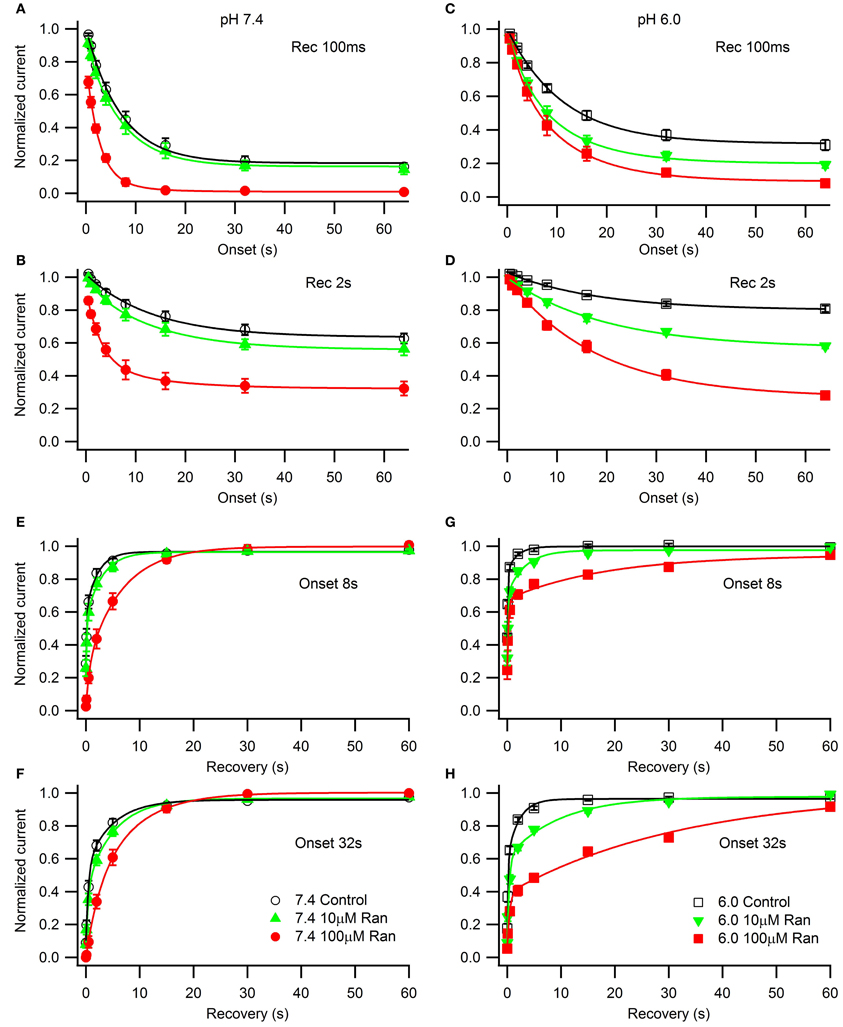
Figure 8. Slow inactivation onset and recovery and effects of ranolazine at pH 7.4 (left) and 6.0 (right). Onset of slow inactivation (A–D) was induced by conditioning pulses to −10 mV ranging from 500 ms to 64 s. Amount of slow-inactivated channels (control, open black symbols) or a combination of slow-inactivated and ranolazine blocked channels (closed symbols) was assessed by 5 ms test pulses to −10 mV after either 100 ms (A,C) or 2 s (B,D) recovery at −130 mV. Onset kinetics were fitted with a single exponential function for control conditions and with a double exponential function for ranolazine conditions. Fit parameters are summarized in Table A7. Slow inactivation and ranolazine block recovery (E–H) was measured with a series of 5 ms test pulses ranged from 20 ms to 60 s after either 8 s (E,G) or 32 s (F,H) onset. Recovery followed double exponential kinetics in all conditions. Fit parameters are summarized in Table A8.
In drug-free conditions, slow inactivation onset followed a single exponential time course that was faster at pH 7.4 (τ1 = 7.0 ± 0.5 s, Figure 8A, open circles) than at pH 6.0 (τ1 = 11.2 ± 0.5 s, Figure 8C, open squares). Ranolazine causes an additional decrease in test pulse current amplitudes (Figures 8A–D, solid symbols). This reduction of channel availability in the presence of the drug was fitted with a double exponential function (Table A7). The onset of ranolazine block can be measured best with the 2 s recovery pulse. This time interval allowed near complete recovery from native slow inactivation but not from ranolazine block (Figures 8B,D). The drug-induced time constant of onset was significantly faster at pH 7.4 (100 μM: 3.0 ± 0.3 s, Figure 8B, solid circles) than at pH 6.0 (100 μM: 18 ± 2 s, Figure 8D, solid squares). We also observed a statistically significant effect of 10 μM ranolazine at pH 6.0 (Figures 8C,D; Table A7).
Recovery from slow inactivation and ranolazine block was observed after 8 or 32 s conditioning pulses (Figures 8E–H). Recovery followed double exponential kinetics in all conditions (Table A8). In drug-free conditions, recovery was faster at low pH. A distinct ultra slow recovery component developed in the presence of ranolazine. Interestingly, this drug-induced component of recovery was 4–5 fold slower at pH 6.0 than at pH 7.4. Thus, acidic pH not only impairs native Nav1.5 slow inactivation but also affects the interaction between the channel and ranolazine, making both onset and recovery of the drug effect considerably slower (Figure 8, Tables A7, A8).
Lastly, we examined the voltage-dependence of steady-state slow inactivation and its modulation by ranolazine (Figure 9). Cells were given a 30 s conditioning pulse to potentials ranging from −150 to +10 mV in 10 mV intervals, then stepped to −130 mV for 20 ms to recover fast-inactivated channels, and finally a test pulse to −10 mV. Channels were allowed to recover for 30 s at −130 mV between the sweeps. Experiments immediately before and after drug perfusion were performed in a limited number of cells to estimate the amount of tonic inhibition by ranolazine at −150 mV (pH 7.4: 10 μM 8 ± 3%, n = 3, 100 μM 33 ± 4%, n = 4; pH 6.0: 10 μM 20 ± 8%, n = 3, 100 μM 39 ± 4%, n = 6). pH did not significantly affect tonic block by ranolazine. Steady-state availability curves normalized to peak availability in the absence of ranolazine are plotted in Figure 9. The insets show curves normalized to the respective current maxima at −150 mV.
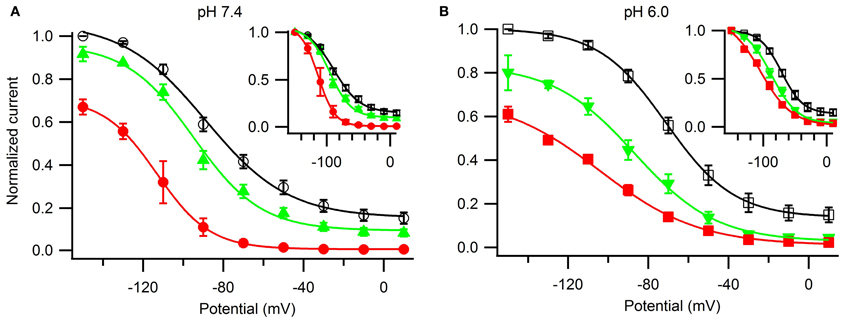
Figure 9. The effects of ranolazine on steady-state slow inactivation and tonic block. Slow inactivation was induced with alternating 30 s conditioning pulses to a range of voltages (−150 mV through +10 mV in 10 mV intervals) followed by a 20 ms recovery period at −130 mV to allow recovery from fast inactivation, and a −10 mV test pulse to measure channel availability. Channels spent additional 30 s at holding potential −130 mV between the sweeps. Matched pair experiments before and after ranolazine perfusion were performed to assess tonic inhibition by the drug at −150 mV (pH 7.4: 10 μM 8 ± 3%, n = 3, p = 0.001; 100 μM 33 ± 4%, n = 4, p < 0.001; pH 6.0: 10 μM 20 ± 8%, n = 3, p = 0.01; 100 μM 39 ± 4%, n = 6, p < 0.001). Currents were normalized to the maximum current in the absence of ranolazine (A,B) or to the maximum value at −150 mV (insets) and plotted as a function of the conditioning potential for control (open black symbols), 10 μM ranolazine (solid green triangles), and 100 μM ranolazine (solid red symbols). Data were fitted with a Boltzmann equation. Fit parameters are listed in Table A9. The differences between control and 10 μM ranolazine at pH 7.4 were not statistically significant (p > 0.05).
In drug-free conditions acidic pH depolarized the steady-state availability by 18 mV (Table A9). 100 μM ranolazine produced dramatic leftward shifts at both pH 7.4 and 6.0 and eliminated the plateaus of the availability curves at depolarized potentials (Figure 9). 10 μM ranolazine at acidic pH significantly left shifted the availability curve by ~16 mV.
Discussion
We examined the effects of ranolazine on Nav1.5 channels transiently expressed in HEK 293 cells at pH 7.4 and 6.0. We found that the kinetics of drug-channel interaction depends on pH conditions. Both the onset of and recovery from ranolazine modulation are slowed by 4–5 fold at pH 6.0 relative to pH 7.4 (Figures 4, 8). Ranolazine significantly reduced late/window sodium current at both pH conditions (Figures 6, 7).
Physiological Effects of Extracellular pH
Extracellular pH plays a significant role in controlling activity of many physiological processes. Normal extracellular pH is ~7.4. Pathological conditions, such as hypoxia and/or ischemia decrease extracellular pH. Acidification decreases peak conductance of Nav channels by protonation of outer vestibule carboxylates (Khan et al., 2002, 2006; Vilin et al., 2012) and also causes a depolarizing shift in the voltage-dependence of gating by surface charge screening (Hille, 1968; Benitah et al., 1997). In Nav1.5, acidosis increases persistent INa (Figure 6), which is considered to be a predisposing factor for cardiac arrhythmias (Amin et al., 2010; Jones et al., 2011). Low extracellular pH affects not only sodium channels. Peak current is reduced and kinetic changes are induced by low extracellular pH in a wide range of potassium channels (Deutsch and Lee, 1989; Kehl et al., 2002; Trapani and Korn, 2003). Similarly, calcium channels and the sodium/calcium exchanger (NCX) are affected by low extracellular pH in ways that are both direct and, in the case of NCX, at least partly related to changes in sodium channel behavior (see Carmeliet, 1999, for review). Our experimental system isolates sodium channels and thus provides only one facet of a complex suite of effects induced by low extracellular pH.
pH Effects on Use-Dependence in Ranolazine
It is well-established that ranolazine at high concentrations (such as 100 μM) causes prominent use-dependent inhibition in various types of sodium channels at physiological extracellular pH (Fredj et al., 2006; Rajamani et al., 2008, 2009; Kahlig et al., 2010; Huang et al., 2011; Hirakawa et al., 2012). Consistent with these previous reports, we found that at extracellular pH 7.4 100 μM ranolazine produced substantial use-dependent block at 10 or 25 Hz (Figures 4A,B). At extracellular pH 6.0 use-dependent block with 100 μM ranolazine is substantially diminished at both frequencies (Figures 4C,D). The kinetics of use-dependence is also greatly slowed at acidic pH (Figure 4, Table A5).
The loss of channel availability during the pulse trains occur due to onset of ranolazine inhibition during the depolarizing pulses, and to slow recovery of channels from ranolazine-induced block between the pulses. In the presence of 100 μM ranolazine at pH 7.4 a pronounced slow component of recovery after 500 ms conditioning is evident (Figure 3A). Such a distinct slow component cannot be observed at pH 6.0 (Figure 3B). We hypothesized that its absence is due to slow kinetics of onset at acidic pH: a 500 ms conditioning pulse is insufficient to cause a detectable degree of inhibition by 100 μM ranolazine.
Prolonged trains of long (300 ms) and low frequency (1 Hz) depolarizations approximating cardiac action potential pace and duration demonstrate that 100 μM ranolazine produces substantial inhibition of sodium current even at pH 6.0 (Figure 5). This inhibition develops at a rate that is ~5 fold slower at pH 6.0 than at pH 7.4 but reaches a comparable magnitude after 500 s. The slow kinetics of onset at pH 6.0 explains the observed lack of effects of 100 μM ranolazine in short pulse duration protocols (Figures 1–3) and the reduced affects on high frequency use-dependence (Figure 4).
However, the effects of ranolazine on use-dependent reduction of peak sodium current at concentrations as high as 100 μM might not be therapeutically relevant. Ranolazine undergoes extensive biotransformation, primarily via CYP3A-mediated pathways of metabolism (Chaitman, 2006). Less than 7% of the parent compound remain un-metabolized (Jerling and Abdallah, 2005). Thus, with now commonly used sustained-release ranolazine, the concentration in patients' blood serum reaches only 2–6 μM (Chaitman, 2006; Antzelevitch et al., 2011). In our experimental conditions a therapeutically relevant concentration (10 μM) did not produce a significant use-dependent block of peak INa at either pH (Figures 4, 5). This finding might be important in the context of cardiac therapy since a prominent use-dependent block in the ischemic condition can be especially pro-arrhythmic (Moreno et al., 2011). The action of ranolazine in acidic conditions suggests the drug as ever more promising in the setting of ischemia due to stabilization of the inactivation state and, critically important, a reduction in the extent of use-dependent block.
Impairment of Inactivation States by Acidic pH and its Restoration by Ranolazine
Acidic pH 6.0 impairs Nav channel inactivation states causing depolarizing shifts in the steady-state profiles, slowing the onset of, and accelerating the recovery from both fast and slow inactivation. Addition of ranolazine selectively offsets the effects of low pH on slow inactivation. Therapeutically relevant ranolazine concentration (10 μM) at pH 6.0 essentially negates the effects of acidic pH (positive 18 mV shift, Figures 9A,B, open symbols) on the steady-state availability curve (pH 7.4: control V1/2 = −89 ± 3 mV; pH 6.0: 10 μM ranolazine V1/2 = −87 ± 2 mV). Although the onset and recovery drug kinetics are 4–5 fold slower at pH 6.0 (Figure 8), the Kd-value at steady-state is expected to be in the similar range in both pH conditions.
In situations where Nav fast inactivation is impaired and unable to shut off the ion flux through the pore slow inactivation can act as a “fail safe mechanism.” Consistent with this view, we suggest the stabilizing effect of ranolazine on slow inactivation reduces channel availability in a voltage-dependent manner (Figure 9) leading to a decrease in persistent (Figure 6) and window (Figure 7) sodium currents at both pH conditions.
On the Mechanism of Action of Ranolazine
Ranolazine action on Nav channels is commonly viewed within the framework of the Modulated Receptor hypothesis (Hille, 1977), and presumed to be similar to local anesthetic drugs that were extensively studied over the past 3 decades (see Fozzard et al., 2005, for review). According to this paradigm, drug affinity is dependent upon the conformational state of the channel. Resting channels display lower affinity while open or inactivated channels have higher drug affinity. Within this framework, ranolazine use-dependent inhibition is often attributed to open channel block (Rajamani et al., 2008; Wang et al., 2008; Huang et al., 2011). This interpretation also assumes that binding of the drug molecule to the channel physically obstructs (“blocks”) ion flow through the channel pore. While this presumption is not an essential element of Modulated Receptor hypothesis and has not been unequivocally proven, it is often taken for granted. An alternative concept, “gating modifier,” suggests that binding of a drug molecule does not block the ion flow through the pore per se, but modulates conformational balance of the channel's intrinsic states. Most of the agents recognized as gating modifiers at present are toxins (Swartz and MacKinnon, 1997; Sokolov et al., 2008; Wang et al., 2011; Zhang et al., 2011) that interact primarily with voltage-sensing domains and modulate activation or fast inactivation gating. Ranolazine, along with lacosamide (Errington et al., 2008), may represent a distinct class of gating modifiers that affect slow inactivation gating of the channel. A crystal structure of the channel drug complex could provide strong evidence in favor of either the blocker or gating modifier hypotheses; however, such a structure is not available at this time.
Our data suggest that ranolazine does not block open Nav channels. Instead, we propose that ranolazine induces a slow inactivation-like state with slow onset kinetics (seconds at pH 7.4, tens of seconds at pH 6.0), slow recovery kinetics (tens of seconds), and sharing the voltage-dependent profile of Nav channel's intrinsic slow inactivation. First, our steady-state slow inactivation protocol featuring 30 s conditioning pulses demonstrates a significant reduction of channel availability at negative voltages where channel openings do not occur or are extremely rare (Figure 9). Second, in our matched ramp experiments, we observed a significant reduction of inward sodium currents during 500 ms ramp depolarizations (Figure 7). This result suggests the drug is already bound at voltages as negative as −130 mV rather than binding to open/inactivating channels during the ramp. Hence, 500 ms is a relatively short time for development of ranolazine block as revealed by the slow onset kinetics described in Figure 8.
Additional arguments for a specific interaction between ranolazine and slow inactivation come from previous studies. Multiple mutations in various Nav channels that show increased persistent current also demonstrated increased sensitivity to ranolazine (Nav1.5 LQT3: ΔKPQ Fredj et al., 2006; Y1767C Huang et al., 2011; R1623Q Rajamani et al., 2009; Nav1.1 GEFS+, SMEI, FHM3: Kahlig et al., 2010). These mutations typically destabilize Nav channel fast inactivation but ranolazine's effectiveness may be owing to its property to boost the “fail safe” slow inactivation mechanism. Recent studies of El-Bizri et al. (2011) in Nav1.4 and Hirakawa et al. (2012) in Nav1.3 demonstrated that ranolazine's effect on steady-state channel availability progressively increases with longer conditioning pulses, consistent with selective interaction between ranolazine and the slow-inactivated states.
Our results suggest that hNav1.5/β1 heterologously expressed in HEK293 cells are affected by ranolazine in a manner similar to ventricular sodium channels as opposed to sodium channels in atria. In canine atrial myocytes, Burashnikov and Antzelevitch (2008) demonstrated substantial use-dependent block by 10 μM ranolazine as well as fast onset kinetics. In our experimental setup, the lack of significant use-dependent block at 10 μM and slow kinetics of drug-channel interaction suggests that atrial sodium channels differ substantially from the heterologously expressed hNav1.5/β 1. Such difference may be attributed to tissue-specific cardiac sodium channel isoforms or differences in the stoichiometry of auxiliary subunits (Burashnikov and Antzelevitch, 2008, 2009, 2010).
Our study was limited to the effects of ranolazine on extracellular changes in pH. Ischemia results a priori in intracellular pH changes. Although intracellular protons are extruded by the sodium/hydrogen exchanger, NHE1, low intracellular pH also affects myocardial contractility and may alter the biophysical properties of ion channels, including Nav1.5 (Clanachan, 2006). Nevertheless, intracellular proton buffering mechanisms may limit the effects of low pHi, relative to those of extracellular protons, on ion channel function, and extracellular pH has been shown to change to a greater extent than intracellular pH (Crampin et al., 2006).
In conclusion, our results demonstrate that ranolazine induces a slow inactivation-like state in hNav1.5/β 1 leading to reduction of late and window Na+ currents. The drug is especially effective at acidic extracellular pH where native slow inactivation is impaired. At pH 6.0 significant effects of the drug on kinetics and voltage-dependence of the drug-induced state can be observed at therapeutically relevant concentrations (10 μM), whereas open state and fast inactivation are not appreciably affected.
Conflict of Interest Statement
S. Rajamani is an employee of Gilead Sciences, Inc, the producer of Ranolazine. This work was funded in part by a grant from Gilead Sciences, Inc. to P.C. Ruben. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David K. Jones for his thoughtful comments on the manuscript.
References
Amin, A., Asghari-Roodsari, A., and Tan, H. (2010). Cardiac sodium channelopathies. Pflugers Arch. 460, 223–237. doi: 10.1007/s00424-009-0761-0
Antzelevitch, C., Burashnikov, A., Sicouri, S., and Belardinelli, L. (2011). Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 8, 1281–1290. doi: 10.1016/j.hrthm.2011.03.045
Belardinelli, L., Shryock, J., and Fraser, H. (2006). Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92(Suppl. 4), iv6–iv14. doi: 10.1136/hrt.2005.078790
Benitah, J., Balser, J., Marban, E., and Tomaselli, G. (1997). Proton inhibition of sodium channels: mechanism of gating shifts and reduced conductance. J. Membr. Biol. 155, 121–131. doi: 10.1007/s002329900164
Burashnikov, A., and Antzelevitch, C. (2008). Atrial-selective sodium channel blockers: do they exist. J. Cardiovasc. Pharmacol. 52, 121–128. doi: 10.1097/FJC.0b013e31817618eb
Burashnikov, A., and Antzelevitch, C. (2009). Atrial-selective sodium channel block for the treatment of atrial fibrillation. Expert Opin. Emerg. Drugs 14, 233–249. doi: 10.1517/14728210902997939
Burashnikov, A., and Antzelevitch, C. (2010). New development in atrial antiarrhythmic drug therapy. Nat. Rev. Cardiol. 7, 139–148. doi: 10.1038/nrcardio.2009.245
Burashnikov, A., Diego, J. D., Zygmunt, A., Belardinelli, L., and Antzelevitch, C. (2007). Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116, 1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890
Carmeliet, E. (1999). Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev. 79, 917–1017.
Chaitman, M. D. (2006). Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation 113, 2462–2472. doi: 10.1161/CIRCULATIONAHA.105.597500
Clanachan, A. S. (2006). Contributions of protons to post-ischemic Na+ and Ca++ overload and left ventricular mechanical dysfunction. J. Cardiovasc. Electrophysiol. 17, 141–148. doi: 10.1111/j.1540-8167.2006.00395.x
Crampin, E. J., Smith, N. P., Langham, A. E., Clayton, R. H., and Orchard, C. H. (2006). Acidosis in models of cardiac ventricular myocytes. Philos. Trans. A Math. Phys. Eng. Sci. 364, 1171–1186. doi: 10.1098/rsta.2006.1763
Deutsch, C., and Lee, S. C. (1989). Modulation of K+ currents in human lymphocytes by pH. J. Physiol. 413, 399–413.
Dobrev, D., and Nattel, S. (2010). New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 375, 1212–1223. doi: 10.1016/S0140-6736(10)60096-7
El-Bizri, N., Kahlig, K., Shyrock, J., George, A. L. J., Belardinelli, L., and Rajamani, S. (2011). Ranolazine block of human Nav1.4 sodium channels and paramyotonia congenita mutants. Channels 5, 161–172. doi: 10.4161/chan.5.2.14851
Errington, A. C., Stoehr, T., Heers, C., and Lees, G. (2008). The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 73, 157–169. doi: 10.1124/mol.107.039867
Estacion, M., Waxman, S. G., and Dib-Hajj, S. D. (2010). Effects of ranolazine on wild-type and mutant hNav1.7 channels and on DRG neuron excitability. Mol. Pain 6, 1–13. doi: 10.1186/1744-8069-6-35
Fozzard, H., Lee, P., and Lipkind, G. (2005). Mechanism of local anesthetic drug action on voltage-gated sodium channels. Curr. Pharm. Des. 11, 2671–2686. doi: 10.2174/1381612054546833
Fredj, S., Sampson, K., Liu, H., and Kass, R. (2006). Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br. J. Pharmacol. 148, 16–24. doi: 10.1038/sj.bjp.0706709
Gould, H., Garrett, C., Donahue, R., Paul, D., Diamond, I., and Taylor, B. (2009). Ranolazine attenuates behavioral signs of neuropathic pain. Behav. Pharmacol. 20, 755–758. doi: 10.1097/FBP.0b013e3283323c90
Hale, S., Leeka, J., and Kloner, R. (2006). Improved left ventricular function and reduced necrosis after myocardial ischemia/reperfusion in rabbits treated with ranolazine, an inhibitor of the late sodium channel. J. Pharmacol. Exp. Ther. 318, 418–423. doi: 10.1124/jpet.106.103242
Hale, S., Shryock, J., Belardinelli, L., Sweeney, M., and Kloner, R. (2008). Late sodium current inhibition as a new cardioprotective approach. J. Mol. Cell. Cardiol. 44, 954–967. doi: 10.1016/j.yjmcc.2008.03.019
Hille, B. (1968). Charges and potentials at the nerve surface. divalent ions and pH. J. Gen. Physiol. 51, 221–236. doi: 10.1085/jgp.51.2.221
Hille, B. (1977). Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69, 497–515. doi: 10.1085/jgp.69.4.497
Hirakawa, R., El-Bizri, N., Shryock, J., Belardinelli, L., and Rajamani, S. (2012). Block of Na+ currents and suppression of action potentials in embryonic rat dorsal root ganglion neurons by ranolazine. Neuropharmacology 62, 2251–2260. doi: 10.1016/j.neuropharm.2012.01.021
Huang, H., Priori, S., Napolitano, C., O'Leary, M., and Chahine, M. (2011). Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am. J. Physiol. Heart Circ. Physiol. 300, H288–H299. doi: 10.1152/ajpheart.00539.2010
Jerling, M., and Abdallah, H. (2005). Effect of renal impairment on multiple-dose pharmacokinetics of extended-release ranolazine. Clin. Pharmacol. Ther. 78, 288–297. doi: 10.1016/j.clpt.2005.05.004
Jones, D. K., Peters, C. H., Tolhurst, S. A., Claydon, T. W., and Ruben, P. C. (2011). Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys. J. 101, 2147–2156. doi: 10.1016/j.bpj.2011.08.056
Kahlig, K. M., Lepist, I., Leung, K., Rajamani, S., and George, A. L. (2010). Ranolazine selectively blocks persistent current evoked by epilepsy—associated Nav1.1 mutations. Br. J. Pharmacol. 161, 1414–1426. doi: 10.1111/j.1476-5381.2010.00976.x
Kehl, S. J., Eduljee, C., Kwan, D. C., Zhang, S., and Fedida, D. (2002). Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn2+. J. Physiol. 541, 9–24. doi: 10.1113/jphysiol.2001.014456
Khan, A., Kyle, J., Hanck, D., Lipkind, G., and Fozzard, H. (2006). Isoform-dependent interaction of voltage-gated sodium channels with protons. J. Physiol. 576, 493–501. doi: 10.1113/jphysiol.2006.115659
Khan, A., Romantseva, L., Lam, A., Lipkind, G., and Fozzard, H. (2002). Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J. Physiol. 543, 71–84. doi: 10.1113/jphysiol.2002.021014
Magyar, J., Kiper, C. E., Dumaine, R., Burgess, D. E., Bányász, T., and Satin, J. (2004). Divergent action potential morphologies reveal nonequilibrium properties of human cardiac Na channels. Cardiovasc Res. 64, 477–487. doi: 10.1016/j.cardiores.2004.07.014
Maruki, Y., Koehler, R. C., Eleff, S. M., and Traystman, R. J. (1993). Intracellular pH during reperfusion influences evoked potential recovery after complete cerebral ischemia. Stroke 24, 697–703. doi: 10.1161/01.STR.24.5.697
Moreno, J. D., Zhu, Z. I., Yang, P. C., Bankston, J. R., Jeng, M. T., Kang, C., et al. (2011). A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci. Transl. Med. 3, 98ra83. doi: 10.1126/scitranslmed.3002588
Moss, A. J., Zareba, W., Schwarz, K. Q., Rosero, S., McNitt, S., and Robinson, J. L. (2008). Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J. Cardiovasc. Electrophysiol. 19, 1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x
Murphy, L., Renodin, D., Antzelevitch, C., Diego, J. D., and Cordeiro, J. (2011). Extracellular proton depression of peak and late Na+ current in the canine left ventricle. Am. J. Physiol. Heart Circ. Physiol. 301, H936–H944. doi: 10.1152/ajpheart.00204.2011
Peters, C., Sokolov, S., Rajamani, S., and Ruben, P. C. (2013). The actions of the antianginal drug, Ranolazine, on the brain sodium channel NaV1.2 and its modulation by extracellular protons. Br. J. Pharmacol. 169, 704–716. doi: 10.1111/bph.12150
Rajamani, S., El-Bizri, N., Shryock, J. C., Makielski, J. C., and Belardinelli, L. (2009). Use-dependent block of cardiac late Na(+) current by ranolazine. Heart Rhythm 6, 1625–1631. doi: 10.1016/j.hrthm.2009.07.042
Rajamani, S., Shryock, J. C., and Belardinelli, L. (2008). Block of tetrodotoxin-sensitive, Nav1.7 and tetrodotoxin-resistant, Nav1.8, Na channels by ranolazine. Channels 2, 449–460. doi: 10.4161/chan.2.6.7362
Reddy, B., Weintraub, H., and Schwartzbard, A. (2010). Ranolazine: a new approach to treating an old problem. Tex. Heart Inst. J. 37, 641–647.
Sokolov, S., Kraus, R., Scheuer, T., and Catterall, W. (2008). Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol. Pharmacol. 73, 1020–1028. doi: 10.1124/mol.107.041046
Sossalla, S., Kallmeyer, B., Wagner, S., Mazur, M., Maurer, U., Toischer, K., et al. (2010). Altered Na+ currents in atrial fibrillation: effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 55, 2330–2342. doi: 10.1016/j.jacc.2009.12.055
Sossalla, S., Wagner, S., Rasenack, E. C., Ruff, H., Weber, S. L., Schondube, F. A., et al. (2008). Ranolazine Improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 45, 32–43. doi: 10.1016/j.yjmcc.2008.03.006
Stone, P. H., Chaitman, B. R., Stocke, K., Sano, J., DeVault, A., and Koch, G. G. (2010). The anti-ischemic mechanism of action of ranolazine in stable ischemic heart disease. J. Am. Coll. Cardiol. 56, 934–942. doi: 10.1016/j.jacc.2010.04.042
Swartz, K., and MacKinnon, R. (1997). Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron 18, 675–682. doi: 10.1016/S0896-6273(00)80307-4
Trapani, J. G., and Korn, S. J. (2003). Effect of external pH on activation of the Kv1.5 potassium channel. Biophys. J. 84, 195–204. doi: 10.1016/S0006-3495(03)74842-5
Undrovinas, A. I., Belardinelli, L., Undrovinas, N. A., and Sabbah, H. N. (2006). Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J. Cardiovasc. Electrophysiol. 17, S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x
Undrovinas, N. A., Maltsev, V. A., Belardinelli, L., Sabbah, H. N., and Undrovinas, A. I. (2010). Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J. Physiol. Sci. 60, 245–257. doi: 10.1007/s12576-010-0092-0
Vilin, Y. Y., Peters, C. H., and Ruben, P. C. (2012). Acidosis differentially modulates inactivation in Nav1.2, Nav1.4, and Nav1.5 channels. Front. Pharmacol. 3:109. doi: 10.3389/fphar.2012.00109
Wang, G. K., Calderon, J., and Wang, S.-Y. (2008). State- and use-dependent block of muscle Nav1.4 and neuronal Nav1.7 voltage-gated Na channel isoforms by ranolazine. Mol. Pharmacol. 73, 940–948. doi: 10.1124/mol.107.041541
Wang, J., Yarov-Yarovoy, V., Kahn, R., Gordon, D., Gurevitz, M., Scheuer, T., et al. (2011). Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 108, 15426–15431. doi: 10.1073/pnas.1112320108
Wasserstrom, J. A., Sharma, R., O'Toole, M. J., Zheng, J., Kelly, J. E., Shryock, J., et al. (2009). Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated intact heart. J. Pharmacol. Exper. Ther. 331, 382–391. doi: 10.1124/jpet.109.156471
Wu, L., Shryock, J. C., Song, Y., Li, Y., Antzelevitch, C., and Belardinelli, L. (2004). Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J. Pharmacol. Exper. Ther. 310, 599–605. doi: 10.1124/jpet.104.066100
Zhang, J., Yarov-Yarovoy, V., Scheuer, T., Karbat, I., Cohen, L., Gordon, D., et al. (2011). Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 86, 33641–33651. doi: 10.1074/jbc.M111.282509
Appendix

Table A1. Conductance.

Table A2. Time constants of fast inactivation.

Table A3. Steady state fast inactivation.
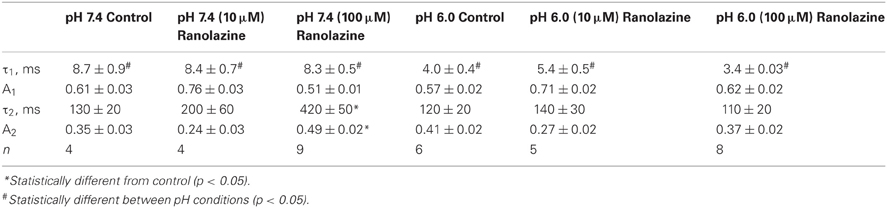
Table A4. Recovery from fast inactivation.
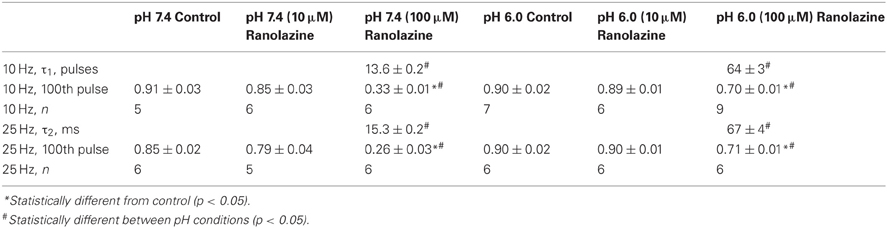
Table A5. Use dependence 10 and 25 Hz.
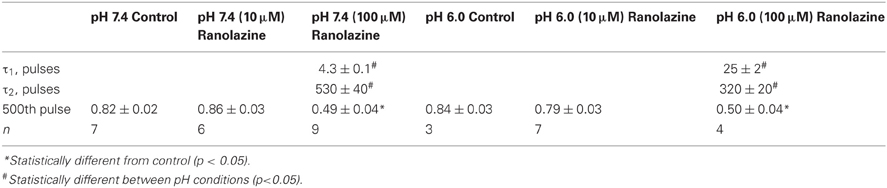
Table A6. Use-dependence 1 Hz.
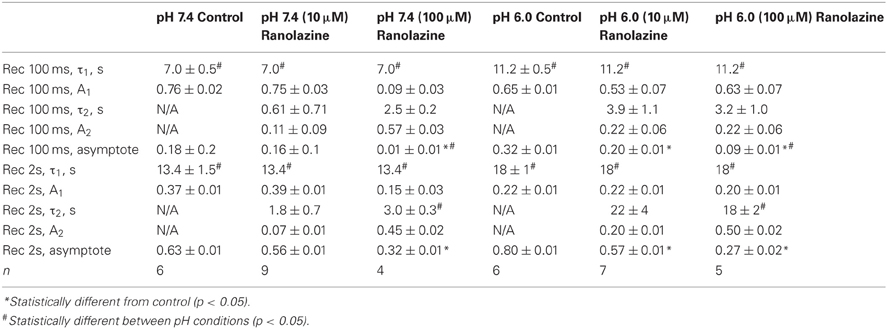
Table A7. Slow inactivation onset.
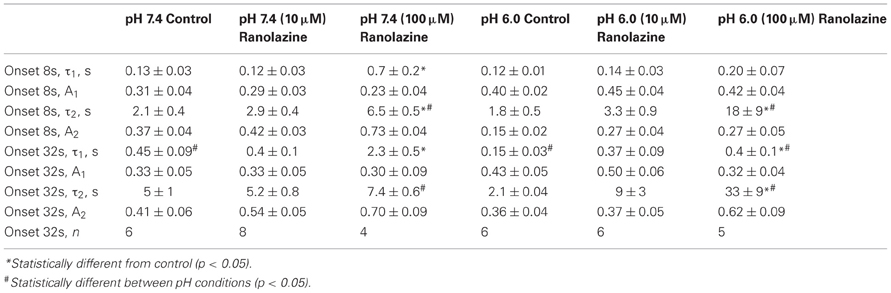
Table A8. Slow inactivation recovery.

Table A9. Steady state slow inactivation.
Keywords: ranolazine, acidosis, cardiac sodium channel, Nav1.5, electrophysiology
Citation: Sokolov S, Peters CH, Rajamani S and Ruben PC (2013) Proton-dependent inhibition of the cardiac sodium channel Nav1.5 by ranolazine. Front. Pharmacol. 4:78. doi: 10.3389/fphar.2013.00078
Received: 02 May 2013; Accepted: 03 June 2013;
Published online: 21 June 2013.
Edited by:
Aurelien Chatelier, Laboratoire Signalisation & Transports Ioniques Membranaires, FranceReviewed by:
Wayne R. Giles, The University of Calgary, CanadaTheodore R. Cummins, Indiana University School of Medicine, USA
Copyright © 2013 Sokolov, Peters, Rajamani and Ruben. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: P. C. Ruben, Department of Biomedical Physiology and Kinesiology, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6 Canada e-mail: pruben@sfu.ca