 Ester Aso1,2*
Ester Aso1,2* Isidre Ferrer1,2*
Isidre Ferrer1,2*- 1Institut de Neuropatologia, Servei d’Anatomia Patològica, Institut d’Investigació Biomèdica de Bellvitge-Hospital Universitari de Bellvitge, Universitat de Barcelona, L’Hospitalet de Llobregat, Spain
- 2Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas, Instituto Carlos III, Spain
The limited effectiveness of current therapies against Alzheimer’s disease (AD) highlights the need for intensifying research efforts devoted to developing new agents for preventing or retarding the disease process. During the last few years, targeting the endogenous cannabinoid system has emerged as a potential therapeutic approach to treat Alzheimer. The endocannabinoid system is composed by a number of cannabinoid receptors, including the well-characterized CB1 and CB2 receptors, with their endogenous ligands and the enzymes related to the synthesis and degradation of these endocannabinoid compounds. Several findings indicate that the activation of both CB1 and CB2 receptors by natural or synthetic agonists, at non-psychoactive doses, have beneficial effects in Alzheimer experimental models by reducing the harmful β-amyloid peptide action and tau phosphorylation, as well as by promoting the brain’s intrinsic repair mechanisms. Moreover, endocannabinoid signaling has been demonstrated to modulate numerous concomitant pathological processes, including neuroinflammation, excitotoxicity, mitochondrial dysfunction, and oxidative stress. The present paper summarizes the main experimental studies demonstrating the polyvalent properties of cannabinoid compounds for the treatment of AD, which together encourage progress toward a clinical trial.
Introduction
Alzheimer is an age-dependent neurodegenerative process distinct from normal aging and characterized morphologically by the presence of senile plaques, mainly composed of different species of fibrillar β-amyloid (Aβ) produced by the cleavage of the Aβ precursor protein (APP) due to β- and γ-secretases, and by the presence of neurofibrillary tangles, mostly composed of various isoforms of hyper-phosphorylated and nitrated tau protein (Duyckaerts and Dickson, 2011; Ferrer, 2012). One tendency of opinion proposes that Aβ triggers plaque formation, tau hyper-phosphorylation, and disease progression (Hardy, 2009). This may happens in a percentage of familial Alzheimer’s disease (AD) cases linked to mutations in genes encoding APP, and presenilin 1 and presenilin 2 which are enzymes involved in the cleavage of APP, or in Down syndrome (Bertram and Tanzi, 2011). However, tau hyper-phosphorylation precedes Aβ deposition in many cerebral regions in sporadic cases of AD (Braak and Braak, 1999).
Recent studies have shown that Aβ acts as a seed of new Aβ production and deposition under appropriate conditions (Frost and Diamond, 2010) and that abnormal tau promotes the production and deposition of hyper-phosphorylated tau under determinate experimental conditions (Clavaguera et al., 2009). Therefore, Aβ and hyper-phosphorylated tau promote the progression of the pathological process in an exponential way once these abnormal proteins are accumulated in the brain (Goedert et al., 2010; Ferrer, 2012).
In addition to these pathological hallmarks, multiple alterations converge in the pathogenesis of AD, including genetic and environmental factors. Vascular factors and concomitant pathologies worsen disease symptoms (Kovacs et al., 2008). Mitochondrial functional defects, increased production of reactive oxygen and nitrogen species (ROS and RNS), and damage to enzymes involved in energy metabolism are causative of nerve cell exhaustion (Pamplona et al., 2005; Ferrer, 2009; Sultana and Butterfield, 2010).
Altered lipid composition of membranes particularly lipid rafts (Martín et al., 2010), inflammatory responses (Akiyama et al., 2000), and altered production of trophic factors, neurotransmitter and neuromodulators, together with impaired function of degradation pathways such as those related to cytoplasmic proteolysis, autophagy, and ubiquitin–proteasome system play crucial roles as well (Keller et al., 2000; Ferrer, 2012; Selkoe, 2012).
Neurofibrillary tangles first appear in middle age in selected nuclei of the brain stem, later in the entorhinal cortex, and then extend to other parts of the brain (Braak and Braak, 1999; Simic et al., 2009). Senile plaques appear first in the orbitofrontal and temporal cortex and then extend to the whole cortex, diencephalic nuclei, and eventually to the cerebellum at terminal stages (Braak and Braak, 1999). Synaptic loss, reduced dendritic arbors, progressive isolation of remaining neurons and nerve cell loss occurs with disease progression, and affects multiple brain regions not only the cerebral cortex but also the amygdala, nuclei of the forebrain including Meynert nucleus, striatum, thalamus, and selected nuclei of the brain stem thus involving multiple neurotransmitter systems (Duyckaerts and Dickson, 2011).
Importantly, the progression from early stages of the neurodegenerative process to symptomatic stages may take decades, whereas once the cognitive impairment and dementia appear the disease progression is much more rapid. Therefore, Alzheimer is a relatively well-tolerated degenerative process during a long period of time, but it may have devastating effects once thresholds are crossed (Ferrer, 2012). These facts highlight the need to search for treatments that act on selective targets during the silent period of the disease, aimed at curbing or retarding disease progression toward dementia (Ferrer, 2012; Selkoe, 2012).
During the last few years, targeting the endogenous cannabinoid system (ECS) has emerged as a potential therapeutic approach to treat Alzheimer in such first stages. Endocannabinoid signaling has been demonstrated to modulate the main pathological processes occurring during the silent period of the neurodegenerative process, including protein misfolding, neuroinflammation, excitotoxicity, mitochondrial dysfunction, and oxidative stress. The present paper summarizes the experimental studies demonstrating the multi-faceted properties of cannabinoid compounds for the treatment of AD.
The Endogenous Cannabinoid System
The last two decades of research have brought a tremendous improvement in knowledge of the endocannabinoid system components and functions under physiological and pathological conditions. This neuromodulatory system consists of cannabinoid receptors, endogenous ligands, and several enzymes responsible for their synthesis and degradation (Piomelli, 2003). To date, two subtypes of cannabinoid Gi/o-coupled receptors, CB1 and CB2, have been fully characterized and cloned. However, cannabinoid compounds may also bind to other receptors, such as GPR55, peroxisome proliferator-activated receptors PPARα and PPARγ, and transient receptor potential vannilloid-1 (TRPV1) channels (Maccarrone et al., 2010; Pertwee et al., 2010). CB1 receptors are the most abundant G protein-coupled receptors in the central neural system, expressed in both neurons and glial cells, where they regulate important brain functions including cognition and memory, emotion, motor control, feeding, and pain perception (Wilson and Nicoll, 2002; Howlett, 2005). CB1 receptors are mostly located at the terminals of neurons of the central and peripheral nervous system where they act as modulators of excitatory and inhibitory neurotransmission. Moreover, CB1 receptors are also found in peripheral tissues, playing an important role in energy balance and metabolism (Silvestri and Di Marzo, 2013). CB2 receptors are localized in cells of the immune system and modulate the immune cell migration and the release of cytokines; within the nervous system CB2 receptors are mainly located in microglia (Cabral and Griffin-Thomas, 2009). Relatively low CB2 receptor expression has also recently been identified in some neurons (Van Sickle et al., 2005; Brusco et al., 2008; Onaivi et al., 2008). Further evidence of CB2 receptor expression in neurons comes from the observation that axonal damage in one cerebellar hemisphere induced the expression of CB2 receptors in contralateral precerebellar neurons; CB2 receptor agonist facilitated neuronal survival, whereas the selective PI3K inhibitor blocked CB2R effects on axotomized neurons (Viscomi et al., 2009). Most of the knowledge acquired about cannabinoid receptor pharmacology was made possible by the study of the mechanisms of action of numerous natural, but also synthetic, cannabinoid compounds. Among the natural cannabinoids, the most well-known are Δ9-tetrahydrocannabinol (Δ9-THC), the main psychoactive compound of the Cannabis sativa plant, and cannabidiol (CBD), which is devoid of any psychoactivity, both differing in cannabinoid receptor affinity and activity (Pertwee, 2008).
The characterization of CB1 and CB2 receptors permitted the discovered of endocannabinoids or cannabinoids produced and released by nerve cells. Endocannabinoids are lipid compounds of the eicosanoid family derived from the degradation of membrane phospholipids (Piomelli, 2003). The most representative are arachidonoylethanolamine (AEA), also named anandamide, and 2-arachidonoylglycerol (2-AG), although several others have also been identified, such as 2-arachidonylglyceryl ether (2-AGE), virodhamine, and N-arachidonyldopamine (Piomelli, 2003). Endocannabinoids act as neurotransmitters since they are synthesized and released by neurons, are able to bind and activate membrane receptors, and are inactivated by reuptake and enzymatic degradation within the cell. However, endocannabinoids have two fundamental characteristics that differentiate them from other neurotransmitters: they act as retrograde messengers and they do not accumulate in the interior of synaptic vesicles (Wilson and Nicoll, 2002). For several years, endocannabinoid compounds have been supposed to be exclusively synthesized on demand to act on cells located near their site of synthesis, and then to be rapidly inactivated by the action of specific degradation enzymes. However, recent studies have shown intracellular stores of anandamide in places other than synaptic vesicles as in adiposomes where it is sequestered and concentrated to higher levels than in the extracellular space (Hillard and Jarrahian, 2000; Maccarrone et al., 2010). Moreover, an active intracellular binding of anandamide to TRPV1 and PPARs suggest an additional role of anandamide as a second messenger in intracellular signaling (Maccarrone et al., 2010). This new scenario contemplates the possibility that anandamide may act as an autocrine/paracrine ligand of CB receptors but also as an intracellular ligand to TRPV1 and PPARs; moreover, the presence of extracellular anandamide transporters would point anandamide as an endocrine messenger (Maccarrone et al., 2010).
Interestingly, neuronal damage can increase the production of endocannabinoids, which may provide a defense mechanism against toxicity (Stella et al., 1997; Marsicano et al., 2003). In the case of AEA, the synthesis is performed from the phosphatidylethanolamine existing in the cell membrane by the successive action of 2 enzymes, the N-acetyltransferase and phospholipase D (PLD). AEA action is determined by two processes that limit their availability, (i) transport from the synaptic cleft into the cell by passive diffusion or by a selective transporter, and (ii) the hydrolysis caused by two enzyme systems, mainly the fatty acid amide hydrolase (FAAH) but also monoacylglycerol lipase (MAGL; Di Marzo et al., 1994). 2-AG is the most abundant endogenous cannabinoid in the brain and its concentration is about 200 times that of AEA (Stella et al., 1997). The formation of 2-AG is mediated by phospholipase C and diacylglycerol lipase (DAGL), and also produced on demand. In contrast to AEA, MAGL seems to be more involved in 2-AG degradation than FAAH (Dinh et al., 2002).
Interest in the role that ECS may play in neurodegenerative processes is based on findings revealing that the augmentation of cannabinoid tone contributes to brain homeostasis and neuron survival, suggesting that may offer protection against the deleterious consequences of pathogenic molecules.
The Endogenous Cannabinoid System in AD Brains
The analysis of human post-mortem samples revealed some alterations in ECS composition and signaling in AD brains, although the bestowal of such modifications in the pathophysiology of the disease remains to be elucidated. The modifications described for CB1 receptors in AD are ambiguous. Whereas some authors have reported a significant reduction in the CB1 levels in cortical areas and in neurons distant from senile plaques (Ramírez et al., 2005; Solas et al., 2013), others have described no changes in the expression, distribution, or availability of CB1 receptors in cortex and hippocampus in AD (Benito et al., 2003; Lee et al., 2010; Mulder et al., 2011; Ahmad et al., 2013) or have failed to dissociate CB1 receptor expression changes from normal aging (Westlake et al., 1994). No correlation between CB1 levels and any AD molecular marker or cognitive status has been found (Solas et al., 2013). In contrast, there is no controversy regarding the significant increase of CB2 levels in AD brains, mainly corresponding to receptors expressed on microglia surrounding senile plaques (Ramírez et al., 2005; Solas et al., 2013). Interestingly, expression levels of CB2 receptors correlates with Aβ42 levels and plaque deposition, although not with cognitive status (Solas et al., 2013), suggesting that such pathogenic events induce CB2 receptor expression. Additionally, both CB1 and CB2 cannabinoid receptors in the AD brain are nitrosylated, and this could contribute to the impaired coupling of these receptors to downstream effector signaling molecules (Ramírez et al., 2005).
A few studies addressed other components of ECS in AD human samples. The first study analyzing endocannabinoid levels reported no differences between AD patients and healthy controls in the plasmatic concentrations of AEA and 2-AG (Koppel et al., 2009). However, a recent lipidomic study in post-mortem brain samples revealed lower AEA levels in midfrontal and temporal cortices in AD compared to control subjects, which inversely correlated with the neurotoxic brain Aβ42 peptide levels and cognitive deficiencies recorded in these patients, suggesting a contribution for Aβ42-dependent AEA impairment to cognitive dysfunction (Jung et al., 2012). Moreover, some alterations have been found in the contents and/or activity of the enzymes related to endocannabinoid synthesis and degradation in AD brains. Thus, the endocannabinoid metabolizing enzyme FAAH is up-regulated in AD both neuritic plaque-associated glia (Benito et al., 2003) and in peripheral blood mononuclear cells (D’Addario et al., 2012), and this could participate in the increase of AEA degradation in the vicinity of the senile plaque. Such FAAH overexpression may have at least two harmful consequences in disease progression, (i) neuronal AEA availability limitation and (ii) increase of pro-inflammatory molecules induced by AEA metabolites such as arachidonic acid (Calder, 2005). An elegant study revealed altered 2-AG signaling during late stages of AD due to the combination of impaired MAGL recruitment and increased DAGL levels, which subsidize synapse silencing in AD (Mulder et al., 2011). The same study failed to detect changes in PLD, FAAH, or TRPV1 protein levels in total hippocampal homogenates.
Clinical and Preclinical Evidence of Therapeutic Properties of Cannabinoids in AD
Most of the evidence accumulated sustaining the potential therapeutic utility of cannabinoids in AD has been obtained by using cellular and animal models that mimic a variety of AD-related changes, and they will be discussed later on in this review. However, it is of note that the scarce clinical data available also support the beneficial effects of cannabinoid compounds for treating some behavioral symptoms related to AD. Only a few clinical trials and one case report are available on the topic so far. In all the cases an analog of Δ9-THC (nabilone or dronabinol) was tested. Interestingly, one clinical trial including 15 AD patients resulted in a decreased severity of altered behavior and an increase in the body weight in AD patients, who were previously refusing food, after 6 weeks of dronabinol treatment. Side effects associated with cannabinoid administration were limited to euphoria, somnolence, and tiredness, but these did not warrant discontinuation of therapy (Volicer et al., 1997). Similarly, two pilot studies including eight patients with dementia concluded with a reduction in night-time agitation and behavioral disturbances, without adverse effects during the trial period with dronabinol (Walther et al., 2006, 2011). In line with these observations, the use of the cannabinoid receptor agonist nabilone correlated with prompt and dramatic improvements in the severe agitation and aggressiveness exhibited by an advanced AD patient who was refractory to anti-psychotic and anxiolytic medications (Passmore, 2008). In spite of the low number of patients included in these trials and the fact that none of them evaluated cognitive or neurodegenerative markers, the positive behavioral results are promising and represent valuable, albeit limited, information, considering that no remarkable side effects were reported. However, the revision in 2009 of the Cochrane Dementia and Cognitive Improvement Group Specialized Register found no evidences of cannabinoid effectiveness in the improvement of behavior and other parameters of dementia, and suggested that more controlled trials are needed to assess the effectiveness of cannabinoids in the treatment of dementia (Krishnan et al., 2009).
Effect of Cannabinoids on Aβ
Several in vitro and in vivo studies demonstrate that certain cannabinoid compounds confer neuroprotection against Aβ, as previously reported elsewhere (Ruiz-Valdepeñas et al., 2010). Some endocannabinoids such as AEA, 2-AG, and noladin ether, directly supplied to the cell culture or augmenting their availability through administration of endocannabinoid reuptake inhibitors, increased the viability of neurons after exposure to different toxic Aβ species (Milton, 2002; Chen et al., 2011; Harvey et al., 2012; Janefjord et al., 2013), and reduced Aβ-induced memory impairment in rats (van der Stelt et al., 2006). Similar positive results in the survival of neuronal cultures exposed to Aβ peptide were obtained with exogenous cannabinoids such as CBD (Iuvone et al., 2004; Janefjord et al., 2013), the selective CB1 receptor agonist arachidonyl-2-chloroethylamide (ACEA; Aso et al., 2012), the CB2 selective agonists JWH-015 and JWH-133, and the mixed CB1/CB2 receptor agonists Δ9-THC, HU-210, and WIN55,212-2 (Ramírez et al., 2005; Janefjord et al., 2013). The neuroprotective properties of exogenous cannabinoids have consistently been demonstrated to prevent memory deficits in Aβ-injected rats and mice for both synthetic CB1 (Haghani et al., 2012) and CB2 selective agonists (Wu et al., 2013), as well as mixed CB1/CB2 receptor agonists (Ramírez et al., 2005; Martín-Moreno et al., 2011; Fakhfouri et al., 2012) and natural CBD (Martín-Moreno et al., 2011). Moreover, chronic treatment with ACEA (Aso et al., 2012), JWH-133 (Martín-Moreno et al., 2012; Aso et al., 2013), or WIN55,212-2 (Martín-Moreno et al., 2012) resulted in cognitive improvement in two different transgenic mouse models of brain amyloidosis. The efficacy of the cannabinoid compounds in curbing the cognitive impairment was inversely proportional to the disease progression stage at the beginning of the treatment in the transgenic animals (Aso et al., 2012, 2013).
The mechanisms of action that underlie the cannabinoid neuroprotection against Aβ, which ultimately may lead to the memory improvement, are multiple and are assumed to act in parallel or interacting within them. Although most of these proposed protective mechanisms are related to the capacity of cannabinoids to indirectly mitigate the harmful effects of Aβ, as we will discuss in later sections of this review (i.e., inflammation, oxidative stress, excitotoxicity, aberrant cellular signaling), some authors also described direct effects of cannabinoids on Aβ processing. Thus, stimulation of CB2 receptors produced Aβ removal by human macrophages (Tolón et al., 2009; Wu et al., 2013) and favored Aβ transport through the choroid plexus (Martín-Moreno et al., 2012). This facilitation of Aβ clearance across the blood–brain barrier (BBB) was also demonstrated for the endocannabinoid 2-AG, a synthetic CB1/CB2 receptor agonist and MAGL inhibitors, but no FAAH, in in vitro an in vivo BBB models (Bachmeier et al., 2013). These findings could explain the reduction in Aβ levels and plaque burden observed in AD mouse models chronically treated with CB2 or CB1/CB2 receptor agonists (Martín-Moreno et al., 2012) and MAGL inhibitors (Chen et al., 2012; Piro et al., 2012). In contrast, no significant contribution of CB1 receptors in Aβ production, aggregation or clearance was reported after chronic treatment with ACEA (Aso et al., 2012) or HU-210 (Chen et al., 2010) in two different transgenic AD models. However, there is a study reporting a regulatory influence of CB1 receptor on APP processing since APP23 transgenic mice deficient for CB1 receptor exhibited reduced APP protein levels and Aβ plaque deposition, likely due to changes in intracellular APP transport, although the animals presented enhanced cognitive deficits (Stumm et al., 2013). Moreover, a recent publication revealed that Δ9-THC significantly increased the expression of neprilysin, an important endopeptidase for Aβ degradation, but not β-site APP cleaving enzyme 1 (BACE1), leading to a remarkable reduction of Aβ plaques in 5xFAD APP transgenic mice (Chen et al., 2013). This study failed, however, to clarify the specific role of CB1 or CB2 receptors in such Δ9-THC effect on Aβ clearance.
Cannabinoids on TAU Hyper-Phosphorylation
A role for cannabinoids in another AD pathological hallmark, tau hyper-phosphorylation, has also been described. First studies performed in cell cultures demonstrated that CBD, ACEA, and WIN55,212-2 inhibited tau protein hyper-phosphorylation in Aβ-stimulated PC12 neuronal cells (Esposito et al., 2006a,b). In the case of CBD, the effect was mediated through the reduction of the phosphorylated active form of glycogen synthase kinase 3β (GSK-3β), one of the known tau kinases (Ferrer et al., 2005), which in turn resulted in Wnt/β-catenin pathway rescue and consequent reduction of neuronal apoptosis (Esposito et al., 2006a). In contrast, the ACEA and WIN55,212-2 effect on tau hyper-phosphorylation was selectively mediated by CB1 receptor through the down-regulation of inducible nitric oxide synthase (iNOS) protein expression and nitric oxide (NO) production in Aβ-stimulated astroglioma cells co-cultured with the PC12 neuronal cells (Esposito et al., 2006b). In line with the described role for CB1 receptor on tau hyper-phosphorylation, chronic treatment with the CB1 selective agonist ACEA reduced the levels of tau phosphorylated at Thr181 site in the area surrounding Aβ plaques in treated APP/PS1 mice, probably through the ACEA-induced reduction in GSK-3β harmful activity (Aso et al., 2012). Moreover, a specific role for CB2 receptor in the modulation of tau phosphorylation was also suggested. Chronic JWH-133 administration reduced tau hyper-phosphorylation in the vicinity of Aβ plaques in APP/PS1 mice, which may be explained by decreased activity of GSK-3β, p38, and stress-activated protein kinase/c-Jun NH(2)-terminal kinase (SAPK/JNK) kinases in the treated animals (Aso et al., 2013).
Confirming these observations, a recent study reported a marked reduction in neurofibrillary tangles in parkin-null, human tau overexpressing (PK-/-/TauVLW) mice, a model of complex frontotemporal dementia, parkinsonism, and lower motor neuron disease, after prolonged exposure to Sativex®, an already approved medicine based on mixed Δ9-THC and CBD natural extracts (Casarejos et al., 2013). The authors suggested the cannabinoid potentiation of autophagy or improvement of redox state as likely mechanisms accounting for the reduction in tau deposition.
Anti-Inflammatory Properties of Cannabinoids
Neuroinflammation, initially manifested as microglial activation, is a prominent feature in AD which contributes to progressive cell damage and neuron loss (Akiyama et al., 2000; Hensley, 2010; Sardi et al., 2011). As CB2 receptors are essentially expressed in the immune system including microglial cells, where they are known to inhibit microglia-mediated neurotoxicity, the main interest in the role of cannabinoids as anti-inflammatory agents in several diseases concurring with inflammation has focused on compounds acting on CB2 receptors (Cabral and Griffin-Thomas, 2009). In the case of AD, several studies reported that activation of CB2 receptors reduced the neuroinflammatory response to Aβ insults in different models of the disease. After the inoculation of Aβ into the rat or mouse brain, selective or mixed CB2 receptor agonists reduced microglial response and pro-inflammatory molecule production in a plethora of studies (Ramírez et al., 2005; van der Stelt et al., 2006; Esposito et al., 2007; Fakhfouri et al., 2012; Wu et al., 2013). Similarly, selective CB2 receptor agonists decreased the number of activated microglial cells surrounding Aβ deposition and the levels of pro-inflammatory cytokines in at least two APP transgenic models (Martín-Moreno et al., 2012; Aso et al., 2013). Moreover, the Δ9-THC and CBD natural mixture present in Sativex® also blunted the microglial reactivity in a genetic tauopathy model (Casarejos et al., 2013), although no evidence of direct implication of CB2 receptors or other receptors in such effects was provided. In fact, other mechanisms related to ECS components distinct from CB2 receptors could explain the anti-inflammatory effects of the Sativex® preparation. As noted above, Δ9-THC is a partial agonist of CB1 receptors, which could also play a role in the AD inflammatory process according to a recent study demonstrating that chronic treatment with the selective agonist ACEA reduced the astrocytic expression of the pro-inflammatory cytokine interferon-γ in APP/PS1 transgenic mice (Aso et al., 2012). Additionally, CBD, which has no affinity for CB1 or CB2 receptors, also presents anti-inflammatory properties in AD models (Esposito et al., 2006a; Martín-Moreno et al., 2011). The precise site at which CBD could exert its neuroinflammatory and neuroprotective effects is still not fully elucidated, but some findings point to the selective involvement of PPARγ in such CBD properties (Esposito et al., 2011).
The enzymes related to AEA and 2-AG degradation also contribute to modulating the inflammatory process in AD models. FAAH is expressed in both neurons and astrocytes, where it may play a role in the response to inflammation. In fact, an astrocyte-specific increase in FAAH expression is markedly maintained in neuroinflammatory conditions including amyloidosis, which was assumed to contribute to the harmful processes induced by toxic insults because of the reduction in endocannabinoid tone (Benito et al., 2003). However, cortical mouse astrocytes genetically modified to lack FAAH exhibited a pro-inflammatory phenotype when exposed to Aβ, characterized by an increase in cytokine concentration and cell death probably due to the modification of signaling cascades involved in cell survival and inflammation, such as extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), p38 mitogen-activated protein kinase (p38MAPK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), as well as to the increase in inflammatory mediators such as iNOS and cyclooxygenase (COX-2; Benito et al., 2012). The authors demonstrated that these processes involved PPAR-α, PPAR-γ, and TRPV1, but not CB1 or CB2 receptors. Yet, the pharmacological blockade of FAAH in cell cultures did not lead astrocytes to a pro-inflammatory phenotype, indicating that the observed effects in astrocytes lacking FAAH could be due to compensatory changes that result from the potentially prolonged enhancement of N-acylethanolamines. These data suggest that an excessively prolonged enhancement of the endocannabinoid tone may have harmful consequences. In contrast, the genetic inactivation of MAGL, an enzyme known to hydrolyze endocannabinoids and generate the primary arachidonic acid pool for neuroinflammatory prostaglandins (Nomura et al., 2011), attenuated neuroinflammation and lowered Aβ levels and plaques in APP/PS1 mice (Piro et al., 2012). These observations were confirmed by the pharmacological blockade of MAGL, which recapitulated the cytokine-lowering effects through reduced prostaglandin production, rather than enhanced endocannabinoid signaling.
Cannabinoid Actions on Mitochondria Activity: Oxidative Stress and Energy Metabolism
Mitochondria are vital cellular components essential for ATP production and calcium homeostasis. The relevance of these organelles in neurons is even greater than in other cell types because neurons are highly demanding energy cells mainly dependent on aerobic oxidative phosphorylation, due to their limited capacity for glycolysis. Long axons require energy transport over long distances, and synaptic transmission depends on calcium signals. Mitochondria are abundant in presynaptic nerve terminals where they provide energy for sustained neurotransmitter release. Therefore, defects in mitochondrial activity can have severe consequences for the cell, including energetic failure associated with decreased ATP production and apoptosis resulting from the release of death factors and impaired calcium-buffering capacity. Moreover, alterations in the protein complexes of the respiratory chain located in the inner mitochondrial membrane lead to electron transport leakage that enables the production of ROS, which may overwhelm the capacity of the anti-oxidant systems existing in cells to counteract free radical damage, with the subsequent oxidative damage produced to proteins, DNA, RNA, and lipids. Numerous studies have linked mitochondrial dysfunction to neurodegenerative diseases, including AD (Ferrer, 2009; Ankarcrona et al., 2010; Burchell et al., 2010). Altered mitochondrial function appears early in time in AD, even preceding the characteristic Alzheimer pathology in mouse models, and ultimately leads to exhausted neurons as a result of the convergence of reduced energy production, increased energy demand, and excessive oxidative stress (Ferrer, 2009).
The anti-oxidant properties of cannabis derivatives, notably CBD, were demonstrated early in cell cultures exposed to toxic glutamate levels (Hampson et al., 1998, 2000). In line with these findings, CBD prevented ROS production and lipid peroxidation in PC12 neuronal cells exposed to Aβ, as well as reduced apoptosis from reduced caspase 3 levels, and in counteracting the Aβ-induced increase in intracellular calcium concentration (Iuvone et al., 2004). CBD also reduced, in similar conditions, the levels of nitrite (NO), a potent oxidant reactive molecule, as well as the expression of iNOS, one of the enzymes responsible for the synthesis of NO (Esposito et al., 2006b, 2011). Moreover, other cannabinoid compounds exhibited anti-oxidant properties in animal models of AD. Thus, the selective CB2 receptor agonist JWH-133 reduced hydroxynonenal adducts, derived from lipid peroxidation, and enhanced the levels of the superoxide dismutases SOD1 and SOD2 in the vicinity of plaques in APP/PS1 mice, indicating the role of CB2 receptors in reducing oxidative stress damage and to promote responses against such damage (Aso et al., 2013). A reduction of free radicals and mitochondrial activity was also suggested in a mouse model of tauopathy exposed to chronic treatment with the Sativex® mixture of Δ9-THC and CBD (Casarejos et al., 2013).
An additional topic deserving attention is the putative role of cannabinoid receptors in the regulation of neuronal energy metabolism. The little information available to date supports the direct control of CB1 over neuronal respiration and energy production. One study, using anti-CB1 receptor antibodies, revealed CB1 receptor protein localization in approximately 30% of neuronal mitochondria, which when activated by exogenous or endogenous cannabinoids reduces the respiratory chain complex I activity and oxygen consumption, likely through cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) signaling (Bénard et al., 2012). These findings are in agreement with previous observations showing that AEA, Δ9-THC, and HU-210, all of them partial CB1 agonists, significantly decreased oxygen consumption and mitochondrial membrane potential (Athanasiou et al., 2007). However, these data must be interpreted with caution as commercial anti-CB1 receptor antibodies also recognize the mitochondrial protein stomatin-like protein 2 and that the formerly described effect of WIN 55,212-2 on mitochondrial complex III is in fact not detectable in isolated mitochondrial preparations (Morozov et al., 2013).
Cannabinoids Modulate Neurotransmission
Nowadays the approved drugs for treating AD are based on acetylcholine esterase (AChE) inhibitors, which increase the ACh availability partially palliating this neurotransmitter deficiency in AD patients, or they are non-competitive antagonists of the N-methyl D-aspartate (NMDA) receptor, which reduce calcium influx and limit excitotoxicity. Interestingly, certain cannabinoid compounds act on the same targets than current medications, resulting in similar or enhanced beneficial effects. For instance, Δ9-THC competitively inhibits AChE, thus increasing ACh levels, as well as preventing AChE-induced Aβ aggregation by binding in the peripheral anionic site of AChE, the critical region involved in amyloidogenesis (Eubanks et al., 2006). The synthetic cannabinoid HU-211 acts as a stereoselective inhibitor of NMDA receptors, and thus protects cells from NMDA induced neurotoxicity (Feigenbaum et al., 1989; Eshhar et al., 1993; Nadler et al., 1993). In the case of HU-211, its neuroprotective activity is due to the direct binding to NMDA receptors, not to cannabinoid receptors, but the broadly accepted cannabinoid-mediated neuroprotection against excitotoxicity can be achieved through a number of other different mechanisms, including inhibition of presynaptic glutamate release (Marsicano et al., 2003), blockage of voltage-dependent calcium channels (Mackie and Hille, 1992; Twitchell et al., 1997) and inhibition of calcium release from ryanodine sensitive stores (Zhuang et al., 2005), which mostly imply the direct or indirect participation of CB1 receptors.
Other Effects of Cannabinoids in AD
Several other mechanisms seem to contribute to the therapeutic properties of cannabinoid compounds in AD, although they have not been fully characterized. Among them, we can note the capacity of endocannabinoids to prevent Aβ-mediated lysosomal destabilization in cultured neurons, reducing in this way the apoptotic signaling, which in turn sustains cell survival (Noonan et al., 2010). Compromised neurogenesis is an early event in AD that limits neuronal replacement once progressive neuronal loss takes place in the brain, contributing to cognitive deterioration (Lazarov and Marr, 2010). Interestingly, AEA and CBD have been described as promoting neurogenesis in response to Aβ insult (Esposito et al., 2011; Tanveer et al., 2012), suggesting an additional beneficial effect of cannabinoids in AD.
Another aspect as yet unexplored is the interaction of cannabinoids with neurotrophic factors in AD. Cannabinoids are capable of increasing brain-derived neurotrophic factor (BDNF; Khaspekov et al., 2004), a neurotrophin reduced in the AD brain (Lee et al., 2005; Peng et al., 2009), which is known to confer protection against excitotoxicity and to promote neurogenesis (Scharfman et al., 2005) and neuronal plasticity; all of these processes play a role in AD. However, there is still no evidence about the implication that such cannabinoid-induced BDNF promotion could have on the cognitive or pathological aspects of AD. Similarly, little is known about the participation of cannabinoid signaling in the impaired function of degradation pathways such as autophagy and ubiquitin–proteasome, which are known to play a relevant role in AD progression. The impairment in these catabolic processes results in accumulation of aggregate-prone proteins, altered mitochondria and other cellular organelles that might exacerbate neurodegenerative process (Nixon et al., 2005; Harris and Rubinsztein, 2011). To date, only one study has reported beneficial effects of cannabinoid-induced autophagy in a model of tauopathy (Casarejos et al., 2013), but this has opened the possibility of exploring in greater detail the involvement of ECS in promoting degradation of toxic components in neurodegenerative diseases. Finally, the effect of cannabinoids on the regulation of cerebral blood flow may contribute to their potential benefits on AD. A number of studies have demonstrated that certain cannabinoids produce vasodilatation of brain blood vessels and increase cerebral blood flow (Ellis et al., 1995; Wagner et al., 2001; Pacher et al., 2005; Iring et al., 2013). Considering that cerebral blood flow in AD contributes to the reduction of oxygen and nutrients in brain (Iadecola, 2004), it can be suggested that treatments improving cerebral perfusion such as cannabinoids are advantageous in AD. Taken together, available information suggest that cannabinoids may have multiple effects on AD by acting not only as anti-oxidant and anti-inflammatory agents, but also modulating a plethora of factors which contribute to the pathogenesis of AD as altered Aβ metabolism, autophagy, trophic factor deficiencies, and impaired blood flow.
Conclusions and Therapeutic Implications
Considering the numerous complex pathological mechanisms involved in the progression of AD, treatments targeting a single causal or modifying factor offer limited benefit. Cannabinoids, however, exhibit pleiotropic activity, targeting in parallel several processes that play key roles in AD, including Aβ and tau aberrant processing, neuroinflammation, excitotoxicity, mitochondrial dysfunction, and oxidative stress. Cannabinoids improve behavioral disturbances, as well. These effects are summarized in Figure 1. Then, because of these widespread properties of cannabinoid compounds, targeting the ECS could represent a unique and reliable opportunity to advance toward an effective therapy against the AD. Moreover, cannabinoids might represent a safe low-cost therapy, with their natural origin and low side effects profile. From our point of view, the success of cannabinoid-based therapy in AD could be increased taking into account two important aspects: (i) the use of a combination of compounds that cover the whole spectrum of therapeutic properties described for cannabinoids, i.e., combination of CB1 and CB2 receptors agonists plus CBD, which presents interesting neuroprotective properties spite of its mechanism of action remaining poorly understood, and (ii) the early initiation of the treatment in the neurodegenerative process, which ensures the integrity of the ECS target components and increases the possibility of curbing the exponential degenerative progression toward dementia.
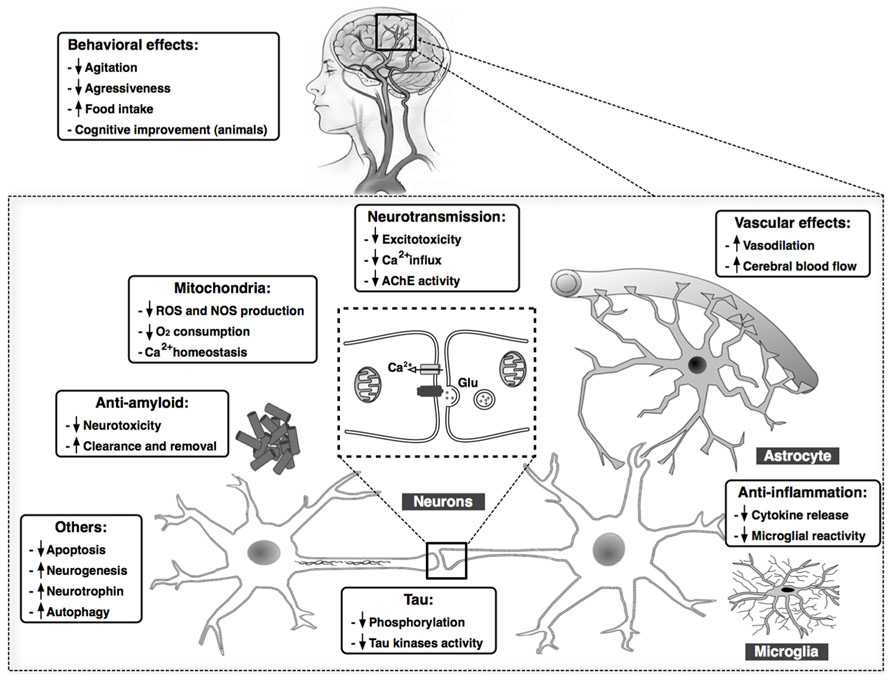
FIGURE 1. Summary of the main findings demonstrating beneficial effects of cannabinoid compounds in AD models. Cannabinoids may target in parallel several processes that play key roles in AD, including Aβ and tau aberrant processing, chronic inflammatory responses, excitotoxicity, mitochondrial dysfunction, and oxidative stress, among others. Clinical data also reveal an improvement in behavioral in patients with AD after treatment with cannabinoids.
The main concerns regarding the use of cannabis derivatives in medicine are related with the psychoactivity of some cannabinoids, especially Δ9-THC, which may disrupt short-term memory, working memory, and attention skills mainly acting through CB1 receptors, as well as with the potential Δ9-THC dependence occurring after long-term use. However, the therapeutic effects of cannabinoids must be clearly dissociated from the risks of abuse and addiction linked to the recreational use of cannabis derivatives. First, the CB1 agonists with potential psychoactivity used in experimental models to demonstrate the therapeutic properties were administered at doses substantially lower than those producing psychoactive effects and cannabis dependence (Maldonado et al., 2011). Second, the preferred therapeutic cannabinoid combination includes CBD, which is known to mitigate the negative consequences on cognition of Δ9-THC administration (Fadda et al., 2004), and therefore insure the avoidance of such undesirable effects. Finally, the brain context in healthy subjects consuming cannabis enriched in Δ9-THC for recreational purposes is completely different from that of AD patients subjected to very determined combinations of cannabinoid species, in terms of ECS organization and neuronal signaling. In conclusion, in light of the polyvalent properties for the treatment of AD and the limited side effects exhibited by these compounds, progress toward a clinical trial to test the capacity of cannabinoids to curb this neurodegenerative disease seems to be fully justified.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank T. Yohannan for editorial assistance. Authors’ work is supported by Agrupació Mútua Foundation, Mutua Madrileña Foundation, and CIBERNED (Institute of Health Carlos III, Spanish Ministry of Economy and Competitiveness).
References
Ahmad, R., Goffin, K., Van den Stock, J., De Winter, F. L., Cleeren, E., Bormans, G., et al. (2013). In vivo type 1 cannabinoid receptor availability in Alzheimer’s disease. Eur. Neuropsychopharmacol. 24, 242–250. doi: 10.1016/j.euroneuro.2013.10.002
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., et al. (2000). Inflammation and Alzheimer disease. Neurobiol. Aging 21, 383–421. doi: 10.1016/S0197-4580(00)00124-X
Ankarcrona, M., Mangialasche, F., and Winblad, B. (2010). Rethinking Alzheimer’s disease therapy: are mitochondria the key? J. Alzheimers Dis. 20(Suppl. 2), S579–S590. doi: 10.3233/JAD-2010-100327
Aso, E., Juvés, S., Maldonado, R., and Ferrer, I. (2013). CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J. Alzheimers Dis. 35, 847–858. doi: 10.3233/JAD-130137
Aso, E., Palomer, E., Juvés, S., Maldonado, R., Muñoz, F. J., and Ferrer, I. (2012). CB1 agonist ACEA protects neurons and reduces the cognitive impairment of AβPP/PS1 mice. J. Alzheimers Dis. 30, 439–459. doi: 10.3233/JAD-2012-111862
Athanasiou, A., Clarke, A. B., Turner, A. E., Kumaran, N. M., Vakilpour, S., Smith, P. A., et al. (2007). Cannabinoid receptor agonists are mitochondrial inhibitors: a unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochem. Biophys. Res. Commun. 364, 131–137. doi: 10.1016/j.bbrc.2007.09.107
Bachmeier, C., Beaulieu-Abdelahad, D., Mullan, M., and Paris, D. (2013). Role of the cannabinoid system in the transit of beta-amyloid across the blood–brain barrier. Mol. Cell. Neurosci. 56, 255–262. doi: 10.1016/j.mcn.2013.06.004
Bénard, G., Massa, F., Puente, N., Lourenço, J., Bellocchio, L., Soria-Gómez, E., et al. (2012). Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 15, 558–564. doi: 10.1038/nn.3053
Benito, C., Núñez, E., Tolón, R. M., Carrier, E. J., Rábano, A., Hillard, C. J., et al. (2003). Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 23, 11136–11141.
Benito, C., Tolón, R. M., Castillo, A. I., Ruiz-Valdepeñas, L., Martínez-Orgado, J. A., Fernández-Sánchez, F. J., et al. (2012). β-Amyloid exacerbates inflammation in astrocytes lacking fatty acid amide hydrolase through a mechanism involving PPAR-α, PPAR-γ and TRPV1, but not CB1 or CB2 receptors. Br. J. Pharmacol. 166, 1474–1489. doi: 10.1111/j.1476-5381.2012.01889.x
Bertram, L., and Tanzi, R. E. (2011). “Genetics of Alzheimer’s disease,” in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd Edn, eds D. Dickson and R. Weller (West Sussex: Wiley-Blackwell), 51–91. doi: 10.1002/9781444341256.ch9
Braak, H., and Braak, E. (1999). “Temporal sequence of Alzheimer’s disease-related pathology,” in Cerebral Cortex, Vol. 14, Neurodegenerative and Age-related Changes in Structure and Function of Cerebral Cortex, eds A. Peters and J. H. Morrison (New York: Kluwer Academic/Plenum Publishers), 475–512. doi: 10.1007/978-1-4615-4885-0_14
Brusco, A., Tagliaferro, P. A., Saez, T., and Onaivi, E. S. (2008). Ultrastructural localization of neuronal brain CB2 cannabinoid receptors. Ann. N. Y. Acad. Sci. 1139, 450–457. doi: 10.1196/annals.1432.037
Burchell, V. S., Gandhi, S., Deas, E., Wood, N. W., Abramov, A. Y., and Plun-Favreau, H. (2010). Targeting mitochondrial dysfunction in neurodegenerative disease: part I. Expert Opin. Ther. Targets 14, 369–385. doi: 10.1517/14728221003652489
Cabral, G. A., and Griffin-Thomas, L. (2009). Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 11:e3. doi: 10.1017/S1462399409000957
Calder, P. C. (2005). Polyunsaturated fatty acids and inflammation. Biochem. Soc. Trans. 33, 423–427. doi: 10.1042/BST0330423
Casarejos, M. J., Perucho, J., Gómez, A., Muñoz, M. P., Fernández-Estévez, M., Sagredo, O., et al. (2013). Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J. Alzheimers Dis. 35, 525–539. doi: 10.3233/JAD-130050
Chen, B., Bromley-Brits, K., He, G., Cai, F., Zhang, X., and Song, W. (2010). Effect of synthetic cannabinoid HU210 on memory deficits and neuropathology in Alzheimer’s disease mouse model. Curr. Alzheimer Res. 7, 255–261. doi: 10.2174/156720510791050948
Chen, R., Zhang. J., Fan, N., Teng, Z. Q., Wu, Y., Yang, H., et al. (2013). Δ(9)-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell 155, 1154–1165. doi: 10.1016/j.cell.2013.10.042
Chen, R., Zhang, J., Wu, Y., Wang, D., Feng, G., Tang, Y. P., et al. (2012). Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2, 1329–1339. doi: 10.1016/j.celrep.2012.09.030
Chen, X., Zhang, J., and Chen, C. (2011). Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neuroscience 178, 159–168. doi: 10.1016/j.neuroscience.2011.01.024
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
D’Addario, C., Di Francesco, A., Arosio, B., Gussago, C., Dell’Osso, B., Bari, M., et al. (2012). Epigenetic regulation of fatty acid amide hydrolase in Alzheimer disease. PLoS ONE 7:e39186. doi: 10.1371/journal.pone.0039186
Di Marzo, V., Fontana, A., Cadas, H., Schinelli, S., Cimino, G., Schwartz, J. C., et al. (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372, 686–691. doi: 10.1038/372686a0
Dinh, T. P., Carpenter, D., Leslie, F. M., Freund, T. F., Katona, I., Sensi, S. L., et al. (2002). Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. U.S.A. 99, 10819–10824. doi: 10.1073/pnas.152334899
Duyckaerts, C., and Dickson, D. (2011). “Neuropathology of Alzheimer’s disease and its variants,” in Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd Edn, eds D. Dickson and R. Weller (West Sussex, Wiley-Blackwell), 62–91.
Ellis, E. F., Moore, S. F., and Willoughby, K. A. (1995). Anandamide and delta 9-THC dilation of cerebral arterioles is blocked by indomethacin. Am. J. Physiol. 269, H1859–H1864.
Eshhar, N., Striem, S., and Biegon, A. (1993). HU-211, a non-psychotropic cannabinoid, rescues cortical neurones from excitatory amino acid toxicity in culture. Neuroreport 5, 237–240. doi: 10.1097/00001756-199312000-00013
Esposito, G., De Filippis, D., Carnuccio, R., Izzo, A. A., and Iuvone, T. (2006a). The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J. Mol. Med. 84, 253–258. doi: 10.1007/s00109-005-0025-1
Esposito, G., De Filippis, D., Steardo, L., Scuderi, C., Savani, C., Cuomo, V., et al. (2006b). CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci. Lett. 404, 342–346. doi: 10.1016/j.neulet.2006.06.012
Esposito, G., Iuvone, T., Savani, C., Scuderi, C., De Filippis, D., Papa, M., et al. (2007). Opposing control of cannabinoid receptor stimulation on amyloid-beta-induced reactive gliosis: in vitro and in vivo evidence. J. Pharmacol. Exp. Ther. 322, 1144–1152. doi: 10.1124/jpet.107.121566
Esposito, G., Scuderi, C., Valenza, M., Togna, G. I., Latina, V., De Filippis, D., et al. (2011). Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 6:e28668. doi: 10.1371/journal.pone.0028668
Eubanks, L. M., Rogers, C. J., Beuscher, A. E. IV, Koob, G. F., Olson, A. J., Dickerson, T. J., et al. (2006). A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol. Pharm. 3, 773–777. doi: 10.1021/mp060066m
Fadda, P., Robinson, L., Fratta, W., Pertwee, R. G., and Riedel, G. (2004). Differential effects of THC- or CBD-rich cannabis extracts on working memory in rats. Neuropharmacology 47, 1170–1179. doi: 10.1016/j.neuropharm.2004.08.009
Fakhfouri, G., Ahmadiani, A., Rahimian, R., Grolla, A. A., Moradi, F., and Haeri, A. (2012). WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 63, 653–666. doi: 10.1016/j.neuropharm.2012.05.013
Feigenbaum, J. J., Bergmann, F., Richmond, S. A., Mechoulam, R., Nadler, V., Kloog, Y., et al. (1989). Nonpsychotropic cannabinoid acts as a functional N-methyl-D-aspartate receptor blocker. Proc. Natl. Acad. Sci. U.S.A. 86, 9584–9587. doi: 10.1073/pnas.86.23.9584
Ferrer, I. (2009). Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 41, 425–431. doi: 10.1007/s10863-009-9243-5
Ferrer, I. (2012). Defining Alzheimer as a common age-related neurodegenerative process not inevitably leading to dementia. Prog. Neurobiol. 97, 38–51. doi: 10.1016/j.pneurobio.2012.03.005
Ferrer, I., Gómez-Isla, T., Puig, B., Freixes, M., Ribé, E., Dalfó, E., et al. (2005). Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2, 3–18. doi: 10.2174/1567205052772713
Frost, B., and Diamond, M. I. (2010). Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159. doi: 10.1038/nrn2786
Goedert, M., Clavaguera, F., and Tolnay, M. (2010). The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325. doi: 10.1016/j.tins.2010.04.003
Haghani, M., Shabani, M., Javan, M., Motamedi, F., and Janahmadi, M. (2012). CB1 cannabinoid receptor activation rescues amyloid β-induced alterations in behaviour and intrinsic electrophysiological properties of rat hippocampal CA1 pyramidal neurones. Cell Physiol. Biochem. 29, 391–406. doi: 10.1159/000338494
Hampson, A. J., Grimaldi, M., Axelrod, J., and Wink, D. (1998). Cannabidiol and (-) D9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. U.S.A. 95, 8268–8273. doi: 10.1073/pnas.95.14.8268
Hampson, A. J., Grimaldi, M., Lolic, M., Wink, D., Rosenthal, R., and Axelrod, J. (2000). Neuroprotective antioxidants from marijuana. Ann. N.Y. Acad. Sci. 899, 274–282. doi: 10.1111/j.1749-6632.2000.tb06193.x
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x
Harris, H., and Rubinsztein, D. C. (2011). Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 8, 108–117. doi: 10.1038/nrneurol.2011.200
Harvey, B. S., Ohlsson, K. S., Mååg, J. L., Musgrave, I. F., and Smid, S. D. (2012). Contrasting protective effects of cannabinoids against oxidative stress and amyloid-β evoked neurotoxicity in vitro. Neurotoxicology 33, 138–146. doi: 10.1016/j.neuro.2011.12.015
Hensley, K. (2010). Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimers Dis. 21, 1–14. doi: 10.3233/JAD-2010-1414
Hillard, C. J., and Jarrahian, A. (2000). The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem. Phys. Lipids 108, 123–134. doi: 10.1016/S0009-3084(00)00191-2
Howlett, A. C. (2005). Cannabinoid receptor signaling. Handb. Exp. Pharmacol. 168, 53–79. doi: 10.1007/3-540-26573-2_2
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iring, A., Ruisánchez, É., Leszl-Ishiguro, M., Horváth, B., Benkõ, R., Lacza, Z., et al. (2013). Role of endocannabinoids and cannabinoid-1 receptors in cerebrocortical blood flow regulation. PLoS ONE 8:e53390. doi: 10.1371/journal.pone.0053390
Iuvone, T., Esposito, G., Esposito, R., Santamaria, R., Di Rosa, M., and Izzo, A. A. (2004). Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 89, 134–141. doi: 10.1111/j.1471-4159.2003.02327.x
Janefjord, E., Mååg, J. L., Harvey, B. S., and Smid, S. D. (2013). Cannabinoid effects on β amyloid fibril and aggregate formation, neuronal and microglial-activated neurotoxicity in vitro. Cell. Mol. Neurobiol. 34, 31–42. doi: 10.1007/s10571-013-9984-x
Jung, K. M., Astarita, G., Yasar, S., Vasilevko, V., Cribbs, D. H., Head, E., et al. (2012). An amyloid β42-dependent deficit in anandamide mobilization is associated with cognitive dysfunction in Alzheimer’s disease. Neurobiol. Aging 33, 1522–1532. doi: 10.1016/j.neurobiolaging.2011.03.012
Keller, J. N., Hanni, K. B., and Markesbery, W. R. (2000). Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 75, 436–439. doi: 10.1046/j.1471-4159.2000.0750436.x
Khaspekov, L. G., Brenz-Verca, M. S., Frumkina, L. E., Hermann, H., Marsicano, G., and Lutz, B. (2004). Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 19, 1691–1698. doi: 10.1111/j.1460-9568.2004.03285.x
Koppel, J., Bradshaw, H., Goldberg, T. E., Khalili, H., Marambaud, P., Walker, M. J., et al. (2009). Endocannabinoids in Alzheimer’s disease and their impact on normative cognitive performance: a case–control and cohort study. Lipids Health Dis. 8, 2. doi: 10.1186/1476-511X-8-2
Kovacs, G. G., Alafuzoff, I., Al-Sarraj, S., Arzberger, T., Bogdanovic, N., Capellari, S., et al. (2008). Mixed brain pathologies in dementia: the BrainNet Europe Consortium experience. Dement. Geriatr. Cogn. Disord. 26, 343–350. doi: 10.1159/000161560
Krishnan, S., Cairns, R., and Howard, R. (2009). Cannabinoids for the treatment of dementia. Cochrane Database Syst. Rev. 2:CD007204. doi: 10.1002/14651858.CD007204.pub2
Lazarov, O., and Marr, R. A. (2010). Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol. 223, 267–281. doi: 10.1016/j.expneurol.2009.08.009
Lee, J., Fukumoto, H., Orne, J., Klucken, J., Raju, S., Vanderburg, C. R., et al. (2005). Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp. Neurol. 194, 91–96. doi: 10.1016/j.expneurol.2005.01.026
Lee, J. H., Agacinski, G., Williams, J. H., Wilcock, G. K., Esiri, M. M., Francis, P. T., et al. (2010). Intact cannabinoid CB1 receptors in the Alzheimer’s disease cortex. Neurochem. Int. 57, 985–989. doi: 10.1016/j.neuint.2010.10.010
Maccarrone, M., Dainese, E., and Oddi, S. (2010). Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem. Sci. 35, 601–608. doi: 10.1016/j.tibs.2010.05.008
Mackie, K., and Hille, B. (1992). Cannabinoids inhibit N-type calcium channels in neuroblastoma–glioma cells. Proc. Natl. Acad. Sci. U.S.A. 89, 3825–3829. doi: 10.1073/pnas.89.9.3825
Maldonado, R., Berrendero, F., Ozaita, A., and Robledo, P. (2011). Neurochemical basis of cannabis addiction. Neuroscience 181, 1–17. doi: 10.1016/j.neuroscience.2011.02.035
Marsicano, G., Goodenough, S., Monory, K., Hermann, H., Eder, M., Cannich, A., et al. (2003). CB1 receptors and on-demand defense against excitotoxicity. Science 302, 84–88. doi: 10.1126/science.1088208
Martín, V., Fabelo, N., Santpere, G., Puig, B., Marín, R., Ferrer, I., et al. (2010). Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimers Dis. 19, 489–502. doi: 10.3233/JAD-2010-1242
Martín-Moreno, A. M., Brera, B., Spuch, C., Carro, E., García-García, L., Delgado, M., et al. (2012). Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation 9, 8. doi: 10.1186/1742-2094-9-8
Martín-Moreno, A. M., Reigada, D., Ramírez, B. G., Mechoulam, R., Innamorato, N., Cuadrado, A., et al. (2011). Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol. Pharmacol. 79, 964–973. doi: 10.1124/mol.111.071290
Milton, N. G. (2002). Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide. Neurosci. Lett. 332, 127–130. doi: 10.1016/S0304-3940(02)00936-9
Morozov, Y. M., Dominguez, M. H., Varela, L., Shanabrough, M., Koch, M., Horvath, T. L., et al. (2013). Antibodies to cannabinoid type 1 receptor co-react with stomatin-like protein 2 in mouse brain mitochondria. Eur. J. Neurosci. 38, 2341–2348. doi: 10.1111/ejn.12237
Mulder, J., Zilberter, M., Pasquare, S. J., Alpar, A., Schulte, G., Ferreira, S. G., et al. (2011). Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 134, 1041–1060. doi: 10.1093/brain/awr046
Nadler, V., Mechoulam, R., and Sokolovsky, M. (1993). The non-psychotropic cannabinoid (+)-(3S,4S)-7-hydroxy-delta 6-tetrahydrocannabinol 1,1-dimethylheptyl (HU-211) attenuates N-methyl-D-aspartate receptor-mediated neurotoxicity in primary cultures of rat forebrain. Neurosci. Lett. 162, 43–45. doi: 10.1016/0304-3940(93)90555-Y
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122.
Nomura, D. K., Morrison, B. E., Blankman, J. L., Long, J. Z., Kinsey, S. G., Marcondes, M. C., et al. (2011). Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334, 809–813. doi: 10.1126/science.1209200
Noonan, J., Tanveer, R., Klompas, A., Gowran, A., McKiernan, J., and Campbell, V. A. (2010). Endocannabinoids prevent β-amyloid-mediated lysosomal destabilization in cultured neurons. J. Biol. Chem. 285, 38543–38554. doi: 10.1074/jbc.M110.162040
Onaivi, E. S., Ishiguro, H., Gong, J. P., Patel, S., Meozzi, P. A., Myers, L., et al. (2008). Functional expression of brain neuronal CB2 cannabinoid receptors are involved in the effects of drugs of abuse and in depression. Ann. N.Y. Acad. Sci. 1139, 434–449. doi: 10.1196/annals.1432.036
Pacher, P., Bátkai, S., and Kunos, G. (2005). Cardiovascular pharmacology of cannabinoids. Handb. Exp. Pharmacol. 168, 599–625. doi: 10.1007/3-540-26573-2_20
Pamplona, R., Dalfó, E., Ayala, V., Bellmunt, M. J., Prat, J., Ferrer, I., et al. (2005). Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation: effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 280, 21522–21530. doi: 10.1074/jbc.M502255200
Passmore, M. J. (2008). The cannabinoid receptor agonist nabilone for the treatment of dementia-related agitation. Int. J. Geriatr. Psychiatry 23, 116–117. doi: 10.1002/gps.1828
Peng, S., Garzon, D. J., Marchese, M., Klein, W., Ginsberg, S. D., Francis, B. M., et al. (2009). Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 29, 9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009
Pertwee, R. G. (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 153, 199–215. doi: 10.1038/sj.bjp.0707442
Pertwee, R. G., Howlett, A. C., Abood, M. E., Alexander, S. P., Di Marzo, V., Elphick, M. R., et al. (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol. Rev. 62, 588–631. doi: 10.1124/pr.110.003004
Piomelli, D. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884. doi: 10.1038/nrn1247
Piro, J. R., Benjamin, D. I., Duerr, J. M., Pi, Y., Gonzales, C., Wood, K. M., et al. (2012). A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 1, 617–623. doi: 10.1016/j.celrep.2012.05.001
Ramírez, B. G., Blázquez, C., Gómez del Pulgar, T., Guzmán M., and de Ceballos, M. L. (2005). Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J. Neurosci. 25, 1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005
Ruiz-Valdepeñas, L., Benito, C., Tolón, R. M., Martínez-Orgado, J. A., and Romero, J. (2010). The endocannabinoid system and amyloid-related diseases. Exp. Neurol. 224, 66–73. doi: 10.1016/j.expneurol.2010.03.024
Sardi, F., Fassina, L., Venturini, L., Inquuscio, M., Guerriero, F., Rolfo, E., et al. (2011). Alzheimer’s disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun. Rev. 11, 149–153. doi: 10.1016/j.autrev.2011.09.005
Scharfman, H., Goodman, J., Macleod, A., Phani, S., Antonelli, C., and Croll, S. (2005). Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 192, 348–356. doi: 10.1016/j.expneurol.2004.11.016
Selkoe, D. J. (2012). Preventing Alzheimer’s disease. Science 337, 1488–1492. doi: 10.1126/science.1228541
Silvestri, C., and Di Marzo, V. (2013). The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 17, 475–490. doi: 10.1016/j.cmet.2013.03.001
Simic, G., Stanic, G., Mladinov, M., Jovanov-Milosevic, N., Kostovic, I., and Hof, P. R. (2009). Does Alzheimer’s disease begin in the brainstem? Neuropathol. Appl. Neurobiol. 35, 532–554. doi: 10.1111/j.1365-2990.2009.01038.x
Solas, M., Francis, P. T., Franco, R., and Ramírez, M. J. (2013). CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 34, 805–808. doi: 10.1016/j.neurobiolaging.2012.06.005
Stella, N., Schweitzer, P., and Piomelli, D. (1997). A second endogenous cannabinoid that modulates long-term potentiation. Nature 388, 773–778. doi: 10.1038/42015
Stumm, C., Hiebel, C., Hanstein, R., Purrio, M., Nagel, H., Conrad, A., et al. (2013). Cannabinoid receptor 1 deficiency in a mouse model of Alzheimer’s disease leads to enhanced cognitive impairment despite of a reduction in amyloid deposition. Neurobiol. Aging 34, 2574–2584. doi: 10.1016/j.neurobiolaging.2013.05.027
Sultana, R., and Butterfield, D. A. (2010). Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimers Dis. 19, 341–353. doi: 10.3233/JAD-2010-1222
Tanveer, R., Gowran, A., Noonan, J., Keating, S. E., Bowie, A. G., and Campbell, V. A. (2012). The endocannabinoid, anandamide, augments Notch-1 signaling in cultured cortical neurons exposed to amyloid-β and in the cortex of aged rats. J. Biol. Chem. 287, 34709–34721. doi: 10.1074/jbc.M112.350678
Tolón, R. M., Núñez, E., Pazos, M. R., Benito, C., Castillo, A. I., Martínez-Orgado, J. A., et al. (2009). The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 1283, 148–154. doi: 10.1016/j.brainres.2009.05.098
Twitchell, W., Brown, S., and Mackie, K. (1997). Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 78, 43–50.
van der Stelt, M., Mazzola, C., Esposito, G., Matias, I., Petrosino, S., De Filippis, D., et al. (2006). Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. 63, 1410–1424. doi: 10.1007/s00018-006-6037-3
Van Sickle, M. D., Duncan, M., Kingsley, P. J., Mouihate, A., Urbani, P., Mackie, K., et al. (2005). Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 310, 329–332. doi: 10.1126/science.1115740
Viscomi, M. T., Oddi, S., Latini, L., Pasquariello, N., Florenzano, F., Bernardi, G., et al. (2009). Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J. Neurosci. 29, 4564–4570. doi: 10.1523/JNEUROSCI.0786-09.2009
Volicer, L., Stelly, M., Morris, J., McLaughlin, J., and Volicer, B. J. (1997). Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 12, 913–919. doi: 10.1002/(SICI)1099-1166(199709)12:9<913::AID-GPS663>3.0.CO;2-D
Wagner, J. A., Járai, Z., Bátkai, S., and Kunos, G. (2001). Hemodynamic effects of cannabinoids: coronary and cerebral vasodilation mediated by cannabinoid CB(1) receptors. Eur. J. Pharmacol. 423, 203–210. doi: 10.1016/S0014-2999(01)01112-8
Walther, S., Mahlberg, R., Eichmann, U., and Kunz, D. (2006). Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl.) 185, 524–528. doi: 10.1007/s00213-006-0343-1
Walther, S., Schüpbach, B., Seifritz, E., Homan, P., and Strik, W. (2011). Randomized, controlled crossover trial of dronabinol, 2.5 mg, for agitation in 2 patients with dementia. J. Clin. Psychopharmacol. 31, 256–258. doi: 10.1097/JCP.0b013e31820e861c
Westlake, T. M., Howlett, A. C., Bonner, T. I., Matsuda, L. A., and Herkenham, M. (1994). Cannabinoid receptor binding and messenger RNA expression in human brain: an in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer’s brains. Neuroscience 63, 637–652. doi: 10.1016/0306-4522(94)90511-8
Wilson, R. I., and Nicoll, R. A. (2002). Endocannabinoid signaling in the brain. Science 296, 678–682. doi: 10.1126/science.1063545
Wu, J., Bie, B., Yang, H., Xu, J. J., Brown, D. L., and Naguib, M. (2013). Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol. Aging 34, 791–804. doi: 10.1016/j.neurobiolaging.2012.06.011
Keywords: cannabinoids, Alzheimer, CB1 receptor, CB2 receptor, β-amyloid, tau, oxidative stress, neuroinflammation
Citation: Aso E and Ferrer I (2014) Cannabinoids for treatment of Alzheimer’s disease: moving toward the clinic. Front. Pharmacol. 5:37. doi: 10.3389/fphar.2014.00037
Received: 14 January 2014; Accepted: 19 February 2014;
Published online: 05 March 2014.
Edited by:
Cesare Mancuso, Catholic University School of Medicine, ItalyReviewed by:
Mauro Maccarrone, Campus Bio-Medico University of Rome, ItalyAntonio Calignano, University of Naples Federico II, Italy
Copyright © 2014 Aso and Ferrer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ester Aso and Isidre Ferrer, Institut de Neuropatologia, Servei d’Anatomia Patològica, Institut d’Investigació Biomèdica de Bellvitge-Hospital Universitari de Bellvitge, Universitat de Barcelona, Carrer Feixa Llarga s/n, 08907 L’Hospitalet de Llobregat, Spain e-mail: aso@bellvitgehospital.cat; 8082ifa@gmail.com