 José A. G. Agúndez
José A. G. Agúndez Gara Esguevillas
Gara Esguevillas Gemma Amo
Gemma Amo Elena García-Martín
Elena García-Martín- 1Department of Pharmacology, University of Extremadura, Cáceres, Spain
- 2ISCIII Research Network of Adverse Reactions to Allergens and Drugs, Madrid, Spain
The development of clinical practice recommendations or guidelines for the clinical use of pharmacogenomics data is an essential issue for improving drug therapy, particularly for drugs with high toxicity and/or narrow therapeutic index such as anticancer drugs. Although pharmacogenomic-based recommendations have been formulated for over 40 anticancer drugs, the number of clinical practice guidelines available is very low. The guidelines already published indicate that pharmacogenomic testing is useful for patient selection, but final dosing adjustment should be carried out on the basis of clinical or analytical parameters rather than on pharmacogenomic information. Patient selection may seem a modest objective, but it constitutes a crucial improvement with regard to the pre-pharmacogenomics situation and it saves patients’ lives. However, we should not overstate the current power of pharmacogenomics. At present the pharmacogenomics of anticancer drugs is not sufficiently developed for dose adjustments based on pharmacogenomics only, and no current guidelines recommend such adjustments without considering clinical and/or analytical parameters. This objective, if ever attained, would require the use of available guidelines, further implementation with clinical feedback, plus a combination of genomics and phenomics knowledge.
Adverse drug reactions (ADRs) constitute a major problem in clinical practice, as these reactions may result in patient risk and/or therapeutic failure. The identification, validation, refinement, and clinical use of biomarkers of drug response are primary objectives in medical research. In this context, pharmacogenomic biomarkers have shown success as predictors of drug response and ADRs, though several limitations and barriers hamper the widespread clinical use of pharmacogenomic biomarkers. These factors have been analyzed in detail elsewhere (Deverka et al., 2007; Haga and Burke, 2008; Agundez, 2009; Agundez et al., 2009; Relling et al., 2010; Fernandez et al., 2012; Agúndez and García-Martín, 2014; Caudle et al., 2014; Quinones et al., 2014; Rossolatos and Aitchison, 2014).
Because anticancer drugs usually show high toxicity and/or narrow therapeutic index, the potential of pharmacogenomic biomarkers in anticancer therapy is particularly high. Pharmacogenomic biomarkers can be used in anticancer therapy for drug selection according to patient’s and/or tumor genomics, and it is expected that – often combined with therapeutic drug monitoring – it will constitute a powerful combination to optimize drug dosing. Some potentially relevant pharmacogenomic biomarkers for anticancer drugs which already have clinical recommendations are summarized in Table 1. In this perspective manuscript we briefly present our viewpoint on the potential of pharmacogenomics in the clinical use of anticancer drugs, as well as some personal insight and opinion on this issue.
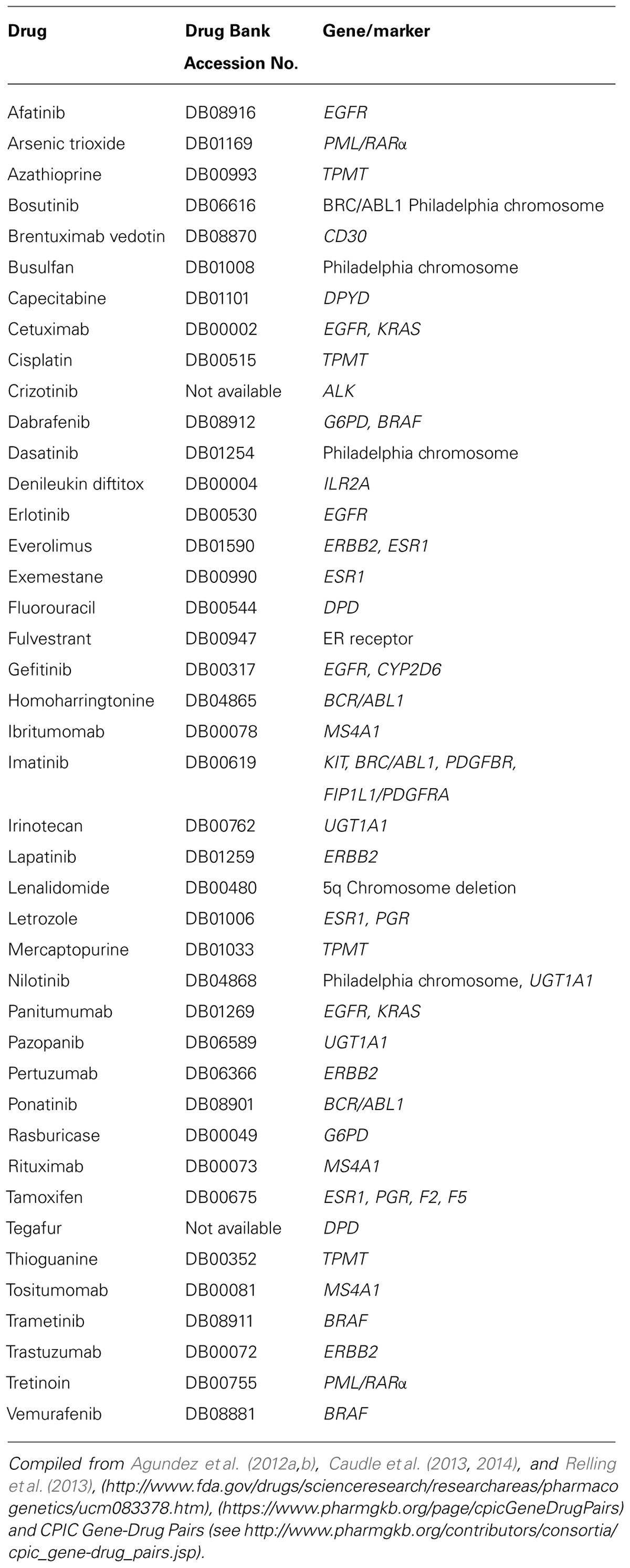
TABLE 1. Examples of anticancer drugs with pharmacogenomic recommendations.
The development of clinical guidelines for implementing pharmacogenomics knowledge in anticancer therapy is still at an early stage. Although Table 1 shows over 40 anticancer drugs that have practice recommendations, clinical guidelines have been developed for a very limited number of these. These clinical guidelines, which are essential to implementing the use of pharmacogenomics (Agundez et al., 2012a,b; Crews et al., 2012; Caudle et al., 2013, 2014; Relling et al., 2013; Quinones et al., 2014), are being drafted for several gene/drug pairs. The Clinical Pharmacogenetics Implementation Consortium (CPIC), the Pharmacogenomics Knowledge Base (PharmGKB1, or the St. Jude Children’s Research Hospital2 as well as other international initiatives, are particularly active in providing guidelines and therapeutic recommendations based on pharmacogenomic testing.
Pharmacogenomic information may be used in clinical practice at diverse levels: the most conservative pharmacogenomics views aim to stratify patient populations (patient selection biomarkers) into those who should or should not receive a given drug (Green and Guyer, 2011). One step further is the use of pharmacogenomic information as a biomarker of clinical response, with drug dose adjustment based on pharmacogenomics tests [see for instance (Swen et al., 2011)]. So far, most guidelines aiming to adjust dose on the basis of pharmacogenomics information also recommend therapeutic drug monitoring and/or close surveillance of the clinical evolution of the patients based, for instance, on analytical biomarkers. A third step would be to substitute therapeutic drug monitoring for pharmacogenomic information. Further steps substituting, for instance, data on clinical evolution for pharmacogenomic data seem unrealistic.
Our personal opinion is that for anticancer drugs pharmacogenomics is as yet insufficiently developed to go beyond the first step, that is, pharmacogenomics for anticancer drug therapy should be limited to predicting whether individuals would respond to a determined drug, or the odds are that a patient will experience adverse effects with a determined drug. Accordingly, CPIC guidelines on anticancer drugs are rather conservative. These guidelines are based on the genetic status for loss of function gene variants, making different (but somewhat overlapping) recommendations for homozygous or heterozygous carriers of defect genes.
For azathioprine and TPMT, for example, the guidelines recommend using alternative drugs for homozygous individuals, or decreasing starting doses, and then tritrating for drug tolerance allowing 2–4 weeks to reach steady-state after each dose adjustment in heterozygous individuals (Relling et al., 2011, 2013). For mercaptopurine and TPMT, recommendations are to consider different drugs or to reduce drastically starting doses for homozygous individuals and to adjust doses based on myelosuppression and disease-specific guidelines, both in homozygous and heterozygous individuals (Relling et al., 2011, 2013). For thioguanine and TPMT, recommendations are to consider other drugs or to start with drastically reduced doses and to adjust doses based on myelosuppression and disease-specific guidelines, both in homozygous and heterozygous individuals (Relling et al., 2011, 2013). For capecitabine, fluorouracil, tegafur and DPYD, recommendations are to consider alternative drugs for homozygous individuals, or to reduce by about a 50% the starting dose and to adjust doses based on the toxicity or the pharmacokinetics [see for instance Caudle et al. (2013) or www.stjude.org/pg4kds].
It should be noted that these recommendations usually fall within one of two categories: to select an alternative drug or to reduce the starting dose by a determined percentage and then titrate the dosage according to the patient’s response. Drug adjustments therefore are not based on pharmacogenomic testing as such. All recommended changes in drug dosage should be supported by clinical or analytical biomarkers. We do not think that dosage recommendations should go beyond this point, that is, at the present stage of pharmacogenomic knowledge the use of clinical or analytical biomarkers should not be substituted by pharmacogenomic testing. The objective of dose adjustment based on pharmacogenomics tests only, if ever reached, would require further implementation of phenotype or pharmacokinetic inference and this would require a combination of genomics and phenomics knowledge at a level that we do not presently have.
When incorporating pharmacogenomics testing into routine clinical practice, we should bear in mind that the genotype is a surrogate biomarker of the metabolic status or the clinical response of a determined individual, with a determined drug, at a determined dose, and in a particular situation. Many variables influence the genotype–phenotype, gene-concentration and gene-dose relationships. For instance, gene variations very often do not cause relevant changes in drug plasma concentration. Another issue is that conventional pharmacogenomic genotyping classify alleles as functional by exclusion, that is, when no common enzyme inactivating mutations are identified. Unless patients are genotyped by using whole gene sequencing, the chances of mistyping are relatively high. Moreover, the analysis of some SNPs may yield ambiguous genotypes making it necessary to use haplotype reconstruction algorithms or diplotype–phenotype conversion tables (Agundez et al., 2008). Another issue is ethnic variability because commonly used genotyping tests may not be well suited to all human populations (Roco et al., 2012; Moreno-Guerrero et al., 2013).
Additional uncertainty in phenotype inference comes from the occurrence of induction and inhibition processes that may modify in vivo activity regardless of the genotype. Moreover, conventional genotyping misses the influence of other gene variations. Even taking all these issues into consideration it is clear that phenotypes may change as a result of several factors including environmental conditions, disease progression, concomitant drug therapy and many other phenomic factors (Houle et al., 2010; Neuraz et al., 2013). So far we are aware of some examples, probably the tip of the iceberg, of elements which can modify genotype–phenotype associations.
When developing guidelines for the clinical use of pharmacogenomic information for the use of anticancer drugs, these factors should be addressed in the guidelines, bearing in mind that the combination of genotyping, drug pharmacokinetics, pharmacodynamics, and phenome-wide studies in large groups of patients with different clinical situations is necessary in order to gain more ground in genotype–phenotype association. Recently the CPIC group released the development process of guidelines designed to provide guidance to clinicians as to how available genetic test results should be interpreted to ultimately improve drug therapy (Caudle et al., 2014). These guidelines are based on the assessment of a known gene–drug relationships, the identification of content experts and the formation of writing committee, retrieval, summarization and presentation of the evidence linking genotype to drug variability, development of therapeutic recommendation and assignment of strength of the recommendation, internal and external review, and periodic review and guideline updates. A crucial point in the development of guidelines for the clinical use of pharmacogenomics information includes caveats to the effect that non-genetic considerations are also important for prescribing decisions. Thiopurine methyltransferase (TPMT) is a good example of this. Whereas a strong phenotype/genotype association exists for TPTM (Ford et al., 2009) most of the azathioprine/mercaptopurine-induced adverse reactions and the efficacy of therapy are not explained by TPMT polymorphisms (Palmieri et al., 2007).
Besides known and extensively discussed barriers and limitations to implementing the use of pharmacogenomic testing, related to institutional support or clinician’s awareness of the usefulness of pharmacogenomics testing, there an important barrier which is attributable to the scientific community, that is, some possible overstating of our findings or overestimating the current power of pharmacogenomics. We believe that one of the best services we can give to pharmacogenomic testing is to recognize its limitations and to work to solve them. In our opinion, part of the disenchantment experienced with pharmacogenomics in recent years may be at least partly attributable to an overoptimistic expectation of making a safe and reliable personalized dose adjustment based on pharmacogenomics tests only. We still have a long way to go before we can do that. In the meantime, the use of pharmacogenomic guidelines such as those already published by CPIC3 and the assessment of their performance in clinical practice would provide a highly valuable feedback that could be used to refine the whole process and eventually to gain more ground on the inference of drug response.
Conflict of Interest Statement
The Guest Associate Editor Luis Quinones declares that, despite having published together with the authors Elena García-Martín and José A. G. Agúndez in the past 2 years, the review process was handled objectively. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Prof. James McCue for assistance in language editing. The work in the author’s laboratory is financed by Grants PI12/00241, PI12/00324 and RETICS RD12/0013/0002, and by Grants GR10068 from Junta de Extremadura, Spain. Financed in part with FEDER funds from the European Union.
Footnotes
References
Agundez, J. A. (2009). Recent advances in drug intolerance. Curr. Drug Metab. 10, 946. doi: 10.2174/138920009790711869
Agúndez, J. A., and García-Martín, E. (2014). “Importance of CYP2D6 genotype/activity testing and applications,” in CYP2D6: Genetics, Pharmacology and Clinical Relevance, ed. P. Baumann (London: Future Medicine Ltd), 118–133.
Agundez, J. A., Garcia-Martin, E., and Martinez, C. (2009). Genetically based impairment in CYP2C8- and CYP2C9-dependent NSAID metabolism as a risk factor for gastrointestinal bleeding: is a combination of pharmacogenomics and metabolomics required to improve personalized medicine? Expert Opin. Drug Metab. Toxicol. 5, 607–620. doi: 10.1517/17425250902970998
Agundez, J. A., Golka, K., Martinez, C., Selinski, S., Blaszkewicz, M., and Garcia-Martin, E. (2008). Unraveling ambiguous NAT2 genotyping data. Clin. Chem. 54, 1390–1394. doi: 10.1373/clinchem.2008.105569
Agundez, J. A. G., Abad-Santos, F., Aldea, A., Alonso-Navarro, H., Bernal, M. L., Borobia, A. M.,et al. (2012a). Toward a clinical practice guide in pharmacogenomics testing for functional polymorphisms of drug-metabolizing enzymes. Gene/drug pairs and barriers perceived in Spain. Front. Genet. 3:273. doi: 10.3389/fgene.2012.00273
Agundez, J. A. G., Del Barrio, J., Padro, T., Stephens, C., Farre, M., Andrade, R. J.,et al. (2012b). Trends in qualifying biomarkers in drug safety. Consensus of the 2011 meeting of the spanish society of clinical pharmacology. Front. Pharmacol. 3:2. doi: 10.3389/fphar.2012.00002
Caudle, K. E., Klein, T. E., Hoffman, J. M., Müller, D. J., Whirl-Carrillo, M., Gong, L.,et al. (2014). Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 15, 209–217. doi: 10.2174/1389200215666140130124910
Caudle, K. E., Thorn, C. F., Klein, T. E., Swen, J. J., Mcleod, H. L., Diasio, R. B.,et al. (2013). Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin. Pharmacol. Ther. 94, 640–645. doi: 10.1038/clpt.2013.172
Crews, K. R., Hicks, J. K., Pui, C. H., Relling, M. V., and Evans, W. E. (2012). Pharmacogenomics and individualized medicine: translating science into practice. Clin. Pharmacol. Ther. 92, 467–475. doi: 10.1038/clpt.2012.120
Deverka, P. A., Doksum, T., and Carlson, R. J. (2007). Integrating molecular medicine into the US health-care system: opportunities, barriers, and policy challenges. Clin. Pharmacol. Ther. 82, 427–434. doi: 10.1038/sj.clpt.6100319
Fernandez, C. A., Smith, C., Yang, W., Lorier, R., Crews, K. R., Kornegay, N.,et al. (2012). Concordance of DMET plus genotyping results with those of orthogonal genotyping methods. Clin. Pharmacol. Ther. 92, 360–365. doi: 10.1038/clpt.2012.95
Ford, L., Kampanis, P., and Berg, J. (2009). Thiopurine S-methyltransferase genotype-phenotype concordance: used as a quality assurance tool to help control the phenotype assay. Ann. Clin. Biochem. 46, 152–154. doi: 10.1258/acb.2008.008167
Green, E. D., and Guyer, M. S. (2011). Charting a course for genomic medicine from base pairs to bedside. Nature 470, 204–213. doi: 10.1038/nature09764
Haga, S. B., and Burke, W. (2008). Pharmacogenetic testing: not as simple as it seems. Genet. Med. 10, 391–395. doi: 10.1097/GIM.0b013e31817701d4
Houle, D., Govindaraju, D. R., and Omholt, S. (2010). Phenomics: the next challenge. Nat. Rev. Genet. 11, 855–866. doi: 10.1038/nrg2897
Moreno-Guerrero, S. S., Ramirez-Pacheco, A., Dorantes-Acosta, E. M., and Medina-Sanson, A. (2013). Analysis of genetic polymorphisms of thiopurine S-methyltransferase (TPMT) in Mexican pediatric patients with cancer. Rev. Invest. Clin. 65, 156–164.
Neuraz, A., Chouchana, L., Malamut, G., Le Beller, C., Roche, D., Beaune, P.,et al. (2013). Phenome-wide association studies on a quantitative trait: application to TPMT enzyme activity and thiopurine therapy in pharmacogenomics. PLoS Comput. Biol. 9:e1003405. doi: 10.1371/journal.pcbi.1003405
Palmieri, O., Latiano, A., Bossa, F., Vecchi, M., D’inca, R., Guagnozzi, D.,et al. (2007). Sequential evaluation of thiopurine methyltransferase, inosine triphosphate pyrophosphatase, and HPRT1 genes polymorphisms to explain thiopurines’ toxicity and efficacy. Aliment. Pharmacol. Ther. 26, 737–745. doi: 10.1111/j.1365-2036.2007.03421.x
Quinones, L., Lavanderos, M., Cayun, J., Garcia-Martin, E., Agundez, J., Caceres, D.,et al. (2014). Perception of the usefulness of drug/gene pairs and barriers for pharmacogenomics in latin america. Curr. Drug Metab. 15, 202–208. doi: 10.2174/1389200215666140202220753
Relling, M. V., Altman, R. B., Goetz, M. P., and Evans, W. E. (2010). Clinical implementation of pharmacogenomics: overcoming genetic exceptionalism. Lancet Oncol. 11, 507–509. doi: 10.1016/S1470-2045(10)70097-8
Relling, M. V., Gardner, E. E., Sandborn, W. J., Schmiegelow, K., Pui, C. H., Yee, S. W.,et al. (2011). Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 89, 387–391. doi: 10.1038/clpt.2010.320
Relling, M. V., Gardner, E. E., Sandborn, W. J., Schmiegelow, K., Pui, C. H., Yee, S. W.,et al. (2013). Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin. Pharmacol. Ther. 93, 324–325. doi: 10.1038/clpt.2013.4
Roco, A., Quinones, L., Agundez, J. A., Garcia-Martin, E., Squicciarini, V., Miranda, C.,et al. (2012). Frequencies of 23 functionally significant variant alleles related with metabolism of antineoplastic drugs in the chilean population: comparison with caucasian and asian populations. Front. Genet. 3:229–229. doi: 10.3389/fgene.2012.00229
Rossolatos, D., and Aitchison, K. J. (2014). Genomics for clinical utility: the future is near. Genome Med. 6, 3. doi: 10.1186/gm522.
Keywords: biomarkers, adverse drug reactions, pharmacogenomics, clinical recommendations, clinical relevance
Citation: Agúndez JAG, Esguevillas G, Amo G and García-Martín E (2014) Clinical practice guidelines for translating pharmacogenomic knowledge to bedside. Focus on anticancer drugs. Front. Pharmacol. 5:188. doi: 10.3389/fphar.2014.00188
Received: 03 February 2014; Paper pending published: 02 March 2014;
Accepted: 24 July 2014; Published online: 19 August 2014.
Edited by:
Luis Abel Quiñones, University of Chile, ChileReviewed by:
Katherine J. Aitchison, King’s College London, UKBeatriz Henriques, University of Alberta, Canada (in collaboration with Katherine J. Aitchison) Luis Abel Quiñones, University of Chile, Chile
Copyright © 2014 Agúndez, Esguevillas, Amo and García-Martín. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José A. G. Agúndez, Department of Pharmacology, University of Extremadura, Avda de la Universidad s/n, E-10071 Cáceres, Spain e-mail: jagundez@unex.es