 Simone Brusco
Simone Brusco Paola Ambrosi
Paola Ambrosi Simone Meneghini
Simone Meneghini Andrea Becchetti
Andrea Becchetti- Department of Biotechnology and Biosciences, University of Milano-Bicocca, Milano, Italy
Regulation of the “neuronal” nicotinic acetylcholine receptors (nAChRs) is implicated in both tobacco addiction and smoking-dependent tumor promotion. Some of these effects are caused by the tobacco-derived N-nitrosamines, which are carcinogenic compounds that avidly bind to nAChRs. However, the functional effects of these drugs on specific nAChR subtypes are largely unknown. By using patch-clamp methods, we tested 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone (NNK) and N'-nitrosonornicotine (NNN) on human α4β2 nAChRs. These latter are widely distributed in the mammalian brain and are also frequently expressed outside the nervous system. NNK behaved as a partial agonist, with an apparent EC50 of 16.7 μM. At 100 μM, it activated 16% of the maximal current activated by nicotine. When NNK was co-applied with nicotine, it potentiated the currents elicited by nicotine concentrations ≤ 100 nM. At higher concentrations of nicotine, NNK always inhibited the α4β2 nAChR. In contrast, NNN was a pure inhibitor of this nAChR subtype, with IC50 of approximately 1 nM in the presence of 10 μM nicotine. The effects of both NNK and NNN were mainly competitive and largely independent of Vm. The different actions of NNN and NNK must be taken into account when interpreting their biological effects in vitro and in vivo.
Introduction
The nAChRs are ligand-gated ion channels permeable to cations, which regulate cell excitability and synaptic transmission. They are formed by different α and β subunits, which assemble to form homo- or heteropentamers (Dani and Bertrand, 2007). The homopentameric receptors, such as the widespread (α7)5, have low sensitivity to ACh and nicotine (with EC50 higher than 100 μM), high permeability to Ca2+ (PCa) and rapid desensitization. The heteromeric nAChRs generally display higher sensitivity to the agonists, lower PCa and slower desensitization (Dani and Bertrand, 2007). The most common high-affinity form in the mammalian brain is the α4β2*, with apparent EC50 values at least 10 times lower than those displayed by (α7)5. In the peripheral nervous system, the prevalent nAChR subtype is α3β4, which shows a more restricted expression in the CNS (Zoli et al., 2015). Nicotinic subunits are also widely expressed in other tissues, where their functions are still debated (Wessler and Kirkpatrick, 2008). In cancer cells, different types of homo- and heteromeric nAChRs cooperate in controlling several aspects of the neoplastic phenotype (Egleton et al., 2008; Schuller, 2009; Ambrosi and Becchetti, 2013; Schaal and Chellappan, 2014). Recently, the nAChR genes CHRNA3, CHRNA5, and CHRNB4 have been implicated in lung cancer susceptibility as well as nicotine addiction (Improgo et al., 2013, and references therein).
The complex pattern of nAChR expression may partly explain the pleiotropic effects of smoking. In the brain, uncontrolled stimulation of nAChRs is thought to mediate the addictive effects of tobacco (Changeux, 2010). In other tissues, smoking can produce toxic as well as carcinogenic effects. These latter depend on long-term exposure to several tobacco metabolites, especially the N-nitrosamines (here simply referred to as “nitrosamines”; Hecht and Hoffmann, 1988). When metabolically activated, these drugs cause DNA mutations, particularly G to T transversions that may lead to mutation of k-ras and p53 (Pfeifer et al., 2002). Nonetheless, many harmful effects of the tobacco-related nitrosamines may be attributed to direct targeting of nAChRs. In fact, the nitrosamines that produce the most potent biological effects are structural analogs of either ACh (e.g., diethylnitrosamine) or nicotine (especially NNK and NNN; Figure 1). All of these compounds can bind to nAChRs (Schuller and Orloff, 1998; Schuller, 2007). Although nicotine is not carcinogenic, engagement of nAChRs with either nicotine or nitrosamines can promote the neoplastic progression in cultured cells, by stimulating cell cycle, migration, angiogenesis, epithelial-to-mesenchymal transition, and inhibiting apoptosis (Schuller, 1989; Maneckjee and Minna, 1990; Heeschen et al., 2001; Arredondo et al., 2006; Guo et al., 2008; Paleari et al., 2008; Song et al., 2008; Al-Wadei and Schuller, 2009; Calleja-Macias et al., 2009; Dasgupta et al., 2009). Similar evidence is slowly accumulating in vivo (Heeschen et al., 2001; Paleari et al., 2009).
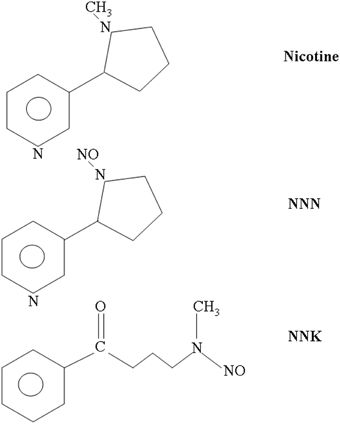
Figure 1. Molecular structures of nicotine, NNK, and NNN. NNN and NNK are the two most potent carcinogenic N-nitrosamines. They derive from nitrosation of nicotine, which occurs during tobacco treatment and storage as well as through metabolic processing in mammals.
The pathophysiological interpretation of these studies requires to clarify the signaling pathways downstream to nAChR binding (West et al., 2003; Tsurutani et al., 2005; Guo et al., 2008; Ambrosi and Becchetti, 2013). However, a general hindrance to full comprehension of the cellular effects of nitrosamines is that the direct functional action of these drugs on nAChRs is largely unknown. The only such study currently available shows that NNN inhibits α3β4 nAChR at high concentrations (IC50 was 1.14 mM, in the presence of 100 μM nicotine), whereas it exerts little effect on the muscle subtype (Nunes-Alves et al., 2012). The lack of direct functional tests is a major lacuna, as binding studies alone cannot distinguish whether a certain compound directly activates or inhibits nAChRs, nor the related kinetics. Moreover, the downstream cell signals could be regulated by conductive as well as non-conductive mechanisms (Becchetti, 2011). Understanding the functional interplay between nitrosamines and ACh or nicotine is also necessary to interpret their effects in the brain. Smokers are exposed to a mixture of tobacco-related nAChR ligands. Compounds such as NNK easily traverse the blood brain barrier (Jorquera et al., 1992; Gerde et al., 1998; Berridge et al., 2010) and have been found to activate microglia in vivo (Ghosh et al., 2009). These molecules are likely to interfere with one another and with the physiological agonist, to alter synaptic transmission and regulate tobacco addiction in currently unpredictable ways.
We studied by patch-clamp methods the effect of NNK and NNN on human α4β2 nAChRs stably expressed in HEK cells. This nAChR subtype is widely distributed in the brain, where it regulates both excitatory and inhibitory transmission (Becchetti et al., 2015) and is implicated in the cognitive and addictive effects of smoking (Changeux, 2010; Faure et al., 2014). It is also commonly expressed in non-nervous tissue, including lung cells (Fu et al., 2009), and tumor cell lines (Egleton et al., 2008). We found that these nitrosamines exert distinct actions, as NNK is a partial agonist, whereas NNN is a pure inhibitor of α4β2 nAChRs.
Materials and Methods
Cell Cultures
HEK 293 cells stably expressing human α4β2 nAChRs were cultured by standard methods (Di Resta et al., 2010). In brief, cultures were maintained in Dulbecco's modified Eagle's medium (Hyclone Laboratories), supplemented with 10% fetal calf serum (Hyclone), 4.5 g/l glutamine, 0.05 ng/ml hygromycin β, and 0.25 μg/ml amphoterycin B. Cells were grown at 37°C and 5% CO2. For patch-clamp experiments, cells were harvested by treatment with trypsin and plated onto 35 mm Petri dishes (Corning Inc.).
Patch-clamp Recording
Cells were voltage-clamped by using the whole-cell configuration of the patch-clamp method. Currents were registered with an Axopatch 200B amplifier (Molecular Devices), at room temperature (20–22°C). Micropipettes (3–4 MΩ) were pulled from borosilicate capillaries (GMBH) with a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument Co.). The cell capacitance and series resistance (up to about 75%) were always compensated. Currents were low-pass filtered at 2 kHz and acquired on-line at 5–10 kHz with Molecular Devices hardware and software (pClamp 8 and Axoscope 8). After patch rupture, we usually allowed 1–2 min for pipette solution exchange and signal stabilization, before applying our stimuli. Extracellular solutions were applied with an RSC-160 Rapid Solution Changer (BioLogic Science Instruments). The solution is delivered to the cells through borosilicate capillaries connected to Tygon tubes. Seven independent lines are available. Each line was generally reserved to one compound at a given concentration. The perfusing line was totally substituted whenever the drug was changed. No corrections for leak or junction potentials was ever applied to any of the displayed results.
Solutions and Drugs
Unless otherwise indicated, chemicals, and drugs were purchased by Sigma-Aldrich Italia Srl. The extracellular solution contained (mM): NaCl 130, KCl 5, CaCl2 2, MgCl2 2, HEPES 10, D-glucose 5 (pH 7.4; adjusted with NaOH). Pipettes contained (mM): K-aspartate 120, NaCl 10, MgCl2 2, CaCl21.3, EGTA-KOH 10, HEPES 10, MgATP 1, (pH 7.3; adjusted with KOH). Stock solutions of nicotine (up to 10 mM) were prepared weekly in our external solution and kept refrigerated. The pH was always checked after nicotine addition. NNK and NNN (20 mM) were dissolved in water and kept refrigerated for no longer than 1 week. Nicotine and/or nitrosamines were added daily to the extracellular solution. The final concentrations were obtained by serial dilution.
Analysis of Data
Data were analyzed with Clampfit 9.2 (Molecular Devices) and OriginPro 9 (OriginLab Co.). Theoretical curves best fitting the data were determined by a nonlinear least-squares method (Levenberg-Marquardt algorithm). For nAChR activation we used both a simple Hill function and the sum of two Hill expressions (Covernton and Connolly, 2000). The single-term expression was:
where Imaxis the maximal current, IL is the peak current at a given concentration of agonist L, EC50 is the concentration of L at which IL/Imax = 0.5, and nH is the Hill coefficient.
The two-terms Hill expression was:
where A is the fraction of receptors in the high-affinity state; EC50high and EC50low are the EC50 values of the components at high and low affinity, respectively; nH1 and nH2 are the corresponding Hill coefficients, and the other symbols are as in Equation (1).
The function used to fit the desensitization data points (Figure 3) was:
where ISS is the average steady state current at a given concentration of agonist L, Ipeakis the corresponding peak current,B is the minimal fractional steady state current, IC50 is the concentration of L at which ISS/Ipeak = 0.5, and nH is the Hill coefficient. ISS was found by fitting the current decay with a single exponential function and taking the steady state value of this function. A similar function was used to fit the fractional inhibition in Figures 4B, 5B.
The function used to fit the concentration-response data for nicotine at a given NNk concentration (Figure 5C; red continuous line) was:
where y is the fractional current in the presence of NNK plus nicotine, αNicand αNNKare the maximal fractional responses produced by nicotine and NNK, respectively (also named intrinsic activities of the agonists; Hogg and Bertrand, 2007); [Nic] and [NNK] are, respectively, the concentrations of nicotine and NNK; EC50 and EC50(NNK) are, respectively, the EC50 values for nicotine and NNK. The model is simplified in that it only considers single EC50's for both the full and the partial agonist.
Statistics
Data are given as mean values ± standard error of the mean, with n indicating the number of determinations (i.e., the number of cells tested). Statistical significance was determined with two-tailed t-test for paired or unpaired samples, as appropriate, at the indicated significance level (p). Normality was tested by the Kolmogorov-Smirnov test.
Results
NNK Activates Human α4β2 nAChRs
We first tested if NNK can activate α4β2 nAChRs, by applying the drug at −80 mV. A representative example is shown in Figure 2A. Consecutive applications of different NNK concentrations were spaced 2–3 min apart, to permit full recovery from nAChR desensitization. NNK concentrations higher than 10 nM elicited inward currents, and the maximal effect was obtained at 100 μM NNK. Nicotine was usually applied at the beginning and at the end of the experiment, to check for possible current rundown. The concentration-response relation for NNK was obtained by plotting the average fractional peak currents obtained at the indicated NNK concentrations (Figure 2B). On average, the current elicited by 100 μM NNK was approximately 14% of the current activated by saturating concentrations of nicotine (300 μM). No significantly higher currents were observed when using 300 μM NNK. For both nicotine and NNK, data points were fitted with Equation 1, which gave apparent EC50's of approximately 28 μM for nicotine and 17 μM for NNK. For all kinetic parameters, detailed statistics are given in the figure legends, which also report the Hill coefficients. For easier consulting, the main results are summarized in Table 1.
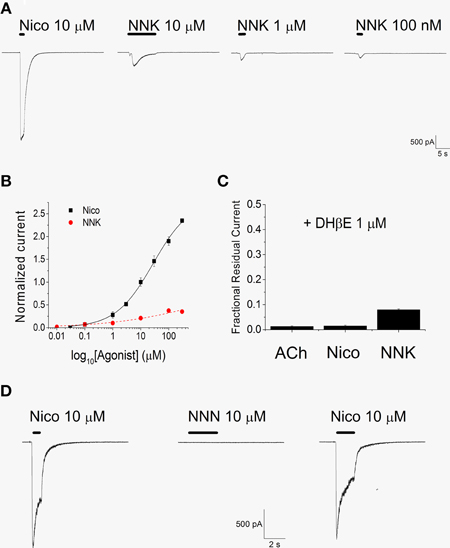
Figure 2. NNK, but not NNN, activates α4β2 nAChRs. (A) Consecutive whole-cell current traces, elicited at -80 mV by the indicated compound. Tests were spaced 2–3 min apart (gaps in the continuous current trace). Continuous bars above the current traces indicate the time of agonist application. Nicotine was repeatedly applied during the experiment to check for possible current rundown. (B) Concentration-response relations for nicotine (black squares) and NNK (red circles). Data points are average peak whole-cell currents recorded at −80 mV, normalized to the current elicited by 10 μM nicotine and plotted against the agonist concentration (in Log10 scale). Each point is the average of at least seven determinations. The concentration-response relation for nicotine (continuous line) and that for NNK (dashed line) were fitted with Equation 1, which gave an EC50 = 27.8 ± 5.6 μM (nH = 0.64) for nicotine and 16.7 ± 20 μM (nH = 0.44) for NNK. The estimated maximal current activated by NNK was approximately 16% of the current elicited by 300 μM nicotine. The measured peak current in the presence of 100 μM NNK was 0.14 ± 0.02 (n = 13) of the value measured in the presence of 300 μM nicotine. (C) DHβE (1 μM) strongly blocked the currents elicited by 300 μM ACh, 300 μM nicotine, and 10 μM NNK, as indicated. Data are averages of at least seven determination for each condition, carried out at −80 mV. Bars display the fractional residual currents (i.e., the peak current measured in the presence of the agonist plus DHβE divided by the peak current activated by the agonist alone). In the presence of 10 μM NNK, DHβE brought the average peak current density (pA/pF) from 5.75 ± 1.22 to 0.53 ± 0.199 (p < 0.01 with paired t-test; n = 9). (D) Comparison of the effects of NNN (10 μM) and nicotine (10 μM) on whole-cell currents from α4β2 nAChRs, at −80 mV. NNN never produced nAChR activation in cells in which functional receptors were shown to be present by nicotine application.

Table 1. Comparison of the effects of nicotine, NNN, and NNK, on α4β2 nAChRs.
The current activated by NNK was strongly blocked by 1 μM dihydro-β-erythroidine (DHβE), which is known to inhibit α4β2, but not homomeric, nAChRs (Buisson et al., 1996; Alkondon et al., 2000). To allow equilibration of DHβE concentration, this compound was perfused in the bath for 30 s before the agonist was added. To generate the data shown in Figure 2C, the peak current measured in the presence of agonist plus inhibitor was divided by the one elicited by the agonist alone. These fractional residual currents were plotted for NNK (10 μM), nicotine (300 μM), and ACh (300 μM). DHβE blocked more than 90% of the currents activated by either agonist, which is consistent with our hypothesis that NNK specifically activated the α4β2 nAChRs expressed in our cells.
A more detailed analysis of the concentration-response relations for nicotine and NNK is shown in Figure 3. Best fitting of the experimental data points was obtained by using the two-terms Hill function (Equation 2). This is usually observed with α4β2 nAChRs (Covernton and Connolly, 2000; Buisson and Bertrand, 2001), and is attributed to the coexistence of two receptor's stoichiometries: (α4)3(β2)2 (with lower affinity) and (α4)2(β2)3 receptors (with higher affinity; Nelson et al., 2003). The fitting parameters were: EC50high = 0.035 μM and EC50low = 12.81 μM, for NNK, and EC50high = 0.14 μM and EC50low = 7.7 μM for nicotine. These results suggest that NNK is particularly effective at stimulating the high affinity nAChR component. Moreover, for both nicotine (e.g., Figure 5A) and NNK (e.g., Figure 2A), the activated current displayed progressive desensitization in the presence of the agonist. To quantify such process, the current decay was fitted with a monoexponential function (e.g., Paradiso and Steinbach, 2003). Next, desensitization curves were generated by plotting the average fractional steady state currents calculated from the fitting procedure, for the indicated agonist concentration (Figure 3). By using Equation 3 (continuous lines through the data points), we estimated IC50 = 1.7 μM for NNK, and IC50 = 2.64 μM for nicotine. In the absence of other nicotinic ligands, the nAChR-dependent biological effect of NNK must depend on the steady state current sustained by this drug. This is proportional to the product of the activation and desensitization curves (sometimes referred to as “window current”), at a given NNK concentration. Figure 3 suggests that the window currents of NNK and nicotine are broadly similar, but that NNK tends to be comparatively more effective at the low concentrations, in line with the range of plasma doses observed in smokers.
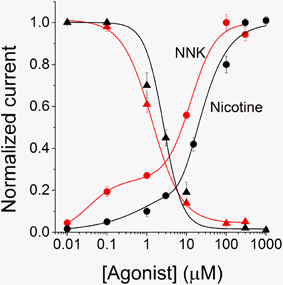
Figure 3. Activation and desensitization profiles for α4β2 nAChRs. The activation curve for NNK (red circles) was generated by using experiments analogous to those shown in Figure 2. The average peak current measured at each concentration of NNK was normalized to the current elicited by 300 μM NNK. Data points are averages of at least seven determinations and were fitted by using Equation 2 (continuous line), which gave EC50high = 0.035 ± 0.012 μM (nH2 = 1.2), and EC50low = 12.81 ± 1.58 μM (nH1 = 1.4). The desensitization curve for NNK was generated by plotting average steady state fractional currents (red triangles), as a function of NNK concentration. At each concentration, the current decay in the presence of the drug was fitted with a single exponential function. The steady state current values thus estimated were divided by the corresponding peak current values. Data points are averages of at least 6 determinations and were fitted by using Equation 3 (continuous line), which gave IC50 = 1.7 μM ± 0.2 (nH = 0.9). The nicotine activation (black circles) and desensitization (black triangles) curves were obtained in a similar way. For activation, data points are averages of at least 6 determinations. They were fitted by using Equation 2 (continuous line), giving EC50high = 0.14 ± 1.03 μM (nH2 = 0.62), and EC50low = 7.7 ± 14.7 μM (nH1 = 1.8). For desensitization, data points were fitted with Equation 3 (continuous line), which gave IC50 = 2.64 ± 0.47 μM (nH = 1.84).
The Inhibitory Effect of NNN
Differently from NNK, NNN did not elicit any whole-cell current even at concentrations (100 μM) much higher than those encountered in physiological conditions. Figure 2D shows a typical experiment at−80 mV, comparing the effects of 10 μM nicotine and 10 μM NNN, in a cell expressing a large nicotinic current. Once again, nicotine was applied before and after NNN, to exclude channel rundown. Given that NNN produced no nAChR activation, we studied its possible inhibitory effect in the presence of nicotine. We tested NNN concentrations between 1 pM and 100 μM on currents activated by concentrations of nicotine ranging between 10 nM and 100 μM. A representative current trace is shown in Figure 4A, in which 10 μM NNN was applied in the presence of 10 μM nicotine, at −80 mV. NNN was generally applied until the effect had reached the steady state. Next, the drug was removed. After the current had recovered from inhibition, nicotine was also washed out. This experimental procedure (Buisson et al., 1996; Palma et al., 1996) was preferred to the alternative procedure of pre-conditioning with NNN and then applying simultaneously the agonist and the antagonist (as we did with DHβE). The former method allows to directly compare the effect of NNN with the one produced by nicotine. In this way, the possible artifacts occurring in repetitive consecutive trials (such as channel rundown, poor solution exchange, precise estimation of the current peak, etc.) are avoided or immediately recognized. Moreover, the blockade kinetics can be directly appreciated. For each concentration, the steady state current in the presence of NNN was divided by the current remaining after NNN was removed. These fractional currents were plotted in Figure 4B, as a function of the NNN concentration, for the indicated nicotine concentrations (0.5, 10, and 100 μM). Notice that higher nicotine concentrations decreased the inhibitory effect of NNN, which suggests a competitive blocking mechanism. For instance, the IC50 value for NNN was >10 μM in the presence of 100 μM nicotine, whereas it was approximately 1 nM in the presence of 10 μM nicotine (full statistics are reported in the figure legend). Conversely, the concentration-response relations for nicotine in the presence of the indicated NNN concentrations are plotted in Figure 4C. Data points were fitted with Equation 1 (continuous lines through the data points). The right-shift of the curves produced by NNN is consistent with the notion that this drug tends to produce competitive block of α4β2 nAChRs. From a pathological point of view, it is worth noticing that NNN can exert significant current block at concentrations normally encountered in smokers' plasma (<100 pM; Schuller, 2007).
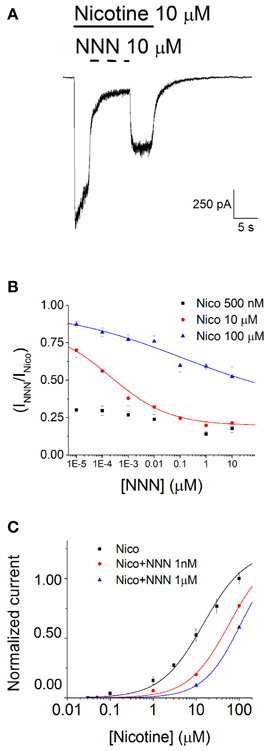
Figure 4. NNN inhibits α4β2 nAChRs. (A) Typical whole-cell current traces elicited by 10 μM nicotine at −80 mV, in the presence and in the absence of 10 μM NNN. Horizontal bars mark time of application of the indicated compound. (B) Steady state inhibition curves were generated by plotting the residual fractional steady state currents as a function of NNN concentration (in Log10 scale). The different data sets were obtained at the indicated concentration of nicotine. Data points are averages of at least nine determinations. Lines through the data points are best fitting curves, obtained with Equation 3. At 100 μM nicotine, IC50 was > 20 μM, whereas at 10 μM nicotine IC50 was 0.21 ± 0.4 nM. (C) Activation curves in the absence (black squares) or in the presence of the indicated concentration of NNN. Data points are peak whole-cell currents normalized to the current elicited by, 100 μM nicotine. Continuous lines through the data points are best fitting curves, obtained with Equation 1. The corresponding parameters were: EC50 = 14.5 ± 1.34 μM (nH = 0.83), for nicotine alone; EC50 = 58.3 ± 4.4 μM (nH = 0.92), in the presence of 1 nM NNN; EC50 = 109.1 ± 0.14 μM (nH = 1), in the presence of 1 μM NNN.
The Effect of NNK in the Presence of Nicotine
NNK is expected to produce effects more complex than those shown by NNN, as partial agonists can produce channel activation or inhibition depending on the concentration of the full agonist (e.g., Hogg and Bertrand, 2007; Rollema et al., 2007). In fact, concentrations of NNK up to 100 nM produced no effect on the currents activated by 10 μM nicotine, whereas higher concentrations progressively inhibited α4β2 nAChRs (Figure 5A). These data were obtained and analyzed as previously illustrated for NNN. Figure 5B plots the average fractional residual currents measured in the presence of 10 μM nicotine, as a function of NNK concentration. Data points were fitted with Equation 3 (continuous line), giving an IC50 > 100 μM. In contrast, the currents activated by low concentrations of nicotine could be potentiated by NNK. An example is given in the bottom panel of Figure 5A, showing that 100 μM NNK increased the current activated by 100 nM nicotine at −80 mV, by approximately 80%. In agreement with the results shown in Figure 3, the current activated by 100 μM NNK also displayed progressive desensitization. The effects of the tested concentrations of NNK on the currents activated by different concentrations of nicotine are summarized in Figure 5C. For comparison, the activation curve for nicotine (black circles) is also reported. With 1 μM NNK (red squares), the potentiation produced on the currents activated by 0.01 μM nicotine was 26%. The concentration of nicotine at which the drug reversed its effect was around 20 nM, as is estimated by fitting the data points with the simplified model expressed by Equation 4 (continuous red line). Saturating concentrations of NNK (100 μM; red circles) potentiated by about 80% the current activated by 0.1 μM nicotine, whereas they inhibited by 40% the current elicited by 10 μM nicotine. In this case, the sign reversal of NNK effect can be estimated to occur at approximately 400 nM nicotine. For clarity, the error bars are not reported in Figure 5C. Instead, the statistics of the current potentiation observed with NNK are reported in detail in Figure 5D and in the figure legend. We conclude that NNK, consistently with its partial agonist nature, can produce either potentiation or inhibition of α4β2 nAChRs, depending on the concomitant concentration of the full agonist.
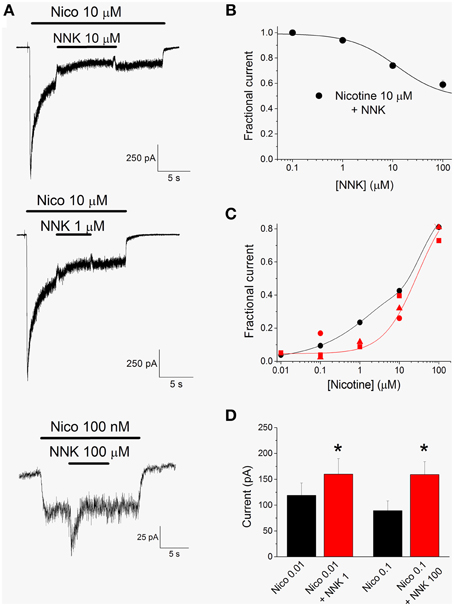
Figure 5. The effect of NNK on α4β2 nAChRs, in the presence of nicotine. (A) Representative current traces elicited at −80 mV by the indicated concentration of nicotine (Nico), in the presence or in the absence of the indicated concentration of NNK. The currents elicited by 10 μM nicotine (top and middle panels) are representative of 11 similar experiments. The bottom panel illustrates the potentiating effect of 100 μM NNK on the current activated by 100 nM nicotine (representative of eight similar experiments). Bars mark the time of application of nicotine and NNK, as indicated. (B) Concentration-response relation for the inhibitory effect of NNK tested on currents activated by 10 μM nicotine, as illustrated in (A). Data points are average steady state currents (n = 11) measured in the presence of a given NNK concentration, divided by the current obtained after NNK was rinsed. Continuous line through the data points is best fitting to Equation 3, giving IC50 > 100 μM (nH = 0.95). (C) Summary of the effects of NNK plus nicotine. All data are normalized to the current measured with 300 μM nicotine. Black circles: concentration-response for nicotine (same as in Figure 3; only concentrations up to 100 μM are shown). Red symbols: average fractional currents in the presence of 1 μM (squares), 10 μM (triangles), or 100 μM (squares) NNK, as a function of [nicotine]. Data were obtained as illustrated in (A). The values obtained with NNK were generally significantly different from those obtained with nicotine alone. Detailed statistics for the potentiation data are given in (D). The red continuous line is the curve best fitting the data points relative to 1 μM NNK, obtained by using Equation 4. The fit parameters were A = 1, B = 0.27, [NNK] = 1 μM, αNIC = 24 μM, αNNK = 5 μM. (D) The peak current measured in the presence of 10 nM nicotine was 119 ± 24 pA, which was brought to 160 ± 30 pA by 1 μM NNK (0.05 < p < 0.01, with t-test for paired samples; n = 11). The effect of 100 μM NNK was tested in a different series of cells, in which the average current elicited by 100 nM nicotine was 89.4 ± 19.4 pA, which was brought to 159.1 ± 25.1 pA by 100 μM NNK (0.05 < p < 0.01, with t-test for paired samples; n = 8). These results are plotted as black and red bars, respectively for nicotine (Nico) and NNK. Statistical significance is indicated by *.
The Block Produced by NNN and NNK was not Voltage-dependent
The voltage dependence of the NNN and NNK effect is illustrated in Figure 6. Current traces (Figure 6A) illustrate typical experiments in which nAChRs were activated by 10 μM nicotine, at the indicated Vm. For briefness, only traces obtained at −80 mV (left) and +80 mV (right) are shown. NNN (10 μM) was applied in the presence of nicotine and removed after inhibition had reached the steady state. These results are summarized in Figure 6B (top panel), showing the current voltage relations in the presence of either nicotine alone (Nico) or nicotine plus NNN (NNN). Data points are average current values normalized to the absolute value of the current measured at −80 mV. The I/V plots displayed the typical inward rectification of α4β2 nAChRs. Analogous results were obtained with NNK (Figure 6C, top panel). The apparent reversal potential (Vrev) was usually between +5 and +20 mV. The fractional block produced by either NNN or NNK at the steady state was independent of the applied Vm. This is better appreciated in the bottom panels of Figures 6B,C, which give the average fractional residual currents as a function of Vm, in the presence of NNN or NNK, respectively. The data points around Vrev were removed as the small current values prevented a reliable measure of the drugs' effect. The quick development and reversal of channel block suggests that the effect was mainly caused by the nitrosamines accessing to the channel from the extracellular milieu (i.e., we assume that intracellular accumulation of the drugs was negligible). Hence, because inhibition was virtually independent of the net direction of ion flow, these nitrosamines are unlikely to exert significant open channel block, at the concentrations we applied (much higher than those observed in vivo).
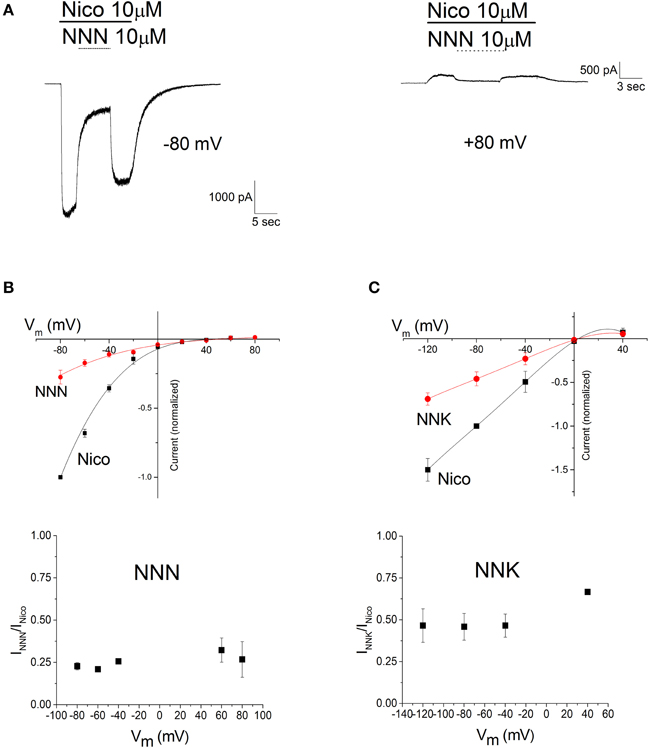
Figure 6. The effects of NNN and NNK are not voltage-dependent. (A) Typical current traces obtained as illustrated in Figure 3, except that trials were carried out at different Vm's. For briefness, only the traces obtained at −80 and +80 mV are displayed. Bars mark time of application of 10 μM nicotine (Nico, continuous) and 10 μM NNN (dashed). (B). Top panel: current-voltage relations obtained in the absence (squares) and in the presence (circles) of NNN. Data points are average steady state current densities measured at the indicated Vm and normalized to the value obtained at -80 mV (with a reversed sign, for consistency with the usual convention of displaying inward current as negative). Data summarize the results of nine independent experiments. Continuous lines are polynomial curves best fitting the data points. No correction was applied for junction potentials. Bottom panel: the fractional block produced by NNN is plotted as a function of Vm. Data points are average steady state currents in the presence of NNN (INNN), divided by the current in the absence of NNN (INico). The data points around Vrev were omitted (see the main text). No significant difference was observed among the results obtained at different Vm's. (C) Top panel: same as in (C), for NNK (1 μM, in the presence of 100 nM nicotine). Bottom panel: the fractional block produced by NNK as a function of Vm was calculated as illustrated for NNN in (B), except that the tested Vm's were: −120/−80/−40/0/+40. Once again, no significant difference was observed among the results obtained at different Vm's.
Discussion
Comparison with Previous Studies
Binding of nitrosamines to nAChRs was previously studied with radioactive ligand competition assays. These were mostly carried out in lung cancer cell lines, by using labeled epibatidine (to target heteromeric nAChRs) or α-bungarotoxin (specific for α7; Schuller and Orloff, 1998; Schuller, 2007). Our results qualitatively agree with these studies in that our nitrosamines compete with the full agonist for binding to nAChRs. However, a detailed quantitative comparison is difficult to draw. First, tumor cell lines generally express multiple nAChR subtypes (Egleton et al., 2008), and these have different pharmacological properties (e.g., Dani and Bertrand, 2007). Second, electrophysiological and binding analyses address different molecular features that can be difficult to reconcile (e.g., Chang and Weiss, 1999). For example, patch-clamp measurements reveal the population of active channels, whereas the binding profile can be affected by silent receptors, whose level of expression may be significant (McNerney et al., 2000). Even with those qualifications, the EC50 we observed for NNK (Figure 3) with the single-term Hill model is in good agreement with the one measured in binding studies carried out on epibatidine-sensitive nAChRs (about 17 μM, in SCLC cells; Schuller, 2007). The highest binding efficacy of NNN on α4β2 nAChRs is also in broad agreement with the binding studies. The fact that NNN inhibited α4β2 nAChRs (Figure 4) is consistent with the functional studies carried out on α3β4 receptors, although in the latter the affinity is lower and the mechanism is mainly non-competitive (Nunes-Alves et al., 2012). Based on our results and the absence of open channel block (Figure 6), we hypothesize that the main site of nitrosamine action on α4β2 nAChRs is close to the orthosteric binding site.
Functional Implications for Peripheral and Cancer Tissue
Work on cell lines showed that nicotine and nitrosamines produce similar effects, particularly stimulation of proliferation and inhibition of apoptosis. In SCLC cells and gastrointestinal cancer, the effects are mainly attributed to the activation of α7 receptors, whereas in other neoplastic cells, such as the NSCLC, both homo- and heteromeric nAChRs contribute to the observed effects (Schuller, 2009). In brief, nAChR activation tends to stimulate exocytosis of autocrine factors (Cattaneo et al., 1993; Jull et al., 2001) as well as other Ca2+-dependent pathways that regulate proliferation and apoptosis (Schuller, 2009). The relative contribution of the different nAChR subtypes is still debated. One general working hypothesis is that, differently from α7 receptors, activation of heteromeric nAChRs stimulates pathways that tend to suppress cancer progression (Schuller, 2009). If this hypothesis is correct, our results suggest that NNN could stimulate tumor progression by inhibiting the protective effects contributed by heteromeric nAChRs, without affecting the less sensitive α7. Alternatively, NNN might exert signaling roles not depending on ion conduction (e.g., Dasgupta et al., 2006), or activate other types of membrane receptors, or both. Evidence is indeed available about the activation produced by NNK on β-adrenergic receptors (Schuller et al., 1999), but not about NNN, to the best of our knowledge.
The partial agonist nature of NNK makes it more difficult to suggest a general interpretation of its cellular effects, particularly because the direct effects of NNK on α7 nAChRs are currently unknown. Considering that the binding studies indicate that homomeric receptors are more sensitive to NNK, a reasonable working hypothesis is that NNK induces both homo- and heteromeric nAChR activation in cultured cells, and that the overall effect on Vm and calcium signals tends to prevail on the tumor suppressing signals specifically induced by α4β2 activation. Moreover, although neither α7 (Kawai and Berg, 2001) nor α4β2 (Buisson and Bertrand, 2001) receptors display long-term inactivation in the presence of nicotine, the long-term effects of nitrosamines on nicotinic currents are unknown.
Even more caution is necessary to interpret the effects of the tobacco-related nitrosamines in vivo. We showed that the typical concentrations of these compounds observed in vivo can produce functional effects on α4β2 nAChRs. Smokers' blood contain steady nitrosamine levels around 30–50 pM, and the peak concentrations can be at least 10 times as high (e.g., Hecht and Hoffmann, 1988; Schuller, 2007). In parallel, smokers are exposed to widely fluctuating levels of plasma nicotine, depending on smoking habits, not to speak of the concomitant physiologically oscillating ACh levels depending on parasympathetic activity. How nitrosamines and the full agonists interfere in single individuals is difficult to predict, given that the typical peak concentration of plasma nicotine in smokers generally vary between 10 and 500 nM (e.g., Russell et al., 1980), but can reach concentrations as high as 10 μM, immediately after smoking (Schaal and Chellappan, 2014). According to our results, this wide range of concentrations allows both potentiating and inhibiting effects of NNK, at least for α4β2 nAChRs. These observations may contribute to explain the individual variability in the response to long-term smoking. In general, our study points to the necessity of carrying out more extensive work in vitro on the combined effects of nitrosamines and the full agonists on specific nAChR subtypes.
Implication for Brain Pathophysiology
Because of their slow desensitization and high sensitivity to ACh, α4β2 nAChRs are major regulators of the overall cerebral excitability, as is also testified by the observation that the nAChR-related epileptogenic mutations known to date are located on genes coding for subunits of heteromeric receptors (Becchetti et al., 2015). In the mammalian neocortex, heteromeric (largely α4β2*) nAChRs regulate excitatory (Vidal and Changeux, 1993; Gioanni et al., 1999; Lambe et al., 2003; Disney et al., 2007; Zolles et al., 2009; Poorthuis et al., 2012; Aracri et al., 2013) as well as inhibitory (Xiang et al., 1998; Porter et al., 1999; Alkondon et al., 2000; Couey et al., 2007; Aracri et al., 2010) neurotransmission at both the pre- and post-synaptic level. These receptors also regulate dopaminergic neurons, which bears implications for tobacco addiction (Faure et al., 2014).
Interpreting the effects of nitrosamines in the brain requires considering their tonic actions in resting conditions as well as the phasic effects during transmitter release. The steady concentration of nicotine in the cerebrospinal fluid of smokers is close to the plasma levels (Berridge et al., 2010). Because the tonic extracellular concentrations of ACh in the brain is thought to be in the nanomolar range (Descarries et al., 1997), the steady effects of nitrosamines in the brain likely compete mostly with nicotine, whose levels depend on the smoking regimen. Considering that α4β2 nAChRs display a higher affinity for NNN, and that the latter tends to have a concentration higher than NNK, our results suggest that the inhibitory effect of NNN should prevail, in steady state conditions, irrespective of the nicotine level.
In contrast, in bona fide cholinergic synapses, on vesicle release, the ACh concentration in the synaptic cleft quickly reaches millimolar levels, thus activating the postsynaptic current within hundreds of μs. The subsequent current decay displays a kinetics that depends on tissue, largely because of the balance between intrinsic nAChR desensitization and ACh removal by acetylcholinesterase, but is generally complete within 5 to 20 ms (e.g., Magleby and Stevens, 1972; Chu et al., 2000). Our results (Figures 4B, 5B) suggest that the typical steady state concentrations of nitrosamines normally observed in blood are probably too low to affect classic postsynaptic currents activated by high ACh levels. However, it is possible that long exposures to nitrosamine compounds could affect the nAChR sensitivity and expression.
Conclusions
The study of the interaction between nitrosamines and nAChRs is still in its infancy. Our results show that NNK behaves as a partial agonist of α4β2 nAChRs, whereas NNN is a receptor's inhibitor, with a relatively high affinity. In light of the above discussion, these properties may explain at least in part the effects produced by nitrosamines on neoplastic cell lines. However, to reach a full biological interpretation of the nitrosamines' roles in vivo, including those in the CNS, further studies are needed along the following lines. First, the effects of NNN and NNK should be tested on other nAChR subtypes, particularly α7. Second, whether nitrosamines induce long-term inactivation of nAChRs should be also analyzed. Finally, it will be necessary to ascertain the presence of different binding sites for these drugs on the receptor's protein.
Funding
The present work was supported by the BML (Banca del Monte di Lombardia) Foundation to AB and the University of Milano-Bicocca (Fondo di Ateneo per la Ricerca to AB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CNS, central nervous system; DHβE, dihydro-β-erythroidine; nAChR, neuronal nicotinic acetylcholine receptor; EC50high and EC50low, EC50 values for two-term Hill equations; HEK, human embryonic kidney; nH, Hill coefficient for one-term Hill equation; nH1, nH2, Hill coefficients for two-term Hill equation; NNK, 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone; NNN, N′-nitrosonornicotine; Vm, membrane potential; Vrev, reversal potential.
References
Alkondon, M., Pereira, E. F., Eisenberg, H. M., and Albuquerque, E. X. (2000). Nicotinic receptor activation in human cerebral cortical interneurons: a mechanism for inhibition and disinhibition of neuronal networks. J. Neurosci. 20, 66–75.
Al-Wadei, H. A., and Schuller, H. M. (2009). Nicotinic receptor-associated modulation of stimulatory and inhibitory neurotransmitters in NNK-induced adenocarcinoma of the lungs and pancreas. J. Pathol. 218, 437–445. doi: 10.1002/path.2542
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ambrosi, P., and Becchetti, A. (2013). Targeting neuronal nicotinic receptors in cancer: new ligands and potential side-effects. Recent Pat. Anticancer Drug Discov. 8, 38–52. doi: 10.2174/1574892811308010038
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aracri, P., Amadeo, A., Pasini, M. E., Fascio, U., and Becchetti, A. (2013). Regulation of glutamate release by heteromeric nicotinic receptors in layer V of the secondary motor region (Fr2) in the dorsomedial shoulder of prefrontal cortex in mouse. Synapse 67, 338–357. doi: 10.1002/syn.21655
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aracri, P., Consonni, S., Morini, R., Perrella, M., Rodighiero, S., Amadeo, A., et al. (2010). Tonic modulation of GABA release by nicotinic acetylcholine receptors in layer V of the murine prefrontal cortex. Cereb. Cortex 20, 1539–1555. doi: 10.1093/cercor/bhp214
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arredondo, J., Chernyavsky, A. I., and Grando, S. A. (2006). Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol. Ther. 5, 511–517. doi: 10.4161/cbt.5.5.2601
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becchetti, A. (2011). Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am. J. Physiol. Cell Physiol. 301, C255–C265. doi: 10.1152/ajpcell.00047.2011
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becchetti, A., Aracri, P., Meneghini, S., Brusco, S., and Amadeo, A. (2015). The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front. Physiol. 6:22. doi: 10.3389/fphys.2015.00022
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berridge, M. S., Apana, S. M., Nagano, K. K., Berridge, C. E., Leisure, G. P., and Boswell, M. V. (2010). Smoking produces rapid rise of [11C]nicotine in human brain. Psychopharmacology 209, 383–394. doi: 10.1007/s00213-010-1809-8
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buisson, B., and Bertrand, D. (2001). Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J. Neurosci. 21, 1819–1829.
Buisson, B., Gopalakrishnan, M., Arneric, S. P., Sullivan, J. P., and Bertrand, D. (1996). Human α4β2 neuronal nicotinic acetylcholine receptor in HEK293 cells: a patch-clamp study. J. Neurosci. 16, 7880–7891.
Calleja-Macias, I. E., Kalantari, M., and Bernard, H. U. (2009). Cholinergic signaling through nicotinic acetylcholine receptors stimulates the proliferation of cervical cancer cells: an explanation for the molecular role of tobacco smoking in cervical carcinogenesis? Int. J. Cancer 124, 1090–1096. doi: 10.1002/ijc.24053
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cattaneo, M. G., Codignola, A., Vicentini, L. M., Clementi, F., and Sher, E. (1993). Nicotine stimulates a serotonergic autocrine loop in human small-cell lung carcinoma. Cancer Res. 53, 5566–5568.
Chang, Y., and Weiss, D. S. (1999). Channel opening locks agonist onto the GABAC receptor. Nat. Neurosci. 2, 219–225. doi: 10.1038/6313
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Changeux, J. P. (2010). Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat. Rev. Neurosci. 11, 389–401. doi: 10.1038/nrn2849
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, Z. G., Zhou, F. M., and Hablitz, J. J. (2000). Nicotinic acetylcholine receptor-mediated synaptic potentials in rat neocortex. Brain Res. 887, 399–405. doi: 10.1016/S0006-8993(00)03076-6
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Couey, J. J., Meredith, R. M., Spijker, S., Poorthuis, R. B., Smit, A. B., Brussaard, A. B., et al. (2007). Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron 54, 73–87. doi: 10.1016/j.neuron.2007.03.006
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Covernton, P. J., and Connolly, J. G. (2000). Multiple components in the agonist concentration-response relationship of neuronal nicotinic acetylcholine receptors. J. Neurosci. Methods 96, 63–70. doi: 10.1016/S0165-0270(99)00185-5
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dani, J. A., and Bertrand, D. (2007). Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 47, 699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dasgupta, P., Rastogi, S., Pillai, S., Ordonez-Ercan, D., Morris, M., Haura, E., et al. (2006). Nicotine induces cell proliferation by beta-arrestin mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Invest. 116, 2208–2217. doi: 10.1172/JCI28164
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dasgupta, P., Rizwani, W., Pillai, S., Kinkade, R., Kovacs, M., Rastogi, S., et al. (2009). Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 124, 36–45. doi: 10.1002/ijc.23894
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Descarries, L., Gisiger, V., and Steriade, M. (1997). Diffuse transmission by acetylcholine in the CNS. Prog. Neurobiol. 53, 603–625. doi: 10.1016/S0301-0082(97)00050-6
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Resta, C., Ambrosi, P., Curia, G., and Becchetti, A. (2010). Effect of carbamazepine and oxcarbazepine on wild type and mutant neuronal nicotinic receptors linked to nocturnal frontal lobe epilepsy. Eur. J. Pharmacol. 643, 13–20. doi: 10.1016/j.ejphar.2010.05.063
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Disney, A. A., Aoki, C., and Hawken, M. J. (2007). Gain modulation by nicotine in macaque V1. Neuron 56, 701–713. doi: 10.1016/j.neuron.2007.09.034
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Egleton, R. D., Brown, K. C., and Dasgupta, P. (2008). Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 29, 151–158. doi: 10.1016/j.tips.2007.12.006
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faure, P., Tolu, S., Valverde, S., and Naudé, J. (2014). Role of nicotinic acetylcholine receptors in regulating dopamine neuron activity. Neuroscience 282, 86–100. doi: 10.1016/j.neuroscience.2014.05.040
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fu, X. W., Lindstrom, J., and Spindel, E. R. (2009). Nicotine activates and up-regulates nicotinic acetylcholine receptors in bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 41, 93–99. doi: 10.1165/rcmb.2008-0352OC
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gerde, P., Muggenburg, B. A., Stephens, T., Lewis, J. L., Pyon, K. H., and Dahl, A. R. (1998). A relevant dose of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is extensively metabolized and rapidly absorbed in the canine tracheal mucosa. Cancer Res. 58, 1417–1422.
Ghosh, D., Mishra, M. K., Das, S., Kaushik, D. K., and Basu, A. (2009). Tobacco carcinogen induces microglial activation and subsequent neuronal damage. J. Neurochem. 110, 1070–1081. doi: 10.1111/j.1471-4159.2009.06203.x
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gioanni, Y., Rougeot, C., Clarke, P. B. S., Lepousè, C., Thierry, A. H., and Vidal, C. (1999). Nicotinic receptors in the rat prefrontal cortex: increase in glutamate release and facilitation of mediodorsal thalamo-cortical transmission. Eur. J. Neurosci. 11, 18–30. doi: 10.1046/j.1460-9568.1999.00403.x
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, J., Ibaragi, S., Zhu, T., Luo, L. Y., Hu, G. F., Huppi, P. S., et al. (2008). Nicotine promotes mammary tumor migration via a signaling cascade involving protein kinase C and CDC42. Cancer Res. 68, 473–8481. doi: 10.1158/0008-5472.CAN-08-0131
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hecht, S. S., and Hoffmann, D. (1988). Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis 9, 875–884. doi: 10.1093/carcin/9.6.875
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heeschen, C., Jang, J. J., Weis, M., Pathak, A., Kaji, S., Hu, R. S., et al. (2001). Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 7, 833–839. doi: 10.1038/89961
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hogg, R. C., and Bertrand, D. (2007). Partial agonists as therapeutic agents at neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 73, 459–468. doi: 10.1016/j.bcp.2006.08.010
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Improgo, M. R., Soll, L. G., Tapper, A. R., and Gardner, P. D. (2013). Nicotinic acetylcholine receptors mediate lung cancer growth. Front. Physiol. 4:251. doi: 10.3389/fphys.2013.00251
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jorquera, R., Castonguay, A., and Schuller, H. M. (1992). Placental transfer of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone instilled intratracheally in Syrian golden hamsters. Cancer Res. 52, 3273–3280.
Jull, B. A., Plummer, H. K. III., and Schuller, H. M. (2001). Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. Clin. Oncol. 127, 707–717.
Kawai, H., and Berg, D. K. (2001). Nicotinic acetylcholine receptors containing the α7 subunits on rat cortical neurons do not undergo long lasting inactivation even when upregulated by chronic nicotine exposure. J. Neurochem. 78, 1367–1378. doi: 10.1046/j.1471-4159.2001.00526.x
Lambe, E. K., Picciotto, M. R., and Aghajanian, G. K. (2003). Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology 28, 216–225. doi: 10.1038/sj.npp.1300032
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Magleby, K. L., and Stevens, C. F. (1972). The effect of voltage on the time course of end-plate currents. J. Physiol. 223, 151–171. doi: 10.1113/jphysiol.1972.sp009839
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maneckjee, R., and Minna, J. D. (1990). Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc. Natl. Acad. Sci. U.S.A. 87, 3294–3298. doi: 10.1073/pnas.87.9.3294
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McNerney, M. E., Pardi, D., Pugh, P. C., Nai, Q., and Margiotta, J. F. (2000). Expression and channel properties of alpha-bungarotoxin-sensitive acetylcholine receptors on chick ciliary and choroid neurons. J. Neurophysiol. 84, 1314–1329.
Nelson, M. E., Kuryatov, A., Choi, C. H., Zhou, Y., and Lindstrom, J. (2003). Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol. Pharmacol. 63, 332–341. doi: 10.1124/mol.63.2.332
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nunes-Alves, A., Nery, A. A., and Ulrich, H. (2012). Tobacco nitrosamine N-nitrosonornicotine as inhibitor of neuronal nicotinic acetylcholine receptors. J. Mol. Neurosci. 49, 52–61. doi: 10.1007/s12031-012-9859-5
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paleari, L., Catassi, A., Ciarlo, M., Cavalieri, Z., Bruzzo, C., Servent, D., et al. (2008). Role of alpha7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif. 41, 936–959. doi: 10.1111/j.1365-2184.2008.00566.x
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paleari, S., Sessa, F., Catassi, A., Servent, D., Mourier, G., Doria-Miglietta, G., et al. (2009). Inhibition of non-neuronal alpha7-nicotinic receptor reduces tumorigenicity in A459 NSCLC xenografts. Int. J. Cancer 125, 199–211. doi: 10.1002/ijc.24299
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palma, E., Bertrand, S., Binzoni, T., and Bertrand, D. (1996). Homomeric neuronal nicotinic α7 receptors present five putative high affinity binding sites for the toxin MLA. J. Physiol. 491, 151–61. doi: 10.1113/jphysiol.1996.sp021203
Paradiso, K. G., and Steinbach, J. H. (2003). Nicotine is highly effective at producing desensitization of rat α4β2 neuronal nicotinic receptors. J. Physiol. 553, 857–871. doi: 10.1113/jphysiol.2003.053447
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeifer, G. P., Denissenko, M. F., Olivier, M., Tretyakova, N., Hecht, S. S., and Hainaut, P. (2002). Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21, 7435–7451. doi: 10.1038/sj.onc.1205803
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poorthuis, R. B., Bloem, B., Schak, B., Wester, J., de Kock, C. P., and Mansvelder, H. D. (2012). Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb. Cortex 23, 148–161. doi: 10.1093/cercor/bhr390
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Porter, J. T., Cauli, B., Tsuzuki, K., Lambolez, B., Rossier, J., and Audinat, E. (1999). Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J. Neurosci. 19, 5228–5235.
Rollema, H., Chambers, L. K., Coe, J. W., Glowa, J., Hurst, R. S., Lebel, L. A., et al. (2007). Pharmacological profile of the α4β2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 52, 985–994. doi: 10.1016/j.neuropharm.2006.10.016
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Russell, M. A. H., Jarvis, M., Iyer, R., and Feyerabend, C. (1980). Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br. Med. J. 280, 972–976. doi: 10.1136/bmj.280.6219.972
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaal, C., and Chellappan, S. P. (2014). Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 12, 14–23. doi: 10.1158/1541-7786.MCR-13-0541
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuller, H. M. (1989). Cell type specific, receptor mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem. Pharmacol. 38, 3439–3442. doi: 10.1016/0006-2952(89)90112-3
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuller, H. M. (2007). Nitrosamines as nicotinic receptor ligands. Life Sci. 80, 2274–2280. doi: 10.1016/j.lfs.2007.03.006
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuller, H. M. (2009). Is cancer triggered by altered signaling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 9, 195–205. doi: 10.1038/nrc2590
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuller, H. M., and Orloff, M. (1998). Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem. Pharmacol. 55, 1377–1384. doi: 10.1016/S0006-2952(97)00651-5
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuller, H. M., Tithof, P. K., Williams, M., and Plummer, H. III. (1999). The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 59, 4510–4515.
Song, P., Sekhon, H. S., Fu, X. W., Maier, M., Jia, Y., Duan, J., et al. (2008). Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res. 68, 4693–4700. doi: 10.1158/0008-5472.CAN-08-0183
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsurutani, J., Sianna Castillo, S., Brognard, J., Granville, C. A., Zhang, C., Gills, J. J., et al. (2005). Tobacco components stimulate Akt-dependent proliferation and NFκB-dependent survival in lung cancer cells. Carcinogenesis 26, 1182–1195. doi: 10.1093/carcin/bgi072
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vidal, C., and Changeux, J. P. (1993). Nicotinic and muscarinic modulations of excitatory synaptic transmission in the rat prefrontal cortex. Neuroscience 56, 23–32. doi: 10.1016/0306-4522(93)90558-W
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wessler, I., and Kirkpatrick, C. J. (2008). Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br. J. Pharmacol. 154, 1558–1571. doi: 10.1038/bjp.2008.185
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
West, K. A., Brognard, J., Clark, A. S., Linnoila, I. R., Yang, X., Swain, S. M., et al. (2003). Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J. Clin. Invest. 111, 81–90. doi: 10.1172/JCI200316147
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiang, Z., Huguenard, J. R., and Prince, D. A. (1998). Cholinergic switching within neocortical inhibitory networks. Science 281, 985–988. doi: 10.1126/science.281.5379.985
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zoli, M., Pistillo, F., and Gotti, C. (2015). Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96, 302–311. doi: 10.1016/j.neuropharm.2014.11.003
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zolles, G., Wagner, E., Lampert, A., and Sutor, B. (2009). Functional expression of nicotinic acetylcholine receptors in rat neocortical layer V. Cereb. Cortex 19, 1079–1091. doi: 10.1093/cercor/bhn158
PubMed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: CHRNA4, CHRNB2, HEK, nAChR, NNK, NNN, partial agonist, patch-clamp
Citation: Brusco S, Ambrosi P, Meneghini S and Becchetti A (2015) Agonist and antagonist effects of tobacco-related nitrosamines on human α4β2 nicotinic acetylcholine receptors. Front. Pharmacol. 6:201. doi: 10.3389/fphar.2015.00201
Received: 26 July 2015; Accepted: 01 September 2015;
Published: 22 September 2015.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
David J. Adams, RMIT University, AustraliaHildegard M. Schuller, University of Tennessee, USA
Copyright © 2015 Brusco, Ambrosi, Meneghini and Becchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Becchetti, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza, 2, 20126 Milano, Italy, andrea.becchetti@unimib.it