 Kentaro Tokudome1Takahiro Okumura1Ryo Terada1
Kentaro Tokudome1Takahiro Okumura1Ryo Terada1 Saki Shimizu1Naofumi Kunisawa1
Saki Shimizu1Naofumi Kunisawa1 Tomoji Mashimo2,3Tadao Serikawa1,2Masashi Sasa4
Tomoji Mashimo2,3Tadao Serikawa1,2Masashi Sasa4 Yukihiro Ohno1*
Yukihiro Ohno1*- 1Laboratory of Pharmacology, Osaka University of Pharmaceutical Sciences, Osaka, Japan
- 2Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University, Kyoto, Japan
- 3Institute of Experimental Animal Sciences, Graduate School of Medicine, Osaka University, Osaka, Japan
- 4Nagisa Clinic, Osaka, Japan
Synaptic vesicle glycoprotein 2A (SV2A) is specifically expressed in the membranes of synaptic vesicles and modulates action potential-dependent neurotransmitter release. To explore the role of SV2A in the pathogenesis of epileptic disorders, we recently generated a novel rat model (Sv2aL174Q rat) carrying a missense mutation of the Sv2a gene and showed that the Sv2aL174Q rats were hypersensitive to kindling development (Tokudome et al., 2016). Here, we further conducted behavioral and neurochemical studies to clarify the pathophysiological mechanisms underlying the seizure vulnerability in Sv2aL174Q rats. Sv2aL174Q rats were highly susceptible to pentylenetetrazole (PTZ)-induced seizures, yielding a significantly higher seizure scores and seizure incidence than the control animals. Brain mapping analysis of Fos expression, a biological marker of neural excitation, revealed that the seizure threshold level of PTZ region-specifically elevated Fos expression in the amygdala in Sv2aL174Q rats. In vivo microdialysis study showed that the Sv2aL174Q mutation preferentially reduced high K+ (depolarization)-evoked GABA release, but not glutamate release, in the amygdala. In addition, specific control of GABA release by SV2A was supported by its predominant expression in GABAergic neurons, which were co-stained with antibodies against SV2A and glutamate decarboxylase 1. The present results suggest that dysfunction of SV2A by the missense mutation elevates seizure susceptibility in rats by preferentially disrupting synaptic GABA release in the amygdala, illustrating the crucial role of amygdalar SV2A-GABAergic system in epileptogenesis.
Introduction
Synaptic vesicle glycoprotein 2A (SV2A) is highly expressed in the brain including the cerebral cortex, limbic regions, and cerebellum, where it modulates action potential-dependent neurotransmitter release (Crowder et al., 1999; Janz et al., 1999; Xu and Bajjalieh, 2001; Custer et al., 2006; Chang and Südhof, 2009). Although the functional mechanisms of SV2A remain to be clarified, it is suggested that SV2A primes synaptic vesicles to fully respond to Ca2+ probably by interacting with the Ca2+ sensor protein synaptotagmin (Xu and Bajjalieh, 2001; Chang and Südhof, 2009; Nowack et al., 2010). In addition, previous studies suggest that SV2A plays an important role in the pathogenesis and treatment of epileptic disorders. This is because (1) SV2A-knockout mice exhibited severe seizures (Crowder et al., 1999; Janz et al., 1999), (2) SV2A serves as a specific binding site for certain antiepileptics (e.g., levetiracetam and its analogs; Lynch et al., 2004; Pollard, 2008; Kaminski et al., 2009; Correa-Basurto et al., 2015; Klitgaard et al., 2016) and (3) the expressional levels of SV2A are reported to be altered in various epileptic conditions both in animals (e.g., chemically- and electrically induced kindling) and humans (e.g., intractable temporal lobe epilepsy and focal cortical dysplasia; Matveeva et al., 2007; Feng et al., 2009; Ohno et al., 2009a, 2012b; Toering et al., 2009; van Vliet et al., 2009; Crèvecoeur et al., 2014; Serajee and Huq, 2015). Furthermore, a recent clinical study reported that a missense mutation (R383Q) in exon 5 of the SV2A gene resulted in intractable epilepsy, involuntary movements, microcephaly and developmental retardation (Serajee and Huq, 2015).
In order to explore the role of SV2A in modulating development of epileptic disorders (epileptogenesis), we recently generated a novel rat model (Sv2aL174Q rat) carrying a missense mutation (L174Q) in the Sv2a gene (Tokudome et al., 2016), using gene-driven ENU mutagenesis/MuT-POWER techniques (Mashimo et al., 2008). Sv2aL174Q rats were susceptible to PTZ seizures and to kindling development associated with repeated PTZ treatments or focal electrical stimulation of the amygdala. In addition, the Sv2aL174Q mutation significantly reduced depolarization-induced GABA release in the hippocampus. These findings suggest that SV2A plays a crucial role in the kindling epileptogenesis possibly by interacting GABAergic neurons. However, the detailed mechanisms underlying the regulation of seizure susceptibility by SV2A remain to be clarified.
In the present study, therefore, we further conducted behavioral and neurochemical studies to clarify the mechanisms (e.g., responsible brain regions and influences on synaptic amino acid release) underlying the seizure vulnerability in Sv2aL174Q rats. The present results show that the Sv2aL174Q mutation elevates excitability of the corticolimbic neural circuit, especially in the amygdala, by preferentially disrupting synaptic GABA release, illustrating the crucial role of amygdalar SV2A-GABAergic system in epileptogenesis.
Materials and Methods
Animals
Male Sv2aL174Q rats (Tokudome et al., 2016) were obtained from the National BioResource Project-Rat (F344-Sv2am1Kyo NBRP-Rat No:0668). The Sv2aL174Q rat, carrying a single nucleotide mutation T521A, was first identified in a gene-driven ENU mutagenesis project in Kyoto University (Mashimo et al., 2008). Thereafter, Sv2aL174Q rats were backcrossed more than five generations on the F344/NSlc inbred background to eliminate mutations potentially induced by ENU mutagenesis elsewhere in the genome. Age-matched male F344 rats (Japan SLC, Shizuoka, Japan) were used as the control animal. The animals were kept in air-conditioned rooms under a 12-h light/dark cycle and allowed ad libitum access to food and water. All animal experiments were approved by the Animal Research Committees of Osaka University of Pharmaceutical Sciences and were conducted according to the Institutional Committees’ regulations on animal experimentation.
Evaluation of Seizure Susceptibility
To evaluate the seizure sensitivity, Sv2aL174Q or F344 rats were treated with an intraperitoneal dose of PTZ (30, 35, and 40 mg/kg for Sv2aL174Q rats; 35, 40, 45, and 50 mg/kg for F344 rats). PTZ-induced seizures were evaluated over 20 min after the drug treatment using a 6-point ranked scale as follows, 0: none response, 1: facial automatisms and twitching of the ears and whiskers, 2: convulsive waves propagating axially along the trunk, 3: myoclonic convulsions with a delay, 4: clonic convulsions, 5: repeated powerful clonic-tonic or lethal convulsions (Racine, 1972; Franke and Kittner, 2001; Kudryashov et al., 2007). The incidence of seizures was judged as positive when the animal showed a seizure score of 3 or more.
Analysis of Fos Protein Expression
To explore causal brain regions for PTZ seizures in Sv2aL174Q rats, expression of Fos protein, a biological marker of neural excitation, by the seizure threshold level of PTZ was analyzed. For this purpose, Sv2aL174Q and F344 rats were cumulatively injected with an increasing dose of PTZ (first dose: 10 mg/kg, second dose: 20 mg/kg, third dose: 30 mg/kg) with 30-min intervals. The incidence of seizures was monitored for 10 min immediately after each PTZ injection using a seizure scale described previously. Since the PTZ-induced seizures were observed only at 30 mg/kg, brain samples were obtained 2 h after the PTZ (30 mg/kg) injection under pentobarbital (80 mg/kg, i.p.) anesthesia.
After fixation with 4% formaldehyde solution, coronal sections (30 μm thickness) were cut from each brain using a Microslicer (DSK-3000, Dosaka, Kyoto Japan). The immunostaining of Fos protein was performed using a previously published method (Ohno et al., 2011, 2012a). Briefly, formalin-fixed sections were immunohistochemically stained with a goat c-Fos antiserum (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) by the ABC method. Fos-immunoreactivity was visualized by the diaminobenzidine–nickel staining method and quantified by counting the number of Fos-positive neurons. Brain regions analyzed includes (1) cerebral cortices: mPFC, CgC, MC, SC, AIC, Pir, AuC, PRh-Ent; (2) basal ganglia and limbic regions: AcC, AcS, dlST, dmST, GP, LS, CA, DG, PMCo, BMA, BLA; (3) diencephalon: LHb, PT, AM, CM, VM, AH, PH, DMH (see Paxinos and Watson, 2007).
In Vivo Microdialysis Study
Sv2aL174Q or F344 rats were anesthetized with pentobarbital (40 mg/kg, i.p.) and fixed in a stereotaxic instrument (Narishige, SR-6, Tokyo, Japan). A guide cannula (1 mm diameter) was inserted into a position 2 mm above the amygdala (P: 2.8 mm, L: 4.8 mm, H: -6.1 mm; Paxinos and Watson, 2007) and fixed to the skull using dental cement. After a recovery period of about 1 week, animals with a chronically implanted guide cannula were subjected to microdialysis experiments. Briefly, a dialysis probe (Eicom, A-I-10-02, Kyoto, Japan) was inserted into the amygdala through a guide cannula and aCSF containing (in mM): NaCl 140, KCl 2.4, MgCl2 1.0, CaCl2 1.2, NaHCO3 5.0, was perfused at a flow rate of 1 μL/min using a microperfusion pump (Eicom, ESP-32, Kyoto, Japan). The dialysate samples were collected into a microtube every 10 min (10 μL/sample). To evaluate the depolarization-evoked synaptic release, high concentration (100 mM) K+-containing aCSF was perfused for 60 min through the dialysis probe.
The dialysate samples were analyzed for GABA and glutamate levels using a HPLC-ECD system. GABA and glutamate were derivatized with o-phthalaldehyde before the HPLC injection and separated on a cation exchange column (Eicom, 3.0φ × 150 mm; Eicompak SC-5ODS, Kyoto, Japan). The mobile phase consisted of 0.1 M phosphate buffer, 5 mg/L EDTA 2Na, pH6.0, with 27% methanol pumped at a flow rate of 500 μL/min. All data were analyzed by using eDAQ Power Chrom (eDAQ Pty Ltd, Denistone East, NSW, Australia). Extracellular GABA and glutamate levels were expressed as a percentage of the basal control level, which was the mean of the three points before the high K+ application, in each animal. The AUC of the high K+-evoked GABA or glutamate release was also estimated by the trapezoidal approximation method.
Immunofluorescence Double Staining
Sv2aL174Q or F344 rats were decapitated under pentobarbital (80 mg/kg, i.p.) anesthesia and brains were removed from the skull. After fixation with 4% paraformaldehyde solution for 24 h, the brain was dehydrated and embedded in paraffin. Formalin-fixed and paraffin-embedded amygdaloid tissues were cut into 4 μm thick sections and the sections were subjected to immunofluorescence double staining with anti-SV2A and anti-Gad1 (Ohno et al., 2012b). Sections were incubated with a goat anti-rat SV2A (dilution 1:500, Santa Cruz Biotechnology, Dallas, TX, USA) and mouse anti-human Gad1 (dilution 1:1000, Santa Cruz Biotechnology, Dallas, TX, USA) for 42 h at 4°C, and then with an FITC (green fluorescence)-conjugated rabbit anti-goat IgG secondary antibody (dilution 1:500, Sigma–Aldrich, St. Louis, MO, USA) and TRITC (red fluorescence)-conjugated rabbit anti-mouse IgG secondary antibody (dilution 1:500, Sigma–Aldrich, St. Louis, MO, USA) to probe SV2A and Gad1, respectively. Immunofluorescence images were obtained with a confocal laser scanning microscope (Carl Zeiss Japan, LSM 700 ZEN, Tokyo, Japan). To quantify SV2A and Gad1 expression, digital images of the amygdala were stored and the integrated optical density was measured by computer analysis with ImageJ software (ver. 1.42, NIH).
Statistical Analysis
Statistical significance of differences between two groups was performed by Mann–Whitney’s U-test (behavioral scores) or Student’s t-test (Fos and SV2A expression). Comparisons of seizure incidence rate were done by X2 test. Differences in GABA and glutamate release (in vivo microdialysis) were analyzed by two-way ANOVA followed by Tukey’s post hoc test. A P-value of less than 0.05 was considered statistically significant.
Results
Seizure Susceptibility of Sv2aL174Q Rats
Sv2aL174Q rats have a missense mutation T521A in the Sv2a gene, which results in the substitution of Leu 174 to Gln (Figure 1A). The mutation site is located in the first transmembrane region of SV2A that was reportedly essential for the normal function and structure of SV2A (Chang and Südhof, 2009). In order to confirm seizure susceptibility of Sv2aL174Q rats, we evaluated the responses of Sv2aL174Q and F344 rats to PTZ injections (30–50 mg/kg, i.p.). While gross behaviors of Sv2aL174Q rats were normal, these animals showed high susceptibility to PTZ seizures, yielding significantly higher seizure scores at 35 mg/kg [U(17) = 20.5, P < 0.05] and 40 mg/kg [U(17) = 12.0, P < 0.01], and a higher seizure incidence at 40 mg/kg (X2 = 6.34, P < 0.05) than F344 rats (Figure 1B).
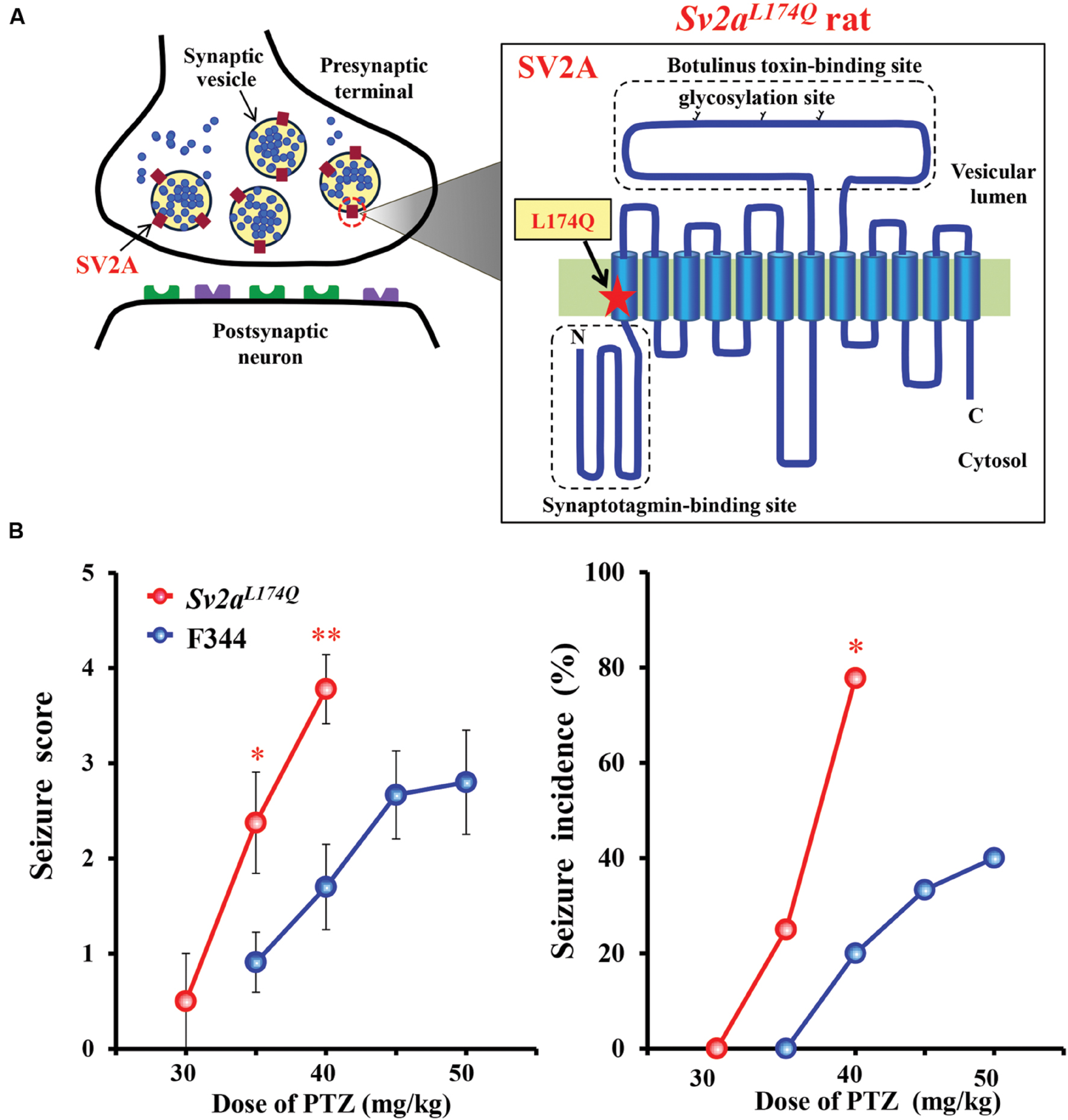
FIGURE 1. Seizure susceptibility of Sv2aL174Q rats. (A) Structure of SV2A showing the mutation site L174Q. SV2A possesses a 12-transmembrane structure which contains a putative binding site for synaptotagmin in the N terminal region and glycosylation sites in a long intravesicular loop between transmembrane regions 7 and 8. (B) Susceptibility of Sv2aL174Q rats to PTZ-induced seizures. Each point represents the mean ± SEM of 8–11 animals. ∗P < 0.05, ∗∗P < 0.01 significantly different from F344 rats.
Fos Expression Analysis
To explore the brain sites most sensitive to the seizure threshold level of PTZ, Sv2aL174Q and F344 rats were cumulatively injected with an increasing dose of PTZ (10, 20, or 30 mg/kg, i.p.) with 30-min intervals. Under these conditions, the first two does did not evoke any seizure either in Sv2aL174Q or F344 rats. However, subsequent 30 mg/kg PTZ induced clonic or tonic-clonic seizures in five out of seven Sv2aL174Q rats while none of seven F344 rats tested exhibited seizures.
We next compared the expression of Fos protein, a biological marker of neural excitation, in various regions of the brain between the Sv2aL174Q (PTZ seizure-positive, N = 5) and F344 (PTZ-negative, N = 7) rats (Figure 2). Sv2aL174Q rats showed considerably higher Fos expression than F344 rats in most of the cerebral cortex, reflecting the generalized seizure property of PTZ seizures (Figure 3A). PTZ-induced Fos expression in Sv2aL174Q rats were statistically significant in the SC and Pir. On the other hand, the seizure threshold level of PTZ region-specifically elevated Fos expression in the amygdala among 21 subcortical regions examined including basal ganglia, limbic regions, and diencephalon (Figures 3B,C).
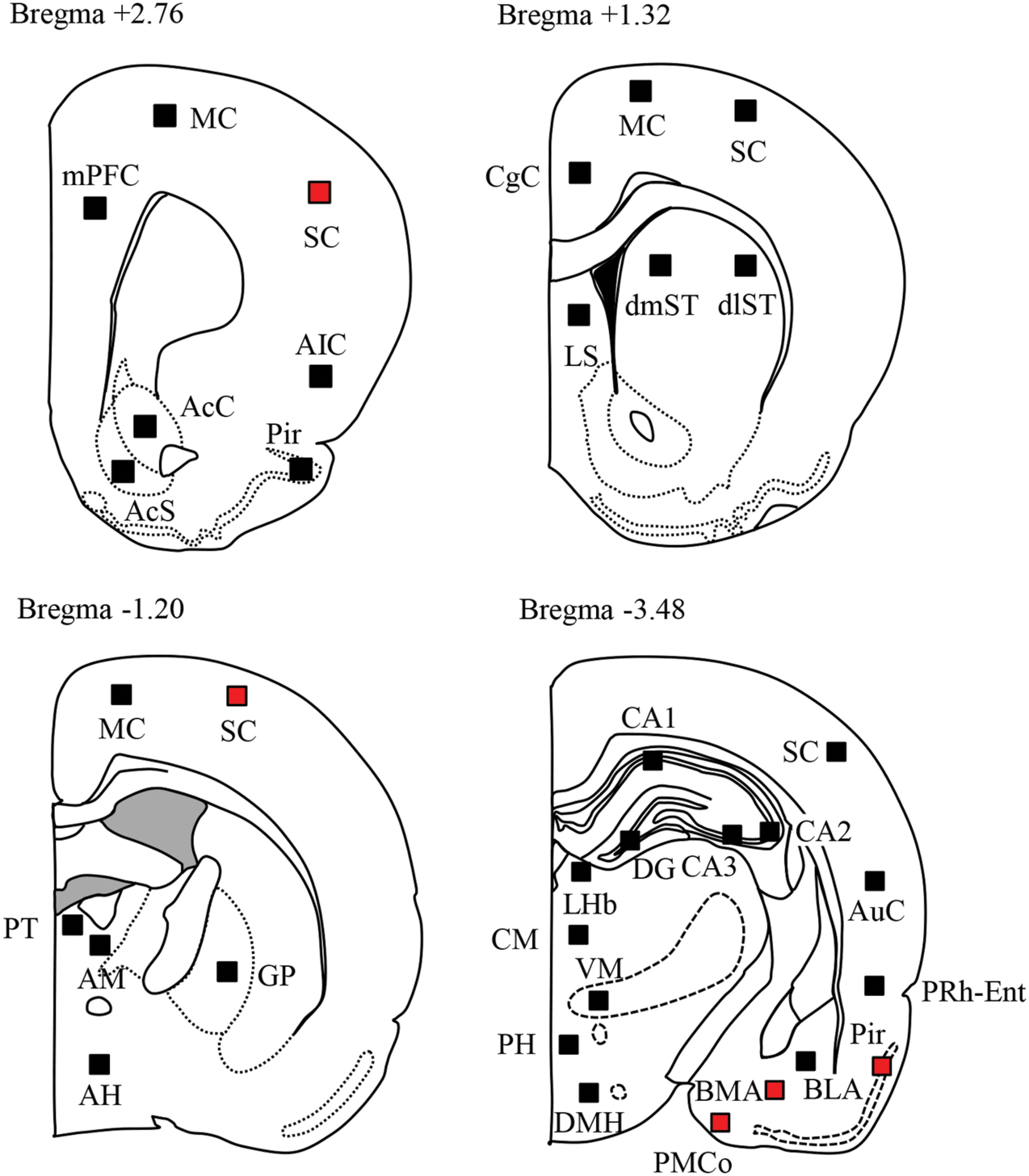
FIGURE 2. Topographical Fos expression analysis following PTZ-induced seizures. Schematic illustrations of the brain sections selected for quantitative analysis of Fos protein expression. Filled boxes in each section indicate the sample areas analyzed. Red boxes represent the regions where Fos expression was significantly elevated by PTZ treatments.
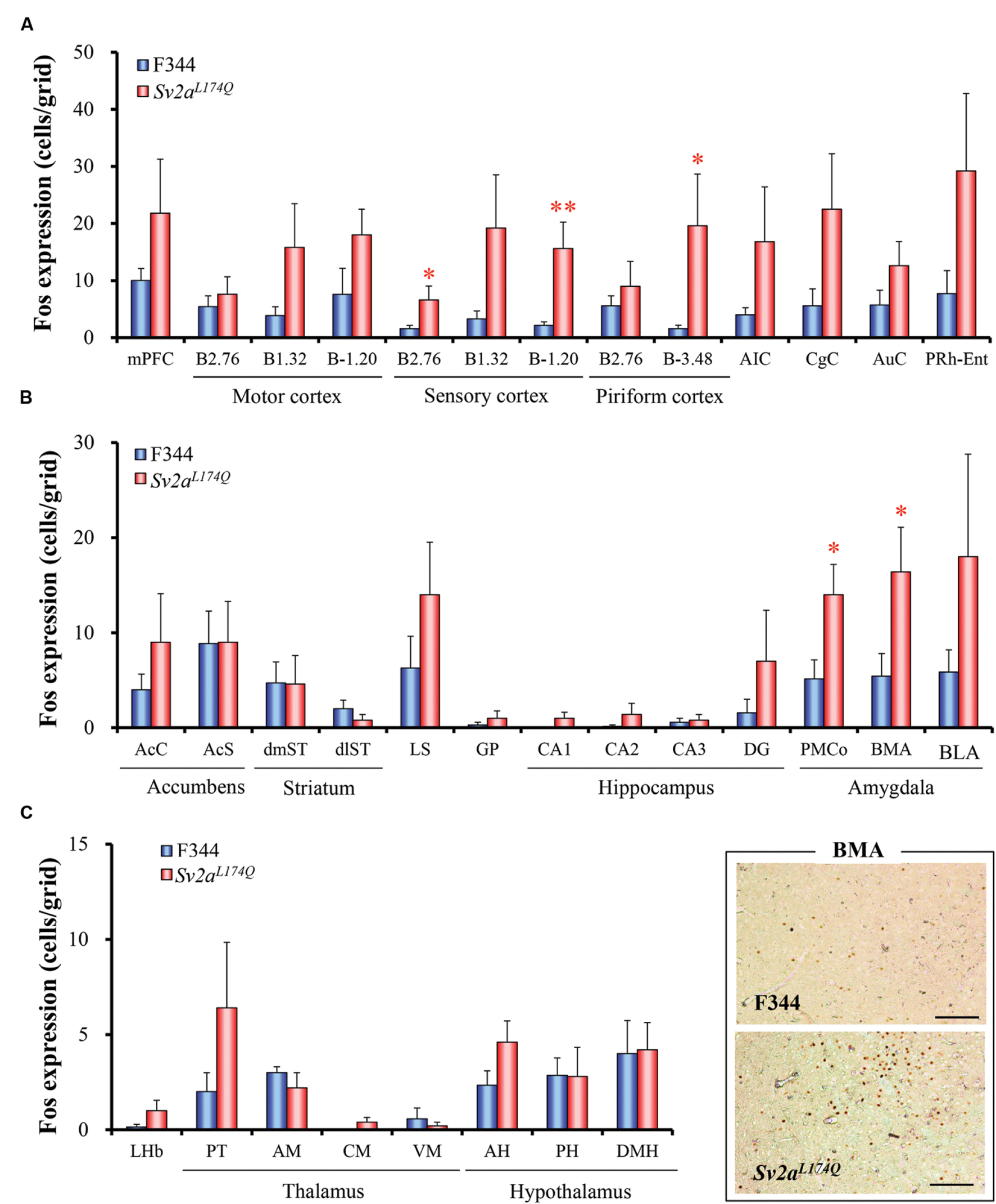
FIGURE 3. Regional changes in Fos expression levels by the seizure threshold level of PTZ. Graphs show Fos expression in the cerebral cortical regions (A), basal ganglia and limbic regions (B), and diencephalon (C). Typical photos of Fos expression in the amygdala are also shown in the right bottom. Each column represents the mean ± SEM of five Sv2aL174Q (PTZ seizure-positive) and seven F344 (PTZ seizure-negative) rats. ∗P < 0.05, ∗∗P < 0.01 significantly different from F344 rats. Scale bar: 100 μm.
GABA and Glutamate Release in the Amygdala
Since Sv2aL174Q rats exhibited a region-specific excitation of the amygdala by PTZ, we conducted in vivo microdialysis studies to evaluate synaptic release of GABA and glutamate in the amygdala. As shown in Figure 4A, high K+ (depolarization) stimuli evoked GABA release both in Sv2aL174Q and F344 rats. However, the depolarization-evoked GABA release was largely diminished by the Sv2aL174Q mutation [F(1,218) = 56.72, P < 0.001] (Figure 4A). A comparison of AUC also revealed a significant reduction in depolarization-evoked GABA release in Sv2aL174Q rats. On the other hand, in contrast to GABA release, high K+-evoked glutamate release was not significantly affected by the Sv2aL174Q mutation (Figure 4B).
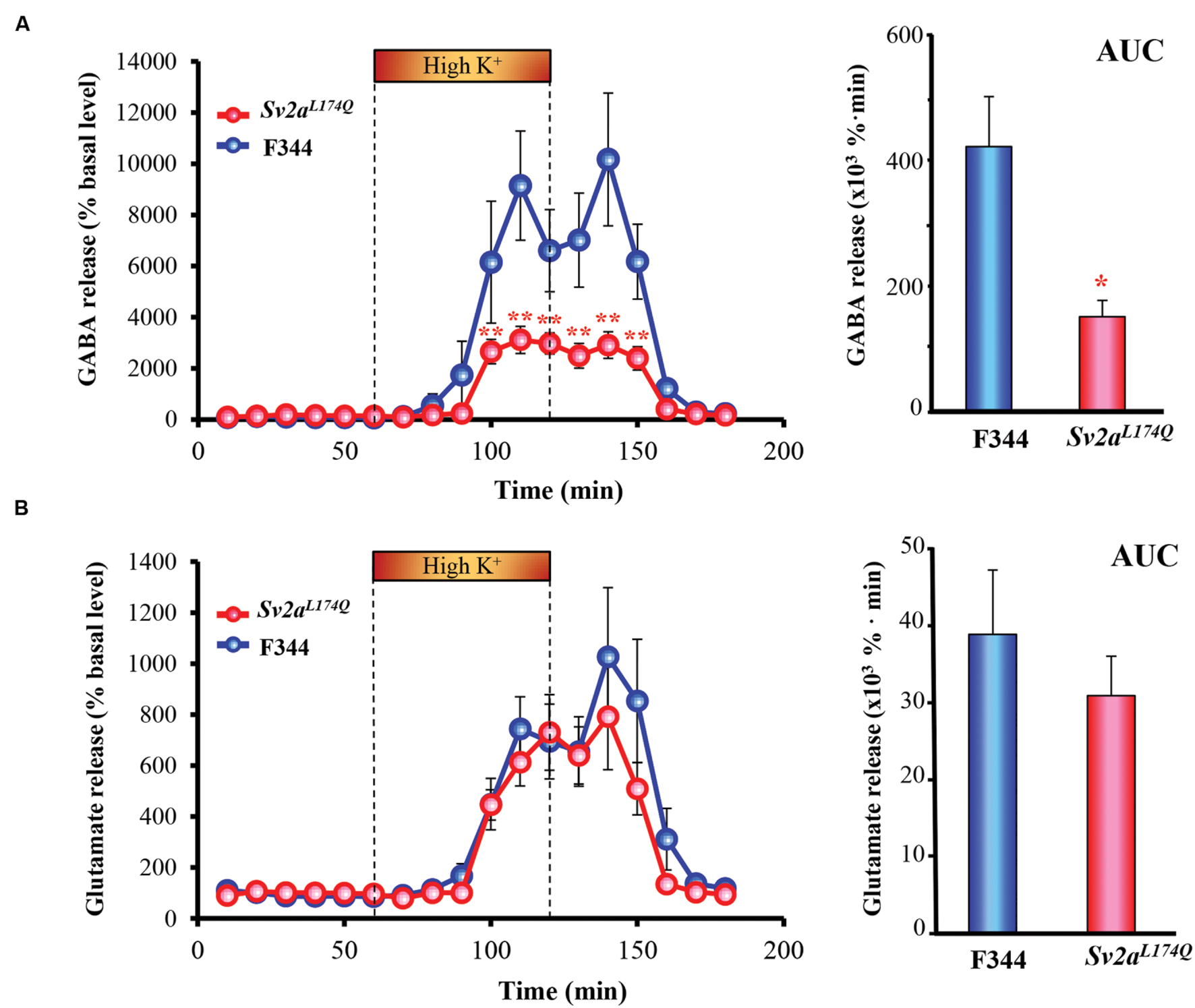
FIGURE 4. Synaptic GABA and glutamate release in the amygdala. (A) GABA release. (B) Glutamate release. Each point represents the mean ± SEM of 10 or 5 animals. Depolarization stimulation was given by applying high concentration (100 mM) K+-containing aCSF (High K+) for 60 min through the dialysis probe. AUC (the area under the curve) comparisons of GABA and glutamate release are shown in the right. ∗P < 0.05, ∗∗P < 0.01 significantly different from F344 rats. Each point represents the mean ± SEM of 10 or 5 animals.
SV2A and Gad1 Double Staining in the Amygdala
We further conducted immunofluorescence double staining of SV2A with Gad1 (also known as GAD67), a marker protein of GABAergic neurons, in the amygdala. As shown in Figure 5A, SV2A (green) was mostly co-stained with Gad1 (red) both in Sv2aL174Q and F344 rats, illustrating a specific expression of SV2A in the amygdalar GABAergic neurons. In addition, there were no significant differences in expressional levels of SV2A and Gad1 between Sv2aL174Q and F344 rats (Figure 5B).
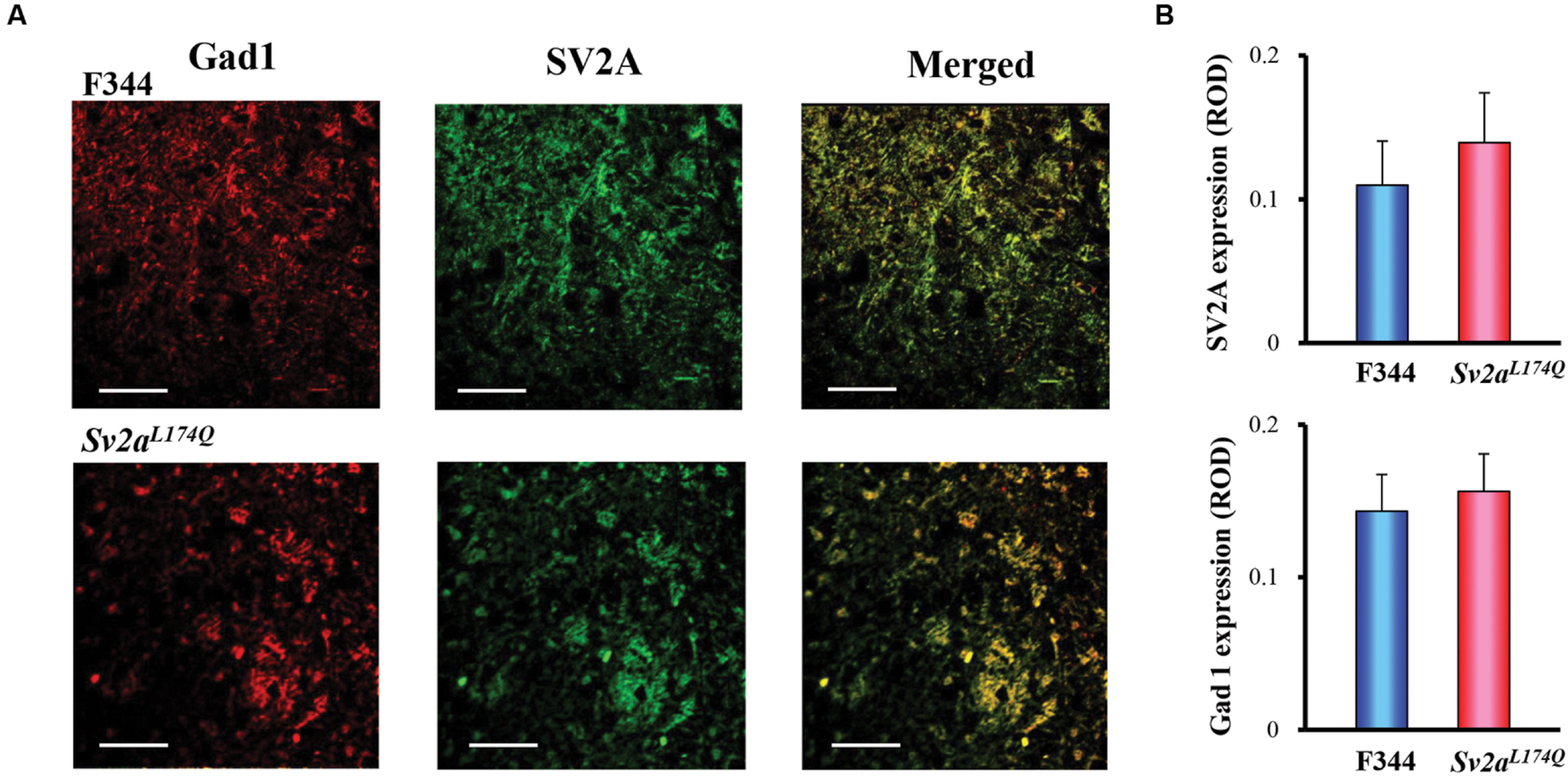
FIGURE 5. SV2A and Gad1 double staining in the amygdala. (A) Photos showing representative double staining of Gad1 (left: red), SV2A (center: green) in the amygdala of Sv2aL174Q and F344 rats. Merged photos (right) revealed a predominant co-expression of SV2A with Gad1 (GABAergic neurons) in the amygdala. (B) Expression levels of SV2A and Gad1 in Sv2aL174Q and F344 rats. ROD, relative optical density. Each column represents the mean ± SEM of four animals. Scale bar: 50 μm.
Discussion
The present study confirmed that Sv2aL174Q rats carrying a missense mutation, L174Q, in the Sv2a gene were highly sensitive to PTZ-induced seizures, supporting the notion that SV2A plays the crucial role in controlling seizure susceptibility. Although a complete deletion of SV2A is known to cause premature death with severe seizure incidence (Crowder et al., 1999; Janz et al., 1999), the behavioral phenotype of Sv2aL174Q rats mimicked those reported in heterozygous SV2A-deficient mice (Kaminski et al., 2009), implying that the Sv2aL174Q mutation causes a partial loss of the SV2A function.
Fos protein is the immediate early gene product and is widely used as a cellular marker of neural excitation. Specifically, brain mapping analysis of Fos expression is useful to identify brain regions related to disease conditions (e.g., epilepsy, emotional disorders and cognitive impairments) or responses to various pathophysiological and pharmacological stimuli (e.g., pain, body temperature, stress, and drug treatments; Morgan and Curran, 1989; Herrera and Robertson, 1996; Ohno et al., 2009b, 2011, 2012a, 2015; Mukai et al., 2013; Fumoto et al., 2014; Iha et al., 2016). In the present study, we treated animals with the seizure threshold dose (30 mg/kg, i.p.) of PTZ which first evoked seizures only in Sv2aL174Q rats. Under these conditions, the PTZ treatment region-specifically elevated Fos expression in the cerebral cortex (e.g., SM and Pir), amygdala. Since PTZ evoked generalized clonic or tonic-clonic seizures associated with a wide-spread excitation of the cerebral cortex (Figure 3A), we could not specifically identify the causative regions in the cortical regions. Nonetheless, the present results clearly illustrates that the amygdala is most sensitive to and potential seizure initiation site for PTZ seizures in Sv2aL174Q rats. In addition, in vivo microdialysis demonstrated that the Sv2aL174Q mutation preferentially impaired depolarization-evoked synaptic release of GABA in the amygdala without affecting glutamate release. These findings provide important information for our understanding of the SV2A function in modulating seizure susceptibility. Although the hippocampus was less sensitive to PTZ seizures than the amygdala, our results do not deny the potential role of hippocampus in seizure vulnerability of Sv2aL174Q rats since it is known that PTZ evokes seizures by activating limbic regions including the hippocampus (Humpel et al., 1993; Löscher and Ebert, 1996; Szyndler et al., 2009) and that, indeed, a sufficient dose (70 mg/kg, i.p.) of PTZ increases Fos expression in the hippocampus (Bastlund et al., 2005). In addition, we previously demonstrated that the Sv2aL174Q mutation also disrupted depolarization-evoked synaptic release of GABA in the hippocampus. Furthermore, the piriform cortex also seems to be partly involved in the seizure vulnerability in Sv2aL174Q rats since the threshold level of PTZ significantly elevated Fos expression in the posterior part of this structure.
Preferential modulation of synaptic GABA release by SV2A was further supported by the SV2A expression pattern in amygdala GABAergic neurons. In that, most of SV2A-immunoreactivity was expressed in the amygdala neurons and dendrites, which were co-stained with antibodies against SV2A and Gad1. These findings are consistent with our previous findings that SV2A was predominantly expressed in GABAergic neurons, but only rarely in glutamatergic neurons in the mouse hippocampus (Ohno et al., 2012b). Therefore, SV2A seems to specifically regulate synaptic GABA release both in the amygdala and hippocampus. Since the amygdala and hippocampus are potential causative sites for various epileptic disorders (Löscher and Ebert, 1996; Morimoto et al., 2004; Avoli and de Curtis, 2011), the SV2A-GABAergic system in these structures is likely to be involved in pathogenesis of SV2A-related epileptic disorders.
The detailed mechanisms underlying SV2A dysfunction by the Sv2aL174Q mutation are currently unknown. Interestingly, a previous study showed that the neighboring missense mutations (D179A and E182A) of the two charged polar amino acids to the non-polar alanine abolished the normal function of SV2A, possibly by disrupting the protein folding and/or trafficking into synaptic membranes (Chang and Südhof, 2009). Thus, the changes in polarization of the first transmembrane region by the substitution of hydrophobic non-polar Leu 174 to polar Gln may impair the integrity of SV2A function. In addition, we previously showed that the Sv2aL174Q mutation specifically reduced the expression level Syt1, the Ca2+ sensor protein modulating synaptic release, among exocytosis regulatory proteins examined (Tokudome et al., 2016). Since SV2A is suggested to prime synaptic vesicles by interacting with the Syt1 (Xu and Bajjalieh, 2001; Chang and Südhof, 2009; Nowack et al., 2010), the disruption of synaptic GABA release by the Sv2aL174Q mutation may result from reduced expression of Syt1. Further studies are required to delineate molecular mechanisms of SV2A dysfunction by the Sv2aL174Q mutation.
The present study supports the clinical view that dysfunction of SV2A is involved in the pathogenesis of epilepsy, including intractable temporal lobe epilepsy and focal cortical dysplasia epilepsy (Feng et al., 2009; Toering et al., 2009; van Vliet et al., 2009; Crèvecoeur et al., 2014). Indeed, a recent study showed that a missense mutation R383Q in the SV2A gene caused intractable epilepsy and involuntary movements, which were accompanied by developmental retardation (Serajee and Huq, 2015). Thus, the Sv2aL174Q rat may be useful for exploring the epileptogenic mechanisms of SV2A-related epileptic disorders. Furthermore, since SV2A is known as a specific binding site for certain antiepileptics (e.g., levetiracetam, brivaracetam, and seletracetam; Lynch et al., 2004; Pollard, 2008; Kaminski et al., 2009; Correa-Basurto et al., 2015; Klitgaard et al., 2016), the Sv2aL174Q rat may also be useful as a novel animal model for analyzing the action mechanisms of the levetiracetam-analogs.
Conclusion
We confirmed high susceptibility of Sv2aL174Q rats to PTZ seizures. Treatment of Sv2aL174Q rats with PTZ at threshold level specifically elevated Fos expression in the amygdala, suggesting that the amygdala is the potential site responsible for seizure vulnerability in Sv2aL174Q rats. In addition, the Sv2aL174Q mutation preferentially reduced depolarization-evoked GABA, but not glutamate, release in the amygdala. The preferential disruption of GABA release due to the Sv2aL174Q mutation was supported by the specific expression of SV2A in GABAergic neurons. The present study suggests that dysfunction of SV2A by the missense mutation elevates seizure susceptibility by disrupting amygdalar synaptic GABA release, illustrating the crucial role of the SV2A-GABAergic system in epileptogenesis.
Author Contributions
YO designed research. KT, TO, SS, RT, NK, TM, and YO performed pharmacological and neurochemical research. KT, TO, SS, RT, NK, TM, and YO analyzed data. KT, TM, TS, MS, and YO wrote the paper.
Funding
This study was supported in part by a research grant from a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (No.26460111 and 15H04892; YO) and from the Japan Agency for Medical Research and Development (15ek0109120s0701; YO).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer TK and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgment
We are also thankful to the National BioResource Project-Rat for providing the Sv2aL174Q rats.
Abbreviations
AcC, core region of nucleus accumbens; AcS, shell region of nucleus accumbens; aCSF, artificial cerebrospinal fluid; AIC, agranular insular cortex; AH, anterior hypothalamus; AM, anteromedial thalamic nucleus; AUC, area under the curve; AuC, auditory cortex; BLA, anterior basolateral amygdaloid nucleus; BMA, anterior basomedial amygdaloid nucleus; CA, Cornu Ammonis area of hippocampus; CgC, cingulated cortex; CM, centromedial thalamic nucleus; DG, dentate gyrus of the hippocampus; dlST, dorsolateral striatum; DMH, dorsomedial hypothalamic nucleus; dmST, dorsomedial striatum; ENU, N-ethyl-N-nitrosourea; FITC, fluorescein isothiocyanate; Gad1, glutamate decarboxylase 1; GP, globus pallidus; LHb, lateral habenular nucleus; LS, lateral septum; MC, motor cortex; mPFC, the medial prefrontal cortex; Pir, piriform cortex; PH, posterior hypothalamus; PMCo, posteromedial cortical amygdaloid nucleus; PRh-Ent, perirhinal-entorhinal cortex; PT, paratenial thalamic nucleus; PTZ, pentylenetetrazole; SC, sensory cortex; SV2A, Synaptic vesicle glycoprotein 2A; Syt1, synaptotagmin1; TRITC, tetramethyrhodamine-5-(and 6)- isothiocyanate; VM, ventromedial thalamic nucleus.
References
Avoli, M., and de Curtis, M. (2011). GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog. Neurobiol. 95, 104–132. doi: 10.1016/j.pneurobio.2011.07.003
Bastlund, J. F., Berry, D., and Watson, W. P. (2005). Pharmacological and histological characterization of nicotine-kindled seizures in mice. Neuropharmacology 48, 975–983. doi: 10.1016/j.neuropharm.2005.01.015
Chang, W. P., and Südhof, T. C. (2009). SV2 renders primed synaptic vesicles competent for Ca2+-induced exocytosis. J. Neurosci. 29, 883–897. doi: 10.1523/JNEUROSCI.4521-08.2009
Correa-Basurto, J., Cuevas-Hernández, R. I., Phillips-Farfán, B. V., Martínez-Archundia, M., Romo-Mancillas, A., Ramírez-Salinas, G. L., et al. (2015). Identification of the antiepileptic racetam binding site in the synaptic vesicle protein 2A by molecular dynamics and docking simulations. Front. Cell. Neurosci. 9:125. doi: 10.3389/fncel.2015.00125
Crèvecoeur, J., Kaminski, R. M., Rogister, B., Foerch, P., Vandenplas, C., Neveux, M., et al. (2014). Expression pattern of synaptic vesicle protein 2 (SV2) isoforms in patients with temporal lobe epilepsy and hippocampal sclerosis. Neuropathol. Appl. Neurobiol. 40, 191–204. doi: 10.1111/nan.12054
Crowder, K. M., Gunther, J. M., Jones, T. A., Hale, B. D., Zhang, H. Z., Peterson, M. R., et al. (1999). Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. U.S.A. 96, 15268–15273. doi: 10.1073/pnas.96.26.15268
Custer, K. L., Austin, N. S., Sullivan, J. M., and Bajjalieh, S. M. (2006). Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J. Neurosci. 26, 1303–1313. doi: 10.1523/JNEUROSCI.2699-05.2006
Feng, G., Xiao, F., Lu, Y., Huang, Z., Yuan, J., Xiao, Z., et al. (2009). Down-regulation synaptic vesicle protein 2A in the anterior temporal neocortex of patients with intractable epilepsy. J. Mol. Neurosci. 39, 354–359. doi: 10.1007/s12031-009-92882
Franke, H., and Kittner, H. (2001). Morphological alterations of neurons and astrocytes and changes in emotional behavior in pentylenetetrazol-kindled rats. Pharmacol. Biochem. Behav. 70, 291–303. doi: 10.1016/S0091-3057(01)006128
Fumoto, N., Mashimo, T., Masui, A., Ishida, S., Mizuguchi, Y., Minamimoto, S., et al. (2014). Evaluation of seizure foci and genes in the Lgi1L385R/+ mutant rat. Neurosci. Res. 80, 69–75. doi: 10.1016/j.neures.2013.12.008
Herrera, D. G., and Robertson, H. A. (1996). Activation of c-fos in the brain. Prog. Neurobiol. 50, 83–107. doi: 10.1016/S0301-0082(96)000214
Humpel, C., Wetmore, C., and Olson, L. (1993). Regulation of brain-derived neurotrophic factor messenger RNA and protein at the cellular level in pentylenetetrazol-induced epileptic seizures. Neuroscience 53, 909–918. doi: 10.1016/0306-4522(93)90476-V
Iha, H. A., Kunisawa, N., Tokudome, K., Mukai, T., Kinboshi, M., Shimizu, S., et al. (2016). “Immunohistochemical analysis of Fos protein expression for exploring brain regions (foci) related to central nervous system (CNS) disorders and drug actions,” in In Vivo Neuropharmacology and Neurophysiology, ed. A. Philippu (New York, NY: Springer).
Janz, R., Goda, Y., Geppert, M., Missler, M., and Südhof, T. C. (1999). SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 24, 1003–1016. doi: 10.1016/S0896-6273(00)810466
Kaminski, R. M., Gillard, M., Leclercq, K., Hanon, E., Lorent, G., Dassesse, D., et al. (2009). Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia 50, 1729–1740. doi: 10.1111/j.1528-1167.2009.02089.x
Klitgaard, H., Matagne, A., Nicolas, J. M., Gillard, M., Lamberty, Y., De Ryck, M., et al. (2016). Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia 57, 538–548. doi: 10.1111/epi.13340
Kudryashov, I. E., Pavlova, T. V., Kudryashova, I. V., Egorova, L. K., and Gulyaeva, N. V. (2007). Kindling in the early postnatal period: effects on the dynamics of age-related changes in electrophysiological characteristics of hippocampal neurons. Neurosci. Behav. Physiol. 37, 765–772. doi: 10.1007/s11055-007-0080-x
Löscher, W., and Ebert, U. (1996). The role of the piriform cortex in kindling. Prog. Neurobiol. 50, 427–481. doi: 10.1016/S0301-0082(96)000366
Lynch, B. A., Lambeng, N., Nocka, K., Kensel-Hammes, P., Bajjalieh, S. M., Matagne, A., et al. (2004). The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. U.S.A. 101, 9861–9866. doi: 10.1073/pnas.0308208101
Mashimo, T., Yanagihara, K., Tokuda, S., Voigt, B., Takizawa, A., Nakajima, R., et al. (2008). An ENU-induced mutant archive for gene targeting in rats. Nat. Genet. 40, 514–515. doi: 10.1038/ng0508-514
Matveeva, E. A., Vanaman, T. C., Whiteheart, S. W., and Slevin, J. T. (2007). Asymmetric accumulation of hippocampal 7S SNARE complexes occurs regardless of kindling paradigm. Epilepsy Res. 73, 266–274. doi: 10.1016/j.eplepsyres.2006.11.003
Morgan, J. I., and Curran, T. (1989). Stimulus-transcription coupling in neurons: role of cellular immediate-early genes. Trends Neurosci. 12, 459–462. doi: 10.1016/0166-2236(89)90096-9
Morimoto, K., Fahnestock, M., and Racine, R. J. (2004). Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog. Neurobiol. 73, 1–60. doi: 10.1016/j.pneurobio.2004.03.009
Mukai, T., Nagao, Y., Nishioka, S., Hayashi, T., Shimizu, S., Ono, A., et al. (2013). Preferential suppression of limbic Fos expression by intermittent hypoxia in obese diabetic mice. Neurosci. Res. 77, 202–207. doi: 10.1016/j.neures.2013.09.013
Nowack, A., Yao, J., Custer, K. L., and Bajjalieh, S. M. (2010). SV2 regulates neurotransmitter release via multiple mechanisms. Am. J. Physiol. Cell Physiol. 299, C960–C967. doi: 10.1152/ajpcell.00259.2010
Ohno, Y., Ishihara, S., Mashimo, T., Sofue, N., Imaoku, T., Shimizu, S., et al. (2011). Scn1a missense mutation causes limbic hyperexcitability and vulnerability to experimental febrile seizures. Neurobiol. Dis. 41, 261–269. doi: 10.1016/j.nbd.2010.09.013
Ohno, Y., Ishihara, S., Terada, R., Kikuta, M., Sofue, N., Kawai, Y., et al. (2009a). Preferential increase in the hippocampal synaptic vesicle protein 2A (SV2A) by pentylenetetrazole kindling. Biochem. Biophys. Res. Commun. 390, 415–420. doi: 10.1016/j.bbrc.2009.09.035
Ohno, Y., Okano, M., Masui, A., Imaki, J., Egawa, M., Yoshihara, C., et al. (2012a). Region-specific elevation of D1 receptor-mediated neurotransmission in the nucleus accumbens of SHR, a rat model of attention deficit/hyperactivity disorder. Neuropharmacology 63, 547–554. doi: 10.1016/j.neuropharm.2012.04.031
Ohno, Y., Okumura, T., Terada, R., Ishihara, S., Serikawa, T., and Sasa, M. (2012b). Kindling-associated SV2A expression in hilar GABAergic interneurons of the mouse dentate gyrus. Neurosci. Lett. 510, 93–98. doi: 10.1016/j.neulet.2012.01.009
Ohno, Y., Shimizu, S., Harada, Y., Morishita, M., Ishihara, S., Kumafuji, K., et al. (2009b). Regional expression of fos-like immunoreactivity following seizures in Noda epileptic rat (NER). Epilepsy Res. 87, 70–76. doi: 10.1016/j.eplepsyres.2009.07.012
Ohno, Y., Shimizu, S., Tatara, A., Imaoku, T., Ishii, T., Sasa, M., et al. (2015). Hcn1 is a tremorgenic genetic component in a rat model of essential tremor. PLoS ONE 10:e123529. doi: 10.1371/journal.pone.0123529
Paxinos, G., and Watson, C. (2007). The Rat Brain in Stereotaxic Coordinates, 6th Edn. Manhattan, NY: Elsevier.
Pollard, J. R. (2008). Seletracetam, a small molecule SV2A modulator for the treatment of epilepsy. Curr. Opin. Investig. Drugs 9, 101–107.
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294. doi: 10.1016/0013-4694(72)90177-0
Serajee, F. J., and Huq, A. M. (2015). Homozygous mutation in synaptic vesicle glycoprotein 2A gene results in intractable epilepsy, involuntary movements, microcephaly, and developmental and growth retardation. Pediatr. Neurol. 52, 642–646. doi: 10.1016/j.pediatrneurol.2015.02.011
Szyndler, J., Maciejak, P., Turzyńska, D., Sobolewska, A., Taracha, E., Skórzewska, A., et al. (2009). Mapping of c-Fos expression in the rat brain during the evolution of pentylenetetrazol-kindled seizures. Epilepsy Behav. 16, 216–224. doi: 10.1016/j.yebeh.2009.07.030
Toering, S. T., Boer, K., de Groot, M., Troost, D., Heimans, J. J., Spliet, W. G., et al. (2009). Expression patterns of synaptic vesicle protein 2A in focal cortical dysplasia and TSC-cortical tubers. Epilepsia 50, 1409–1418. doi: 10.1111/j.1528-1167.2008.01955.x
Tokudome, K., Okumura, T., Shimizu, S., Mashimo, T., Takizawa, A., Serikawa, T., et al. (2016). Synaptic vesicle glycoprotein 2A (SV2A) regulates kindling epileptogenesis via GABAergic neurotransmission. Sci. Rep. 6:27420. doi: 10.1038/srep27420
van Vliet, E. A., Aronica, E., Redeker, S., Boer, K., and Gorter, J. A. (2009). Decreased expression of synaptic vesicle protein 2A, the binding site for levetiracetam, during epileptogenesis and chronic epilepsy. Epilepsia 50, 422–433. doi: 10.1111/j.1528-1167.2008.01727.x
Keywords: synaptic vesicle glycoprotein 2A (SV2A), seizure susceptibility, GABA release, glutamate release, amygdala, pentylentetrazole
Citation: Tokudome K, Okumura T, Terada R, Shimizu S, Kunisawa N, Mashimo T, Serikawa T, Sasa M and Ohno Y (2016) A Missense Mutation of the Gene Encoding Synaptic Vesicle Glycoprotein 2A (SV2A) Confers Seizure Susceptibility by Disrupting Amygdalar Synaptic GABA Release. Front. Pharmacol. 7:210. doi: 10.3389/fphar.2016.00210
Received: 22 May 2016; Accepted: 30 June 2016;
Published: 14 July 2016.
Edited by:
Tomoyuki Kuwaki, Kagoshima University Graduate School of Medical and Dental Sciences, JapanReviewed by:
Takashi Kurihara, Kagoshima University, JapanYoshitoshi Kasuya, Chiba University, Japan
Copyright © 2016 Tokudome, Okumura, Terada, Shimizu, Kunisawa, Mashimo, Serikawa, Sasa and Ohno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukihiro Ohno, yohno@gly.oups.ac.jp