 Jennifer C. Wong
Jennifer C. Wong Stacey B. B. Dutton
Stacey B. B. Dutton Stephen D. Collins3
Stephen D. Collins3 Steven Schachter
Steven Schachter Andrew Escayg
Andrew Escayg- 1Department of Human Genetics, Emory University, Atlanta, GA, USA
- 2Department of Biology, Agnes Scott College, Atlanta, GA, USA
- 3Biscayne Pharmaceuticals, Miami, FL, USA
- 4Department of Neurology, Harvard Medical School, Beth Israel Deaconess Medical Center, and Massachusetts General Hospital, Boston, MA, USA
De novo loss-of-function mutations in the voltage-gated sodium channel (VGSC) SCN1A (encoding Nav1.1) are the main cause of Dravet syndrome (DS), a catastrophic early-life encephalopathy associated with prolonged and recurrent early-life febrile seizures (FSs), refractory afebrile epilepsy, cognitive and behavioral deficits, and a 15–20% mortality rate. SCN1A mutations also lead to genetic epilepsy with febrile seizures plus (GEFS+), which is an inherited disorder characterized by early-life FSs and the development of a range of adult epilepsy subtypes. Current antiepileptic drugs often fail to protect against the severe seizures and behavioral and cognitive deficits found in patients with SCN1A mutations. To address the need for more efficacious treatments for SCN1A-derived epilepsies, we evaluated the therapeutic potential of Huperzine A, a naturally occurring reversible acetylcholinesterase inhibitor. In CF1 mice, Hup A (0.56 or 1 mg/kg) was found to confer protection against 6 Hz-, pentylenetetrazole (PTZ)-, and maximal electroshock (MES)-induced seizures. Robust protection against 6 Hz-, MES-, and hyperthermia-induced seizures was also achieved following Hup A administration in mouse models of DS (Scn1a+/−) and GEFS+ (Scn1aRH/+). Furthermore, Hup A-mediated seizure protection was sustained during 3 weeks of daily injections in Scn1aRH/+ mutants. Finally, we determined that muscarinic and GABAA receptors play a role in Hup A-mediated seizure protection. These findings indicate that Hup A might provide a novel therapeutic strategy for increasing seizure resistance in DS and GEFS+, and more broadly, in other forms of refractory epilepsy.
Introduction
Epilepsy is a common neurological disorder that affects 0.5–1% of the population and is characterized by recurrent seizures that often manifest during childhood. Despite a growing number of available antiepileptic drugs (AEDs), the efficacy of pharmacological intervention for epilepsy has not improved substantially in the last 30 years (Kwan and Brodie, 2006), highlighting the critical need to develop alternative treatments, while minimizing unwanted side effects. Towards this goal, the use of appropriate genetic models of human epilepsy to evaluate potential AEDs might provide a better predictor of clinical efficacy (Loscher and Schmidt, 1994).
Genetic factors play an important role in the etiology of epilepsy, and the neuronal voltage-gated sodium channels (VGSCs) have emerged as an important family of epilepsy genes (Catterall et al., 2010; Escayg and Goldin, 2010; O'Brien and Meisler, 2013). De novo loss-of-function mutations in the VGSC SCN1A (encoding Nav1.1 channels) are the main cause of Dravet syndrome (DS), a catastrophic early-life encephalopathy associated with prolonged and recurrent early-life febrile seizures (FSs), refractory afebrile epilepsy, cognitive and behavioral deficits, and a 15–20% mortality rate (Claes et al., 2001, 2009; Wallace et al., 2003; Lossin, 2009; Escayg and Goldin, 2010). Current AEDs used to treat DS include stiripentol, valproate, and benzodiazepines, as well as the ketogenic diet (Chiron and Dulac, 2011). Unfortunately, most DS patients do not achieve adequate seizure control, nor do they demonstrate sufficient improvements in behavior or cognitive function (Dravet et al., 2005). SCN1A mutations also lead to genetic epilepsy with febrile seizures plus (GEFS+), which is an inherited disorder characterized by FSs that persist beyond 6 years of age and the development of adult epilepsy (Escayg et al., 2000, 2001). SCN1A mutations account for at least 80 and 10% of DS and GEFS+ cases, respectively (Claes et al., 2009; Lossin, 2009; Escayg and Goldin, 2010).
We previously described the generation of a mouse model of GEFS+ by knock-in of the human SCN1A GEFS+ mutation, R1648H (Martin et al., 2010). Heterozygous Scn1aR1648H/+ mutants (Scn1aRH/+) exhibit spontaneous seizures, increased seizure susceptibility, and behavioral deficits (Martin et al., 2010; Papale et al., 2013; Purcell et al., 2013). Homozygous mutants exhibit frequent spontaneous seizures and typically die 3–4 weeks after birth (Martin et al., 2010). Heterozygous knockout mice (Scn1a+/−), a model of DS, exhibit frequent spontaneous seizures and behavioral deficits, whereas homozygous DS mice exhibit spontaneous seizures, ataxia, and have a lifespan of ~15 days (Yu et al., 2006; Ogiwara et al., 2007). The hippocampus has been proposed as the site of spontaneous seizure generation in DS (Liautard et al., 2013), and dissociated cortical and hippocampal neurons from GEFS+ and DS mutants show decreased GABAergic interneuron excitability (Yu et al., 2006; Ogiwara et al., 2007; Martin et al., 2010). In addition, we and others recently showed that the selective deletion of Scn1a from parvalbumin interneurons in the cortex and hippocampus is sufficient to increase seizure susceptibility and generate spontaneous seizures (Dutton et al., 2012; Tai et al., 2014).
Huperzine A (Hup A), originally isolated from the Chinese club moss Huperzia serrata (Ma et al., 2007), is a naturally occurring sesquiterpene Lycopodium alkaloid (Ma et al., 2007). Hup A is a potent, highly specific reversible inhibitor of acetylcholinesterase (AChE) that can cross the blood-brain barrier to significantly increase brain acetylcholine levels (Ma et al., 2007). Both isomers of Huperzine A, the naturally occurring form ([−]-Hup A) and the synthetic isomer ([+]-Hup A), can also act as dose-dependent NMDA receptor antagonists, reducing NMDA- and glutamate-induced neurotoxicity (Ved et al., 1997; Gordon et al., 2001). However, the naturally occurring isomer, [−]-Hup A, is 38 times more potent at inhibiting AChE compared to [+]-Hup A (McKinney et al., 1991) and does not bind to the NMDA receptor at affinities that would be attainable for clinical use. [−]-Hup A also protects against apoptosis following ischemia/reperfusion (Ye et al., 2010) and in neuronal cultures (Hemendinger et al., 2008), attenuates mitochondrial dysfunction in vivo (Yang et al., 2012) and in isolated brain mitochondria (Gao and Tang, 2006), and reduces cholinergic pathway-mediated inflammatory responses (Wang et al., 2008).
Hup A has proved beneficial in several animal and cell culture models of neurological disease (Hemendinger et al., 2008; Wang et al., 2008). Following administration to healthy volunteers and patients, Hup A demonstrated clinically acceptable safety and tolerability (Wang et al., 2009). There are also reports of its efficacy in neurological disorders, such as Alzheimer's disease (Wang et al., 2009), benign senescent forgetfulness (Wang et al., 2006), vascular dementia (Xu et al., 2012), and schizophrenia (Zhang et al., 2007). Adverse events are infrequent, generally mild, and transient; these include dizziness, gastric discomfort, insomnia, and sweating (Zangara, 2003; Yang et al., 2013).
In the current study, we evaluated the anticonvulsant potential of the naturally occurring [−]-Hup A isomer in mouse models of SCN1A-derived GEFS+ and DS. Given the underlying reduction in neuronal inhibition (Yu et al., 2006; Ogiwara et al., 2007; Martin et al., 2010) and the role of the hippocampus in seizure generation in SCN1A-acquired epilepsy (Liautard et al., 2013), we hypothesize that Hup A would restore a more normal balance between neuronal inhibition and excitation in epilepsy subtypes that are caused by mutations in SCN1A. Although not assessed in this study, additional biological properties of Hup A, including protection against cell death (Hemendinger et al., 2008) and inflammation (Wang et al., 2012), would also be of expected benefit in the treatment of epilepsy.
Materials and Methods
Animals
Heterozygous mice expressing the human SCN1A R1648H GEFS+ mutation (Scn1aRH/+) and heterozygous Scn1a knock-out mice (Scn1a+/−) were generated as previously described (Yu et al., 2006; Martin et al., 2010) and maintained by backcrossing to the C57BL/6J and FVB backgrounds, respectively. Experiments were conducted at the N13 generation for Scn1aRH/+ mutants and on a mixed FVB X C57BL/6J background for Scn1a+/− mutants. Scn1a mutants and their respective wild-type (WT) littermates at P21-23 were used in the febrile seizure (FS) induction paradigm. Scn1aRH/+ (3–5 months old) and Scn1a+/− (4–6 weeks old) mutants and their respective age-matched WT littermates, as well as 6–8-week-old male CF1 mice (Charles River), were used for all other experiments. WT littermates were used as controls for all experiments to minimize variation due to differences in genetic background and rearing conditions. All mice were housed on a 12-h light/dark cycle, with food and water available ad libitum. All experiments were conducted during the light cycle prior to 4:00 p.m. and were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Emory University. In experiments where both mutants and WT littermates were used, the experimenter was blinded to genotype. However, since Hup A administration results in visible mild side effects, it was not possible for the experimenter to be blinded to treatment.
Huperzine A Administration
Huperzine A (Hup A; Biscayne Pharmaceuticals) was suspended in a 10% solution of cyclodextrin (CD, 2-hydroxypropyl-β-cyclodextrin, Sigma-Aldrich) dissolved in sterile saline (0.9%). Hup A was administered via intraperitoneal (i.p.) injection (10 ml/kg) 1 h prior to seizure induction, based on previous studies that demonstrated maximum brain AChE inhibition at 1 h following Hup A administration (Tang et al., 1989). Control mice were handled similarly but were only administered vehicle (Veh, 10% CD).
Acetylcholinesterase Activity
Male CF1 mice (N = 3–4/dose) were administered either Hup A (doses based on a ¼ logarithmic scale: 0.10, 0.18, 0.32, 0.56, 1, or 1.8 mg/kg) or vehicle 1 h prior to sacrifice. The right hemisphere of the brain was used for quantification of protein (Pierce BCA Protein Assay, Thermo Scientific) and AChE activity using the standard spectrophotometric method (Ellman et al., 1961; Padilla et al., 1999).
6 Hz Psychomotor Seizure Induction
Seizures were induced by the 6 Hz paradigm as previously described (Barton et al., 2001; Gilchrist et al., 2014). Briefly, 1 h prior to seizure induction, mice were administered either Hup A or vehicle (i.p.), and a topical anesthetic (0.5% tetracaine hydrochloride) was applied to the cornea. Each mouse was manually restrained during corneal stimulation (6 Hz, 0.2-ms pulse, 3 s) using a constant current device (ECT Unit 57800; Ugo Basile, Comerio, Italy), and then immediately placed in a clean cage for behavioral observations. Seizures were scored based on a modified Racine Scale (RS; Gilchrist et al., 2014): RS0, no abnormal behavior; RS1, immobile ≥ 3 s; RS2, forelimb clonus, paw waving; RS3, rearing and falling.
To determine the relationship between Hup A dose and susceptibility to 6 Hz-induced seizures, a ¼ logarithmic dose-response curve was generated following Hup A (0.10, 0.18, 0.32, 0.56, 1 mg/kg) or vehicle administration in male CF1 mice (N = 12). The 6 Hz paradigm was performed at a current of 44 mA, which was previously found to be twice the convulsive current at which 97% of CF1 mice seize (2xCC97; Barton et al., 2001). Over a 6-week experimental period, seizure induction was performed once per week on each mouse following the randomized administration of either vehicle or one of the Hup A doses, ensuring that all mice received each Hup A dose.
The effect of Hup A (1 mg/kg) on 6 Hz-induced seizures was evaluated further in a separate cohort of male CF1 mice (44 mA, N = 15). We also examined the effect of Hup A (0.5 and 1 mg/kg) on 6 Hz-induced seizures in male Scn1aRH/+ (N = 9–12) and male Scn1a+/− (N = 8–11) mice and their respective WT littermates. CF1 mice and Scn1a mutants and their WT littermates were randomized into 2 groups (0.5 mg/kg Hup A or Veh) over 2 trials (1 week apart). Mice that received Hup A during the first trial subsequently received vehicle during the second trial, and vice versa. The same experimental design was applied to separate cohorts of CF1 mice, Scn1a mutants and WT littermates using 1 mg/kg Hup A and vehicle. Scn1aRH/+ and Scn1a+/− mice were first tested at currents of 24 and 20 mA, respectively. The experiment was subsequently repeated in separate groups of mice at twice the initial current intensity (Scn1aRH/+ = 48 mA, Scn1a+/− = 40 mA).
To determine the contribution of different classes of neurotransmitter receptors to Hup A-mediated seizure protection, male CF1 mice (N = 5–12/group) were co-administered Hup A (1 mg/kg) and either a muscarinic receptor antagonist (scopolamine hydrobromide (SH), 30 mg/kg, Sigma-Aldrich), a GABAA receptor antagonist (pentylenetetrazole, 25 mg/kg, Sigma-Aldrich), or a nicotinic receptor antagonist (bupropion hydrochloride (BH), 5 mg/kg, Sigma-Aldrich) 1 h prior to assessing the body temperature of each mouse and 6 Hz seizure induction (44 mA). All compounds were dissolved in sterile saline (0.9%). Similarly handled mice administered vehicle (cyclodextrin) or saline were used as controls.
Maximal Electroshock Seizure Induction
Maximal electroshock seizures were induced as previously described (Swinyard, 1972). One hour prior to seizure induction, a topical anesthetic (0.5% tetracaine hydrochloride) was applied to the cornea, and Hup A (0.5 or 1 mg/kg) or vehicle was administered (i.p.) to male CF1 mice (N = 10/treatment), and male and female Scn1a+/− and Scn1aRH/+ mutants and their respective age- and sex-matched WT littermates (N = 5–13/genotype/treatment). Each mouse was manually restrained, subjected to corneal stimulation (60 Hz, 50 mA, 0.2 s; ECT Unit 57800 Ugo Basile, Comerio, Italy), and observed for the presence or absence of a seizure.
In a separate cohort of male CF1 mice (N = 6), body temperature was maintained at 37.5°C during the 1-h period following the administration of Hup A (1 mg/kg) prior to MES induction. Body temperature was maintained by the use of a rectal temperature probe connected to a heat lamp, with a thermostat set at 37.5°C.
Pentylenetetrazole Seizure Induction
Pentylenetetrazole (PTZ) seizures were induced as previously described (Loscher et al., 1991). Hup A (1 mg/kg) or vehicle was administered (i.p.) to male CF1 mice (N = 6/group) 1 h prior to subcutaneous PTZ injection (85 mg/kg). PTZ (Sigma-Aldrich) was dissolved in sterile saline (0.9%). Latencies to the first myoclonic jerk (MJ) and generalized tonic-clonic seizure (GTCS) were recorded over a 30-min observation period.
Flurothyl Seizure Induction
The effect of Hup A (1 mg/kg) or vehicle on seizures induced by the chemiconvulsant flurothyl (Prichahd et al., 1969; Dutton et al., 2011) was evaluated in male CF1 mice (N = 13–15/treatment). One hour prior to seizure induction, Hup A or vehicle was administered (i.p.) to each mouse. Next, each mouse was placed in a clear acrylic chamber and flurothyl (2,2,2-trifluroethylether; Sigma-Aldrich) was introduced at a constant rate of 20 μL/min. Latencies to the first MJ and generalized tonic-clonic seizure with hindlimb extension (GTCS-HLE) were recorded.
Hyperthermia Seizure Induction
Susceptibility to hyperthermia-induced seizures were evaluated in male and female Scn1aRH/+ (N = 4–6) and Scn1a+/− (N = 9) mutants and their respective WT littermates (N = 3–7) at P21-23 as previously described (Oakley et al., 2009; Dutton et al., 2013). One hour prior to seizure induction, mice were administered Hup A (1 mg/kg) or vehicle and body temperature was maintained at 37.5°C. Hyperthermia seizure induction was then conducted (Dutton et al., 2013). Briefly, the body temperature of each mouse was elevated by 0.5°C every 2 min until the first GTCS or 42.5°C is reached. The temperature at which the seizure occurred was recorded.
Chronic Hup A Treatment
The effect of chronic Hup A (0.5 and 1 mg/kg) administration on susceptibility to 6 Hz-induced seizures was first examined in male CF1 mice (N = 10/dose). On Day 0, all mice were administered vehicle (i.p.) 1 h prior to 6 Hz seizure induction (44 mA) to establish baseline seizure susceptibility. The mice were then divided into 2 cohorts based on the dose of Hup A administered. Cohort 1 mice received one injection per day of Hup A (0.5 mg/kg, i.p.) for 7 consecutive days and were subjected to 6 Hz seizure induction 1 h after Hup A administration on Day 7. Cohort 2 mice received Hup A (1 mg/kg, i.p.) for 12 consecutive days and were subjected to 6 Hz seizure induction 1 h after Hup A administration on Days 7 and 12. Cohort 2 mice then received Hup A (1.8 mg/kg, i.p.) for the next 5 consecutive days (Days 13–17), and 6 Hz seizures were induced 1 h after Hup A administration on Day 17. To determine the effect of chronic Hup A administration on AChE activity, separate groups of mice (N = 3/group) were administered Hup A (0.5 or 1 mg/kg) as described above for Cohorts 1 and 2, and AChE activity was quantified on Day 7 (to correspond with Cohort 1 mice) and Days 7 and 12 (to correspond with Cohort 2 mice).
We next determined the effect of chronic Hup A (1 mg/kg) administration on susceptibility to 6 Hz-induced seizures in male Scn1aRH/+ mutants (N = 9). On Day 0, all mice were administered vehicle (i.p.) 1 h prior to seizure induction (24 mA) to establish baseline seizure susceptibility. The mice were then administered Hup A (i.p.) daily for 21 consecutive days and subjected to 6 Hz seizure induction on Days 7, 14, and 21. The mice were also subjected to 6 Hz seizure induction on Day 22 to determine whether Hup A conferred lasting protection.
Statistical Analyses
A one-way repeated measures ANOVA (rANOVA) followed by the Dunn's multiple comparisons test (for non-parametric data) was used to compare Racine scores (RS) following 6 Hz seizure induction in CF1 mice, Scn1a mutants, and their respective WT littermates administered Hup A (0.10–1 mg/kg) or vehicle. A one-way ANOVA followed by the Holm-Šídák's multiple comparisons test was used to compare latencies to flurothyl-induced MJ and GTCS-HLE, percent AChE activity, and temperature in CF1 mice administered vehicle or Hup A (0.1–1.8 mg/kg). The chi-squared test was used to compare the percent of deaths between vehicle- and Hup-A treated mice following flurothyl seizure induction. An unpaired student's t-test was used to compare latencies to the MJ and GTCS following PTZ administration. A two-way ANOVA was used to compare the average temperature at seizure occurrence between Hup A- and vehicle-treated Scn1a mutants and their respective WT littermates during hyperthermia. A one-way rANOVA followed by the Dunn's multiple comparisons test was used to compare RS following 6 Hz seizure induction in CF1 and Scn1aRH/+ mice during chronic Hup A administration. All data are presented as the mean ± SEM; p < 0.05 was considered statistically significant.
Results
Hup A Protects against Induced Seizures in CF1 Mice
We first examined the relationship between Hup A dose (0.0–1.0 mg/kg) and resistance to seizures induced by the 6 Hz paradigm at 44 mA. The administration of Hup A was randomized with respect to the order in which each mouse received each dose of Hup A and vehicle. Each mouse was subjected to 6 Hz seizure induction once per week during the 6-week experimental period. Behavioral seizure responses were consistent for each Hup A dose, indicating that repeated seizure induction over the 6-week period did not influence seizure susceptibility or severity. Seizures were observed in all 12 vehicle-treated mice (1 RS1, 10 RS2, 1 RS3; Figure 1A), with most mice exhibiting forelimb clonus and paw waving. However, seizure occurrence and severity were significantly reduced following the administration of 0.56 and 1 mg/kg Hup A, with seizures observed in only 4/12 and 1/12 mice, respectively (Figure 1A). This robust Hup A-mediated protection against 6 Hz seizures was reproduced in a separate group of CF1 mice that were administered Hup A (1 mg/kg) or vehicle (N = 15/treatment, Figure 1B). As shown in Figure 1B, RS2 and RS3 seizures were observed in all vehicle-treated mice; however, 11/15 (74%) Hup A-treated CF1 mice were completely protected (RS0).
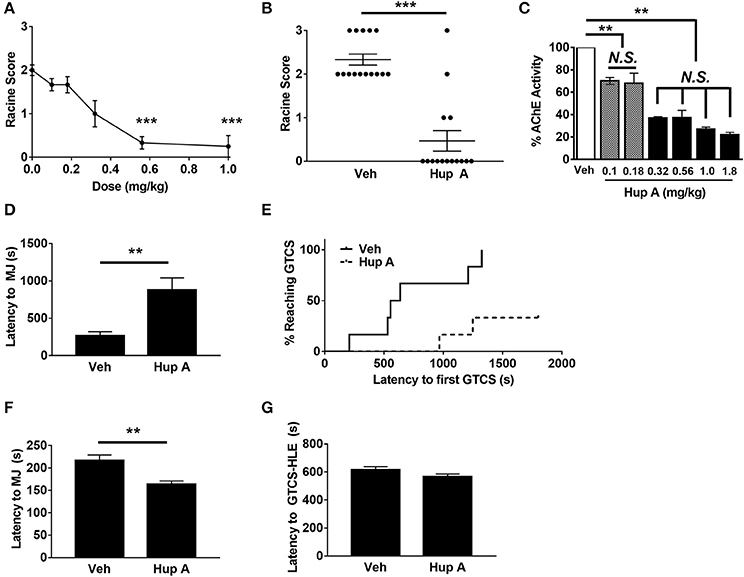
Figure 1. Hup A protects against induced seizures in CF1 mice. (A) A ¼ log dose-response curve was generated to determine the relationship between Hup A dose and resistance to 6 Hz-induced seizures in male CF1 mice (N = 12/dose). Hup A (0.10, 0.18, 0.32, 0.56, 1 mg/kg) or vehicle (10% CD) was administered (i.p.) 1 h prior to seizure induction at 44 mA. The greatest protection was observed with 0.56 and 1 mg/kg Hup A. (B) Robust protection against 6 Hz-induced seizures at 44 mA was observed in male CF1 mice administered Hup A (1 mg/kg) compared to vehicle (10% CD). One-way rANOVA followed by Dunn's multiple comparisons post-hoc analyses. ***p < 0.001. (C) A ¼ log dose-response curve was generated to examine AChE inhibition following Hup A administration. Hup A (0.10, 0.18, 0.32, 0.56, 1.0, 1.8 mg/kg) or vehicle (10% CD) was administered (i.p.) to male CF1 mice (N = 3–4/dose) 1 h prior to sacrifice. The two lowest doses of Hup A (0.1 and 0.18 mg/kg) resulted in 25% reduction in AChE activity compared to vehicle. Hup A (0.32–1.8 mg/kg) caused 63–78% reduction in AChE activity. One-way ANOVA followed by Holm-Šídák's multiple comparisons post-hoc analyses. **p < 0.01. (D) Hup A (1 mg/kg) significantly increased latency to the first PTZ-induced MJ in CF1 mice (N = 6/dose). (E) Only 2/6 Hup A-treated mice experienced a GTCS due to PTZ, whereas a GTCS was observed in all vehicle-treated mice. (F) Hup A (1 mg/kg) significantly decreased the latency to the flurothyl-induced MJ compared to vehicle treated CF1 mice (unpaired Student's t-test, p < 0.01, N = 13–15/treatment). (G) The average latency to the GTCS-HLE was not significantly altered by Hup A.
We also evaluated the effect of Hup A (0.1–1.8 mg/kg) on brain AChE activity 1 h after administration (Figure 1C). The two lowest Hup A doses (0.1 and 0.18 mg/kg) resulted in ~25% reduction in AChE activity when compared to vehicle-injected mice (p < 0.01). Hup A doses of 0.32–1.8 mg/kg resulted in comparable reductions of AChE activity (63–78%; Figure 2). Hup A doses of 0.56 or 1.0 mg/kg were selected for subsequent analyses based on the magnitude of their effect on AChE activity and the robust increase in seizure resistance associated with these doses.
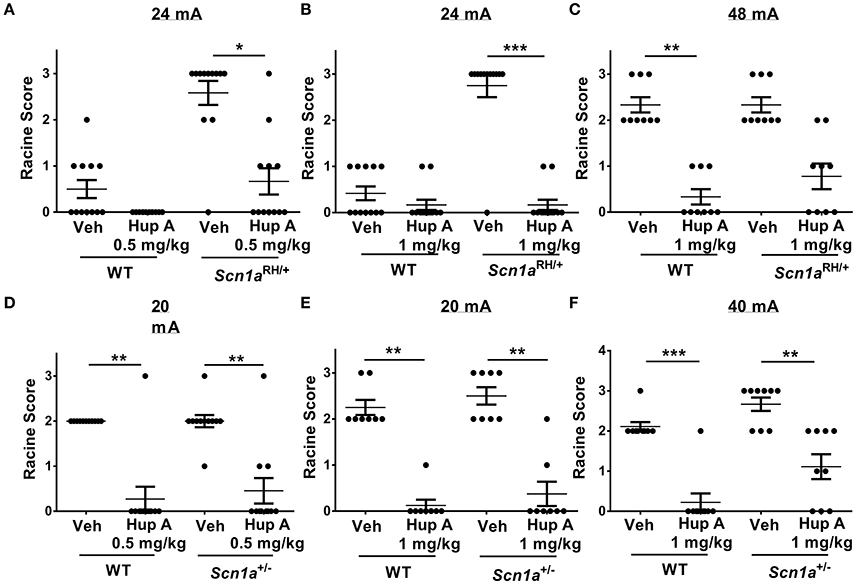
Figure 2. Hup A provides robust protection against 6 Hz-induced seizures in Scn1a mutants and wild-type (WT) littermates. (A,B) Hup A conferred robust protection against 6 Hz-induced seizures (24 mA) in male Scn1aRH/+ mutants (N = 9–12/treatment) when administered at 0.5 mg/kg (A) and 1 mg/kg (B). (C) Although not statistically significant, when the current was doubled to 48 mA, Hup A (1 mg/kg) reduced the number of Scn1aRH/+ mutants that seized and decreased seizure severity when compared to vehicle-treated mutants. (D) Hup A (0.5 mg/kg) conferred robust seizure protection to male Scn1a+/− mutants and WT littermates at 20 mA (N = 8–11/treatment/genotype). (E,F) Hup A (1 mg/kg) effectively increased resistance to 6 Hz-induced seizures in Scn1a+/− mutants and WT littermates at 20 mA (E) and 40 mA (F). One-way rANOVA followed by Dunn's multiple comparisons post-hoc analyses. *p < 0.05, **p < 0.01, ***p < 0.001.
The ability of Hup A to protect against MES-induced seizures was also examined in male CF1 mice. As expected, all vehicle-treated mice experienced a maximal seizure. In contrast, all CF1 mice treated with either 0.5 mg/kg or 1 mg/kg Hup A were protected. We next determined whether Hup A (1 mg/kg) was capable of increasing resistance to PTZ-induced seizures in CF1 mice (Figures 1D,E). Average latencies to the first PTZ-induced myoclonic jerk were significantly higher in Hup A-treated mice compared to vehicle-treated mice (p < 0.01, Figure 1D). Furthermore, only 2/6 Hup A-treated mice exhibited a GTCS, whereas all (6/6) vehicle-treated mice had a GTCS (Figure 1E). Of the mice that exhibited a GTCS, the latency to the initial GTCS was significantly higher in the Hup A-treated mice compared to mice administered vehicle (p < 0.01).
Contrary to the robust Hup A-mediated protection against 6 Hz, MES, and PTZ-induced seizures, average latencies to the flurothyl-induced MJ was significantly lower in Hup A-treated CF1 mice compared to vehicle-treated mice (Figure 1F, p < 0.01). Hup A administration did not significantly change the average latency to the GTCS-HLE (Figure 1G); however, 11/13 Hup A-treated mice died after the GTCS-HLE while all vehicle-treated mice survived.
Transient dose-dependent side effects (e.g., hypothermia, lethargy, fasiculations) were observed following acute Hup A administration. Since hypothermia has been shown to be neuroprotective and increases seizure resistance (Essman and Sudak, 1964), we examined whether it might contribute to Hup A-mediated seizure protection. To prevent the Hup A-induced hypothermia, we maintained the body temperature of male CF1 mice at 37.5°C during the 1-h interval between Hup A administration and seizure induction. Complete protection against MES-induced seizures was still observed in these mice, indicating that hypothermia does not contribute to Hup A-conferred seizure protection (data not shown).
Hup A Significantly Increases Resistance to 6 Hz-Induced Seizures in Scn1a Mutants
We next examined whether Hup A was capable of reducing the occurrence or severity of 6 Hz-induced seizures in Scn1aRH/+ and Scn1a+/− mutants and their respective WT littermates. As previously described, mice were administered vehicle or Hup A (0.5 and 1 mg/kg) 1 h prior to seizure induction.
Scn1aRH/+ Mutants
We first examined susceptibility to 6 Hz-induced seizures at a current of 24 mA in Scn1aRH/+ mutants and WT littermates administered either vehicle or Hup A (0.5 mg/kg). Seizures were observed in 5/12 (4 RS1, 1 RS2) and 11/12 (2 RS2, 9 RS3) vehicle-administered WT littermates and Scn1aRH/+ mutants, respectively (Figure 2A). Following the administration of 0.5 mg/kg Hup A, all WT littermates were protected, while seizures were observed in 5/12 (3 RS1, 1 RS2, 1 RS3) Scn1aRH/+ mutants (Figure 2A). We next determined the effect of administering 1 mg/kg Hup A or vehicle under the same conditions (Figure 2B). Seizures were observed in 5/12 (5 RS1) WT littermates and 11/12 (11 RS3) mutants that received vehicle. Following the administration of 1 mg/kg Hup A, mild seizures (RS1) were observed in 2/12 WT littermates and 2/12 Scn1aRH/+ mutants. When the current was doubled (48 mA; Figure 2C), seizures were observed in all vehicle-treated WT littermates and Scn1aRH/+ mutants (6 RS2, 3 RS3, both genotypes). In contrast, only mild seizures (RS1) were observed in 3/9 WT littermates that were treated with Hup A (1 mg/kg). Similarly, Hup A-treated Scn1aRH/+ mutants had fewer and less severe seizures (3 RS1, 2 RS2) compared to those injected with vehicle, but this difference was not statistically significant (Figure 2C).
Scn1a+/− Mutants
Susceptibility to 6 Hz-induced seizures was first examined in Scn1a+/− mutants and WT littermates at 20 mA (Figure 2D) following the administration of vehicle or Hup A (0.5 mg/kg). Seizures were observed in all vehicle-treated WT littermates (11 RS2) and Scn1a+/− mutants (1 RS1, 9 RS2, 1 RS3). Following the administration of 0.5 mg/kg Hup A, seizures were only observed in 1/11 (1 RS3) WT littermates and 3/11 (2 RS1, 1 RS3) Scn1a+/− mutants. We next determined the effect of administering 1 mg/kg Hup A or vehicle under the same conditions (Figure 2E). All vehicle-treated mice seized (WT, 6 RS2, 2 RS3; Scn1a+/−, 4 RS2, 4 RS3). In contrast, among the Hup A-treated mice, seizures were only observed in 1/8 WT littermates (1 RS1) and 2/8 Scn1a+/− mutants (1 RS1, 1 RS2). When the current was doubled to 40 mA (Figure 2F), seizures were again observed in all vehicle-treated WT littermates (8 RS2, 1 RS3) and Scn1a+/− mutants (3 RS2, 6 RS3); however, seizure severity was significantly reduced in the Hup A-treated WT littermates (1 RS2) and Scn1a+/− mutants (2 RS1, 4 RS2).
Hup A Protects against Maximal Electroshock Seizures (MES)
The ability of Hup A to protect against MES-induced seizures was examined in both sexes of Scn1a mutants and their respective WT littermates (Table 1). As expected, all vehicle-treated mice experienced a maximal seizure. Following the administration of 0.5 mg/kg Hup A, protection was observed in 5/9 male and 1/6 female Scn1aRH/+ mutants and 10/11 male and 2/6 female WT littermates. Similarly, 5/10 male and 5/11 female Scn1a+/− mutants and 8/9 male and 8/9 female WT littermates were protected. More robust seizure protection was observed among the mutant mice following the administration of 1 mg/kg Hup A regardless of sex and genotype. Specifically, 6/7 male and 6/8 female Scn1aRH/+ mutants and all sex-matched WT littermates were protected. Similarly, all Hup A-treated male Scn1a+/− mutants and 6/7 male WT littermates were protected. Protection was also observed in 6/9 Hup A-treated female Scn1a+/− mutants and 9/10 female WT littermates.
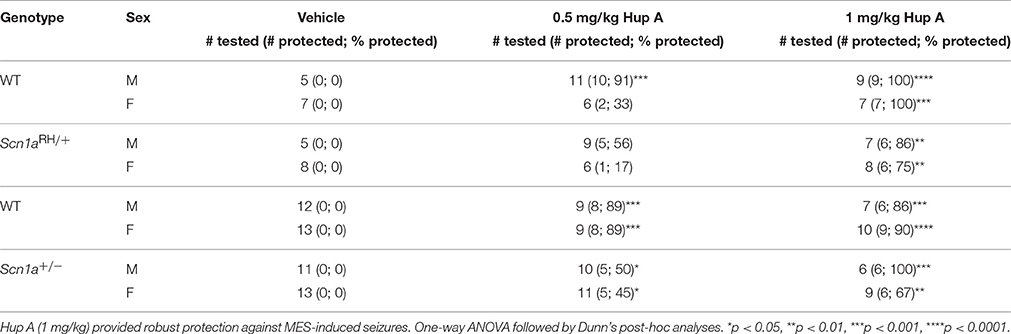
Table 1. Maximal electroshock seizure (MES) induction in both sexes of Scn1a mutant mice and their respective WT littermates.
Hup A Provides Robust Protection against Hyperthermia-Induced Seizures in Scn1a Mutant Mice
The ability of Hup A to protect against hyperthermia-induced seizures was evaluated in Scn1a mutant mice and their respective WT littermates. All vehicle-treated Scn1a mutants exhibited a seizure. In contrast, WT littermates administered vehicle did not exhibit a seizure in this paradigm. Hup A-treated WT littermates responded similarly to the vehicle-treated WT mice. Although all Hup A-treated Scn1aRH/+ and Scn1a+/− mutant mice exhibited a seizure, the average temperature at seizure occurrence was significantly higher than the temperature at which the vehicle-treated mutants seized (Scn1aRH/+, p < 0.01; Scn1a+/−, p < 0.001; Figure 3).
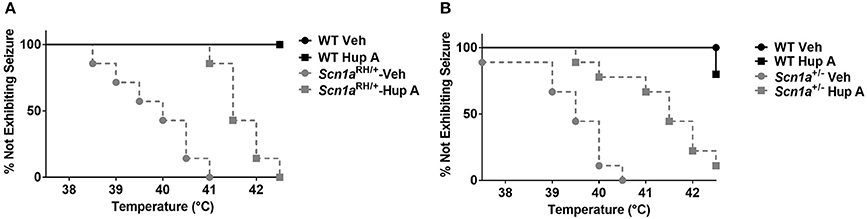
Figure 3. Hup A increases resistance to hyperthermia-induced seizures in Scn1a mutant mice. Hup A (1 mg/kg) or vehicle was administered (i.p.) 1 h prior to seizure induction to Scn1aRH/+ (A) and Scn1a+/− (B) mutant mice and their respective WT littermates (N = 3–9/treatment/genotype).
Resistance to 6 Hz-Induced Seizures Is Maintained in Scn1aRH/+ Mice during Chronic Hup A Administration
Having established that acute Hup A administration confers robust seizure protection, we next investigated whether protection would be maintained during chronic Hup A delivery. We first examined the effect of daily Hup A administration (0.5 or 1 mg/kg) on susceptibility to 6 Hz-induced seizures in CF1 mice (Figures 4A,B). As previously observed, all mice exhibited RS2 seizures prior to initiating Hup A treatment (Day 0). Seizure response was next examined after 7 days of Hup A administration. In contrast to observations following acute treatment, no protection was observed in the mice that received 0.5 mg/kg Hup A (Figure 4A). At the higher dose of Hup A (1 mg/kg), robust protection was still seen after 7 days; however, protection was not observed on Day 12 (Figure 4B). To determine whether seizure protection in these mice could be restored by increasing the daily dose of Hup A, 1.8 mg/kg Hup A was administered for the next 5 consecutive days (Days 13–17). On Day 13, we observed a statistically significant increase in seizure protection when compared to Day 0; however, protection was not observed on Day 17.
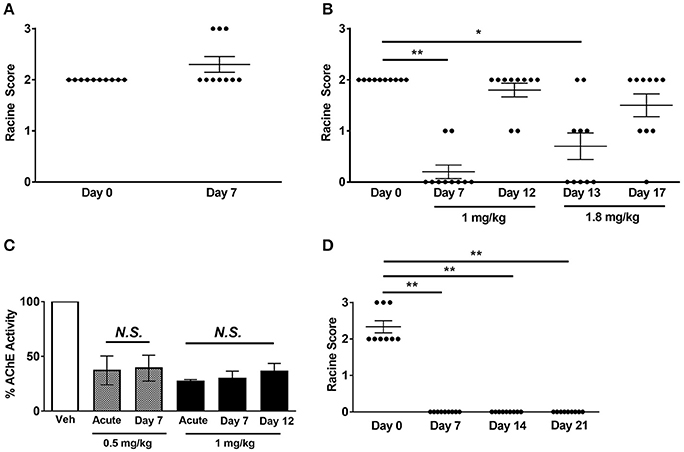
Figure 4. Effect of chronic Hup A administration on susceptibility to 6 Hz-induced seizures and AChE activity in CF1 and Scn1aRH/+ mutant mice. (A) We did not observe protection against 6 Hz-induced seizures in male CF1 mice after 7 consecutive days of 0.5 mg/kg/day Hup A administration. (B) Protection against 6 Hz-induced seizures was observed after 7 days of 1 mg/kg Hup A administration; however, protection was not observed on Day 12. We again observed significant seizure protection on Day 13, when the dose of Hup A was increased to 1.8 mg/kg; however, protection was not seen on Day 17 following 5 days of 1.8 mg/kg Hup A administration (N = 10/Hup A dose). One-way rANOVA followed by Dunn's multiple comparisons post-hoc analyses. *p < 0.05, **p < 0.01. (C) AChE activity was comparable after acute and seven days of chronic Hup A administration (0.5 mg/kg). Similarly, there were no statistically significant differences between AChE activity following acute, 7 and 12 days of chronic Hup A (1 mg/kg). (D) Baseline susceptibility to 6 Hz-induced seizures in Scn1aRH/+ mutant mice (N = 9) was determined on Day 0. Hup A (1 mg/kg) was administered (i.p.) daily for 21 consecutive days in the Scn1aRH/+ mutant mice. Hup A conferred robust protection against 6 Hz-induced seizures following 7, 14, and 21 days of daily administration. One-way rANOVA followed by Dunn's multiple comparisons post-hoc analyses. **p < 0.01.
Chronic Hup A administration did not alter its effect on AChE activity, with approximately a 60% reduction observed after 7 days of 0.5 mg/kg and a 70% reduction after 7 and 12 days of 1 mg/kg (Figure 4C). Furthermore, the transient side effects, including fasiculations, hypothermia, and lethargy, diminished with continued administration. We also examined the effect of chronic Hup A administration (1 mg/kg) on susceptibility to 6 Hz-induced seizures in Scn1aRH/+ mice. At baseline (Day 0), all vehicle-treated mutants (9/9) experienced a severe seizure (6 RS2, 3 RS3, Figure 4D). However, we observed complete protection against 6 Hz-induced seizures after 7, 14, and 21 consecutive days of Hup A administration (p < 0.01). To determine whether seizure protection would be maintained after cessation of Hup A administration, 6 Hz seizures were induced 24 h after the last Hup A injection (Day 22); however, we found no significant difference between the seizure responses on Days 0 and 22, indicating a lack of maintained seizure protection.
Central Muscarinic and GABAA Receptors Are Important for Hup A-Mediated Protection against 6 Hz-Induced Seizures
To gain further insight into the mechanism of action of Hup A, we examined the effect of blocking muscarinic receptors, nicotinic receptors, or GABAA receptors on Hup A-conferred resistance to 6 Hz seizures in CF1 mice. Mice were co-administered Hup A (1 mg/kg) and each antagonist 1 h prior to seizure induction, and seizure response was compared to control mice that were injected with vehicle plus saline, vehicle plus antagonist, and Hup A plus saline (Figure 5). Seizure responses were comparable between control mice administered vehicle plus saline and vehicle plus each antagonist, confirming that the doses of the antagonists used did not alter seizure response (Figure 5).
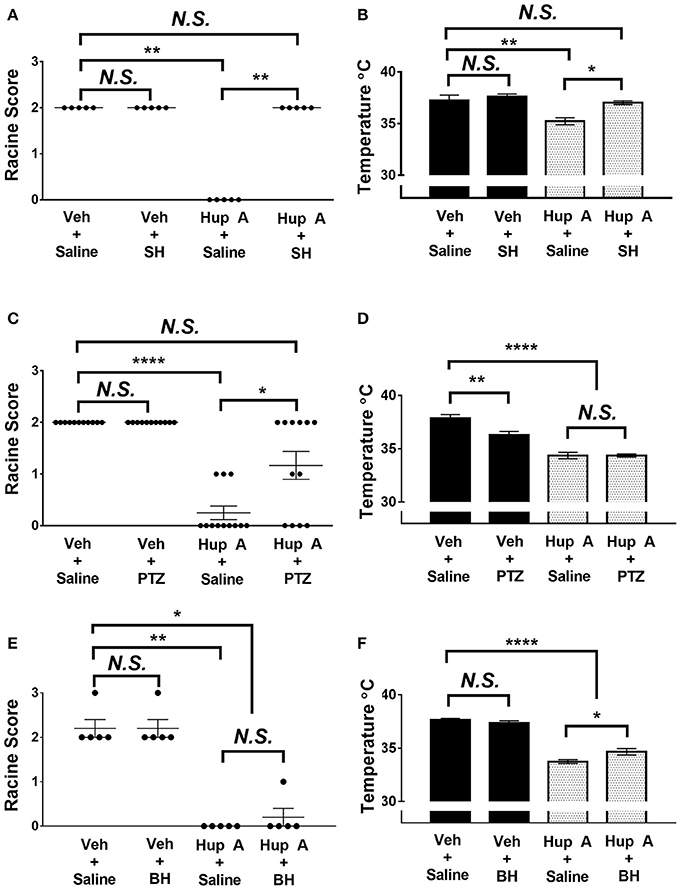
Figure 5. Effect of co-administration of neurotransmitter receptor antagonists on the protective effects of Hup A in the 6 Hz seizure paradigm (44 mA). (A,C) Co-administration of either the central-acting muscarinic receptor antagonist scopolamine hydrobromide (SH) or the GABAA receptor antagonist PTZ blocked the protective effect of Hup A. One-way ANOVA followed by Dunn's multiple comparisons post-hoc analyses. *p < 0.05, **p < 0.01, ****p < 0.0001. (E) Co-administration of the nicotinic receptor antagonist bupropion hydrochloride (BH) does not block the protective effect of Hup A. (B,F) Co-administration of SH or BH reduces Hup A-induced hypothermia. (D) Co-administration of PTZ does not influence Hup A-induced hypothermia. (N = 5–12/group). One-way ANOVA followed by Holm-Šídák's multiple comparisons post-hoc analyses. *p < 0.05, **p < 0.01, ****p < 0.0001.
Block of central muscarinic receptors was achieved by injection of the muscarinic receptor antagonist, scopolamine hydrobromide (SH; Figure 5A). All control mice that received vehicle and saline or vehicle and SH exhibited RS2 seizures. These results also demonstrate that SH alone does not worsen or ameliorate the behavioral seizure response. In contrast to the seizure protection observed in the Hup A plus saline group, all mice co-administered Hup A and SH exhibited RS2 seizures, demonstrating a role for muscarinic receptors in Hup A-mediated seizure protection. The seizure response of mice administered Hup A and the GABAA receptor antagonist PTZ were comparable to control mice that received vehicle and saline or vehicle plus PTZ, indicating that GABAA receptors also contribute to Hup A-mediated seizure protection (Figure 5C). However, 4/12 mice administered Hup A plus PTZ did not exhibit a seizure and 2/12 mice had mild seizures (RS1), raising the possibility that blocking GABAA receptors only partially affects Hup A activity (Figure 5C). In contrast, the nicotinic receptor antagonist bupropion hydrochloride (BH) had no effect on Hup A activity, as demonstrated by the comparable seizure response of the Hup A plus saline and Hup A plus BH groups (Figure 5E).
To determine whether these receptors were involved in the Hup A-induced hypothermia, we also measured the body temperature of the mice 1 h after drug administration. The average body temperature of mice administered Hup A plus SH was comparable to the control mice and was significantly higher than the Hup A plus saline group (Figure 5B). Although the higher body temperature observed in mice co-administered Hup A and BH was statistically different from the Hup A plus saline group (p < 0.05), the average body temperature in the presence of BH was still significantly lower than the control mice (Figure 5F). Average body temperature of mice co-administered Hup A and PTZ was comparable to mice given Hup A and saline, suggesting that GABAA receptors are not involved in the Hup A-mediated hypothermia (p < 0.0001, Figure 5D).
Discussion
A role for reversible AChE inhibitors in the treatment of epilepsy has focused largely on seizures that are caused by exposure to organophosphates (e.g., soman) that bind irreversibly to AChE. Hup A has been shown to protect against seizures in rat (Tonduli et al., 2001), guinea pig (Wang et al., 2013), and non-human primate models (Lallement et al., 2002) of soman toxicity. In addition, Coleman et al. (2008) reported that Hup A (the [+] isomer) protects against NMDA-induced status epilepticus in rats, and there is one case of Hup-mediated suppression of complex partial seizures in a dog (Schneider et al., 2009). Hup A (0.6 mg/kg) was also shown to protect against PTZ-induced seizures in rats (Gersner et al., 2015). In a recent antiepileptic drug screen using a zebrafish model of DS, Hup A (100–1000 μM) did not protect against behavioral seizures; however, it did protect against PTZ-induced seizures without toxic effects (Dinday and Baraban, 2015).
We hypothesize, based on potential mechanisms of action of Hup A and our findings, that it might be particularly efficacious in epilepsy subtypes like DS that are caused by reduced neuronal inhibition. Hup A administration directly inhibits AChE, leading to an increase in brain acetylcholine levels. Acetylcholine is known to act directly on muscarinic receptors on GABAergic interneurons (Gonzalez et al., 2011), thereby increasing neuronal inhibition and suppression of hippocampal excitation (Pitler and Alger, 1992). Furthermore, Hup A was found to increase intracortical inhibition as measured by paired-pulse transcranial magnetic stimulation, suggesting that it works, in part, by increasing cortical GABAergic activity (Gersner et al., 2015). Hup A is also known to have primarily central effects (Gersner et al., 2015), which makes it a more attractive reversible AChE inhibitor compared to others with peripheral side effects or reduced penetrance across the blood-brain barrier (e.g., pyridostigmine; Philippens et al., 1996). Thus, given the underlying reduction in neuronal inhibition (Yu et al., 2006; Ogiwara et al., 2007; Martin et al., 2010) and the known role of the hippocampus in seizure generation in DS (Liautard et al., 2013), the administration of Hup A would be predicted to enhance endogenous GABAergic tone, thereby normalizing the balance between neuronal inhibition and excitation in patients with SCN1A mutations. Importantly, likely because it augments endogenous GABA rather than being a direct GABA agonist, Hup A has not been associated with the side effects and risks of benzodiazepines, such as clonazepam, including addiction and respiratory suppression, and therefore may have advantages over GABA agonists and related drugs. Furthermore, we expect the additional biological properties of Hup A, including protection against inflammation (Wang et al., 2008) and cell death (Hemendinger et al., 2008), and increasing neurotrophic (Tang et al., 2005) and antioxidant (Xiao et al., 2000) activity, will also be beneficial in the treatment of epilepsy.
Huperzine A Provides Protection against 6 Hz-Induced Seizures
The 6 Hz seizure induction paradigm uses a 3-s, low-frequency (6 Hz) current to produce a seizure that is less intense and more focal compared to seizures generated by MES and PTZ (Brown et al., 1953). In conjunction with c-Fos immunohistochemistry, Barton et al. (2001) found that different regions of the brain could be activated by varying the intensity of the current used in the 6 Hz paradigm. Lower intensities (22 and 32 mA; CC97 and 1.5xCC97 for CF1 mice, respectively) activate the amygdala, cortices, and entorhinal cortex, while a higher current (44 mA; 2xCC97) activates the dentate gyrus of the hippocampus. Based on these findings, the 6 Hz seizure induction paradigm has been used as a model of therapy-resistant limbic seizures (Barton et al., 2001).
We found that 0.56 and 1 mg/kg of Hup A provided robust protection against 6 Hz-induced seizures (44 mA) in CF1 mice. Consistent with these results, we found that both Hup A doses also provided seizure protection in the Scn1a mutant mice. Interestingly, robust seizure protection was still achieved in Scn1a mutants when the current was doubled (48 mA for Scn1aRH/+ and 40 mA for Scn1a+/−), suggesting that Hup A might also provide protection against spontaneous seizures in DS that may initiate in the hippocampus (Liautard et al., 2013).
Daily administration of Hup A (1 mg/kg) provided protection against 6 Hz-induced seizures in CF1 mice for 7–12 days. More importantly, we demonstrated in the Scn1aRH/+ mutants that Hup A could be provided daily for at least 21 days without loss of seizure protection. However, we did not observe protection 24 h after cessation of treatment. Two potential reasons for the observed differences in the effect of chronic Hup A administration between the CF1 mice and the Scn1aRH/+ mutants are: (1) the different genetic backgrounds of each strain and (2) the predicted increase in neuronal inhibition attributable to Hup A might confer greater seizure protection in the mutants due to the effect of the Scn1a mutation on the excitability of inhibitory interneurons. These results make Hup A an attractive therapeutic option for epilepsy since chronic treatment is often necessary.
Huperzine A Provides Protection against Maximal Electroshock Seizures
The MES paradigm uses a high-frequency current of short duration (60 Hz, 50 mA, 0.2 ms) to produce a seizure that can spread throughout the brain (Toman, 1951) as reflected in c-Fos expression in distal structures, such as the midbrain and brainstem (Barton et al., 2001). Drugs that can inhibit seizure propagation are highly effective in the MES paradigm (Woodbury and Esplin, 1959). Furthermore, compounds that are effective against MES-induced seizures are found to clinically block or mitigate generalized tonic-clonic seizures (GTCSs; White et al., 1995). Valproate and topiramate, two AEDs currently used in the treatment of DS (Shi et al., 2015), also protect against MES-induced seizures. Our findings that Hup A protects against MES-induced seizures in Scn1a mutants suggest that it may provide protection by blocking seizure propagation.
Huperzine A Provides Protection against PTZ but Does Not Protect against Flurothyl-Induced Seizures
Similar to 6 Hz and MES, we observed robust protection against PTZ-induced seizures in CF1 mice. This was reflected by the absence of a GTCS in 4 of 6 Hup A-treated mice and an increase in average latencies to the MJ and GTCS (in the mice that did exhibit a GTCS). These results are consistent with the recent findings by Gersner et al. (2015), where Hup A (0.6 mg/kg) was found to protect against PTZ-induced seizures in rats. Similar to our results, a significantly lower number of Hup A-treated rats (only 30%) exhibited a GTCS compared to saline-treated rats (~75%). PTZ seizure induction is routinely used for screening potential anticonvulsants in part because protection against PTZ-induced seizures is predictive of efficacy in treating absence epilepsy (Krall et al., 1978).
In contrast, following flurothyl exposure, we observed a decrease in the average latency to the MJ and higher mortality following the GTCS-HLE in Hup A-treated mice. Seizure behavior and c-Fos immunoreactivity patterns have been found to be similar following flurothyl and PTZ seizure induction (Jensen et al., 1993), which are both mediated, at least in part, by inhibition of GABAA receptors. The ability of Hup A to provide protection against PTZ but not flurothyl-induced seizures suggests the contribution of a non-GABAergic component to the mechanism of action of flurothyl. Finally, it is possible, since flurothyl is inhaled, there may be peripheral effects elicited by both flurothyl and Hup A that, when combined, worsens the seizure phenotype.
Huperzine A Confers Robust Protection against Hyperthermia-Induced Seizures in Scn1a Mutant Mice
Infants with SCN1A mutations often experience severe and prolonged febrile seizures (FSs) that can have a negative impact on long-term prognosis (Wolff et al., 2006; Akiyama et al., 2010). Currently, there are no routine clinical interventions that can protect against early-life FSs. We and others have demonstrated increased susceptibility to hyperthermia-induced seizures in Scn1a mutant mice (Oakley et al., 2009; Dutton et al., 2013). Although the mechanism underlying febrile seizure generation is not fully understood, it has been demonstrated that febrile seizures originate from the limbic region of the brain, notably the amygdala and hippocampus (Dube et al., 2000; Brewster et al., 2002). Consistent with the robust Hup A-mediated protection against 6 Hz-induced seizures (a model of limbic epilepsy), we observed a significant increase in the temperature at which hyperthermia-induced seizures occurred in Hup A-treated Scn1a mutant mice. Cao and others have suggested that the temperature at seizure occurrence is a good measure of drug efficacy as this index is reproducible within the same mouse (Cao et al., 2012). Other drugs, such as stiripentol, that have demonstrated clinical efficacy in the treatment of SCN1A-derived epilepsy have also been shown to increase resistance to hyperthermia-induced seizures in Scn1a rodent models (Hayashi et al., 2011; Cao et al., 2012).
Huperzine A and Inhibition of AChE Activity
Hup A-mediated inhibition of brain AChE is known to be dose dependent and comparable across routes of administration. Tang et al. (1989) found that Hup A (0.5–1 mg/kg) provided the greatest inhibition of AChE throughout the entire brain (up to 42%), with the fewest side effects. Laganiere et al. (1991) found that 0.5 mg/kg Hup A significantly reduced AChE activity in the hippocampus, striatum, and septum (20–45%), whereas 0.1 mg/kg Hup A only slightly reduced AChE activity in these brain regions. In Rhesus monkeys, Myers et al. (2010) also found that Hup A (5–40 μg/kg) resulted in a dose-dependent inhibition of AChE activity (31–74%) without adverse cognitive effects. Although we found that Hup A doses of 0.32–1 mg/kg produced similar reductions in AChE activity, which may indicate a floor effect on AChE activity, more robust seizure protection was observed with 0.56 and 1 mg/kg Hup A. We also observed similar reductions in AChE activity regardless of whether Hup A was administered acutely or chronically (Figure 5C). These findings suggest that Hup A-mediated seizure protection is not dependent solely on the level of AChE activity. In addition, previous studies demonstrated that Hup A inhibition of AChE occurs significantly faster than the corresponding increases in acetylcholine (Tang et al., 1989).
Recently, focal seizures (as a result of hippocampal stimulation) were found to result in decreased subcortical cholinergic neurotransmission both during and after a seizure (Motelow et al., 2015). Decreased acetylcholine levels in the thalamus have also been noted in partial seizures and slow-wave sleep (Williams et al., 1994). Our findings that Hup A protects against 6 Hz-induced seizures, a model of focal limbic seizures, and the noted decreased cholinergic neurotransmission in focal seizures (Motelow et al., 2015), suggest that Hup A may also be efficacious in the treatment of TLE.
Potential Mechanisms for Hup A-Mediated Seizure Protection
Elevated body temperature is known to induce epileptic activity and seizures in animals, while hypothermia (29–34.5°C) has been shown to be anticonvulsant (Essman and Sudak, 1964). We observed that upon acute administration of Hup A, mice displayed transient side effects, including hypothermia. To determine whether Hup A-induced hypothermia contributes to seizure protection, we held the body temperature of mice at 37.5°C for the 1 h interval between Hup A administration and MES induction. Under these conditions, we found that Hup A-induced hypothermia does not contribute to seizure protection.
To evaluate potential neurotransmitter receptors that may be involved in Hup A-mediated seizure protection, we co-administered Hup A and receptor antagonists 1 h prior to 6 Hz seizure induction. Atropine, a competitive muscarinic receptor antagonist that targets receptors in both the central and peripheral nervous systems, has been demonstrated (at 30 mg/kg) to block the protective effects of Hup A in the 6 Hz paradigm (Bialer et al., 2015). However, Gersner et al. (2015) found that co-administration of atropine (30 mg/kg) and Hup A did not block the magnitude of paired-pulse inhibition, suggesting that muscarinic receptors are not involved in Hup A-mediated cortical interneuron activation. We found that the co-administration of scopolamine hydrobromide, a centrally acting muscarinic receptor antagonist, blocked Hup A-mediated protection against 6 Hz-induced seizures. We also found that the co-administration of PTZ, a GABAA receptor antagonist, also partially blocks Hup A-mediated protection in the 6 Hz paradigm. However, the co-administration of a nicotinic receptor antagonist did not block Hup A-mediated seizure protection. These results suggest that Hup A may provide seizure protection through the activation of several pathways, including the muscarinic and GABAA receptors.
Increases in acetylcholine have been shown to decrease body temperature in both humans (Cushing, 1931) and rodents (Everett, 1956) by acting upon interneurons in the hypothalamic thermoregulatory zone. Our results demonstrate that Hup A-induced hypothermia involves both muscarinic and nicotinic cholinergic receptors. Co-administration of Hup A and the muscarinic receptor antagonist, SH, resulted in the maintenance of normal body temperature. Although co-administration of the nicotinic receptor antagonist, BH, partially prevented the decrease in body temperature observed in Hup A plus saline-injected mice, the average body temperature of Hup A plus BH mice was still significantly lower than control mice. Our findings suggest that Hup A-induced hypothermia is mediated primarily by the muscarinic cholinergic receptor.
Conclusion
We demonstrated that Hup A increased the resistance of Scn1a+/− and Scn1aRH/+ mutant mice to seizures induced by MES, 6 Hz, hyperthermia, and PTZ. In addition, seizure protection conferred by Hup A is maintained during chronic administration. Taken together, our findings highlight the therapeutic potential of Huperzine A in increasing seizure resistance in SCN1A-derived epilepsy and potentially other forms of refractory epilepsy. Further studies on the ability of Hup A to reduce spontaneous seizure frequency and severity are therefore warranted.
Author Contributions
All authors made substantial contributions to the conception and design of the work and interpretation of the data. JW and SD collected and analyzed the data. JW, AE, and SD wrote the manuscript. All authors reviewed and approved the manuscript.
Conflict of Interest Statement
SC has a financial interest in Biscayne Pharmaceuticals, which is the holder of all commercial rights to Huperzine A. SS is an inventor on a patent for the use of Huperzine A for the treatment of epilepsy, which is licensed by Harvard Medical School to Biscayne Pharmaceuticals, in which he holds less than 5% equity and for which he serves as chair of the scientific advisory board.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge Dr. William Catterall for providing the Scn1a+/− mutant mice. The authors would also like to thank Cheryl Strauss for editorial assistance. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health [R01NS072221, R21NS098776 (AE)] and the Training in Translational Research in Neurology T32 [2T32NS00748016 (JW)]. The content is solely the responsibility of the authors and does not necessarily represent the views of the NIH.
References
Akiyama, M., Kobayashi, K., Yoshinaga, H., and Ohtsuka, Y. (2010). A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 51, 1043–1052. doi: 10.1111/j.1528-1167.2009.02466.x
Barton, M. E., Klein, B. D., Wolf, H. H., and White, H. S. (2001). Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 47, 217–227. doi: 10.1016/S0920-1211(01)00302-3
Bialer, M., Johannessen, S. I., Levy, R. H., Perucca, E., Tomson, T., and White, H. S. (2015). Progress report on new antiepileptic drugs: a summary of the Twelfth Eilat Conference (EILAT XII). Epilepsy Res. 111, 85–141. doi: 10.1016/j.eplepsyres.2015.01.001
Brewster, A., Bender, R. A., Chen, Y., Dube, C., Eghbal-Ahmadi, M., and Baram, T. Z. (2002). Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J. Neurosci. 22, 4591–4599.
Brown, W. C., Schiffman, D. O., Swinyard, E. A., and Goodman, L. S. (1953). Comparative assay of an antiepileptic drugs by psychomotor seizure test and minimal electroshock threshold test. J. Pharmacol. Exp. Ther. 107, 273–283.
Cao, D., Ohtani, H., Ogiwara, I., Ohtani, S., Takahashi, Y., Yamakawa, K., et al. (2012). Efficacy of stiripentol in hyperthermia-induced seizures in a mouse model of Dravet syndrome. Epilepsia 53, 1140–1145. doi: 10.1111/j.1528-1167.2012.03497.x
Catterall, W. A., Kalume, F., and Oakley, J. C. (2010). NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859. doi: 10.1113/jphysiol.2010.187484
Chiron, C., and Dulac, O. (2011). The pharmacologic treatment of Dravet syndrome. Epilepsia 52(Suppl. 2), 72–75. doi: 10.1111/j.1528-1167.2011.03007.x
Claes, L., Del-Favero, J., Ceulemans, B., Lagae, L., Van Broeckhoven, C., and De Jonghe, P. (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 68, 1327–1332. doi: 10.1086/320609
Claes, L. R., Deprez, L., Suls, A., Baets, J., Smets, K., Van Dyck, T., et al. (2009). The SCN1A variant database: a novel research and diagnostic tool. Hum. Mutat. 30, E904–E920. doi: 10.1002/humu.21083
Coleman, B. R., Ratcliffe, R. H., Oguntayo, S. A., Shi, X., Doctor, B. P., Gordon, R. K., et al. (2008). [+]-Huperzine A treatment protects against N-methyl-D-aspartate-induced seizure/status epilepticus in rats. Chem. Biol. Interact. 175, 387–395. doi: 10.1016/j.cbi.2008.05.023
Cushing, H. I. I. (1931). The similarity in the response to posterior lobe extract (Pituitrin) and to pilocarpine when injected into the cerebral ventricles. Proc. Natl. Acad. Sci. U.S.A. 17, 171–177. doi: 10.1073/pnas.17.4.171
Dinday, M. T., and Baraban, S. C. (2015). Large-scale phenotype-based antiepileptic drug screening in a zebrafish model of Dravet syndrome. eNeuro 2:ENEURO.0068-15.2015. doi: 10.1523/ENEURO.0068-15.2015
Dravet, C., Bureau, M., Oguni, H., Fukuyama, Y., and Cokar, O. (2005). Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv. Neurol. 95, 71–102.
Dube, C., Chen, K., Eghbal-Ahmadi, M., Brunson, K., Soltesz, I., and Baram, T. Z. (2000). Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann. Neurol. 47, 336–344. doi: 10.1002/1531-8249(200003)47:3<336::AID-ANA9>3.0.CO;2-W
Dutton, S. B., Makinson, C. D., Papale, L. A., Shankar, A., Balakrishnan, B., Nakazawa, K., et al. (2012). Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 49C, 211–220. doi: 10.1016/j.nbd.2012.08.012
Dutton, S. B., Makinson, C. D., Papale, L. A., Shankar, A., Balakrishnan, B., Nakazawa, K., et al. (2013). Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 49, 211–220. doi: 10.1016/j.nbd.2012.08.012
Dutton, S. B., Sawyer, N. T., Kalume, F., Jumbo-Lucioni, P., Borges, K., Catterall, W. A., et al. (2011). Protective effect of the ketogenic diet in Scn1a mutant mice. Epilepsia 52, 2050–2056. doi: 10.1111/j.1528-1167.2011.03211.x
Ellman, G. L., Courtney, K. D., Andres, V. Jr., and Feather-Stone, R. M. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95. doi: 10.1016/0006-2952(61)90145-9
Escayg, A., and Goldin, A. L. (2010). Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia 51, 1650–1658. doi: 10.1111/j.1528-1167.2010.02640.x
Escayg, A., Heils, A., MacDonald, B. T., Haug, K., Sander, T., and Meisler, M. H. (2001). A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus–and prevalence of variants in patients with epilepsy. Am. J. Hum. Genet. 68, 866–873. doi: 10.1086/319524
Escayg, A., MacDonald, B. T., Meisler, M. H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., et al. (2000). Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24, 343–345. doi: 10.1038/74159
Essman, W. B., and Sudak, F. N. (1964). Audiogenic seizure in genetically susceptible mice: relation of hypothermia to onset and susceptibility. Exp. Neurol. 9, 228–235. doi: 10.1016/0014-4886(64)90019-6
Gao, X., and Tang, X. C. (2006). Huperzine A attenuates mitochondrial dysfunction in beta-amyloid-treated PC12 cells by reducing oxygen free radicals accumulation and improving mitochondrial energy metabolism. J. Neurosci. Res. 83, 1048–1057. doi: 10.1002/jnr.20791
Gersner, R., Ekstein, D., Dhamne, S. C., Schachter, S., and Rotenburg, A. (2015). Huperzine A prophylaxis against pentylenetetrazole-induced seizures in rats is associated with increased cortical inhibition. Epilepsy Res. 117, 97–103. doi: 10.1016/j.eplepsyres.2015.08.012
Gilchrist, J., Dutton, S., Diaz-Bustamante, M., McPherson, A., Olivares, N., Kalia, J., et al. (2014). Nav1.1 modulation by a novel triazole compound attenuates epileptic seizures in rodents. ACS Chem. Biol. 9, 1204–1212. doi: 10.1021/cb500108p
Gonzalez, J. C., Albinana, E., Baldelli, P., Garcia, A. G., and Hernandez-Guijo, J. M. (2011). Presynaptic muscarinic receptor subtypes involved in the enhancement of spontaneous GABAergic postsynaptic currents in hippocampal neurons. Eur. J. Neurosci. 33, 69–81. doi: 10.1111/j.1460-9568.2010.07475.x
Gordon, R. K., Nigam, S. V., Weitz, J. A., Dave, J. R., Doctor, B. P., and Ved, H. S. (2001). The NMDA receptor ion channel: a site for binding of Huperzine A. J. Appl. Toxicol. 21(Suppl. 1), S47–S51. doi: 10.1002/jat.805
Hayashi, K., Ueshima, S., Ouchida, M., Mashimo, T., Nishiki, T., Sendo, T., et al. (2011). Therapy for hyperthermia-induced seizures in Scn1a mutant rats. Epilepsia 52, 1010–1017. doi: 10.1111/j.1528-1167.2011.03046.x
Hemendinger, R. A., Armstrong, E. J. III. Persinski, R., Todd, J., Mougeot, J. L., Volvovitz, F., et al. (2008). Huperzine A provides neuroprotection against several cell death inducers using in vitro model systems of motor neuron cell death. Neurotox. Res. 13, 49–61. doi: 10.1007/BF03033367
Jensen, F. E., Firkusny, I. R., and Mower, G. D. (1993). Differences in c-fos immunoreactivity due to age and mode of seizure induction. Brain Res. Mol. Brain Res. 17, 185–193. doi: 10.1016/0169-328X(93)90001-6
Krall, R. L., Penry, J. K., White, B. G., Kupferberg, H. J., and Swinyard, E. A. (1978). Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia 19, 409–428. doi: 10.1111/j.1528-1157.1978.tb04507.x
Kwan, P., and Brodie, M. J. (2006). Refractory epilepsy: mechanisms and solutions. Expert Rev. Neurother. 6, 397–406. doi: 10.1586/14737175.6.3.397
Laganiere, S., Corey, J., Tang, X. C., Wulfert, E., and Hanin, I. (1991). Acute and chronic studies with the anticholinesterase Huperzine A: effect on central nervous system cholinergic parameters. Neuropharmacology 30, 763–768. doi: 10.1016/0028-3908(91)90184-D
Lallement, G., Demoncheaux, J. P., Foquin, A., Baubichon, D., Galonnier, M., Clarencon, D., et al. (2002). Subchronic administration of pyridostigmine or huperzine to primates: compared efficacy against soman toxicity. Drug Chem. Toxicol. 25, 309–320. doi: 10.1081/DCT-120005893
Liautard, C., Scalmani, P., Carriero, G., de Curtis, M., Franceschetti, S., and Mantegazza, M. (2013). Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia 54, 1251–1261. doi: 10.1111/epi.12213
Loscher, W., Honack, D., Fassbender, C. P., and Nolting, B. (1991). The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 8, 171–189. doi: 10.1016/0920-1211(91)90062-K
Loscher, W., and Schmidt, D. (1994). Strategies in antiepileptic drug development: is rational drug design superior to random screening and structural variation? Epilepsy Res. 17, 95–134. doi: 10.1016/0920-1211(94)90012-4
Lossin, C. (2009). A catalog of SCN1A variants. Brain Dev. 31, 114–130. doi: 10.1016/j.braindev.2008.07.011
Ma, X., Tan, C., Zhu, D., Gang, D. R., and Xiao, P. (2007). Huperzine A from Huperzia species–an ethnopharmacolgical review. J. Ethnopharmacol. 113, 15–34. doi: 10.1016/j.jep.2007.05.030
Martin, M. S., Dutt, K., Papale, L. A., Dube, C. M., Dutton, S. B., de Haan, G., et al. (2010). Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 285, 9823–9834. doi: 10.1074/jbc.M109.078568
McKinney, M., Miller, J. H., Yamada, F., Tuckmantel, W., and Kozikowski, A. P. (1991). Potencies and stereoselectivities of enantiomers of huperzine A for inhibition of rat cortical acetylcholinesterase. Eur. J. Pharmacol. 203, 303–305. doi: 10.1016/0014-2999(91)90730-E
Motelow, J. E., Li, W., Zhan, Q., Mishra, A. M., Sachdev, R. N., Liu, G., et al. (2015). Decreased subcortical cholinergic arousal in focal seizures. Neuron 85, 561–572. doi: 10.1016/j.neuron.2014.12.058
Myers, T. M., Sun, W., Saxena, A., Doctor, B. P., Bonvillain, A. J., and Clark, M. G. (2010). Systemic administration of the potential countermeasure huperzine reversibly inhibits central and peripheral acetylcholinesterase activity without adverse cognitive-behavioral effects. Pharmacol. Biochem. Behav. 94, 477–481. doi: 10.1016/j.pbb.2009.10.011
Oakley, J. C., Kalume, F., Yu, F. H., Scheuer, T., and Catterall, W. A. (2009). Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. U.S.A. 106, 3994–3999. doi: 10.1073/pnas.0813330106
O'Brien, J. E., and Meisler, M. H. (2013). Sodium channel (Na1.6): properties and mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 4:213. doi: 10.3389/fgene.2013.00213
Ogiwara, I., Miyamoto, H., Morita, N., Atapour, N., Mazaki, E., Inoue, I., et al. (2007). Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007
Padilla, S., Lassiter, T. L., and Hunter, D. (1999). Biochemical measurement of cholinesterase activity. Methods Mol. Med. 22, 237–245.
Papale, L. A., Makinson, C. D., Christopher Ehlen, J., Tufik, S., Decker, M. J., Paul, K. N., et al. (2013). Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+). Epilepsia 54, 625–634. doi: 10.1111/epi.12060
Philippens, I. H., Wolthuis, O. L., Busker, R. W., Langenberg, J. P., and Melchers, B. P. (1996). Side effects of physostigmine as a pretreatment in guinea pigs. Pharmacol. Biochem. Behav. 55, 99–105. doi: 10.1016/0091-3057(96)83115-7
Pitler, T. A., and Alger, B. E. (1992). Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. J. Physiol. 450, 127–142. doi: 10.1113/jphysiol.1992.sp019119
Prichahd, J. W., Gallagher, B. B., and Glaser, G. H. (1969). Experimental seizure-threshold testing with flurothyl. J. Pharmacol. Exp. Ther. 166, 170–178.
Purcell, R. H., Papale, L. A., Makinson, C. D., Sawyer, N. T., Schroeder, J. P., Escayg, A., et al. (2013). Effects of an epilepsy-causing mutation in the SCN1A sodium channel gene on cocaine-induced seizure susceptibility in mice. Psychopharmacology (Berl) 228, 263–270. doi: 10.1007/s00213-013-3034-8
Schneider, B. M., Dodman, N. H., Faissler, D., and Ogata, N. (2009). Clinical use of an herbal-derived compound (Huperzine A) to treat putative complex partial seizures in a dog. Epilepsy Behav. 15, 529–534. doi: 10.1016/j.yebeh.2009.06.011
Shi, X. Y., Tomonoh, Y., Wang, W. Z., Ishii, A., Higurashi, N., Kurahashi, H., et al. (2015). Efficacy of antiepileptic drugs for the treatment of Dravet syndrome with different genotypes. Brain Dev. 38, 40–46. doi: 10.1016/j.braindev.2015.06.008
Swinyard, E. A. (1972). “Electrically induced seizures,” in Experimental Models of Epilepsy: A Manual for the Laboratory Worker, eds D. Purpura, J. K. Penry, D. B. Tower, D. M. Woodbury, and R. D. Walter (New York, NY: Raven Press), 433–458.
Tai, C., Abe, Y., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U.S.A. 111, E3139–E3148. doi: 10.1073/pnas.1411131111
Tang, L. L., Wang, R., and Tang, X. C. (2005). Effects of huperzine A on secretion of nerve growth factor in cultured rat cortical astrocytes and neurite outgrowth in rat PC12 cells. Acta Pharmacol. Sin. 26, 673–678. doi: 10.1111/j.1745-7254.2005.00130.x
Tang, X. C., De Sarno, P., Sugaya, K., and Giacobini, E. (1989). Effect of huperzine A, a new cholinesterase inhibitor, on the central cholinergic system of the rat. J. Neurosci. Res. 24, 276–285. doi: 10.1002/jnr.490240220
Toman, J. E. (1951). Neuropharmacologic considerations in psychic seizures. Neurology 1, 444–460. doi: 10.1212/WNL.1.11-12.444
Tonduli, L. S., Testylier, G., Masqueliez, C., Lallement, G., and Monmaur, P. (2001). Effects of Huperzine used as pre-treatment against soman-induced seizures. Neurotoxicology 22, 29–37. doi: 10.1016/S0161-813X(00)00015-2
Ved, H. S., Koenig, M. L., Dave, J. R., and Doctor, B. P. (1997). Huperzine A, a potential therapeutic agent for dementia, reduces neuronal cell death caused by glutamate. Neuroreport 8, 963–968. doi: 10.1097/00001756-199703030-00029
Wallace, R. H., Hodgson, B. L., Grinton, B. E., Gardiner, R. M., Robinson, R., Rodriguez-Casero, V., et al. (2003). Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology 61, 765–769. doi: 10.1212/01.WNL.0000086379.71183.78
Wang, B. S., Wang, H., Wei, Z. H., Song, Y. Y., Zhang, L., and Chen, H. Z. (2009). Efficacy and safety of natural acetylcholinesterase inhibitor huperzine A in the treatment of Alzheimer's disease: an updated meta-analysis. J. Neural Transm. 116, 457–465. doi: 10.1007/s00702-009-0189-x
Wang, J., Chen, F., Zheng, P., Deng, W., Yuan, J., Peng, B., et al. (2012). Huperzine A ameliorates experimental autoimmune encephalomyelitis via the suppression of T cell-mediated neuronal inflammation in mice. Exp. Neurol. 236, 79–87. doi: 10.1016/j.expneurol.2012.03.024
Wang, R., Yan, H., and Tang, X. C. (2006). Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol. Sin. 27, 1–26. doi: 10.1111/j.1745-7254.2006.00255.x
Wang, Y., Wei, Y., Oguntayo, S., Doctor, B. P., and Nambiar, M. P. (2013). A combination of [+] and [−]-Huperzine A improves protection against soman toxicity compared to [+]-Huperzine A in guinea pigs. Chem. Biol. Interact. 203, 120–124. doi: 10.1016/j.cbi.2012.10.016
Wang, Z. F., Wang, J., Zhang, H. Y., and Tang, X. C. (2008). Huperzine A exhibits anti-inflammatory and neuroprotective effects in a rat model of transient focal cerebral ischemia. J. Neurochem. 106, 1594–1603. doi: 10.1111/j.1471-4159.2008.05504.x
White, H. S., Johnson, M., Wolf, H. H., and Kupferberg, H. J. (1995). The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital. J. Neurol. Sci. 16, 73–77. doi: 10.1007/BF02229077
Williams, J. A., Comisarow, J., Day, J., Fibiger, H. C., and Reiner, P. B. (1994). State-dependent release of acetylcholine in rat thalamus measured by in vivo microdialysis. J. Neurosci. 14, 5236–5242.
Wolff, M., Casse-Perrot, C., and Dravet, C. (2006). Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 47(Suppl. 2), 45–48. doi: 10.1111/j.1528-1167.2006.00688.x
Woodbury, D. M., and Esplin, D. W. (1959). Neuropharmacology and neurochemistry of anticonvulsant drugs. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 37, 24–56.
Xiao, X. Q., Wang, R., Han, Y. F., and Tang, X. C. (2000). Protective effects of huperzine A on beta-amyloid(25-35) induced oxidative injury in rat pheochromocytoma cells. Neurosci. Lett. 286, 155–158. doi: 10.1016/S0304-3940(00)01088-0
Xu, Z. Q., Liang, X. M., Juan, W., Zhang, Y. F., Zhu, C. X., and Jiang, X. J. (2012). Treatment with Huperzine A improves cognition in vascular dementia patients. Cell Biochem. Biophys. 62, 55–58. doi: 10.1007/s12013-011-9258-5
Yang, G., Wang, Y., Tian, J., and Liu, J. P. (2013). Huperzine A for Alzheimer's disease: a systematic review and meta-analysis of randomized clinical trials. PLoS ONE 8:e74916. doi: 10.1371/journal.pone.0074916
Yang, L., Ye, C. Y., Huang, X. T., Tang, X. C., and Zhang, H. Y. (2012). Decreased accumulation of subcellular amyloid-beta with improved mitochondrial function mediates the neuroprotective effect of huperzine A. J. Alzheimers. Dis. 31, 131–142. doi: 10.3233/JAD-2012-120274
Ye, W., Gong, X., Xie, J., Wu, J., Zhang, X., Ouyang, Q., et al. (2010). AChE deficiency or inhibition decreases apoptosis and p53 expression and protects renal function after ischemia/reperfusion. Apoptosis 15, 474–487. doi: 10.1007/s10495-009-0438-3
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Zangara, A. (2003). The psychopharmacology of huperzine A: an alkaloid with cognitive enhancing and neuroprotective properties of interest in the treatment of Alzheimer's disease. Pharmacol. Biochem. Behav. 75, 675–686. doi: 10.1016/S0091-3057(03)00111-4
Keywords: huperzine A, Scn1a, Dravet syndrome, genetic epilepsy with febrile seizures plus, seizure
Citation: Wong JC, Dutton SBB, Collins SD, Schachter S and Escayg A (2016) Huperzine A Provides Robust and Sustained Protection against Induced Seizures in Scn1a Mutant Mice. Front. Pharmacol. 7:357. doi: 10.3389/fphar.2016.00357
Received: 18 August 2016; Accepted: 20 September 2016;
Published: 17 October 2016.
Edited by:
Pascal Bonaventure, Janssen Research & Development, USAReviewed by:
Larry Baum, The Chinese University of Hong Kong, Hong KongJeremy Daniel Slater, University of Texas Medical School at Houston, USA
Copyright © 2016 Wong, Dutton, Collins, Schachter and Escayg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew Escayg, aescayg@emory.edu
†These authors have contributed equally to this work.