 Radhakrishnan Nagarajan1* Sujay Datta2 Marco Scutari3 Marjorie L. Beggs4 Greg T. Nolen5 Charlotte A. Peterson6
Radhakrishnan Nagarajan1* Sujay Datta2 Marco Scutari3 Marjorie L. Beggs4 Greg T. Nolen5 Charlotte A. Peterson6
- 1 Division of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR, USA
- 2 Statistical Center for HIV/AIDS Research and Prevention, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
- 3 Department of Statistical Sciences, University of Padova, Padova, Italy
- 4 College of Public Health, University of Arkansas for Medical Sciences, Little Rock, AR, USA
- 5 Department of Pediatrics, University of Arkansas for Medical Sciences, Little Rock, AR, USA
- 6 College of Health Sciences, University of Kentucky, Lexington, KY, USA
Aging is accompanied by considerable heterogeneity with possible co-expression of differentiation pathways. The present study investigates the interplay between crucial myogenic, adipogenic, and Wnt-related genes orchestrating aged myogenic progenitor differentiation (AMPD) using clonal gene expression profiling in conjunction with Bayesian structure learning (BSL) techniques. The expression of three myogenic regulatory factor genes (Myogenin, Myf-5, MyoD1), four genes involved in regulating adipogenic potential (C/EBPα, DDIT3, FoxC2, PPARγ), and two genes in the Wnt signaling pathway (Lrp5, Wnt5a) known to influence both differentiation programs were determined across 34 clones by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). Three control genes were used for normalization of the clonal expression data (18S, GAPDH, and B2M). Constraint-based BSL techniques, namely (a) PC Algorithm, (b) Grow-shrink (GS) algorithm, and (c) Incremental Association Markov Blanket (IAMB) were used to model the functional relationships (FRs) in the form of acyclic networks from the clonal expression profiles. A novel resampling approach that obviates the need for a user-defined confidence threshold is proposed to identify statistically significant FRs at small sample sizes. Interestingly, the resulting acyclic network consisted of FRs corresponding to myogenic, adipogenic, Wnt-related genes and their interaction. A significant number of these FRs were robust to normalization across the three house-keeping genes and the choice of the BSL technique. The results presented elucidate the delicate balance between differentiation pathways (i.e., myogenic as well as adipogenic) and possible cross-talk between pathways in AMPD.
Introduction
Classical gene expression analysis on mass cultures implicitly assumes homogeneity in expression across a given population of cells. However, there has been accumulating evidence of considerable heterogeneity in expression (Loeffler and Roeder, 2002; Madras et al., 2002; Taylor-Jones et al., 2002; Beggs et al., 2004; Nagarajan et al., 2004; Bahar et al., 2006; Somel et al., 2006). Some of these studies have also emphasized the nexus between aging and heterogeneity in expression (Taylor-Jones et al., 2002; Beggs et al., 2004; Bahar et al., 2006; Somel et al., 2006). Several factors can contribute to heterogeneity in expression, these include (i) noise: stochastic mechanisms at the dynamical and measurement levels inherent in gene expression estimation (Kaern et al., 2005) (ii) plasticity: commitment toward a specific phenotype re-directed to another phenotype by the activation and/or repression of certain crucial genes (Hu et al., 1995). (iii) de-differentiation: differentiated cells revert to a less-differentiated (i.e., more proliferative) phase, resulting in considerable overlap between proliferation and differentiation (McGann et al., 2001), (iv) co-expression of differentiation pathways: gene programs corresponding to multiple differentiation pathways expressed simultaneously (Taylor-Jones et al., 2002), (v) microenvironment: expression across sub-populations of cells governed by their immediate surrounding (Allinen et al., 2004) and (vi) multiple pathways to achieve the same end phenotype (Madras et al., 2002). Probabilistic techniques such as Bayesian structure learning (BSL) accommodate the uncertainty and noisiness in gene expression (Friedman et al., 2000), hence a natural choice for modeling functional relationships (FRs) from heterogeneous populations. FRs represent regulatory connections between the genes inferred from their expression profiles (Gardner et al., 2003). Understanding FRs can provide insight into the concerted working of genes as a system as opposed to independent entities.
Transcriptional regulatory networks controlling both myogenic and adipogenic differentiation are continuously being expanded and refined. The myogenic regulatory factors, MyoD, myogenin, Myf-5 and MRF4 clearly are involved in controlling myogenic progenitor specification and subsequent differentiation both during development and during muscle regeneration postnatally (Chargé and Rudnicki, 2003). Although under different experimental settings, each factor can transactivate the expression of the other, they are expressed in unique patterns in muscle and do not have completely overlapping functions. Similarly, the C/EBP family of transcription factors and PPARγ cross-activate expression to control adipocyte gene expression (Rosen et al., 2000). Typically myogenic and adipogenic differentiation pathways are considered non-overlapping. However, we have shown that myogenic progenitors from aged mice co-express some aspects of both myogenic and adipogenic gene programs (Taylor-Jones et al., 2002), with the balance apparently regulated by Wnt signaling (Vertino et al., 2005). However, to our knowledge there has been limited effort in understanding their interaction. Wnt ligands have been shown to promote myogenic commitment in adult muscle progenitor cells (Polesskaya et al., 2003) and appear to be essential for differentiation of epaxial muscles during development, potentially through regulating MyoD and Myf5 gene expression (Tajbakhsh et al., 1998). However, different Wnts have distinct effects during limb myogenic differentiation (Anakwe et al., 2003) and may activate myogenesis via canonical, β-catenin-dependent and non-canonical pathways (Brunelli et al., 2007). By contrast, Wnt signaling appears to inhibit adipogenic differentiation, potentially by downregulating PPARγ activity (Ross et al., 2000). More recently Wnt signaling has also been implicated in regulating myogenic progenitor fate during aging that may contribute to fibrosis (Brack et al., 2007; Hidestrand et al., 2008).
Bayesian structure learning (Pearl, 1988; Glymour and Cooper, 1999) represents the joint distribution of a given set of random variables as product of conditional probability distributions. Several articles have used BSL for modeling FRs (Friedman et al., 2000) from gene expression profiles. Recent studies (Sachs et al., 2005) also used BSL successfully to model FRs from translational and post-translational expression profiles of single cells. It is important to note that BSL implicitly models the FRs in the form acyclic network, hence does not accommodate feedback structure. Nevertheless, these abstractions have been useful in obtaining preliminary insights and system-level understanding. BSL can also be useful in capturing the cause and effect relationships between the variables of interest under certain implicit assumptions such as the causal-Markov assumption and faithfulness (Glymour and Cooper, 1999).
Materials and Methods
Clonal Gene Expression Data
The clonal gene expression data were generated from RNA isolated from 34 clones of myogenic progenitors obtained from 24-month-old mice, cultured to confluence and allowed to differentiate for 24 h. Real-time RT-PCR was used to quantify expression of 12 genes (nine genes of interest and three controls) across the 34 clones. The 12 genes consisted of: Myogenic regulatory factors: MyoD1, Myogenin and Myf-5; Adipogenesis related genes: FoxC2 (Forkhead box C2), DDIT3 (DNA damage inducible transcript 3), C/EBP (CCAAT/enhancer binding protein alpha), PPAR (Peroxisome proliferator-activated receptor gamma); Wnt signaling related genes: Wnt5a (wingless-type mmtv integration site family member 5A) and Lrp5 (Low-density lipoprotein receptor-related protein 5) and Control genes: GAPDH (Glyceraldehyde-3-phosphate dehydrogenase), 18S (Ribosomal RNA) and B2M (Beta-2 microglobulin).
Myogenic progenitor data generation protocol
Myogenic progenitors were isolated from the tibialis anterior of four aged (23 months) DBA/2JNIA mice as described (11). For isolation of clones, proliferating progenitors were seeded at low density, between 200 and 500 cells per collagen-coated 150 mm plate (Sigma, St Louis, MO, USA), and maintained in growth media containing Ham’s F-10 (BioWhittaker, Walkersville, MD, USA) supplemented with 20% fetal bovine serum (FBS) (BioWhittaker), 0.5% pen-strep (Invitrogen, Carlsbad, CA, USA) and 5 ng/ml basic fibroblast growth factor (Promega, Madison, WI, USA) at 37°C in a humidified 5% CO2-90% air atmosphere. After 12–14 days the clones were large enough to pick. An average of 6 clones from each of 10 plates for aged myoblasts were picked and transferred to collagen-coated 24 well plates for expansion. As each clone reached the optimum density (approximately 90% confluence), they were switched to differentiation media consisting of Dulbecco’s modified Eagle media (DMEM) (Invitrogen) with 2% horse serum (Hyclone, Logan, UT, USA) and 0.5% pen-strep. After 24 h, each clone was observed morphologically and harvested in lysis buffer from the RNAqueous Kit (Ambion, Austin, TX, USA) and stored at −20°C for later RNA isolation as described below. Total RNA was extracted with the Ambion RNAqueous Kit following the manufactures’ instructions. Residual DNA was removed by treatment with RNase-free DNase I (Roche, Mannheim, Germany). RNA quality was assessed on the Agilent Bioanalyzer (Agilent, Palo Alto, CA, USA). For real-time, quantitative reverse transcription-polymerase chain reaction (RT-PCR), cDNA was synthesized from 1 μg RNA using random hexamers and TaqMan RT Reagents for real-time PCR [Applied Biosystems, Inc. (ABI), Foster City, CA, USA] in the ABI 7900HT Fast Real-time PCR System. The ABI Ready-to-Go Assays for real-time PCR were used to capture the expression of the mouse genes MyoD1 (Mm00440387), Myogenin (Mm00446194, Myogenin), Wnt5a (Mm00437347, wingless-type mmtv integration site family member 5A), Lrp5 (Mm00493179, low-density lipoprotein receptor-related protein 5), Myf-5 (Mm00435125, myogenic factor 5), FoxC2 (Mm00546194, forkhead box C2), DDIT3 (Mm00492097, DNA damage inducible transcript 3), C/EBPα (Mm00514283, CCAAT/enhancer binding protein alpha), PPARγ (Mm00440945, peroxisome proliferator-activated receptor gamma). GAPDH (Mm99999915, Glyceraldehyde-3-phosphate dehydrogenase), 18S (Hs99999901, ribosomal RNA), and B2M (Mm03003532, beta-2 microglobulin) were used as controls. The reactions (25 μl) were performed in ABI Fast Master Mix and the default ABI PCR program (60°C anneal). Equal amounts of cDNA from each sample were pooled to use for standard curve reactions with each primer set. Standard curves were generated for each gene using pooled real-time RT-PCR reactions at 20, 5, 1.25, and 0.31 ng/μl. No template controls (NTC) were incorporated for every assay. The relative standard curve method was used to calculate the amplification difference between the samples for each primer set. This is performed by correcting for each signal concentration with the concentration of the controls.
Bayesian Structure Learning
Constraint-based algorithms are all based on the seminal work by Verma and Pearl (1991). The present study uses three constraint-based BSL algorithms [PC, Grow-Shrink (GS), and Incremental Association Markov Blanket (IAMB)] to learn the structure from the given clonal expression data. The Inductive Causation (IC) algorithm (Verma and Pearl, 1991), which provides the theoretical framework for learning the structure of Bayesian networks as a form of causal model, can be summarized in three steps: (i) learn the undirected graphical structure underlying the Bayesian network; arcs correspond to direct probabilistic dependencies between the incident nodes, (ii) set the direction of the arcs that are part of a v-structure (a triplet of nodes incident on a converging connection, i.e., B → A ← C), which is identified by the fact that the parent nodes are never independent given their child (i.e., B and C are dependent conditional on every subset S including A), and (iii) set the direction of the other arcs as needed to obtain a directed acyclic graph. The PC algorithm (Spirtes et al., 2000) is a straightforward implementation of the IC algorithm. It performs independence tests on all pairs of variables conditional on all possible subsets of the remaining variables to complete the first step. Conditioning sets are considered in order of increasing dimension, so that in many cases a small number of low-order tests is sufficient to establish whether two variables are independent. The computational complexity of the algorithm still increases rapidly with the number of variables, to the point that PC can only be applied to small or very sparse networks. The GS algorithm (Margaritis, 2003) improves on PC by learning the Markov Blanket of each variable (which includes the parents, the children and the variables which share a child with that particular variable) first. This preliminary selection limits both the number and the size of the subsets considered in the conditional tests, and results in a lower computational complexity without compromising the accuracy of the resulting network. The IAMB algorithm (Tsamardinos et al., 2003) is similar to GS, but uses the heuristic of the same name which consists of a forward selection instead of a simple sequential selection to learn the Markov Blankets. This approach leads to fewer false positives being included in the Markov Blankets, thus improving the quality of the network structure and further reducing the number of conditional tests. The algorithms used in the present study are implemented in open-source R (R Development Core Team, 2009) and publicly available (Chavan et al., 2009; Kalisch and Maechler, 2009; Scutari, 2009).
A Resampling Approach to Determine Statistically Significant FRs
In Friedman et al. (1999) and Friedman et al. (2000), statistically significant FRs in the learned acyclic graph was chosen as those whose confidence is greater than a pre-defined threshold (0 ≤ θ ≤ 1). Confidence was defined as the frequency of a given FR across independent acyclic networks learned from samples generated from the given empirical sample by bootstrapping with replacement (Friedman et al., 2000). Related studies (Friedman et al., 1999; Husmeier, 2003) have also emphasized the impact of the pre-defined threshold on the conclusions and the network structure. The choice of a user-defined threshold becomes especially challenging at small sample sizes. In the present study, we propose a straightforward approach that obviates the need for a pre-defined threshold in identifying statistically significant FRs in the acyclic network.
Algorithm
Given: Empirical sample Xm×n, representing the expression of m independent clones (replicates) of n genes, Bayesian structure learning technique (B).
Step 1: Generate 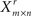 by row-wise resampling of Xm × n with replacement (i.e., each time sample a row from Xm × n with replacement). Learn the network from
by row-wise resampling of Xm × n with replacement (i.e., each time sample a row from Xm × n with replacement). Learn the network from 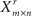 using BSL technique (B). Determine the corresponding partially directed acyclic graph (PDAG)Πr (Chickering, 1995).
using BSL technique (B). Determine the corresponding partially directed acyclic graph (PDAG)Πr (Chickering, 1995).
Step 2: Generate 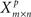 by randomly permuting the values in each column in Xm×n. Learn the network structure from Xp using the BSL technique (B). Determine the corresponding partially directed acyclic graph (PDAG)Πp (Chickering, 1995).
by randomly permuting the values in each column in Xm×n. Learn the network structure from Xp using the BSL technique (B). Determine the corresponding partially directed acyclic graph (PDAG)Πp (Chickering, 1995).
Step 3: Repeat Steps 1 and 2 ns times resulting in partially directed acyclic graphs 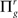 and
and 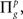 g = 1…ns.
g = 1…ns.
Step 4: Determine the frequency of the edges (i.e., confidence, Friedman et al., 2000) in the resampled graphs in Step 3 from Πr, 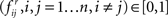 and in the randomly permuted counterparts Πp,
and in the randomly permuted counterparts Πp, 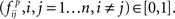 Here, the distribution
Here, the distribution 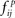 essentially represents the noise-floor.
essentially represents the noise-floor.
Step 5: An edge i → j with frequency 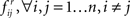 is deemed significant if
is deemed significant if 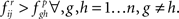 This has to be contrasted to the ad hoc threshold approach where i → j is deemed significant if
This has to be contrasted to the ad hoc threshold approach where i → j is deemed significant if 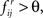 ∀i,j, i ≠ j for a statistically motivated choice of θ, 0 ≤ θ ≤ 1.
∀i,j, i ≠ j for a statistically motivated choice of θ, 0 ≤ θ ≤ 1.
The proposed algorithm is essentially a non-parametric bootstrap (Efron and Tibshirani, 1993) that estimates the joint empirical distribution of the edge frequencies from the given data and compares it to the null distribution of edge frequencies obtained from the randomly permuted counterpart. The correlation structure across the replicates is destroyed in the random permuted counterparts, hence the edge frequencies 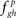 essentially represent the noise-floor (Step 4). It should be noted that the cumulative distribution function of
essentially represent the noise-floor (Step 4). It should be noted that the cumulative distribution function of 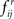 is stochastically larger than that of
is stochastically larger than that of 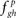 (Lehmann and Romano, 2005) since the latter corresponds to the absence of FRs under the null hypothesis. Therefore, large values of
(Lehmann and Romano, 2005) since the latter corresponds to the absence of FRs under the null hypothesis. Therefore, large values of 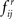 should result in rejection of the null hypothesis and existence of a significant FR, i → j. The use of random permutations does not require additional assumptions since the gene expression measurement across the replicate clones is generated independently. In such a setting, inference is also exact conditionally on the observed sample, which is always a sufficient statistic for the underlying joint distribution (Pesarin, 2001). Alternatively, this conditioning on the empirical distribution of the observed data makes the tests invariant to underlying statistical distribution of the data which may be partially or even completely unknown and thus obviates the need of asymptotic or approximate solutions. Therefore, the performance of the tests can be solely judged by their statistical properties (i.e., power, consistency, unbiasedness etc.) and the number of permutations. No assumption on the distribution of the population is required.
should result in rejection of the null hypothesis and existence of a significant FR, i → j. The use of random permutations does not require additional assumptions since the gene expression measurement across the replicate clones is generated independently. In such a setting, inference is also exact conditionally on the observed sample, which is always a sufficient statistic for the underlying joint distribution (Pesarin, 2001). Alternatively, this conditioning on the empirical distribution of the observed data makes the tests invariant to underlying statistical distribution of the data which may be partially or even completely unknown and thus obviates the need of asymptotic or approximate solutions. Therefore, the performance of the tests can be solely judged by their statistical properties (i.e., power, consistency, unbiasedness etc.) and the number of permutations. No assumption on the distribution of the population is required.
The proposed approach is also faster compared to incorporating permutation tests directly in the learning algorithm (Edwards, 2000; Legendre, 2000), since in general it’s not feasible to reuse the same permutations of the data across different data sets. Thus the latter approach requires on an average 0(n2).ns permutations while the former requires only ns permutations overall.
Results
Synthetic Data
Prior to the analysis of the experimental gene expression data we tested the proposed algorithm on data sampled from the ASIA network (Lauritzen and Spiegelhalter, 1988). The choice of ASIA network can be attributed to its number of nodes (8 nodes), which is comparable to that of the myogenic progenitor differentiation data (i.e., nine genes). The false-positive and false-negative rates (FPR, FNR) were used as performance indicators. The (FPR, FNR) obtained using the proposed algorithm were compared to those obtained using ad hoc threshold, Figure 1. This was accomplished as follows:
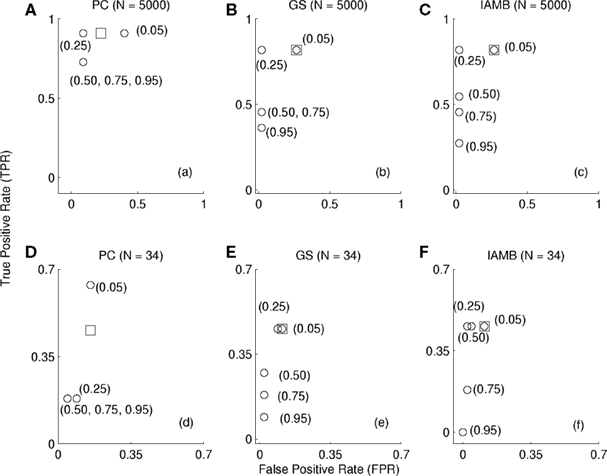
Figure 1. (FPR, TPR) obtained using ad hoc thresholds (circles) and those using the proposed algorithm (squares) across three BSL techniques (PC, GS, IAMB) and across two sample sizes [N = 5000, (A–C)] and [N = 34, (D–F)] for the ASIA network. The value of the ad hoc threshold (θ = 0.05, 0.25, 0.50, 0.75, 0.95) are shown adjacent to the circles for clarity. The circle and the square are stacked one behind the other in (B, C, E, F) around (θ = 0.05). Multiple values in the parentheses imply the circles are stacked one behind the other.
(i) Generate the PDAG (Chickering, 1995) of the true ASIA network (Σo).
(ii) Identify statistically significant edges (Σ1) from the given empirical sample using the proposed algorithm.
(iii) Identify statistically significant edges (Σ2) from the given empirical sample using a statistically motivated threshold θ, (0 ≤ θ ≤ 1).
(iv) Compute (FPR, TPR) from (Σo, Σ1). Compute (FPR, TPR) from (Σo, Σ2).
Repeat steps (ii–v) separately using three BSL techniques (PC, GS, IAMB) with correlation metric across samples sizes (N = 5000, N = 34) and (ns = 1000). In the present study we used ad hoc thresholds (θ = 0.05, 0.25, 0.50, 0.75, 0.95). The results obtained for (N = 5000) samples using (PC, GS, IAMB) with Pearson χ2 test for conditional independence and α = 0.05 are shown in Figures 1A–C respectively. Of interest is to note that the results obtained using the proposed algorithm are considerably better than the ones for several choices of threshold (θ = 0.50, 0.75, and 0.95). The performance of the threshold (θ = 0.05, 0.25) were comparable to that of the proposed algorithm with low FPR and high TPR. Thus it is possible a statistically motivated choice of threshold may perform well. However, given the infinite possibilities θ ∈ [0, 1], the chances of picking the correct threshold are slim. Subsequently, we repeated the analysis using a reduced sample size (N = 34) similar to that of the myogenic progenitor differentiation data (i.e., N = 34). It should be noted that this is a (∼147 = 5000/34) fold reduction in the sample size. The frequency (confidence, Friedman et al., 1999, 2000) is likely to reduce considerably. Therefore, the ad hoc threshold chosen for (N = 5000) may not be appropriate for (N = 34). We also compared the (FPR, TPR) obtained using the proposed algorithm and the ad hoc threshold, Figures 1D–F. As expected, the overall TPR for (N = 34), Figures 1D–F was found to be lower than that for (N = 5000). Interestingly, the FPR for (N = 34) was also lesser than that of (N = 5000). This may be due to the small number of nodes in the network. More importantly, the proposed algorithm seemed to have relatively higher TPR than the ad hoc threshold (θ = 0.50, 0.75, and 0.95), Figures 1D–F. There was no appreciable change in the FPR between the various threshold choices.
Osteoprogenitor Differentiation
A recent study (Madras et al., 2002), established the inherent probabilistic mechanism underlying osteoprogenitor differentiation by investigating the expression of eight genes including bone-related genes and cytokine receptors (COLL1, OCN, ALP, BSP, FGFR1, PTH1R, PTHrP, and PDGFRα) across 99 colonies. Subsequently, we had used BSL techniques in conjunction with ad hoc threshold to identify critical FRs from this data (Nagarajan et al., 2004). There are two reasons why we chose to re-investigate this data (i) The experimental design of the myogenic progenitor differentiation is similar to that of osteoprogenitor differentiation (ii) we eliminate the need for ad hoc threshold using the proposed algorithm minimizing ambiguity in the conclusion and may possibly identify biologically relevant and novel FRs. Since the sample size of the myogenic progenitor differentiation data is 34, we present the results on the osteoprogenitor differentiation at samples sizes 99 as well as 34.
Functional relationships deemed as statistically significant across (PC, GS, IAMB) with Pearson χ2 test for conditional independence and α = 0.05 for sample sizes (N = 99) and (N = 34) are shown in Figures 2A,B respectively. The reduced sample size (34) was generated by row-wise resampling without replacement from the given 99 colonies. Of interest is to note that three of the FRs (COLL-ALP; BSP-ALP; ALP-OCN) reported in the earlier study using an ad hoc threshold (Nagarajan et al., 2004) were deemed as significant using the proposed algorithm across (N = 99), Figure 2A, as well as (N = 34), Figure 2B. In addition, four new FRs (COLL1-FGFR1; FGFR1-BSP; FGFR1-PTH1R; PTH1R-PDGFRα) were also identified across (N = 99) and (N = 34), Figures 2A,B.
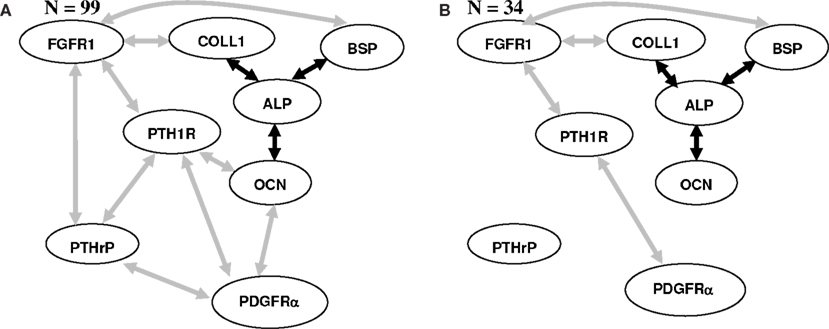
Figure 2. Statistically significant FRs identified using three BSL techniques (PC, GS, IAMB) from the clonal gene expression data generated during osteoprogenitor differentiation with (N = 99) colonies (A) and (N = 34) colonies (B). FRs identified in an earlier study (Nagarajan et al., 2004) using a statistically motivated threshold and new FRs identified using the proposed algorithm are shown in black and gray respectively.
Myogenic Progenitor Differentiation
The expression of nine genes (n = 9) across 34 clones (m = 34) was generated during myogenic progenitor differentiation. These nine genes, Figure 3, consisted of myogenic related genes (Myogenin, MyoD1, Myf-5), adipogenic related genes (CEBPα, PPARγ, DDIT3, FoxC2), and Wnt-related genes (Wnt5a, LRP5). In order to assess reproducibility of the results, normalization was carried out independently across three control genes (GAPDH, 18S, and B2M). The proposed algorithm in conjunction with three BSL techniques (PC, GS, and IAMB) and linear correlation to test for conditional independence (α = 0.05) were used to model the network structure. Only those FRs that were deemed as significant across all the BSL techniques (PC, GS and IAMB) and Pearson correlation metric are shown in Figure 3. The results obtained by normalization across the three control genes (GAPDH, 18S, and B2M) are shown separately in Figures 3A–C respectively. Of interest, is to note the considerable similarity in the results obtained across GAPDH and B2M. FRs that was persistent across the three BSL techniques and across the three control genes are shown in Figure 3D and consists of genes related to myogenic as well as adipogenic pathway and their interaction. The undirected nature of the network, Figure 3, can be attributed to the small sample size (34 clones) and equivalent classes (Chickering, 1995), an inherent feature of BSL techniques. Nevertheless, the results presented un-equivocally supports the co-expression of differentiation pathways and their cross-talk in AMPD. On a related note, equivalence classes may also imply possible existence of multiple networks that may be biologically significant.
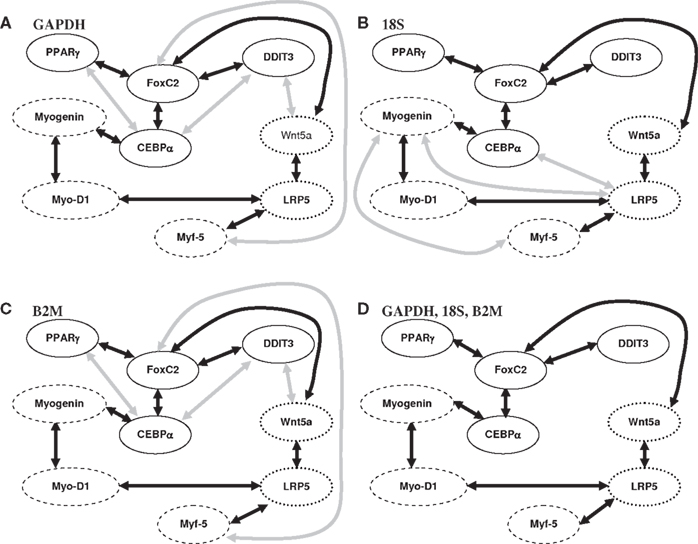
Figure 3. Statistically significant FRs identified as significant across each of the BSL techniques (PC, GS, and IAMB) from the myogenic progenitor clonal gene expression data. Myogenic related genes, adipogenic related and Wnt-related genes are shown by dashed, solid, and dotted circles in (A–D). Subplots (A–C) represent the results upon normalization with respect to the control genes (GAPDH, 18S, and B2M). Edges that were identified as significant by the three BSL techniques as well as normalization with respect to the three control genes are shown in (D). These edges are also represented by dark arrows in the subplots (A–C).
FRs corresponding to myogenic related genes
The FR MyoD1-Myogenin validates the approach as these two genes transactivate the expression of the other and promote muscle-specific gene expression (reviewed in Chargé and Rudnicki, 2003). The FR Myogenin-C/EBPα is unexpected given that overexpression of C/EBPα in muscle cell lines results in down-regulation of Myogenin (Hu et al., 1995). The aged muscle cell environment appears to alter the regulation of these transcription factors resulting in co-expression of some aspects of both myogenic and adipogenic differentiation. This FR warrants further study.
FRs corresponding to Wnt-related genes
The FR Lrp5-Wnt5a may be indicative of activation of the canonical Wnt/β-catenin pathway through binding of Wnt5a to Frizzled 4 (Fzd4) receptor and Lrp5 co-receptors (Mikels and Nusse, 2006). Coordinate regulation of Wnt5a/Fzd4/Lrp5 has been demonstrated during the first phase of hematopoiesis suggesting that these genes are involved in early hematopoietic differentiation (Corrigan et al., 2009). Canonical Wnt signaling has clearly been shown to activate both Myf-5 and MyoD expression during development (Tajbakhsh et al., 1998) and to promote myogenic differentiation during regeneration (Brack et al., 2007), which may justify the FRs MyoD1-LRP5 and Myf-5-LRP5. During aging, Wnt signaling also appears to regulate adipogenic gene expression in myogenic progenitors (Taylor-Jones et al., 2002; Vertino et al., 2005) and to mediate conversion of myogenic progenitors to a fibroblast phenotype (Brack et al., 2007; Hidestrand et al., 2008). We hypothesize that the distinct function of Wnt signaling in aged muscle may be mediated via Lrp5. Whereas Lrp5 function in muscle has been under-studied, it has been well-characterized in regulation of bone mass, where loss of Lrp5 results in low bone mineral density, presumably by decreasing the bone-forming activity of osteoblasts (reviewed in Krishnan et al., 2006).
FRs corresponding to adipogenic related genes
These included FoxC2-C/EBPα, FoxC2-DDIT3, and FoxC2-PPARγ, Figure 3D. Normalization with respect to the control genes GAPDH and B2M also revealed the widely documented FR across adipogenic transcription factors C/EBPα and PPARγ. FoxC2 is a member of the forkhead family of transcription factors that has been most extensively characterized in adipose tissue. It has been proposed that FoxC2 may control white versus brown adipose differentiation, promoting the latter through inhibition of PPARγ transcription (Davis et al., 2004). FoxC2 has also been shown to inhibit the expression of a subset of PPARγ downstream genes that may prevent the acquisition of the mature fat cell phenotype. Further, reduced expression of FoxC2 has been reported to be associated with reduced expression of brown adipogenic genes in humans (Yang et al., 2003) On the other hand, adipocyte-specific overexpression of FoxC2 in transgenic mice resulted in increased adipocyte expression of PPARγ (Kim et al., 2005). Thus, results in adipose tissue are equivocal and difficult to extrapolate to muscle tissue. FoxC2 is detectable in human muscle, but at low levels compared to adipose (Di Gregorio et al., 2005). The dependency of FoxC2-C/EBPα and DDIT3, a gene regulated by C/EBPα, in aged myogenic progenitors, suggests that regulation of adipocyte-specific gene expression may be altered at the molecular level as a function of age. FoxC2 may modulate the relative balance of adipogenic transcriptional activity such that, as proposed for adipocytes, some adipogenic genes are expressed in myogenic progenitors but differentiation does not progress to a fully mature adipocyte (Guan et al., 2002).
Discussion
The present study investigated possible co-expression of differentiation pathways in aged myogenic progenitor differentiation (AMPD) using clonal gene expression profiling and BSL techniques. FRs between critical myogenic, adipogenic, Wnt-related genes and their interaction were identified. While the FRs identified in the present study may not necessarily represent direct relationship, they clearly establish the orchestration of differentiation pathways in AMPD and their interaction. The study also demonstrates the feasibility of a focused small-throughput clonal gene expression profiling for obtaining preliminary insights into co-expression of distinct differentiation markers and their interaction. The proposed resampling approach obviates the need for ad hoc threshold, hence ideally suited from the perspective of pragmatic systems biology that can demand modeling FRs from small sample sizes. A more detailed study and validation is necessary in order to establish the cross-talk between the various pathways and their variation with age. This would include accommodating multiple testing corrections in the structure learning algorithm to control for family-wise error rate and false-discovery rate and comparing the network structure obtained on the aged myoblasts to those obtained on adult myoblasts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by funds from American Federation for Aging Research and R03-LM008853 from the National Library of Medicine to RN and R01-AG20941 to CAP. The authors would also like to thank Professor Fortunato Pesarin, University of Padova, for his comments and suggestions.
References
Allinen, M., Beroukhim, R., Cai, L., Brennan, C., Lahti-Domenici, J., Huang, H., Porter, D., Hu, M., Chin, L., Richardson, A., Schnitt, S., Sellers, W. R., and Polyak, K. (2004). Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6, 17–32.
Anakwe, K., Robson, L., Hadley, J., Buxton, P., Church, V., Allen, S., Hartmann, C., Harfe, B., Nohno, T., Brown, A. M., Evans, D. J., and Francis-West, P. (2003). Wnt signalling regulates myogenic differentiation in the developing avian wing. Development 130, 3503–3514.
Bahar, R., Hartmann, C. H., Rodriguez, K. A., Denny, A. D., Busuttil, R. A., Dollé, M. E., Calder, R. B., Chisholm, G. B., Pollock, B. H., Klein, C. A., and Vijg, J. (2006). Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 1011–1014.
Beggs, M. L. Nagarajan, R., Taylor-Jones, J. M., Nolen, G., Macnicol, M., and Peterson, C. A. (2004). Altered TGFβ signaling pathway in aging myoblasts. Aging Cell 3, 353–361.
Brack, A. S. Conboy, M. J., Roy, S., Lee, M., Kuo, C. J., Keller, C., and Rando, T. A. (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810.
Brunelli, S., Relaix, F., Baesso, S., Buckingham, M., and Cossu, G. (2007). Beta catenin-independent activation of MyoD in presomitic mesoderm requires PKC and depends on Pax3 transcriptional activity. Dev. Biol. 304, 604–614.
Chargé, S., and Rudnicki, M. A. (2003). Fusion with the fused: a new role for interleukin-4 in the building of muscle. Cell 113, 422–423.
Chavan, S. S., Bauer, M. A., Scutari, M., Nagarajan, R. (2009). NATbox: a network analysis toolbox in R. BMC Bioinformatics. 10(Suppl 11), S14.
Chickering, D. M. (1995). “A transformational characterization of equivalent Bayesian network structures,” in Proceedings of 11th Conference on Uncertainty in Artificial Intelligence. Morgan Kauffmann, 87–98.
Corrigan, P. M., Dobbin, E., Freeburn, R. W., and Wheadon, H. (2009). Patterns of Wnt/Fzd/LRP gene expression during embryonic hematopoiesis. Stem Cells Dev. 18, 759–772.
Davis, K. E. Moldes, M., and Farmer, S. R. (2004). The forkhead transcription factor FoxC2 inhibits white adipocyte differentiation. J. Biol. Chem. 279, 42453–42461.
Di Gregorio, G. B., Yao-Borengasser, A., Rasouli, N., Varma, V., Lu, T., Miles, L. M., Ranganathan, G., Peterson, C. A., McGehee, R. E., and Kern, P. A. (2005). Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes 54, 2305–2313.
Efron, B., and Tibshirani, R. (1993). An Introduction to the Bootstrap. Boca Raton FL: Chapman and Hall.
Friedman, N. Goldszmidt, M., and Wyner, A. (1999). “Data analysis with Bayesian networks: a bootstrap approach,” in Proceedings of the 15th Conference on Uncertainty in Artificial Intelligence. UAI, 196–205.
Friedman, N., Goldszmidt, M., and Wyner, A. (2000). Using Bayesian networks to analyze expression data. J. Comput. Biol. 7, 601–620.
Gardner, T. S., di Bernardo, D., Lorenz, D., and Collins, J. J. (2003). Inferring genetic networks and identifying compound of action via expression profiling. Science 301, 102–1005.
Glymour, C., and Cooper, G. F. (1999). Computation, Causation and Discovery. Menlo Park, CA: AAAI Press.
Guan, Y., Taylor-Jones, J. M., Peterson, C. A., and McGehee, R. E. Jr. (2002). p130/p107 expression distinguishes adipogenic potential in primary myoblasts based on age. Biochem. Biophys. Res. Commun. 296, 1340–1345.
Hidestrand, M., Richards-Malcolm, S., Gurley, C. M., Nolen, G., Grimes, B., Waterstrat, A., Zant, G. V., and Peterson, C. A. (2008). Sca-1-expressing nonmyogenic cells contribute to fibrosis in aged skeletal muscle. J. Gerontol. A 63, 566–579.
Hu, E., Tontonoz, P., and Spiegelman, B. M. (1995). Transdifferentiation of myoblasts by the adipogenic transcription factors PPARγ and C/EBPα. Proc. Natl. Acad. Sci. U.S.A. 92, 9856–9860.
Husmeier, D. (2003). Sensitivity and specificity of inferring genetic regulatory interactions from microarray experiments with dynamic Bayesian networks. Bioinformatics 19, 2271–2282.
Kaern, M., Elston, T. C., Blake, W. J., and Collins, J. J. (2005). Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6, 451–464.
Kalisch, M., and Maechler, M. (2009). pcalg: Estimating the skeleton and equivalence class of a DAG. R Package Version 0.1-8 (CRAN).
Kim, J. K., Kim, H. J., Park, S. Y., Cederberg, A., Westergren, R., Nilsson, D., Higashimori, T., Cho, Y. R., Liu, Z. X., Dong, J., Cline, G. W., Enerback, S., and Shulman, G. I. (2005). Adipocyte-specific overexpression of FOXC2 prevents diet-induced increases in intramuscular fatty acyl CoA and insulin resistance. Diabetes 54, 1657–1663.
Krishnan, V., Bryant, H. U., and Macdougald, O. A. (2006). Regulation of bone mass by Wnt signaling. J. Clin. Invest. 116, 1202–1209.
Lauritzen, S. L., and Spiegelhalter, D. J. (1988). Local computations with probabilities on graphical structures and their application to expert systems. J. R. Stat. Soc. B 5, 157–224.
Legendre, P. (2000). Comparison of permutation methods for the partial correlation and partial Mantel tests. J. Stat. Comput. Simul. 67, 37–73.
Lehmann, E. L., and Romano, J. P. (2005). Testing Statistical Hypotheses, 3rd Edn. New York: Springer.
Loeffler, M., and Roeder, I. (2002). Tissue stem cells: definition, plasticity, heterogeneity, self-organization and models – a conceptual approach. Cells Tissues Organs (Print) 1717, 8–26.
Madras, N. Gibbs, A. L., Zhou, Y., and Zandstra, P. W. (2002). Modeling stem cell development by retrospective analysis of gene expression profiles in single progenitor-derived colonies. Stem Cells 20, 230–240.
Margaritis, D. (2003). Learning Bayesian Network Model Structure from Data. Ph.D. thesis, School of Computer Science, Carnegie-Mellon University, Pittsburg. Technical Report CMU-CS-03-153.
McGann, C. J., Odelberg, S. J., and Keating, M. T. (2001). Mammalian myotube dedifferentiation induced by newt regeneration extract. Proc. Natl. Acad. Sci. U.S.A. 98, 13699–13704.
Mikels, A. J., and Nusse, R. (2006). Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 4, e115. doi: 10.1371/journal.pbio.0040115.
Nagarajan, R., Aubin, J. E., and Peterson, C. A. (2004). Modeling genetic networks from clonal analysis. J. Theor. Biol. 230, 359–373.
Pearl, J. (1988). Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. San Francisco, CA: Morgan Kauffmann.
Pesarin, F. (2001). Multivariate Permutation Tests with Applications in Biostatistics. Chichester: Wiley.
Polesskaya, A., Seale, P., and Rudnicki, M. A. (2003). Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell 113, 841–852.
R Development Core Team. (2009). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rosen, E. D., and Walkey, C. J. (2000). Transcriptional regulation of adipogenesis. Genes Dev. 14, 1293–1307.
Ross, S. E. Hemati, N., Longo, K. A., Bennett, C. N., Lucas, P. C., Erickson, R. L., and MacDougald, O. A. (2000). Inhibition of adipogenesis by Wnt signaling. Science 289, 950–953.
Sachs, K., Perez, O., Pe'er, D., Lauffenburger, D. A., and Nolan, G. P. (2005). Causal protein-signaling networks derived from multiparameter single-cell data. Science 308, 523–529.
Scutari, M. (2009). bnlearn: Bayesian network structure learning. R package version 1.5 (CRAN). Available at http://www.bnlearn.com/
Somel, M., Khaitovich, P., Bahn, S., Pääbo, S., and Lachmann, M. (2006). Gene expression becomes heterogeneous with age. Curr. Biol. 16, R359–R360.
Spirtes, P., Glymour, C., and Scheines, R. (2000). Causation, Prediction, and Search, 2nd Edn. Cambridge, MA MIT Press.
Tajbakhsh, S., Borello, U., Vivarelli, E., Kelly, R., Papkoff, J., Duprez, D., Buckingham, M., and Cossu, G. (1998). Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development 125, 4155–4162.
Taylor-Jones, J. M., McGehee, R. E., Rando, T. A., Lecka-Czernik, B., Lipschitz, D. A., and Peterson, C. A. (2002). Activation of an adipogenic program in adult myoblasts with age. Mech. Ageing Dev. 123, 649–661.
Tsamardinos, I., Aliferis, C., Statnikov, A., and Statnikov, Er., (2003). “Algorithms for large scale Markov blanket discovery,” in Proceedings of the Sixteenth International Florida Artificial Intelligence Research Society Conference. AAAI Press, 376–381.
Verma, T. S., and Pearl, J. (1991). Equivalence and synthesis of causal models. Uncertain. Artif. Intell. 6, 255–268.
Vertino, A. M., Taylor-Jones, J. M., Longo, K. A., Bearden, E. D., Lane, T. F., McGehee, R. E. Jr, MacDougald, O. A., and Peterson, C. A. (2005). Wnt10b deficiency promotes coexpression of myogenic and adipogenic programs in myoblasts. Mol. Biol. Cell 16, 2039–2048.
Keywords: functional relationships, aged myogenic progenitor differentiation, Bayesian structure learning
Citation: Nagarajan R, Datta S, Scutari M, Beggs ML, Nolen GT and Peterson CA (2010) Functional relationships between genes associated with differentiation potential of aged myogenic progenitors. Front. Physiology 1:21. doi: 10.3389/fphys.2010.00021
Received: 23 March 2010;
Paper pending published: 03 May 2010;
Accepted: 23 June 2010;
Published online: 09 September 2010.
Edited by:
Michael L. Johnson, University of Virginia, USAReviewed by:
Aaron J. Mackey, The Ohio State University, USAGuanghui Hu, Hong Kong Baptist University, Hong Kong
Copyright: © 2010 Nagarajan, Datta, Scutari, Beggs, Nolen and Peterson. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Radhakrishnan Nagarajan, Division of Biomedical Informatics, University of Arkansas for Medical Sciences, 4301 W. Markham, Slot 781, Little Rock, AR 72205, USA. e-mail: rnagarajan@uams.edu