 Dominga Lapi1* Lina Sabatino2 Giovanna Giuseppina Altobelli3 Paolo Mondola1 Vincenzo Cimini3 Antonio Colantuoni1*
Dominga Lapi1* Lina Sabatino2 Giovanna Giuseppina Altobelli3 Paolo Mondola1 Vincenzo Cimini3 Antonio Colantuoni1*
- 1 Department of Neuroscience, “Federico II” University Medical School of Naples, Naples, Italy
- 2 Department of Biochemistry, Sannio University, Benevento, Italy
- 3 Department of Biomorphological and Functional Sciences, “Federico II” University Medical School of Naples, Naples, Italy
Background and purpose Propionyl-L-carnitine (pLc) exerts protective effects in different experimental models of ischemia–reperfusion (I/R). The aim of the present study was to assess the effects of intravenously and topically applied pLc on microvascular permeability increase induced by I/R in the hamster cheek pouch preparation. Methods The hamster cheek pouch microcirculation was visualized by fluorescence microscopy. Microvascular permeability, leukocyte adhesion to venular walls, perfused capillary length, and capillary red blood cell velocity (VRBC) were evaluated by computer-assisted methods. E-selectin expression was assessed by in vitro analysis. Lipid peroxidation and reactive oxygen species (ROS) formation were determined by thiobarbituric acid-reactive substances (TBARS) and 2′-7′-dichlorofluorescein (DCF), respectively. Results In control animals, I/R caused a significant increase in permeability and in the leukocyte adhesion in venules. Capillary perfusion and VRBC decreased. TBARS levels and DCF fluorescence significantly increased compared with baseline. Intravenously infused pLc dose-dependently prevented leakage and leukocyte adhesion, preserved capillary perfusion, and induced vasodilation at the end of reperfusion, while ROS concentration decreased. Inhibition of nitric oxide synthase prior to pLc caused vasoconstriction and partially blunted the pLc-induced protective effects; inhibition of the endothelium-derived hyperpolarizing factor (EDHF) abolished pLc effects. Topical application of pLc on cheek pouch membrane produced the same effects as observed with intravenous administration. pLc decreased the E-selectin expression. Conclusions pLc prevents microvascular changes induced by I/R injury. The reduction of permeability increase could be mainly due to EDHF release induce vasodilatation together with NO. The reduction of E-selectin expression prevents leukocyte adhesion and permeability increase.
Introduction
L-Carnitine has been proven to induce beneficial effects on ischemic tissues, including myocardium and skeletal muscle. One of the most utilized derivates of L-carnitine is propionyl-L-carnitine (pLc), a naturally short-chain fatty ester of carnitine (3-hydroxy-4-N-trimethyl-aminobutyric acid). pLc increases the intracellular pool of L-carnitine that has a crucial function in the transport of long-chain fatty acids into mitochondria for oxidation, particularly in the heart and skeletal muscles. It is able to chaperon activated fatty acids into mitochondrial matrix for metabolism and prevents accumulation of intermediate compounds in the mitochondrial matrix to facilitate energy production (Wiseman and Brogden, 1988).
Several previous studies indicate that administration of pLc ameliorates damage due to ischemia and subsequent reperfusion in different organs (Shug et al., 1991; Mister et al., 2002).
Two studies carried out on rabbit and rat isolated hearts, subjected to ischemia and reperfusion (I/R), demonstrated that pLc improves the functional recovery of the myocardium. Such protective effects are proposed mediated by a reduction in oxidative stress due to oxygen-derived free radicals (ROS) (Ferrari et al., 1989; Packer et al., 1991). Recent studies demonstrated that the pLc administration to human patients suffering from chronic ischemic heart disease improves the left ventricular function, decreases systemic and pulmonary resistance, and enhances cardiac efficiency (Chiddo et al., 1991). Tarantini et al. (2006) showed that pLc reduces early mortality of patients with anterior acute myocardial infarction.
pLc exhibits hemodynamic effects, improving peripheral vascular blood flow in patients with peripheral artery obstructive disease (PAOD), reducing the plasma levels of adhesion molecules (E, L, and P-selectin, ICAM-1 and VCAM-1) that are known to increase damage in ischemic tissues (Signorelli et al., 2001; Silvestro et al., 2006; Loffredo et al., 2007).
Several studies have been carried out to clarify the mechanism by which pLc prevents the I/R injuries. It has been reported that pLc increases NO availability in isolated small mesenteric arteries from spontaneously hypertensive rats (Alvarez de Sotomayor et al., 2007). This effect has been confirmed in experiments carried out in rat livers subjected to warm I/R showing an increase in hepatic blood flow, measured by laser Doppler technique, after pLc treatment (Yonezawa et al., 2005).
Although all these clinical and experimental data demonstrate that pLc exerts beneficial effects in different pathologies involving several organ systems, to date there is no direct evidence to determine the effects of pLc on the microcirculation during I/R damage. In particular, there is no consensus concerning the mechanisms through which pLc prevents the microvascular permeability increase and improves the supply of blood flow to tissues.
Therefore, the present study was carried out to assess the main effects of intravenously and topically applied pLc on microvascular alterations induced by I/R. The hamster cheek pouch was chosen as the experimental model that permits in vivo observation of the microvascular changes during ischemia and subsequent reperfusion.
The objective of this paper was to evaluate the microvascular permeability increase and the role of leukocytes into I/R-induced damage. These events are strictly correlated because leukocyte activation and successive adhesion in post-capillary and collecting venules represents the background for the consequent vascular endothelial damage. In particular, we tried to assess the effects of inhibiting nitric oxide release or endothelium-derived hyperpolarizing factor (EDHF) effect on microvascular permeability. Therefore, we tested the effects of NG-nitro-L-arginine-methyl ester (L-NAME), inhibitor of nitric oxide synthase (NOS), and Apamin plus Charybdotoxin (inhibitors of both BKCa and voltage-sensitive potassium channels) on microvascular changes resulting from I/R injury.
Finally, to clarify the role of leukocyte activation on permeability increase (Carlos and Harlan, 1994; Billups et al., 1995; Ley et al., 1998) during pLc treatment, we determined the expression of E-selectin, known to mediate leukocyte rolling and to participate in the conversion of rolling to firmly adhering leukocytes.
Materials and Methods
Experimental Groups
Studies were performed on male Syrian hamsters (80–100 g) (Charles River, Calco, Italy) that were randomly assigned to seven groups: (1) Sham-operated group (Group S, n = 36): hamsters subjected to the same procedures as the experimental groups except for clamping of the cheek pouch. (2) Control hamsters (Group C, n = 40) were treated with topically applied vehicle and subjected to 30 min of ischemia and 30 min of reperfusion. (3) The third group of animals (sub-group P1a and sub-group P1b, n = 40 for each sub-group) received intravenously 2 mg/100 g body wt or 5 mg/100 g body wt of pLc in 2 min, starting 10 min before ischemia and at the beginning of reperfusion. (4) The fourth group of hamsters (sub-group P2a and sub-group P2b, n = 40 hamsters for each group) was treated with topically applied 1.5 or 2.5 mM pLc in 2 min, starting 10 min before ischemia and for 2 min at the beginning of reperfusion. (5) The fifth group of animals (group P3, n = 40 hamsters) was treated with topically applied 1 mM L-NAME in 2 min prior to the topical administration of 2.5 mM pLc in 2 min, starting 20 min before ischemia and at the beginning of reperfusion, with a time interval of 8 min between each drug administration. (6) The sixth group of hamsters (P4, n = 40) was treated with topically applied Apamin (0.3 μM) plus Charybdotoxin (0.1 μM) in 2 min, 10 min prior to pLc (2.5 mM), starting 20 min before ischemia and at the beginning of reperfusion, with a time interval of 10 min after each drug administration, as previously described. (7) The seventh group of animals (P5, n = 40) was treated with topical L-NAME (1 mM) plus Apamin (0.3 μM) and Charybdotoxin (0.1 μM) prior to pLc (2.5 mM), starting 30 min before ischemia and at the beginning of reperfusion, with a time interval of 8 min after each drug administration before ischemia, while this time interval was 2 min at the beginning of reperfusion.
For each experimental group nine animals (five for group S) were used for microvascular studies, 15 for the assessment of E-selectin expression, 10 animals for quantitative determination of oxidative stress by evaluation of thiobarbituric acid-reactive substances (TBARS) and six hamsters were used for measurement of 2′-7′-dichlorofluorescein (DCF) fluorescence.
Cheek Pouch Preparation
Animals were anesthetized (pentobarbital sodium 5 mg/100 g body weight i.p.) and tracheostomized. The right carotid artery and femoral vein were cannulated to measure blood pressure and administer additional anesthesia and fluorescent tracers. The cheek pouch was surgically prepared as previously reported (Bertuglia and Colantuoni, 2000). In brief, the membrane was gently everted and fixed to a special stage adapted to the microscope; a thin black blade was inserted through a small incision between the upper and lower layers of the pouch, thus preserving the epithelial cells. The membrane was suffused with Ringer solution (in mM: 137 NaCl, 4.7 KCl, 1.2 MgSO4, 2.0 CaCl2, 18.0 NaHCO3) at 36.0 ± 0.5°C with 5% CO2–95% N2 adjusted to pH 7.35.
Atraumatic microvascular clips were placed for 30 min on the proximal part of the cheek pouch to achieve complete ischemia. After removing clamp, the microcirculation was observed for 30 min (reperfusion).
All experiments conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85.23, revised 1996) and to institutional rules for the care and handling of experimental animals.
Fluorescence Microscopy
Observations of microcirculation were made with a fluorescence microscope (Leitz Orthoplan) with objectives 4×, numerical aperture (NA) 0.14; 20×, NA 0.25; 32×, NA 0.40; 40×, NA 0.30, a 10× eyepiece and a filter block (Ploemopak, Leitz). Epi-illumination was provided by a 100-W mercury lamp using the appropriate filters for fluorescein isothiocyanate (FITC) bound to dextran (molecular weight 150 kDa, 50 mg/100 g body weight i.v. as 5% wt/vol solution in 5 min) (FD 150) (Sigma Chemical), for rhodamine 6G and a heat filter (Leitz KG1). The animals received an intravenous injection of 1 mg rhodamine 6G/100 g body weight in 0.3 ml (Sigma Chemical) and a supplemental injection (final volume 0.3 ml × 100 per g per × h) throughout the experiment to label leukocytes for adhesion evaluation.
The area of interest was televised with a DAGE MTI 300 low-light level digital camera and observed from a Sony PVM 122 CE monitor. Video images were videotaped and microvascular measurements were made off-line using a computer-assisted imaging software system (MIP Image, CNR, Institute of Clinical Physiology, Pisa, Italy).
Microvascular observations were carried out for 1 min every 5 min during application of drugs; then, every 10 min during I/R.
Microvascular Parameters Evaluation
Macromolecular leakage was manifested by extravasation of FITC–dextran, which appeared as fluorescent spots outside microvessels. Therefore, the increase in permeability was calculated and reported as normalized gray levels (NGL): NGL = (I − Ir)/Ir, where Ir is the average baseline gray level at the end of vessel filling with fluorescence (average of five windows located outside the blood vessels with the same windows being used throughout the experimental procedure), and I is the same parameter after ischemia or reperfusion. Gray levels ranging from 0 to 255 were determined by the MIP Image program in five regions of interest (ROI) measuring 50 × 50 μm.
The endothelial dysfunction causes a reduction of tissue perfusion that involves mainly the capillary network. The perfused capillary length was measured by MIP Image in an area of 550 × 550 μm. In this experimental system, the length of perfused capillaries is easily established by the automated process because it is outlined by dextran.
Adherent leukocytes highlighted with rhodamine 6G (i.e., cells on vessel walls that did not move over a 30-s observation period) were quantified in terms of numbers/100 μm venular length/30 s.
Vessel diameters and capillary red blood cell velocity (VRBC) were measured with a computer-assisted method (frame by frame). The results of diameter measurements were in accord with those obtained by shearing method (±0.5 μm). To avoid bias due to single operator measurements, two independent “blinded” operators determined all the measured parameters. Their measurements overlapped in all cases.
Under baseline conditions, the cheek pouch arterioles were classified according to centrifugal ordering scheme from the largest (A1) to the smallest (A4) arterioles. We investigated the responses of each arteriolar order to the experimental conditions: at the end of ischemia (EI) and at the end of reperfusion (ER) compared with the baseline conditions. In each hamster, two A2, two A3, and two A4 arterioles were studied during each experiment; because of homogenous responses in all arteriolar orders, we chose to present only the data regarding A2 larger vessels.
Mean arterial blood pressure (MABP) (Viggo-Spectramed P10E2 transducer – Oxnard, CA, USA – connected to a catheter in the carotid artery) and heart rate were monitored with a Gould Windograf recorder (model 13-6615-10S, Gould, OH, USA). Data were recorded and stored in a computer.
Immunofluorescence
For immunocytochemistry, hamsters (n = 5 for each experimental group) were anaesthetized with pentobarbital (5 mg/100 g body weight), perfused with 0.5% sodium nitrite and then with 10% buffered formalin. Cheek pouches were carefully removed and kept for further 2–3 h in the same fixative. Specimens were then left under refrigeration at 4°C in 30% sucrose in phosphate buffered saline (PBS) for 48 h and embedded in OCT medium before freezing and cryostat sectioning.
Cryostat sections (10-μm-thick) were processed for the indirect immunofluorescence method (Coons, 1971). Goat anti-rat E-selectin antibody (R&D Systems, Inc.; dilution 15 μg/ml) was used to localize the antigen. As a negative control, the primary antiserum was substituted by 1/20 normal goat serum or PBS. FITC (Jackson ImmunoResearch) was used to reveal the antigens as needed.
The sections were then mounted with phenylenediamine/glycerine/PBS medium and viewed with a Leitz Diaplan fluorescence microscope.
RNA Extraction and RT-PCR Assay
Five hamsters were used for each experimental group. RNA was extracted from tissues by Trizol™ (Invitrogen). Random primed double-strand cDNA was synthesized using Superscript III (Invitrogen). The specificity of each oligonucleotide pair used was verified with the BLAST program.
The amplification conditions for each primer pair were experimentally determined. From 20 to 40 amplification cycles were run to avoid saturating conditions. E-selectin was analyzed by amplifying 1 μl of the resulting cDNA with the following oligonucleotides based on the murine sequence: forward, 5′-CCTCGACAGAGGAAGCTCAGAACT-3′; reverse, 5′-TCCACTCTCCAGAGGACGTACACCG-3′ (Yao et al., 1991; Milstone et al., 2000).
The PCR conditions used were the following: 35 cycles at 94°C for 1 min, 68°C for 1 min, and 72°C for 1 min. The amplification product of 374 bp was run on 2.0% agarose gel in TAE buffer and visualized by ethidium bromide staining. The intensity of the amplified bands was quantified by densitometry and referred to that obtained with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). These last specific primers were: forward: 5′-GACCCCTTCATTGACCTCAACTACATG-3′, and reverse: 5′-GTCCACCACCCTGTTGCTGTAGCC-3′. The PCR condition for GAPDH were as follows: 25 cycles at 94°C for 1 min, 60°C for 1 min, and 72°C for 1 min.
Western Blot and Antibodies
Protein extracts were obtained from tissues (n = 5 hamster per group) after homogenization at 4°C in RIPA buffer (Radioimmuno precipitation assay) containing protease inhibitors (Roche Applied Science), using an Ultra-Turrax (IKA; Labortechnik) (Yao et al., 1991). Protein concentration was determined by Bradford assay (Rio-Rad). Protein extracts (50–70 μg) were run on a 7% SDS–PAGE and transferred onto polyvinylidene difluoride membranes. Immunodetection was performed with the primary antibodies (dilution: 1:500) followed by incubation with peroxidase-conjugated secondary antibody, using the enhanced chemiluminescence detection reagent (Amersham Biosciences).
Protein loading was normalized by incubating the same filters with anti-β-actin antibody.
The band intensity was quantified by densitometry (Bio-Rad). The antibodies used in Western blot analysis were: m E-selectin (Santa Cruz Biotechnology) and β-actin A1978 (Sigma).
TBARS Measurements
The tissue content of thiobarbituric acid reactive substances, as an index of lipid peroxidation, was determined according to the method previously reported (Granger, 1999). Briefly, 0.25 g of cheek pouch membrane were homogenized in 5% trichloroacetic acid, in presence of butylated hydroxytoluene (0.01%) and 1 mM EDTA to avoid lipid peroxidation during assay. The precipitate was pelleted by centrifugation (3000 rpm for 15 min) and an aliquot of the supernatant was reacted with an equal volume of 0.67% (w/v) thiobarbituric acid. Subsequently, the samples were heated at 80°C for 30 min and after cooling the absorption at 532 nm was measured. TBARS were expressed as malondialdehyde (MDA) amount using freshly diluted MDA bisdimethylacetal as standard. TBARS in cheek pouch membrane was evaluated at different times: at the end of ischemia and at 20 min of reperfusion.
2′-7′ Dichlorofluorescein (DCF) Fluorescence Measurements
Ringer solution containing 12 μM 2′-7′-dichlorofluorescein-diacetate (DCFH-DA) was superfused for 20 min on the cheek pouch prior to ischemia. The lipophilic DCFH-DA diffuses across the cell membranes and is retained into the intracellular space, converted to membrane impermeable non-fluorescent DCFH. This substance is oxidized by intracellular hydrogen peroxide (H2O2) to yield DCF, a high fluorescent molecule. This reaction is reversible and the intensity of DCF fluorescence is proportional to the intracellular ROS levels. The fluorescence intensity was determined by using an appropriate filter (522 nm); then NGL were evaluated, comparing the fluorescence at the end of ischemia (n = 3) or reperfusion (n = 3) with the baseline represented by fluorescence of the cheek pouch just superfused with DCFH-DA.
Statistical Analysis
All reported values are means ± SEM. The data were analyzed for their statistical significance. The Gaussian distribution of data was examined by Kolmogorov–Smirnov (KS) test. Paired and unpaired Student’s t-test, ANOVA, and Bonferroni’s post hoc test were used in the case of normal distribution; when the data were not normally distributed Mann–Whitney U test and Kruskal–Wallis test were utilized for non-parametric testing of null hypothesis. The changes in diameter and DCF fluorescence were analyzed by non-parametric tests, because KS test indicated non-Gaussian distribution of these data. Diameter data were presented as median (minimum and maximum diameter). Statistical significance was set at p < 0.05.
Results
In all experimental groups the cheek pouch arterioles were classified according to centrifugal scheme, starting from the largest arterioles A1 (median diameter: 30.0 μm; minimum and maximum 26.0–34.0 μm, n = 77), to the smaller A2 (22.5 μm; 19.3–25.7 μm, n = 154), and A3 arterioles (15.0 μm; 10.5–19.5 μm, n = 154) to the smallest ones A4 (7.9 μm; 6.8–9.4 μm, n = 154) in the preparation.
Under baseline conditions in the cheek pouch microvascular networks of sham-operated animals there was no leakage of fluorescence dextran nor leukocyte adhesion, while all capillaries were perfused and capillary VRBC was 0.200 ± 0.005 mm/s (Table 1).
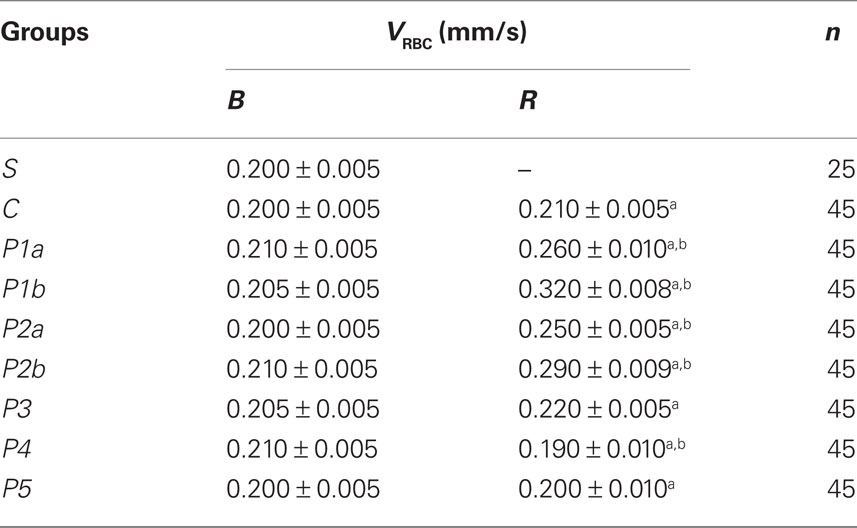
Table 1. Capillary red blood cell velocity (VRBC) under baseline conditions (B) and after reperfusion (R) in all experimental groups.
I/R in Control Hamsters
In control hamsters 30 min of ischemia caused an increase in FD 150 permeation (0.31 ± 0.03 NGL, p < 0.01 vs. baseline) (Figures 1 and 2). A2 arterioles (median diameter: 23.0 μm, minimum and maximum: 20.6–25.7 μm, n = 18) showed a significant decrease in diameter (Figure 3). At ER, there was a marked leakage of FD 150 (Figure 2) and leukocyte adhesion was pronounced in post capillary and connecting venules (11.0 ± 1.2/100 μm of venular length/30 s, p < 0.01 vs. baseline: 1 ± 0.5/100 μm of venular length/30 s) (Figure 4). The perfused capillary length was reduced by 63.0 ± 1.1% of baseline (p < 0.01 vs. baseline) (Table 2) and capillary VRBC was 0.210 ± 0.005 mm/s (p < 0.01 vs. baseline) (Table 1). A2 arteriolar diameter significantly decreased compared with the baseline (Figure 3).
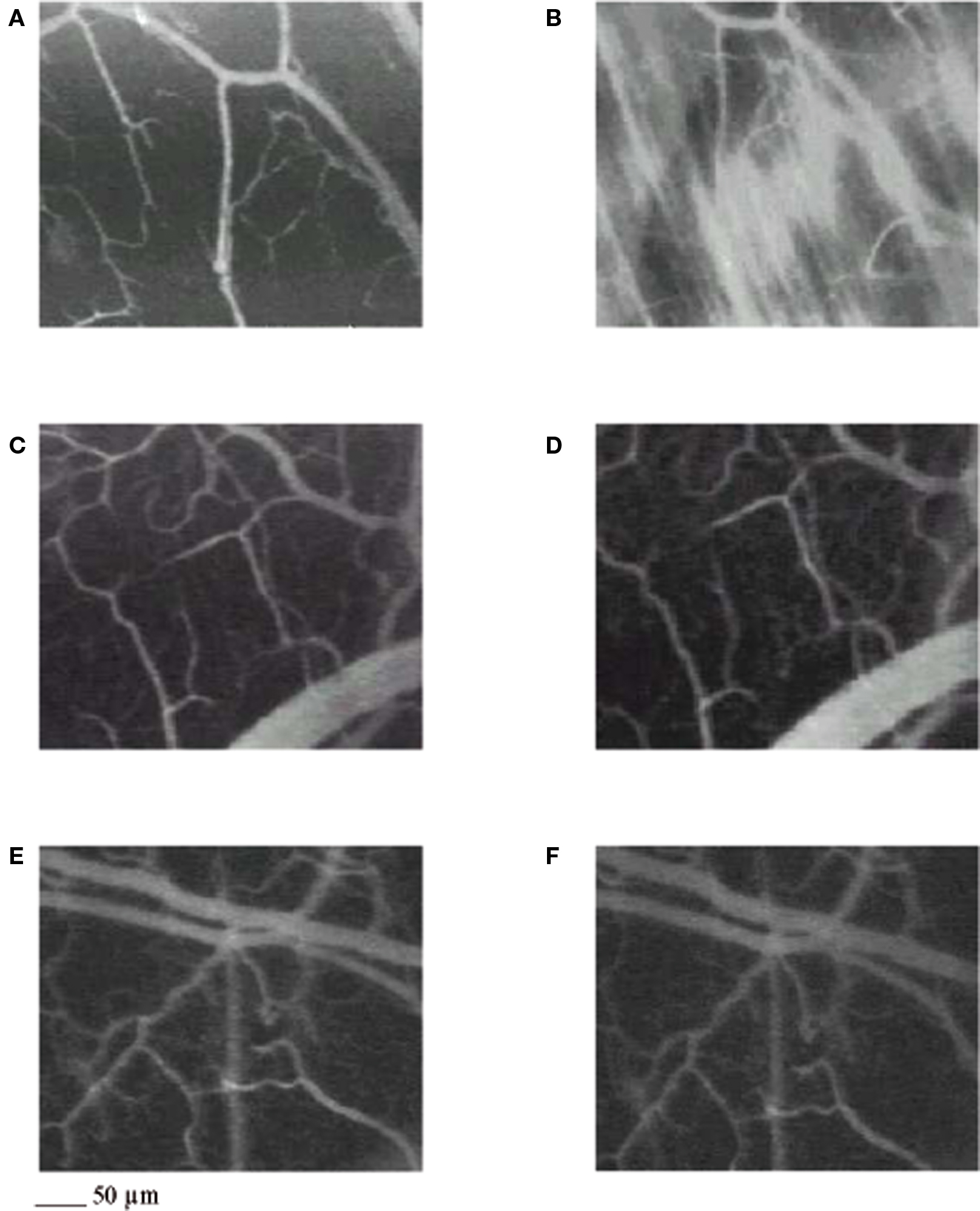
Figure 1. Computer-assisted images of a microvascular networks in control hamster under baseline conditions (A) and at the end of reperfusion (B); in hamster treated with pLc, 5 mg/100 g body wt intravenously injected (C,D), and in hamster treated with pLc 2. 5 mM/l topically applied (E,F), at the same time points as in control. The increase in permeability is apparent by the marked changes in the color of interstitium (from black to white). pLc prevented the increase in permeability because the interstitium did not significantly change in color (black). Scale bar = ___ 50 μm.
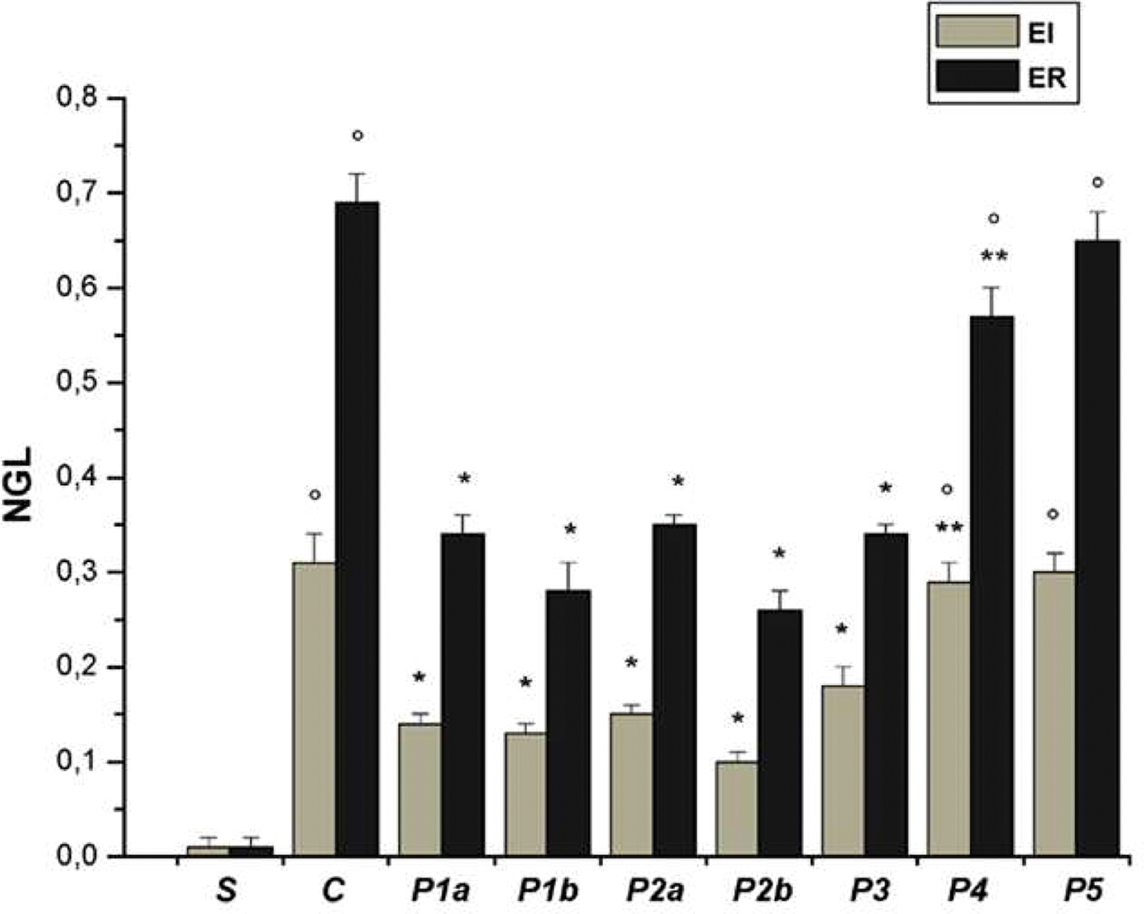
Figure 2. Permeability increase after ischemia (I) (left) and reperfusion (R) (right) in the experimental groups: S, sham-operated group; C, control group; P1a, hamsters treated with propionyl-L-carnitine (pLc) intravenously infused, lower concentration; P1b, hamsters treated with pLc intravenously infused, higher concentration; P2a, animals treated with pLc topically applied, lower concentration and P2b, animals treated with pLc topically applied, higher concentration; P3a, animals treated with topical application of L-NAME prior to pLc higher concentration; P4, hamsters treated with topical application of Apamin and Charybdotoxin prior to pLc higher concentration; P5, hamsters treated with topical application of L-NAME plus Apamin and Charybdotoxin prior to pLc higher concentration. In S group, data were evaluated at 45 and 75 min of observation. NGL, normalized gray levels. °p < 0.01 vs. sham-operated group, *p < 0.01 vs. C group, **p < 0.01 vs. P2b group.
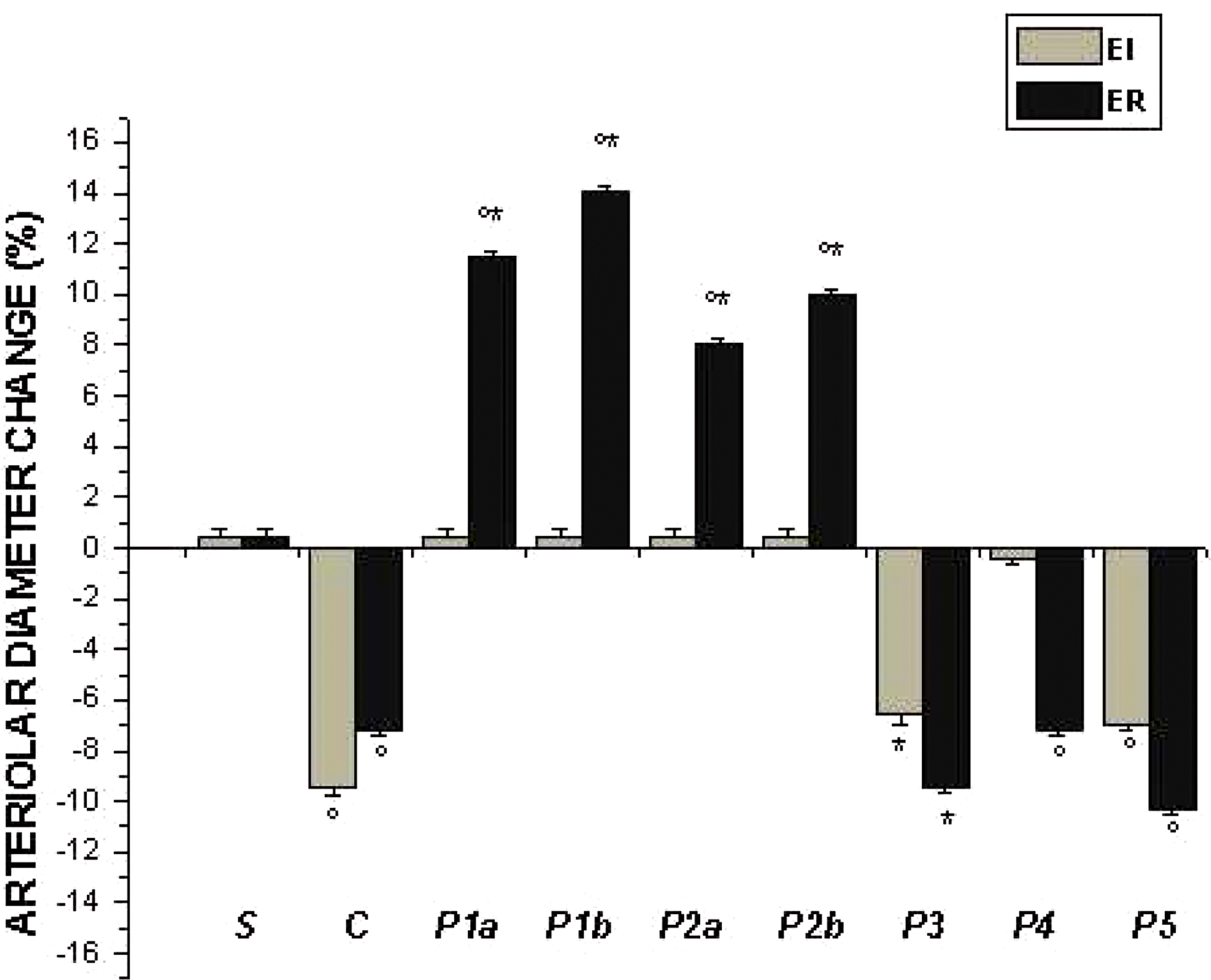
Figure 3. Percent diameter changes of A2 arterioles, at the end of ischemia (left column, EI) and at the end of reperfusion (right column, ER) in the experimental groups, as reported in Figure 2 legend. °p < 0.01 vs. sham-operated group, *p < 0.01 vs. C group.
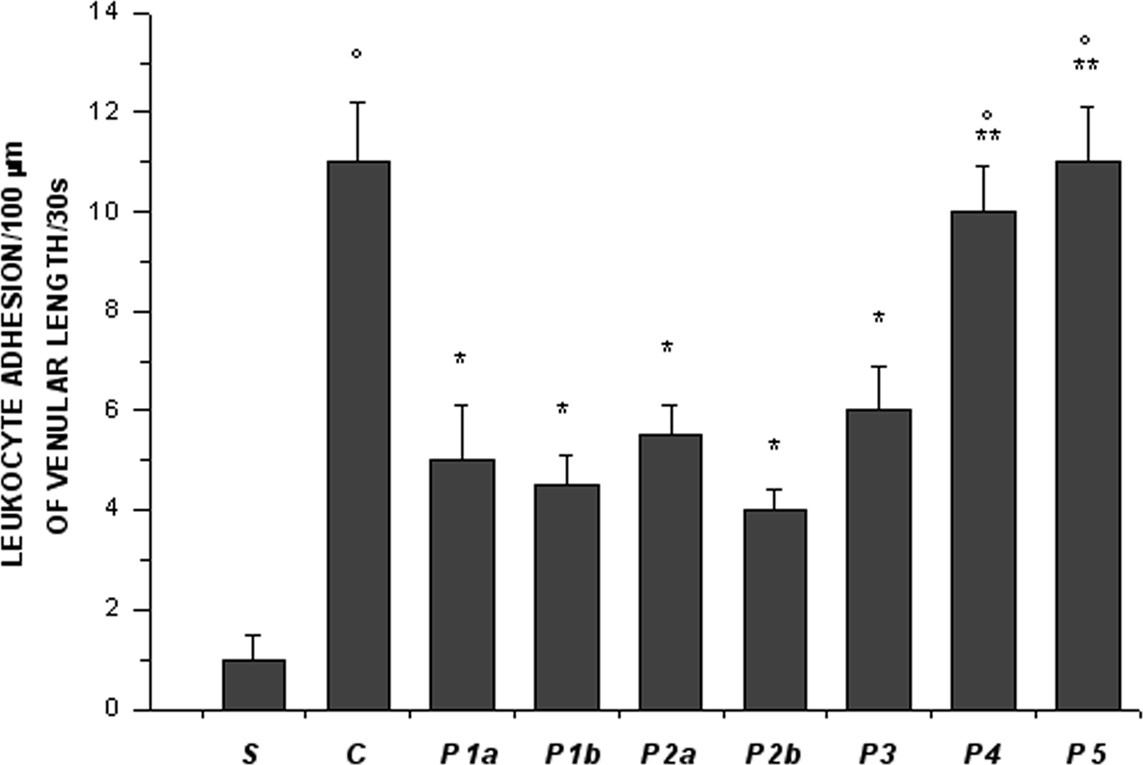
Figure 4. Leukocyte adhesion to venular walls at the end of reperfusion in the experimental groups, as reported in Figure 2 legend. °p < 0.01 vs. sham-operated group, *p < 0.01 vs. C group, **p < 0.01 vs. P2b group.
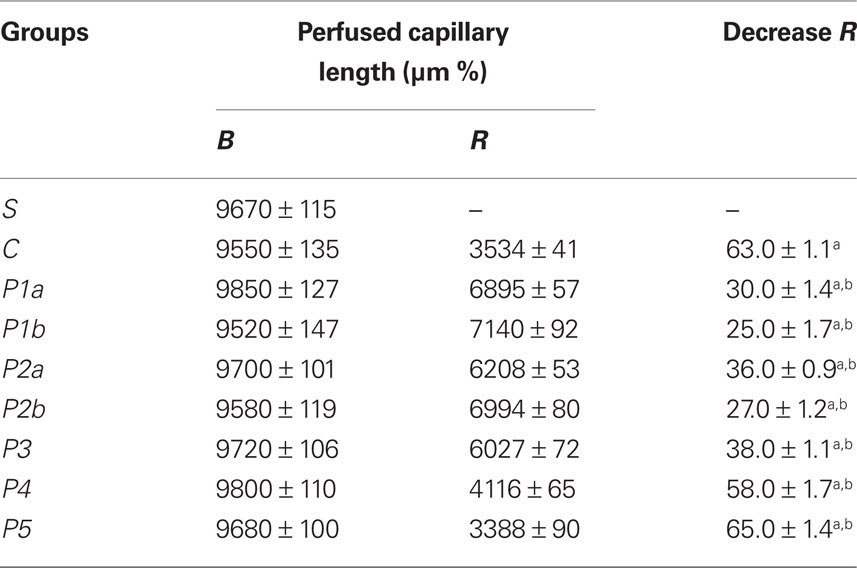
Table 2. The perfused capillary length under baseline conditions (B) and percent changes at the end of reperfusion (R) in the experimental groups.
pLc-Treated Animals
Intravenously infused pLc (2 and 5 mg/100 g body wt) dose-dependently protected microvascular networks. At EI, leakage was prevented (0.14 ± 0.01 and 0.13 ± 0.01 NGL, p < 0.01 vs. controls, respectively) (Figures 1 and 2). A2 arteriolar diameter (P1a–P1b sub-groups diameter: 21.7 μm, 19.7–24.3 μm, and 23.0 μm, 20.5–25.3 μm; n = 18 for each group, respectively) did not significantly change compared with baseline at both dosages (Figure 3). At ER, microvascular leakage was reduced compared with control (0.34 ± 0.02 and 0.28 ± 0.03 NGL, respectively, p < 0.01 vs. controls) (Figures 1 and 2). Adhesion of leukocytes was prevented (5.0 ± 1.1 and 4.5 ± 0.6/100 μm of venular length/30 s, respectively, p < 0.01 vs. controls) (Figure 4), while the perfused capillary length was reduced by 30.0 ± 1.4% and 25.0 ± 1.7% of baseline, according to the doses (p < 0.01 vs. controls) (Table 2). Capillary VRBC was 0.260 ± 0.010 and 0.320 ± 0.008 mm/s with both dosages, respectively (p < 0.01 vs. baseline and controls) (Table 1). Finally, A2 arteriolar diameter significantly increased compared with baseline (Figure 3).
pLc (1.5 and 2.5 mM/l) topically applied on cheek pouch membrane determined the same protective effects as observed with intravenous administration. At EI, pLc dose-dependently prevented permeability increase (0.15 ± 0.01 and 0.10 ± 0.01 NGL, respectively, p < 0.01 vs. controls). A2 arteriolar diameter (P2a–P2b sub-groups diameter: 23.1 μm, 20.2–25.7 μm, and 22.3 μm, 19.3–25.2 μm; n = 18 for each group, respectively) did not significantly change compared with baseline at both dosages (Figure 3). At ER, the leakage was prevented compared with control group (0.35 ± 0.01 and 0.26 ± 0.02 NGL, respectively, p < 0.01 vs. controls) (Figures 1 and 2). The number of adherent leukocytes significantly decreased (5.5 ± 0.6 and 4.0 ± 0.4/100 μm of venular length/30 s, respectively, p < 0.01 vs. controls) (Figure 4). Moreover, the perfused capillary length diminished by 36.0 ± 0.9% and 27.0 ± 1.2% of baseline, respectively (p < 0.01 vs. controls) and capillary VRBC was 0.250 ± 0.005 and 0.290 ± 0.009 mm/s with both dosages, respectively (p < 0.01 vs. baseline and controls) (Tables 1 and 2). A2 arteriolar diameter significantly increased compared with baseline (Figure 3).
NOS Inhibition Plus pLc
The topical application of L-NAME (1 mM) prior to pLc (2.5 mM/l) (P3 group) caused a slightly increase in leakage (0.18 ± 0.02 NGL, p < 0.01 vs. controls), higher when compared with hamsters treated only with pLc (P2b sub-group) at EI (Figure 2). A2 arteriolar diameter (23.7 μm, 20.8–25.7 μm, n = 18) significantly decreased compared with baseline (Figure 3). At ER, microvascular permeability further increased and was significantly different compared with that observed in P2b group (0.34 ± 0.01 NGL, p < 0.01 vs. control) (Figure 2). The leukocyte adhesion decreased (6.0 ± 0.9/100 μm of venular length/30 s, p < 0.01 vs. controls) as well as the perfused capillary length (by 38.0 ± 1.1% of baseline, p < 0.01 vs. controls) (Figure 4 and Table 2).The capillary VRBC was 0.220 ± 0.005 mm/s (p < 0.01 vs. baseline) (Table 1) and A2 arteriolar diameter was significantly reduced compared with the baseline (Figure 3).
The topical application of L-NNA (1 mM) prior to pLc (2.5 mM/l) caused the same microvascular effects as L-NAME treatment, as observed in preliminary experiments.
Apamin and Charybdotoxin Plus pLc
Apamin (0.3 μM) and Charybdotoxin (0.1 μM) prior to pLc (2.5 mM) topically applied on cheek pouch membrane abolished the pLc-induced effects. At EI, NGL increased up to 0.29 ± 0.02 (p < 0.01 vs. baseline and P2b sub-group) (Figure 2), while arteriolar diameter (22.5 μm, 19.3–25.5 μm, n = 18) did not change compared with that detected in P2b sub-group (Figure 3). At ER, NGL increased up to 0.57 ± 0.03 (p < 0.01 vs. baseline and P2b sub-group); the leukocyte adhesion was pronounced (10.0 ± 0.9/100 μm of venular length/30 s, p < 0.01 vs. baseline and P2b sub-group) (Figure 3). Moreover, the perfused capillary length decreased by 58.0 ± 1.7% of baseline (p < 0.01 vs. baseline and P2b sub-group) as well as capillary VRBC (0.190 ± 0.010 mm/s) (p < 0.01 vs. baseline and controls) (Tables 1 and 2). A2 arteriolar diameter did not significantly change compared with control group (Figure 3).
NOS Inhibition, Apamin and Charybdotoxin Plus pLc
Finally, the last group of hamsters treated with L-NAME (1 mM) plus Apamin (0.3 μM) and Charybdotoxin (0.1 μM) prior to pLc (2.5 mM), topically administered, abolished the pLc-induced effects. At EI, the leakage of FD 150 was marked (0.30 ± 0.02 NGL, p = NS vs. controls) and A2 arterioles (22.8 μm, 19.8–25.6 μm, n = 18), significantly constricted compared with baseline (Figure 3). At ER, the microvascular permeability was pronounced (0.65 ± 0.03 NGL, p = NS vs. controls), the leukocyte adhesion was marked (11.0 ± 1.1/100 μm of venular length/30 s, p < 0.01 vs. baseline) and the perfused capillary length decreased by 65.0 ± 1.4% of baseline (p = NS vs. controls) (Figures 2 and 4, Table 2). Capillary VRBC was 0.200 ± 0.010 mm/s (p < 0.01 vs. baseline) (Table 1). A2 arteriolar diameter significantly decreased compared with baseline (Figure 3).
Moreover, there were no significant changes in MABP and heart rate (HR) among the different groups in baseline conditions, at EI and at ER.
In Vitro Analysis
E-selectin expression was assessed by immunocytochemistry in either sham-operated or -control or -treated hamsters. In these last animals, pLc was administered either intravenously or topically at the highest dosage. E-selectin was not detected in cheek pouch preparations in sham-operated hamsters under baseline conditions. At ER in control hamsters, instead, E-selectin was detected along the vessel walls, as accumulated fluorescent spots. Interestingly, administration of pLc was effective in reducing accumulation of protein along the vessel walls. The topical administration of L-NAME or Apamin and Charybdotoxin or L-NAME plus Apamin and Charybdotoxin prior to pLc (groups P3, P4, and P5) increased fluorescent spots indicating E-selectin expression on vessel walls.
To further assess E-selectin gene expression in the experimental conditions used, RT-PCR and Western blot analyses were carried out. Under baseline conditions in sham-operated hamsters we could not detect a specific product by RT-PCR, indicating that the E-selectin gene was not actively transcribed. Conversely, at RE, a specific amplified product of E-selectin gene was obtained in hamsters subjected to I/R, as shown in Figure 5. This band was reduced in intensity by the higher dosage of pLc applied both systemically or topically (sub-groups P1b and P2b). On the contrary, the topical administration of L-NAME or Apamin and Charybdotoxin or L-NAME plus Apamin and Charybdotoxin prior to pLc (groups P3, P4, and P5) increased the band intensity corresponding to E-selectin expression.
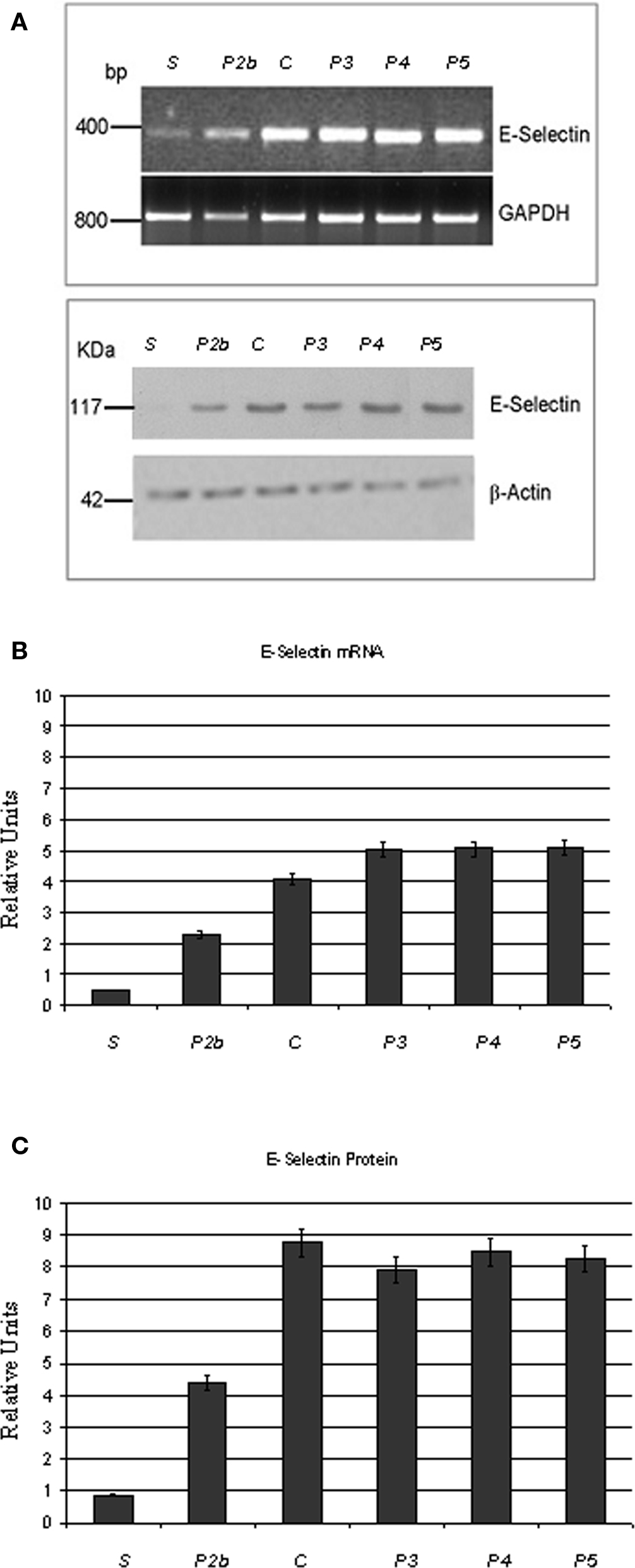
Figure 5. Treatment with topically applied pLc (2. 5 mM/l) reduces the formation of mRNA for E-selectin (upper) and the expression of E-selectin (lower) (A). S, sham-operated; P2b (pLc, 2.5 mM/l, topically applied treated animal); C (I/R control hamster); P3 (L-NAME plus pLc treated hamster), P4 (Apamin- and Charibdotoxin-treated hamster); P5 (L-NAME and Apamin and Charibdotoxin plus pLc-treated hamster. The intensity of mRNA amplified bands was referred to that obtained with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Changes in the E-selectin mRNA concentration expressed as relative units (B). E-selectin protein loading was normalized by incubating the same filters with anti-β-actin antibody. Changes in the E-selectin protein concentration expressed as relative units (C). *p < 0.01 vs. control and the other experimental groups. °p < 0.01 vs. sham-operated group.
These results were confirmed by Western blot analysis on protein extracts from sham-operated or control or pLc-treated cheek pouch preparations. Under basal conditions in sham-operated animals, we could not detect any E-selectin. After reperfusion a band of the expected molecular weight was clearly detectable, whose intensity was significantly reduced by the higher dosage of pLc applied both systemically or topically. Cheek pouch preparations topically treated with L-NAME or Apamin and Charybdotoxin or L-NAME plus Apamin and Charybdotoxin prior to pLc (groups P3, P4, and P5) showed a band corresponding to E-selectin production (Figure 5).
TBARS
The sequence of events that cause lipoperoxidation in the cheek pouch membrane was monitored by determination of TBARS at the end of ischemia and during reperfusion on the homogenate of the cheek pouch. Pilot studies carried out in animals submitted to ischemia–reperfusion indicated that TBARS peaked at 15–20 min of reperfusion. In sham-operated animals there were no significant amounts of TBARS.
At EI in control hamsters (C group) TBARS increased by 57.0 ± 3.2% of baseline MDA, as quantified in contralateral cheek pouch, while at 20 min of reperfusion increased by 60.5 ± 3.5% of baseline MDA (Figure 6). The higher dosage of pLc applied both systemically or topically caused a marked reduction in TBARS compared with controls: at EI and at 20 min of reperfusion TBARS did not significantly change compared with contralateral cheek pouch MDA (1.5 ± 0.2% MDA and 1.3 ± 0.1% MDA, respectively, p < 0.01 vs. control hamsters) (Figure 6).
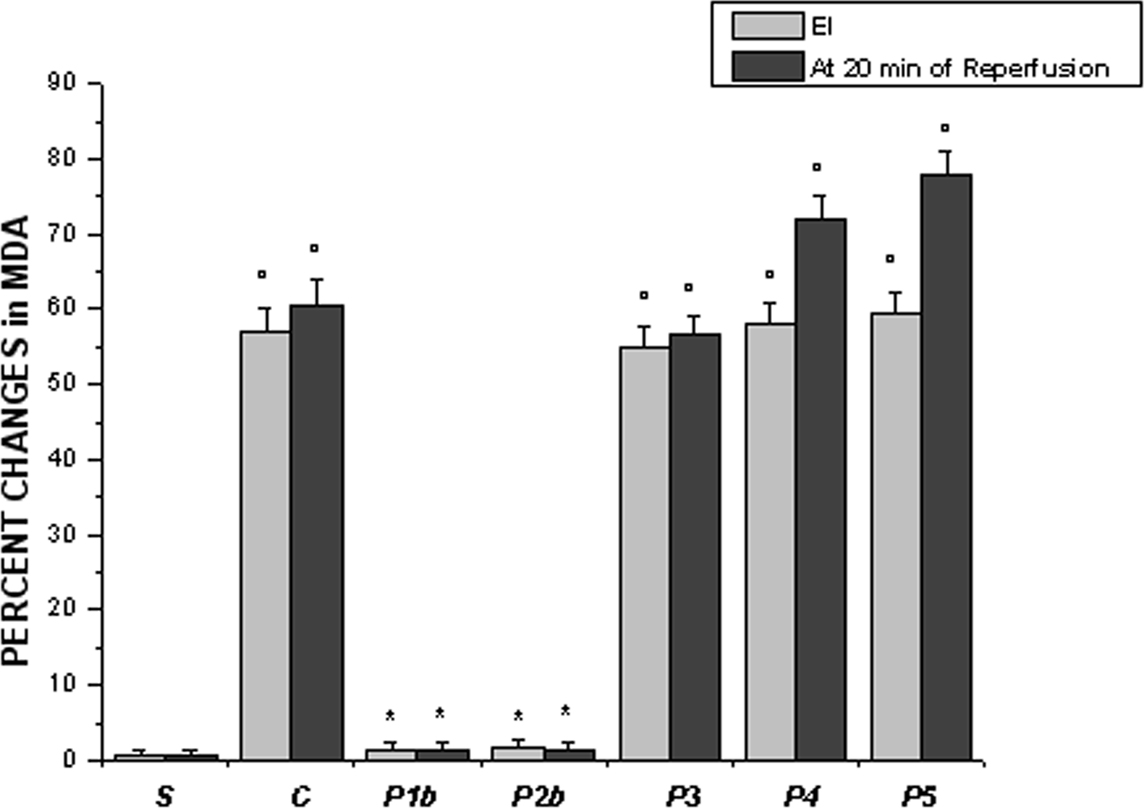
Figure 6. Changes in thiobarbituric acid reactive substances (TBARS) in homogenate of the cheek pouch expressed as percent changes of malondialdehyde (MDA) at the end of ischemia (left column, EI) and after 20 min of reperfusion (right column) in the experimental groups, as reported in Figure 2 legend. °p < 0.01 vs. sham-operated group, *p < 0.01 vs. C group.
L-NAME prior to pLc, topically administered, abolished the pLc protective effects because MDA amount increased by 55.0 ± 2.6% and 56.7 ± 2.3%, at EI and at 20 min of reperfusion, respectively (p = NS vs. controls). The same effects were induced by topical administration of Apamin and Charybdotoxin or L-NAME plus Apamin and Charybdotoxin prior to pLc (P4 and P5 groups). At EI, TBARS increased by 58.0 ± 2.9% MDA and 59.5 ± 2.7% MDA, respectively (p = NS vs. controls); after 20 min of reperfusion TBARS increased by 72.0 ± 3.2% and 78.0 ± 2.9% MDA compared with contralateral cheek pouch, respectively (p = NS vs. controls) (Figure 6).
DCF Fluorescence
Finally, the superfusion of the cheek pouch with DCFH-DA in animals submitted to I/R induced an increase in fluorescence in control animals, especially at ER (1.01 ± 0.04 NGL, p < 0.01 vs. baseline) (Figure 7). Higher dosage of pLc, intravenously and topically applied (P1b and P2b sub-groups) significantly reduced fluorescence of membrane at ER (0.23 ± 0.03 NGL and 0.22 ± 0.04 NGL, respectively, p < 0.01 vs. controls). The fluorescence intensity was amplified in the cheek pouch preparations treated with L-NAME or Apamin and Charybdotoxin or L-NAME, Apamin, and Charybdotoxin prior to pLc (groups P3, P4, and P5) (0.90 ± 0.01 NGL, 0.95 ± 0.02 NGL and 1.08 ± 0.02 NGL, respectively, p = NS vs. controls).
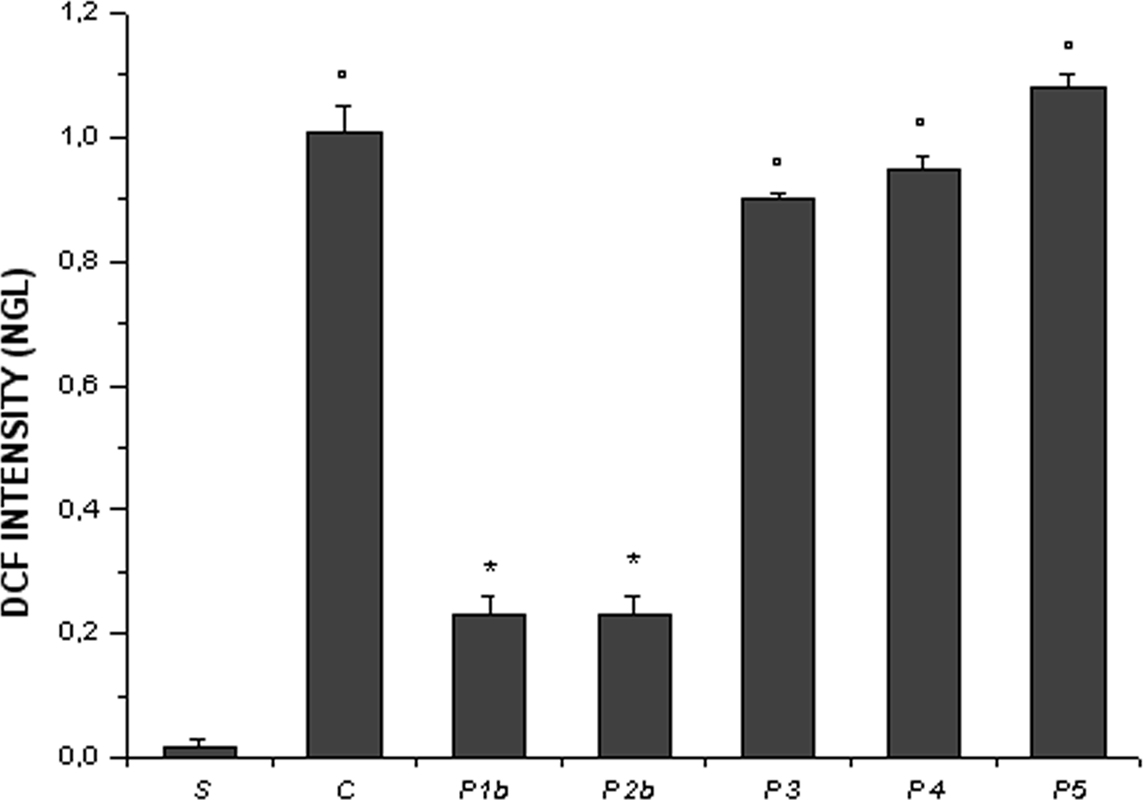
Figure 7. Changes in DCF fluorescence intensity correlated to the intracellular reactive oxygen species (ROS) levels at the end of reperfusion (ER) in the experimental groups, as reported in Figure 2 legend. °p< 0.01 vs. sham-operated group, *p < 0.01 vs. C group.
Discussion
The results of the present study indicate that I/R caused the microcirculation of hamster cheek pouch to be severely compromised because of an increase in leakage following I/R. Moreover, compared to baseline there was pronounced leukocyte adhesion in post capillary and connecting venules; the capillary blood flow was markedly impaired and the arterioles constricted. By immunohistochemical analysis microvessels presented an increased expression of E-selectin as well as an increase in mRNA transcript and protein.
pLc both topically or intravenously administered, exerted dose-dependent protective effects on the cheek pouch microcirculation during I/R. pLc prevented leakage during ischemia. The same pattern was observed at the end of reperfusion, where pLc blunted leukocyte adhesion and preserved capillary perfusion inducing arteriolar dilation when compared with controls. Moreover, E-selectin expression was reduced, as observed with immunohistochemical analysis, RT-PCR assay, and Western blot.
The features of I/R injury are marked reduction in blood flow and leukocyte adhesion. Our data indicate that most cheek pouch capillaries were devoid of blood flow at the end of reperfusion. The permeability increase indicates that the vessel wall function was impaired. pLc protected vessel walls probably by NO and EDHF release. However, Apamin and Charybdotoxin administration prior to pLc caused marked leakage, higher than after L-NAME administration prior to pLc.
Leukocyte adherence to the endothelium represents a key event in I/R injury. E-selectin has been shown to mediate leukocyte rolling and participate in the rolling conversion to ones firmly adhered. This molecule, indeed, occurs in the first phase of I/R damage (Carlos and Harlan, 1994; Billups et al., 1995; Ley et al., 1998). E-selectin expression in the vascular bed appears to be inhomogeneous, because of differences between endothelium in large versus small vessels in their ability to express these proteins. Our data show that E-selectin expression increased during reperfusion and was confined to endothelial cells lining post-capillary venules.
The increase of adhesion molecules expression accompanied by adherence of leukocytes to peripheral circulation endothelium may contribute to changes in vascular permeability. Our data support the hypothesis that E-selectin biosynthesis and expression are associated with ischemia and re-oxygenation because E-selectin expression was absent in hamsters not subjected to I/R.
Interestingly, as reported in previous studies, pLc shows O2− and ·OH scavenging activity. Our results indicate that pLc reduced formation of ROS, as indicated by the decrease in TBARS at the end of ischemia and at 20-min reperfusion. The percent increase in TBARS compared with baseline in control hamsters was higher at 20-min reperfusion, but it is worth noting at the end of ischemia appeared also an increase in TBARS. However, pLc counterbalanced the increase in TBARS both at the end of ischemia and at 20-min reperfusion. Our data are the first results reported on the cheek pouch tissue TBARS, measured at the end of ischemia and at 20-min reperfusion. Therefore, the increase in ROS and E-selectin production indicate that ROS might increase E-selectin expression with higher formation of mRNA and gene products. Our results support previous studies showing that E-selectin expression is triggered by ROS (Mondola et al., 1998).
The effect on ROS formation exerted by pLc was accompanied by preservation of NO release and the protection of NO effects on vascular smooth muscle cells and platelets. The summation of these effects could facilitate preservation of microvascular networks assuring tissue oxygenation. Our data indicate that capillary blood flow was preserved, with an increased capillary perfusion, when compared with control, and increased RBC velocity in capillaries. Previous data indicate that pLc improves arterial perfusion (Alvarez de Sotomayor et al., 2007). The effects on arterial diameter likely due to preserved NO and EDHF release, as shown also by Kajiya et al. (2007) and Karmakar (2001) and the reduction of E-selectin expression, may be the basis for increased capillary perfusion and capillary RBC velocity as indicated by our results.
The effect of pLc on ROS is supported by our results on DCF fluorescence that was significantly decreased by pLc at the end of reperfusion. Therefore, E-selectin reduction expression appears to be due to the decreased formation of ROS caused by pLc, with consequent blunted formation of mRNA for the protein and reduction of protein adduct. These results support previous data on the reduction of circulating adhesion molecules observed under different pathophysiological conditions (Signorelli et al., 2001; Silvestro et al., 2006).
Our results confirm the recovery of the capillary blood flow that has been reported in hepatic warm ischemia–reperfusion treated with pLc. Yonezawa et al. reported that blood flow decreased by 45–50% of baseline at reperfusion, while pLc was able to reduce the damage to 30–35% of baseline. However, the blood flow preservation, measured by the laser Doppler technique, was not accompanied by functional recovery of hepatocytes (Yonezawa et al., 2005). The laser Doppler recordings are influenced by probe dimension, because the signal is produced by red blood cell motion in all vessels in the scanned tissue. Therefore, laser Doppler signal does not reveal changes in flow distribution in the microvessels, because of summation in blood flow of hepatic arterioles, sinusoids, and portal venules. Previous data indicate that 30 min of hepatic ischemia and 60 min of reperfusion are able to reduce by 10–15% sinusoidal blood flow, when monitoring hepatic lobules by intravital microscopy, measuring obstruction of individual capillary in the liver (Eipel et al., 2004). These differences may be due to the measurement methods. Our data, in a different experimental model, indicate that pLc is very powerful in its ability to preserve the microvascular network from I/R injury.
The increase in capillary blood flow may be due to the vascular resistance reduction related to the release of NO and EDHF and subsequent changes in vascular smooth muscle cells, as previously reported (Pola et al., 1991; Signorelli et al., 2001). However, the increase in capillary perfusion may also be due to the leakage reduction and consequent accumulation of extravascular fluid able to compress vascular networks and reduce blood flow velocity. Therefore, during I/R injury pLc shows protective effects on microvascular networks promoting an adequate blood supply to tissues through the release of two different molecules derived from vascular endothelial cells. Our results indicate that NO together with EDHF mediate vasodilatation, while EDHF release primarily acts to prevent the permeability increase and the leukocyte adhesion (Figure 8).
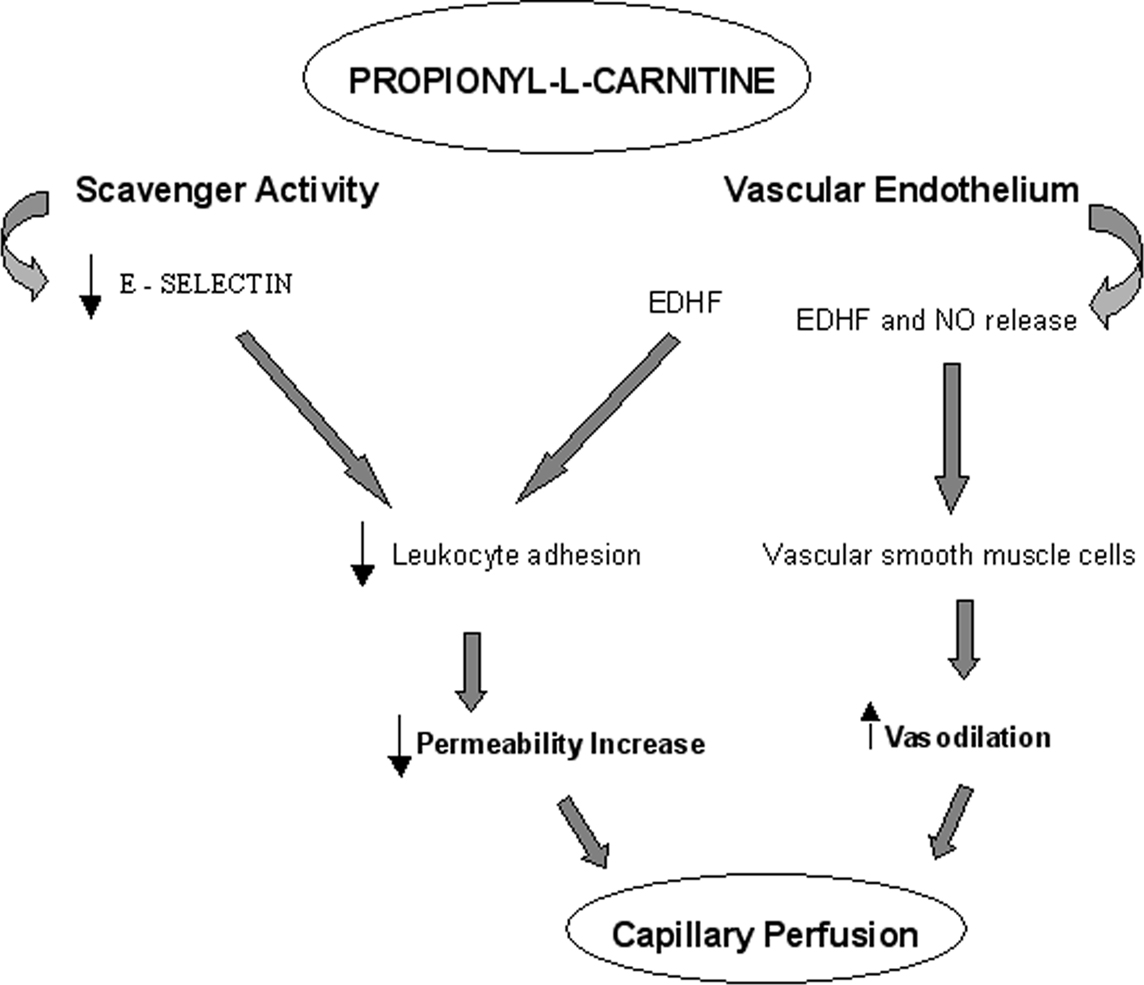
Figure 8. Scheme of mechanisms triggered by pLc during ischemia–reperfusion injury.
In conclusion, the present data indicate that the scavenger activity of pLc is very important in opposing deleterious action of ROS and thus protects the vascular endothelium from leukocyte adhesion and leakage. To the end we can consider these effects maintain the capillary viability and perfusion. The protection exerted by pLc emphasizes the potential therapeutic use of antioxidants in ischemic and post-ischemic vascular disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alvarez de Sotomayor, M., Bueno, R., Pérez-Guerrero, C., and Herrera, M. D. (2007). Effect of L-Carnitine and Propionyl-L-Carnitine on endothelial function of small mesenteric arteries from SHR. J. Vasc. Res. 44, 354–364.
Bertuglia, S., and Colantuoni, A. (2000). Protective effects of leukopenia and tissue plasminogen activator in microvascular ischemia-reperfusion injury. Am. J. Physiol. 278, H755–H761.
Billups, K. L., Palladino, M. A., Hinton, B. T., and Sherley, J. L. (1995). Expression of E-selectin mRNA during ischemia/reperfusion injury. J. Lab. Clin. Med. 125, 626–633.
Carlos, T. M., and Harlan, J. M. (1994). Leukocyte-endothelial adhesion molecules. Blood 84, 2068–2101.
Chiddo, A., Gaglione, A., Musci, S., Troito, G., Grimaldi, N., Lo Curatolo, N., and Rizzon, P. (1991). Hemodynamic study of intravenous propionyl-L-carnitine in patients with ischemic heart disease and normal left ventricular function. Cardiovasc. Drugs Ther. 5, 107–111.
Eipel, C., Bordel, R., Nickels, R. M., Menger, M. D., and Vollmar, B. (2004). Impact of leukocytes and platelets in mediatine hepatocyte apoptosis in a rat model of systemic endotoxemia. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G769–G779.
Ferrari, R., Ceconi, C., Curello, S., Pasini, E., and Visioli, O. (1989). Protective effect of propionyl-L-carnitine against ischemia and reperfusion-damage. Mol. Cell Biochem. 88, 161–168.
Granger, D. N. (1999). Ischemia-reperfusion: mechanisms of microvascular dysfuction and the influence of risk factors for cardiovascular disease. Microcirculation 6, 167–178.
Kajiya, M., Hirota, M., Inai, Y., Kiyooka, T., and Morimoto, T. (2007). Impaired NO-mediated vasodilation with increased superoxide but robust EDHF function in right ventricular arterial microvessels of pulmonary hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 292, H2737–H2744.
Karmakar, N. (2001). Interaction of transmural pressure and shear stress in the transport of albumin across the rabbit aortic wall. Atherosclerosis 156, 321–327.
Ley, K., Allietta, M., Bullard, D. C., and Morgan, S. (1998). Importance of E-selectin for firm leukocyte adhesion in vivo. Circ. Res. 83, 287–294.
Loffredo, L., Marcoccia, A., Pignatelli, P., Andreozzi, P., Borgia, M. C., Cangemi, R., Chiaretti, F., and Violi, F. (2007). Oxidative-stress-mediated arterial dysfunction in patients with peripheral arterial disease. Eur. Hearth J. 28, 608–612.
Milstone, D. S., Redline, R. W., O’Donnell, P. E., Davis, V. M., and Stavrakis, G. (2000). E-selectin expression and function in a unique placental trophoblast population at the fetal-maternal interface: regulation by a trophoblast-restricted transcriptional mechanism conserved between humans and mice. Dev. Dyn. 219, 63–76.
Mister, M., Noris, M., Szymczuk, J., Azzollini, N., Aiello, S., Abbate, M., Trochimowicz, L., Gagliardini, E., Arduini, A., Perico, N., and Remuzzi, G. (2002). Propionyl-L-carnitine prevents renal function deterioration due to ischemia/reperfusion. Kidney Int. 61, 1064–1078.
Mondola, P., Annella, T., Serù, R., Santangelo, F., Iossa, S., Gioielli, A., and Santillo, M. (1998). Secretion and increase of intracellular CuZn superoxide dismutase content in human neuroblastoma SK-NBE cells subjected to oxidative stress. Brain Res. Bull. 45, 517–520.
Packer, L., Valenza, M., Serbinova, E., Starke-Reed, Frost, K., and Kagan, V. (1991). Free radical scavenging is involved in the protective effect of L-propyonil-carnitine against ischemia-reperfusion injury of heart. Arch. Biochem. Biophys. 288, 533–537.
Pola, P., Flore, R., Tondi, P., and Nolfe, G. (1991). Rheological activity of propionyl L-carnitine. Drugs Expt. Clin. Res. 27, 191–196.
Shug, A., Paulson, D., Subramanian, R., and Regitz, V. (1991). Protective effects of propionyl-L-carnitine during ischemia and reperfusion. Cardiovasc. Drugs Ther. 5, 77–83.
Signorelli, S. S., Malaponte, G., Di Pino, L., Digrandi, D., Pennini, G., and Mazzarino, M. C. (2001). Effects of ischemic stress on leukocyte activation processes in patients with chronic peripheral occlusive arterial disease: role of L-propionyl carnitine administration. Pharmacol. Res. 44, 305–309.
Silvestro, A., Schiano, V., Bucur, R., Brevetti, G., Scopacasa, F., and Chiariello, M. (2006). Effect of propionylcarnitine on changes in endothelial function and plasma levels of adhesion molecules induced by acute exercise in patients with intermittent claudication. Angiology 57, 145–154.
Tarantini, G., Scrutinio, D., Bruzzi, P., Boni, L., Rizzon, P., and Iliceto, S. (2006). Metabolic treatment with L-carnitine in acute anterior ST segment elevation myocardial infarction. A randomized controlled trial. Cardiology 106, 215–223.
Yao, L., Setiadi, H., Xia, L., Laszik, Z., Taylor, F. B., and McEver, R. P. (1991). Divergent inducible expression of P-Selectin and E-Selectin in mice and primates. Blood 94, 3820–3828.
Keywords: ischemia–reperfusion, propionyl-L-carnitine, microcirculation, nitric oxide synthase, endothelium-derived hyperpolarizing factor (EDHF), E-selectin, lipid peroxides, 2′-7′-dichlorofluorescein (DCF)
Citation: Lapi D, Sabatino L, Altobelli GG, Mondola P, Cimini V and Colantuoni A (2010) Effects of propionyl-L-carnitine on ischemia–reperfusion injury in hamster cheek pouch microcirculation. Front. Physio. 1:132. doi: 10.3389/fphys.2010.00132
Received: 22 March 2010;
Paper pending published: 20 May 2010;
Accepted: 23 August 2010;
Published online: 19 October 2010
Edited by:
Angela C. Shore, University of Exeter, UKReviewed by:
Richard John Rivers, The Johns Hopkins University School of Medicine, USAEliete Bouskela, State University of Rio de Janeiro, Brazil
Copyright: © 2010 Lapi, Sabatino, Altobelli, Mondola, Cimini and Colantuoni. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Antonio Colantuoni and Dominga Lapi, Department of Neuroscience, “Federico II” University Medical School, Via S. Pansini, 5 80131 Naples, Italy. e-mail: d.lapi@dfb.unipi.it