
- Department of Biomedical Sciences, and Department of Medical Pharmacology and Physiology, Dalton Cardiovascular Research Center, University of Missouri-Columbia, Columbia, MO, USA
In contrast to the “programmed” nature of apoptosis and autophagy, necrotic cell death has always been believed to be a random, uncontrolled process that leads to the “accidental” death of the cell. This dogma, however, is being challenged and the concept of necrosis also being “programmed” is gaining ground. In particular, mitochondria appear to play a pivotal role in the mediation of programmed necrosis. The purpose of this review, therefore, is to appraise the current concepts regarding the signaling mechanisms of programmed necrosis, with specific attention to the contribution of mitochondria to this process.
Cell Death Comes in Three Flavors
There are three recognized types of cell death: apoptosis, autophagy, and necrosis (Kroemer et al., 2009). Much attention has been paid to apoptosis and, especially at the moment, autophagy. This is primarily because of their “programmed’ nature, i.e., there are specific, genetically determined pathways that mediate both these processes. In stark contrast, until recently it was believed that necrosis was a random, uncontrolled process that leads to the “accidental” death of the cell. Consequently, necrosis as a death modality was not well studied, which is unfortunate as necrosis probably plays a larger role in the pathogenesis of disease than either apoptosis or autophagy. This dogma, however, is changing. With the discovery of key mediators of necrotic death such RIP kinases and PARP, the concept of programmed necrosis is gaining ground. Here will go over our current knowledge as to how programmed necrosis is signaled, and then focus on the emerging role of the mitochondrion in this process.
Programmed Necrosis: The Story so Far
Although the concept of “programmed necrosis” (sometimes inappropriately referred to as necroptosis) is a relatively recent one, already a considerable body of literature has amassed delineating some key mediators of this death process. Specifically, receptor interacting protein (RIP) kinases, poly(ADP-ribose) polymerase-1 (PARP1), NADPH oxidases, and calpains have been identified as potential signaling components of a necrotic program(s) (Figure 1).
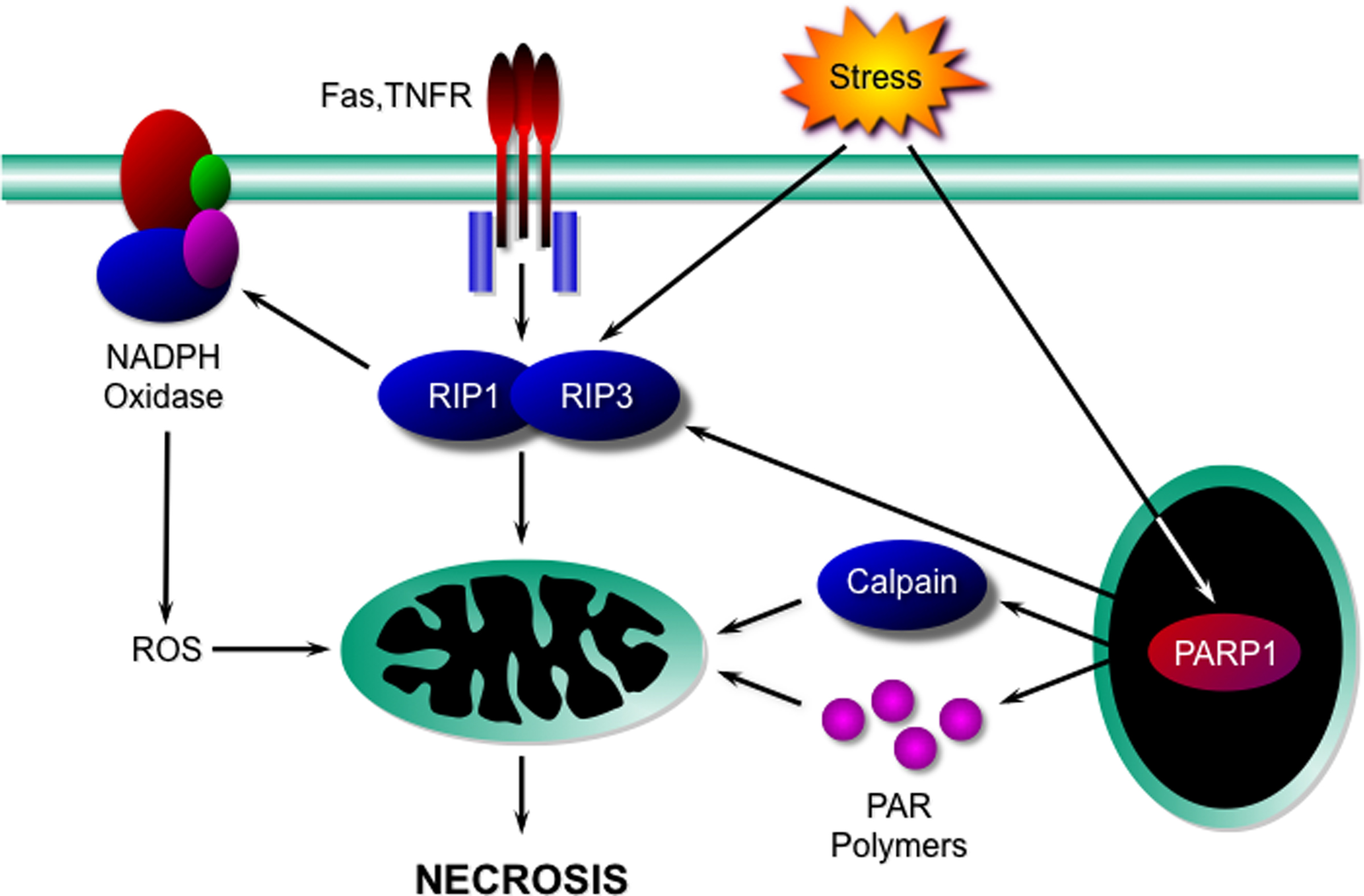
Figure 1. Signaling components of the necrotic programme. Activation of death receptors such as Fas and TNFR, or cellular stresses, induces the interaction and activation of the kinases RIP1 and RIP3. These in turn affect mitochondria either directly, or indirectly through NADPH oxidase-derived ROS to induce necrosis. Necrotic death stimuli can also activate PARP1, which can potentially induce necrosis either through activation of the RIP kinases, activation of calpains, or through the production of PAR polymers. TNFR, TNFα receptor; RIP, receptor interacting protein; ROS, reactive oxygen species; PAR, poly(ADP-ribose); PARP1, poly(ADP-ribose) polymerase-1.
RIP Kinases
It had been known for sometime that inhibition of TNFα-induced apoptosis by pan-caspase inhibitors such as zVAD-FMK could drive some cell types into necrosis instead (Vandenabeele et al., 2006). It was subsequently found that this switch of death modalities was blocked in cells lacking the serine/threonine kinase RIP1 (Holler et al., 2000; Zheng et al., 2006; Degterev et al., 2008). Under normal circumstances, RIP1 mediates TNF-receptor-induced NF-κB and MAPK signaling (Festjens et al., 2007), i.e., pathways normally associated with cell survival. However, under certain conditions (such as caspase inhibition) RIP1’s Dr. Jekyll turns into Mr. Hyde and induces necrotic death. From an important clinical perspective, the identification of specific chemical inhibitors of RIP1, the necrostatins, has confirmed a pathological role for RIP1 in ischemia/reperfusion-induced necrosis in the heart, brain, and eye (Degterev et al., 2005; Smith et al., 2007; Rosenbaum et al., 2010).
RIP1’s contribution to necrosis has perhaps been the most studied of any candidate to date. In particular, several systems biology approaches have been conducted in attempts to identify downstream signaling molecules that are part of RIP1’s necrotic mechanism. Using the L929 cell line where necrosis can be induced simply by treating with zVAD-FMK or NIH3T3 cells treated with TNFα plus zVAD, Hitomi et al. (2008) used a genome-wide siRNA screen to identify 25 “core” components of the necrotic signaling pathway. Not surprisingly, the TNFR1 and RIP1 were identified as critical mediators, as was Cyld, which regulates RIP1 ubiquitination. More surprising was the identification of the Bcl2 family protein, Bcl2 modifying protein (Bmf, which we will discuss later), junctophilin-3, poliovirus receptor (CD155), and the transcription factor Foxi1.
Three other genomic screens simultaneously identified RIP3, a sister kinase of RIP1, as also being essential for programmed necrosis (Cho et al., 2009; He et al., 2009; Zhang et al., 2009). Although, the exact relationship between the two RIPs remains to be clarified, they do form a complex through interaction with their homotypic RHIM motifs (Sun et al., 2002). Moreover, the majority of the data suggest that RIP1 phosphorylates and activates RIP3 in the progression of necrotic signaling (He et al., 2009; Zhang et al., 2009). RIP3-deficient cells were less sensitive to TNFα- and Smac-mimetic-induced necrosis (in the presence of a caspase inhibitor), and Ripk3−/− mice were resistant to necrotic pancreatitis and vaccinia virus-induced hepatic necrosis (Cho et al., 2009; He et al., 2009).
PARP1
Another well-studied necrotic candidate is the DNA repair enzyme PARP1. Genotoxic stresses such as oxidants and alkylating agents have long been known to cause necrotic cell death that is associated with an overstimulation of PARP1 (Hassa, 2009). Moreover, ischemia/reperfusion-induced myocardial and cerebral necrosis is markedly attenuated by genetic inhibition of PARP1 (Zingarelli et al., 2003; Li et al., 2010). It was originally thought that necrosis induced by PARP1 hyperactivation was simply due to metabolic catastrophe, where the overactive PARP1 used up the cell’s supply of NAD+, and subsequently ATP (van Wijk and Hageman, 2005). However, inhibition of specific proteins, such as calpains or CypD, can block PARP1-induced cell death even in the face of severe NAD+ depletion (Alano et al., 2004, 2010; Moubarak et al., 2007), suggesting that a more discrete signaling network is responsible for PARP1-mediated cell death (although NAD+ deletion could still play a role in this signaling network). Susin’s group very elegantly demonstrated that PARP1-induced necrosis was dependent on activation of calpain, which in turn impinged upon the mitochondrion (Moubarak et al., 2007). In contrast, Xu et al. (2006) suggested a PARP1–RIP1–JNK1 sequence of events, whereas another study proposed that PAR polymers themselves were responsible for cell death (Yu et al., 2006). Whether these represent distinct pathways, or are simply different components of the same pathway is something we are currently investigating.
It should be pointed out that, although a pro-necrotic action was thought to be restricted to PARP1, PARP2 might also contribute to this cell death mechanism. PARP2-null mice are similarly resistant to cerebral ischemia as PARP1-deficient mice (Li et al., 2010), and PARP2 was also one of the 25 “core” necrotic genes identified by Hitomi et al. (2008).
NADPH Oxidases
Several studies have reported that reactive oxygen species (ROS) scavengers can abrogate TNFα-induced necrosis in various cell types (Goossens et al., 1995; Lin et al., 2004; Kim et al., 2007). The site of ROS generation during death receptor-induced necrosis is controversial but one postulate is that activation of NADPH oxidases accounts for the increased oxidant production. Kim et al. (2007) demonstrated that TNFα-induced necrosis involved the recruitment of the nox1 and NOXO1 subunits of NADPH oxidase to the receptor complex in a RIP1-dependent manner. Moreover, inhibition of nox1, either by siRNA or rottlerin, blocked the necrotic actions of TNFα (Kim et al., 2007; Byun et al., 2008). In contrast, the nox4 isoform has been implicated in oxidized-LDL-induced necrosis (Lee et al. 2010). Importantly, genetic deletion of gpPhox, nox1, nox2, and/or or nox4 can reduce ischemia/reperfusion-induced necrosis in the heart, liver, and brain (Hoffmeyer et al., 2000; Lehnert et al., 2003; Jackman et al., 2009; Kahles et al., 2010). However, it should be noted that some cell types appear to undergo programmed necrosis independent of ROS production. In particular, necrosis in macrophages, Jurkat cells and HT-29 carcinoma cells has been shown to be ROS-independent (Moquin and Chan, 2010).
Calpains
Just as proteases (i.e., caspases) are critical mediators of apoptosis, it appears that proteolytic enzymes, in the shape of the calpains, may also mediate programmed necrosis. Calpains are a family of cysteine proteases that are activated by Ca2+, and inhibition of calpains has been shown to be anti-necrotic in a number of disease models (Golstein and Kroemer, 2007; Kennedy et al., 2009; Hernando et al., 2010). Molecularly, Susin’s laboratory demonstrated that PARP1-induced necrosis was dependent on activation of calpains, and deletion of the Capn4 gene, which encodes an essential regulatory subunit for μ- and m-calpain, completely prevented cell death (Moubarak et al., 2007). TNFα-induced necrosis has also been reported to be dependent of calpain activation (Chen et al., 2006), although whether this is dependent on RIP1 and/or NADPH oxidases has yet to be tested. The cellular events that occur distal to calpains during necrosis are still being elucidated but include cleavage and activation of pro-death Bcl2 proteins such as Bax (Moubarak et al., 2007), cytoskeletal degradation (Lebart and Benyamin, 2006), and lysosomal rupture (Yamashima and Oikawa, 2009).
Role of Mitochondria in Programmed Necrosis
The establishment of proteins such as RIP kinases and PARPs as mediators of programmed necrosis has given us the beginnings of a framework upon which to build in terms of novel components. In particular, as in apoptosis, it appears that mitochondria play a pivotal role in the propagation of the necrotic signal. Indeed, the pioneering work of Nicotera’s laboratory demonstrated that mitochondria control whether a cell dies by apoptosis or necrosis long before the term “programmed necrosis” was even coined (Ankarcrona et al., 1995). In this section, we shall review our current understanding of the function that mitochondria play in necrosis and the molecular mechanisms by which they do so (Figure 2).
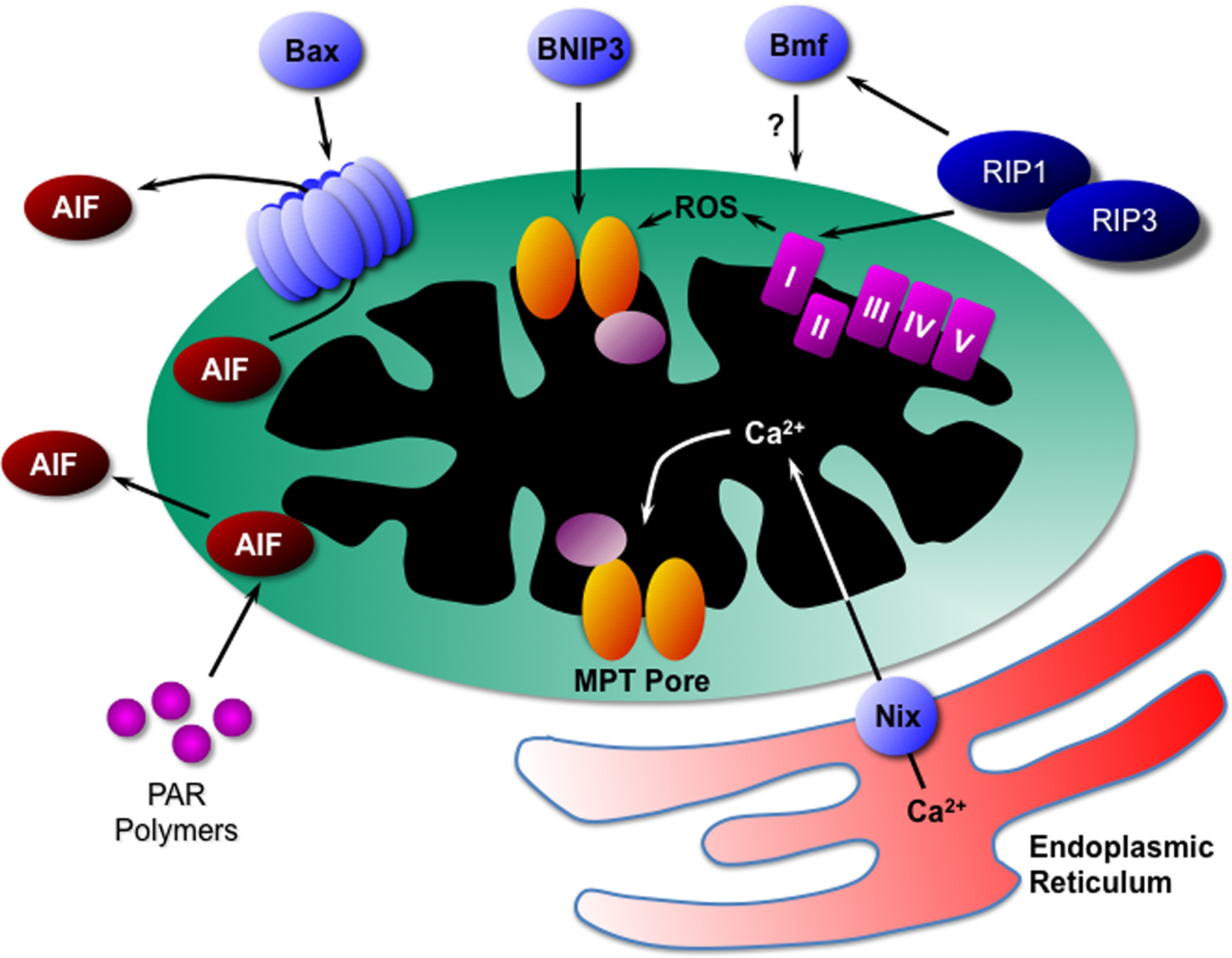
Figure 2. Mitochondrial mediators of programmed necrosis. Bax can induce necrotic cell death through the release of AIF, as can PAR polymers and calpain. Another Bcl2 family protein, BNIP3, appears to act directly through activation of the MPT pore. RIP kinases, possibly through translocation to the mitochondrion itself, can induce ROS production from complex-I of the electron transport chain. In addition, RIPs may act through the Bcl2 family protein Bmf, although the exact mechanism is unknown. Finally the Bcl2 family protein Nix induces release of Ca2+ from the endoplasmic reticulum that is in close proximity to the mitochondrion. The Ca2+ is then taken up into the mitochondrial matrix where it induced opening of the MPT pore and necrosis. AIF, apoptosis-inducing factor; PAR, poly(ADP-ribose); MPT, mitochondrial permeability transition; RIP, receptor interacting protein; ROS, reactive oxygen species.
Mitochondria-Derived ROS
As discussed above, several reports have implicated that NADPH oxidase-derived superoxide is required (at least under some circumstances) for programmed necrosis. However, others have suggested that it is mitochondria that are the source of the ROS required for necrotic cell death. Schulze-Osthoff et al. (1992) demonstrated that TNFα-induced necrosis in L929 cells was dependent on complex-I derived ROS. Similar findings have since been reported by other labs (Goossens et al., 1999; Festjens et al., 2007). Most recently, Davis et al. (2010) showed that the pro-necrotic actions of nitric oxide (NO) involved the nitration of complex-I subunits, with a subsequent increase in superoxide generation by that complex.
Mechanistically, induction of mitochondria-derived ROS by death receptor activation and/or caspase inhibition appears to be dependent on the RIP/RIP3 kinase complex (Cho et al., 2009; Zhang et al., 2009). Similarly, the increase in complex-I generated ROS in response to NO was also mediated by RIP1 and RIP3 (Davis et al., 2010). How the RIP kinases are influencing complex-I (or other mitochondrial functions for that matter) is still up for debate. Several papers have reported that RIP1 or RIP3 can localize to mitochondria in response to a necrotic stimulus (Kasof et al., 2000; Temkin et al., 2006; Davis et al., 2010). In addition, Zhang et al. (2009) found that RIP3 could interact with the mitochondrial protein glutamate dehydrogenase 1 (GLUD1) and that depletion of GLUD1 at least partially blocked TNFα/zVAD-induced ROS production and necrosis in NIH 3T3 cells.
Activation of PARP1 has also been described as influencing mitochondrial respiratory complexes and ROS production. Mary Gray’s laboratory showed that ischemia/reperfusion-induced complex-I defects were abrogated in PARP1-deficient mice (Zhou et al., 2006). Szabo’s group found an increase in the poly(ADP)-ribosylation of several mitochondrial proteins in response to H2O2 or NO (Pankotai et al., 2009), including α-ketoglutarate dehydrogenase which itself can act as a source of ROS (Starkov et al., 2004). The same group has also reported poly(ADP)-ribosylation of multiple components of the electron transport chain, which may also influence ROS production (Lai et al., 2008). Interestingly, GLUD1 can also be ADP-ribosylated (Herrero-Yraola et al., 2001). Similar to the RIP story, it’s not entirely apparent how PARP1 influences mitochondrial function, but PARP1 has been reported in the mitochondrion itself (Rossi et al., 2009) and PARP activators could directly affect the MPT pore (see below).
Cyclophilin-D and the Mitochondrial Permeability Transition Pore
The mitochondrial permeability transition (MPT) pore is a large, non-specific channel in the inner mitochondrial membrane that is opened in response to excessive production of ROS and to Ca2+ overload of the mitochondrial matrix (Halestrap, 2009; Baines, 2010), both of which occur during necrosis. This sudden increase in inner membrane permeability dissipates the proton electrochemical gradient (ΔΨm), leading to ATP depletion, further ROS production, and ultimately swelling and rupture of the organelle (Halestrap, 2009; Baines, 2010). The MPT pore was originally believed to consist of the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane, adenine nucleotide translocase (ANT) in the inner membrane, and the peptidylprolyl isomerase cyclophilin-D (CypD) in the mitochondrial matrix (Hunter and Haworth, 1979; Crompton et al., 1988; Szabó et al., 1993). However, mitochondria lacking either VDAC or ANT still undergo MPT and cells deficient in these proteins are still sensitive to cell death stimuli (Kokoszka et al., 2004; Krauskopf et al., 2006; Baines et al., 2007). In contrast, deletion of the CypD-encoding gene renders mitochondria and cells more resistant to MPT induction and the resultant cell mortality (Baines et al., 2005; Basso et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005).
For many years the MPT pore was believed to contribute to both apoptotic and necrotic cell death. However, recent data, especially from the CypD-deficient mice has suggested that the MPT pore may be more pro-necrotic than pro-apoptotic. Reduction in ischemia/reperfusion-induced necrosis in the heart by inhibitors of CypD was first demonstrated nearly 20 years ago (Griffiths and Halestrap, 1993; Duchen et al., 1993). Since then pharmacological inhibition of the MPT pore has been shown to be protective in many necrotic syndromes, including muscular dystrophy, acetaminophen-induced liver toxicity, and doxorubicin cardiotoxicity (Al-Nasser, 1998; Masubuchi et al., 2005; Angelin et al., 2007; Tiepolo et al., 2009). Importantly, these findings have been corroborated in the CypD-null mice (Baines et al., 2005; Nakagawa et al., 2005; Nakayama et al., 2007; Millay et al., 2008). What was interesting was that the pro-apoptotic effects of Bcl2 proteins such as Bax and Bid, and stimuli such as staurosporine and etoposide were unaffected in the CypD-deficient mitochondria and cells (Baines et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005). In contrast, necrotic cell death induced by oxidative stress was significantly attenuated in the knockout cells. One note of caution: although the data from the CypD-null cells and animals is suggestive of the MPT pore being involved in necrosis, MPT can still occur independent of CypD under some conditions (Basso et al., 2008). Therefore the results described above cannot yet be completely ascribed to the MPT pore, and may rather indicate a role for CypD in programmed necrosis that is independent of MPT.
How the MPT pore fits into our current model of programmed necrosis is the subject of ongoing investigation. Xu et al. (2006) demonstrated that PARP1 activation by MNNG led to abolition of ΔΨm in a RIP1-dependent manner, suggesting that both PARP1 and RIP1 could potentially induce necrosis via the MPT pore. MPT inhibition by cyclosporine-A prevented both the loss of ΔΨm and necrosis. In another study MNNG was suggested to induce MPT through PARP1-mediated depletion of NAD+ (Alano et al., 2004). These reports would place the MPT pore distal to PARP1 activation. However, Dodoni et al. (2004) reported that the PARP1 activator MNNG could directly induce MPT independent of PARP1 suggesting that the MPT pore may in fact be upstream of PARP1 activation and NAD+ depletion. TNFα/smac-mimetic/zVAD-induced necrosis was also partially rescued in CypD-null MEFs (He et al., 2009). Most recently, we have found that depletion of RIP1 could significantly reduce H2O2-induced MPT and necrosis, and that RIP1-induced necrotic death was attenuated in CypD-deficient fibroblasts. Taken together, all these data point to the MPT pore as being a member of the necrotic programme.
Pro-Death Bcl2 Proteins
Previously believed to be purely involved in mediation of the intrinsic apoptotic pathway, pro-death Bcl2 proteins may also contribute to necrotic death as well. In particular, there is evidence implicating Bax, Bmf, BNIP3, and Nix as being part of the necrotic programme. Pharmacological or genetic inhibition of Bax reduced infarcts in response to cardiac, hepatic, and cerebral ischemia/reperfusion (Hochhauser et al., 2003; Hetz et al., 2005; Ben-Ari et al., 2007). Bax-deficient mice were also protected against acetaminophen-induced liver necrosis (Bajt et al., 2008). The fact that in each case the observed protection was considerably greater than expected from purely inhibiting apoptotic signaling, strongly suggests that Bax may also be contributing to the necrotic cell death. Indeed, PARP1-mediated programmed necrosis has been reported to be dependent on mitochondrial translocation of Bax, but not Bak (Moubarak et al., 2007). Bax in turn induced necrosis by releasing apoptosis-inducing factor (AIF) from the mitochondria (see Section Apoptosis-Inducing Factor).
Members of the BH3 family of Bcl2 proteins have also been implicated in programmed necrosis. Unlike Bax, Bmf is one of the least studied Bcl2 proteins, although it is known to be essential for proper lymphocyte homeostasis (Labi et al., 2008; Frenzel et al., 2010). Regarding programmed necrosis, Bmf was one of the surprising candidates discovered as a mediator of TNFα-induced necrosis by Hitomi et al. (2008). Specifically, they found that knockdown of Bmf could prevent TNF and zVAD-induced necrotic death in L929 cells. Whether Bmf is also responsible for programmed necrosis in other contexts, and how it couples to other necrotic signaling molecules such as RIP1 and RIP3 remains to be seen. Moreover, the mechanisms by which Bmf induces necrosis remain unknown.
A BH3-related protein, BNIP3, can also induce necrosis. Under normal conditions BNIP3 is expressed at low levels, but is rapidly upregulated in response to cell stress (Burton and Gibson, 2009). Greenberg’s group, who discovered BNIP3, demonstrated that direct overexpression of BNIP3 could induce MPT and subsequent necrosis in 293T cells (Vande Velde et al., 2000). Similar results were subsequently reported in MEFs and cardiac myocytes (Kubasiak et al., 2002; Kubli et al., 2007). Moreover, in all these studies the BNIP3-induced necrosis could be blocked by inhibition of the MPT pore with cyclosporine-A. In the whole organ, treatment with a BNIP3 inhibitory peptide could reduce infarct size in isolated hearts (Hamacher-Brady et al., 2007). However, it should be pointed out that ischemia/reperfusion-induced myocardial necrosis was no different in BNIP3 knockout mice compared to wildtype controls (Diwan et al., 2007a).
Last, but not least, is the BNIP3-related protein Nix. Nix is constitutively expressed and, like Bmf, appears to play a role in hematopoesis (Diwan et al., 2007b). Elegant work from Dorn’s laboratory has demonstrated that Nix can induce both apoptosis and necrosis, and the nature of the death modality is dependent on the subcellular localization of the Nix protein (Diwan et al., 2009; Chen et al. 2010). Nix that is targeted to the mitochondria induces apoptosis through activation of the canonical intrinsic pathway. In contrast, ER-targeted Nix induces necrosis through calcium-dependent activation of the MPT pore. Again, how BNIP3 and Nix relate to more proximal necrotic signals such as RIP and PARP (or if they are activated more directly in response to stress) has yet to be ascertained.
Apoptosis-Inducing Factor
In addition to Bcl2 proteins moonlighting as necrotic mediators, other canonical apoptotic molecules may also be involved in programmed necrosis. During apoptosis, the mitochondrial flavoprotein AIF is released from the intermembrane space and translocates the nucleus where it cooperates with its fellow apoptogen endonucleus G to mediate DNA fragmentation (Boujrad et al., 2007). However, there is now considerable evidence that AIF plays a role in necrotic cell death too. Kroemer’s group, who identified AIF, first demonstrated that AIF release and nuclear translocation occurred during both staurosporine-induced apoptosis and ATP depletion-induced necrosis (Daugas et al., 2000). Since then many groups have demonstrated that necrosis induced by many stimuli (e.g., oxidative stress, ischemia/reperfusion, alkylating agents) is associated with AIF release and translocation (Xiao et al., 2004; Sancho et al., 2006; Moubarak et al., 2007; Zhu et al., 2007). Interestingly, Harlequin mutant mice, which only have 20% of normal levels of AIF (Klein et al., 2002), are more resistant against cerebral ischemia (Zhu et al., 2007). However, this is complicated by the fact that physiologically AIF is required for complex-I function and suppression of ROS (Vahsen et al., 2004; van Empel et al., 2006).
From a mechanistic perspective, both Susin’s and Dawson’s laboratories have extensively demonstrated that PARP activation-induced necrosis is dependent on mitochondrial release of AIF (Yu et al., 2002, 2006; Moubarak et al., 2007; Artus et al., 2010). However, they diverge on the mechanism(s) by which PARP induces AIF release. Susin’s group (Moubarak et al., 2007) demonstrated that the sequential activation of calpains and Bax were required for AIF liberation. In contrast, Dawson’s group failed to find a role for calpain in PARP-induced AIF release (Wang et al., 2009), and has suggested that it is the PAR polymer itself that induces mitochondrial discharge of AIF (Yu et al., 2006). The reasons for this discrepancy are still not clear. How either (or both) of these mechanisms relate to necrosis induced by other stimuli remains to be tested, although one study has suggested that cell death induced by the hexokinase-2 inhibitor, 3-bromopyruvate, is dependent on PARP activation and AIF release (Kim et al., 2008).
The mechanisms by which AIF contributes to necrotic death are beginning to be delineated (Baritaud et al., 2010). In particular, a recent study by Artus et al. (2010) demonstrated that AIF, complexed with cyclophilin-A, interacts with the histone H2AX in the nucleus and that this interaction is essential for AIF’s ability to induce DNA fragmentation and cell death. Supporting this paradigm was their finding that H2AX-deficient MEFs are resistant to necrotic death induced by PARP activators.
Future Directions
Many studies over the past decade have amassed a considerable list of “pro-necrotic” mediators. While this has been an important and essential first step in our understanding of programmed necrosis, it is still only just that: a first step. What needs to be done now is to try and put all of these pieces of the jigsaw puzzle together. Do they form one common necrotic pathway? Are there separate, parallel pathways, perhaps resembling the extrinsic and intrinsic pathways of apoptosis? Moreover, the vast majority of studies have only looked at programmed necrosis in the context of death receptor activation and/or caspase inhibition. We need to move beyond these settings and ask whether programmed necrosis is involved in the pathogenesis of diseases such as ischemia/reperfusion injury, neurodegenerative diseases, diabetes, and muscular dystrophy; i.e., is programmed necrosis truly clinically relevant? I firmly believe it is, but we need hard evidence.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the author’s laboratory is supported by National Institutes of Health grants HL092327, HL094404, and HL052490.
References
Alano, C. C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T. M., and Swanson, R. A. (2010). NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30, 2967–2978.
Alano, C. C., Ying, W., and Swanson, R. A. (2004). Poly(ADP-ribose) Polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 279, 18895–18902.
Al-Nasser, I. A. (1998). In vivo prevention of adriamycin cardiotoxicity by cyclosporin A or FK506. Toxicology 131, 175–181.
Angelin, A., Tiepolo, T., Sabatelli, P., Grumati, P., Bergamin, N., Golfieri, C., Mattioli, E., Gualandi, F., Ferlini, A., Merlini, L., Maraldi, N. M., Bonaldo, P., and Bernardi, P. (2007). Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc. Natl. Acad. Sci. U.S.A. 104, 991–996.
Ankarcrona, M., Dypbukt, J. M., Bonfoco, E., Zhivotovsky, B., Orrenius, S., Lipton, S. A., and Nicotera, P. (1995). Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15, 961–973.
Artus, C., Boujrad, H., Bouharrour, A., Brunelle, M. N., Hoos, S., Yuste, V. J., Lenormand, P., Rousselle, J. C., Namane, A., England, P., Lorenzo, H. K., and Susin, S. A. (2010). AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 29, 1585–1599.
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., Brunskill, E. W., Sayen, M. R., Gottlieb, R. A., Dorn, G. W., Robbins, J., and Molkentin, J. D. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662.
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., and Molkentin, J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555.
Bajt, M. L., Farhood, A., Lemasters, J. J., and Jaeschke, H. (2008). Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 324, 8–14.
Baritaud, M., Boujrad, H., Lorenzo, H. K., Krantic, S., and Susin, S. A. (2010). Histone H2AX: The missing link in AIF-mediated caspase-independent programmed necrosis. Cell Cycle 9, 3166–3173.
Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M. A., and Bernardi, P. (2005). Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 280, 18558–18561.
Basso, E., Petronilli, V., Forte, M. A., and Bernardi, P. (2008). Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J. Biol. Chem. 283, 26307–26311.
Ben-Ari, Z., Pappo, O., Cheporko, Y., Yasovich, N., Offen, D., Shainberg, A., Leshem, D., Sulkes, J., Vidne, B. A., and Hochhauser, E. (2007). Bax ablation protects against hepatic ischemia/reperfusion injury in transgenic mice. Liver Transpl. 13, 1181–1188.
Boujrad, H., Gubkina, O., Robert, N., Krantic, S., and Susin, S. A. (2007). AIF-mediated programmed necrosis: a highly regulated way to die. Cell Cycle 6, 2612–2619.
Burton, T. R., and Gibson, S. B. (2009). The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 16, 515–523.
Byun, H. S., Won, M., Park, K. A., Kim, Y. R., Choi, B. L., Lee, H., Hong, J. H., Piao, L., Park, J., Kim, J. M., Kweon, G. R., Kang, S. H., Han, J., and Hur, G. M. (2008). Prevention of TNF-induced necrotic cell death by rottlerin through a Nox1 NADPH oxidase. Exp. Mol. Med. 40, 186–195.
Chen, H. C., Wang, C. J., Chou, C. L., Lin, S. M., Huang, C. D., Lin, T. Y., Wang, C. H., Lin, H. C., Yu, C. T., Kuo, H. P., and Liu, C. Y. (2006). Tumor necrosis factor-α induces caspase-independent cell death in human neutrophils via reactive oxidants and associated with calpain activity. J. Biomed. Sci. 13, 261–273.
Chen, Y., Lewis, W., Diwan, A., Cheng, E. H., Matkovich, S. J., and Dorn, G. W. (2010). Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 107, 9035–9042.
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., and Chan, F. K. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123.
Crompton, M., Ellinger, H., and Costi, A. (1988). Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 255, 357–360.
Daugas, E., Susin, S. A., Zamzami, N., Ferri, K. F., Irinopoulou, T., Larochette, N., Prévost, M. C., Leber, B., Andrews, D., Penninger, J., and Kroemer, G. (2000). Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 14, 729–739.
Davis, C. W., Hawkins, B. J., Ramasamy, S., Irrinki, K. M., Cameron, B. A., Islam, K., Daswani, V. P., Doonan, P. J., Manevich, Y., and Madesh, M. (2010). Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic. Biol. Med. 48, 306–317.
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., Abbott, D., Cuny, G. D., Yuan, C., Wagner, G., Hedrick, S. M., Gerber, S. A., Lugovskoy, A., and Yuan, J. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321.
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., Cuny, G. D., Mitchison, T. J., Moskowitz, M. A., and Yuan, J. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119.
Diwan, A., Krenz, M., Syed, F. M., Wansapura, J., Ren, X., Koesters, A. G., Li, H., Kirshenbaum, L. A., Hahn, H. S., Robbins, J., Jones, W. K., and Dorn, G. W. (2007a). Inhibition of ischemic cardiomyocytes apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Invest. 117, 2825–2833.
Diwan, A., Koesters, A. G., Odley, A. M., Pushkaran, S., Baines, C. P., Spike, B. T., Daria, D., Jegga, A. G., Geiger, H., Aronow, B. J., Molkentin, J. D., Macleod, K. F., Kalfa, T. A., and Dorn, G. W. (2007b). Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 104, 6794–6799.
Diwan, A., Matkovich, S. J., Yuan, Q., Zhao, W., Yatani, A., Brown, J. H., Molkentin, J. D., Kranias, E. G., and Dorn, G. W. (2009). Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J. Clin. Invest. 119, 203–212.
Dodoni, G., Canton, M., Petronilli, V., Bernardi, P., and Di Lisa, F. (2004). Induction of the mitochondrial permeability transition by the DNA alkylating agent N-methyl-N’-nitro-N-nitrosoguanidine. Sorting cause and consequence of mitochondrial dysfunction. Biochim. Biophys. Acta Bioenerg. 1658, 58–63.
Duchen, M. R., McGuinness, O., Brown, L. A., and Crompton, M. (1993). On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc. Res. 27, 1790–1794.
Festjens, N., Vanden Berghe, T., Cornelis, S., and Vandenabeele, P. (2007). RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 14, 400–410.
Frenzel, A., Labi, V., Chmelewskij, W., Ploner, C., Geley, S., Fiegl, H., Tzankov, A., and Villunger, A. (2010). Suppression of B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood. 115, 995–1005.
Golstein, P., and Kroemer, G. (2007). Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37–43.
Goossens, V., Grooten, J., De Vos, K., and Fiers, W. (1995). Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 92, 8115–8119.
Goossens, V., Stangé, G., Moens, K., Pipeleers, D., and Grooten, J. (1999). Regulation of tumor necrosis factor-induced, mitochondria- and reactive oxygen species-dependent cell death by the electron flux through the electron transport chain complex I. Antioxid. Redox Signal. 1, 285–295.
Griffiths, E. J., and Halestrap, A. P. (1993). Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell Cardiol. 25, 1461–1469.
Halestrap, A. P. (2009). What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 46, 821–831.
Hamacher-Brady, A., Brady, N. R., Logue, S. E., Sayen, M. R., Jinno, M., Kirshenbaum, L. A., Gottlieb, R. A., and Gustafsson, A. B. (2007). Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 14, 146–157.
Hassa, P. O. (2009). The molecular “Jekyll and Hyde” duality of PARP1 in cell death and cell survival. Front. Biosci. 14, 72–111.
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., and Wang, X. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNFα. Cell 137, 1100–1111.
Hernando, V., Inserte, J., Sartório, C. L., Parra, V. M., Poncelas-Nozal, M., and Garcia-Dorado, D. (2010). Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell. Cardiol. 49, 271–279.
Herrero-Yraola, A., Bakhit, S. M., Franke, P., Weise, C., Schweiger, M., Jorcke, D., and Ziegler, M. (2001). Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. EMBO J. 20, 2404–2412.
Hetz, C., Vitte, P. A., Bombrun, A., Rostovtseva, T. K., Montessuit, S., Hiver, A., Schwarz, M. K., Church, D. J., Korsmeyer, S. J., Martinou, J. C., and Antonsson, B. (2005). Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem. 280, 42960–42970.
Hitomi, J., Christofferson, D. E., Ng, A., Yao, J., Degterev, A., Xavier, R. J., and Yuan, J. (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323.
Hochhauser, E., Kivity, S., Offen, D., Maulik, N., Otani, H., Barhum, Y., Pannet, H., Shneyvays, V., Shainberg, A., Goldshtaub, V., Tobar, A., and Vidne, B. A. (2003). Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 284, H2351–H2359.
Hoffmeyer, M. R., Jones, S. P., Ross, C. R., Sharp, B., Grisham, M. B., Laroux, F. S., Stalker, T. J., Scalia, R., and Lefer, D. J. (2000). Myocardial ischemia/reperfusion injury in NADPH oxidase-deficient mice. Circ. Res. 87, 812–817.
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., Bodmer, J. L., Schneider, P., Seed, B., and Tschopp, J. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495.
Hunter, D. R., and Haworth, R. A. (1979). The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 195, 453–459.
Jackman, K. A., Miller, A. A., De Silva, T. M., Crack, P. J., Drummond, G. R., and Sobey, C. G. (2009). Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br. J. Pharmacol. 156, 680–688.
Kahles, T., Kohnen, A., Heumueller, S., Rappert, A., Bechmann, I., Liebner, S., Wittko, I. M., Neumann-Haefelin, T., Steinmetz, H., Schroeder, K., and Brandes, R. P. (2010). NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol. Dis. 40, 185–192.
Kasof, G. M., Prosser, J. C., Liu, D., Lorenzi, M. V., and Gomes, B. C. (2000). The RIP-like kinase, RIP3, induces apoptosis and NF-kappaB nuclear translocation and localizes to mitochondria. FEBS Lett. 473, 285–291.
Kennedy, C. L., Smith, D. J., Lyras, D., Chakravorty, A., and Rood, J. I. (2009). Programmed cellular necrosis mediated by the pore-forming α-toxin from Clostridium septicum. PLoS Pathog. 5, e1000516. doi: 10.1371/journal.ppat.1000516.
Kim, J. S., Ahn, K. J., Kim, J. A., Kim, H. M., Lee, J. D., Lee, J. M., Kim, S. J., and Park, J. H. (2008). Role of reactive oxygen species-mediated mitochondrial dysregulation in 3-bromopyruvate induced cell death in hepatoma cells : ROS-mediated cell death by 3-BrPA. J. Bioenerg. Biomembr. 40, 607–618.
Kim, Y. S., Morgan, M. J., Choksi, S., and Liu, Z. G. (2007). TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 26, 675–687.
Klein, J. A., Longo-Guess, C. M., Rossmann, M. P., Seburn, K. L., Hurd, R. E., Frankel, W. N., Bronson, R. T., and Ackerman, S. L. (2002). The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419, 367–374.
Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., MacGregor, G. R., and Wallace, D. C. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465.
Krauskopf, A., Eriksson, O., Craigen, W. J., Forte, M. A., and Bernardi, P. (2006). Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim. Biophys. Acta 1757, 590–595.
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., El-Deiry, W. S., Golstein, P., Green, D. R., Hengartner, M., Knight, R. A., Kumar, S., Lipton, S. A., Malorni, W., Nuñez, G., Peter, M. E., Tschopp, J., Yuan, J., Piacentini, M., Zhivotovsky, B., and Melino, G., and Nomenclature Committee on Cell Death 2009. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11.
Kubasiak, L. A., Hernandez, O. M., Bishopric, N. H., and Webster, K. A. (2002). Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. U.S.A. 99, 12825–12830.
Kubli, D. A., Ycaza, J. E., and Gustafsson, A. B. (2007). Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 405, 407–415.
Labi, V., Erlacher, M., Kiessling, S., Manzl, C., Frenzel, A., O’Reilly, L., Strasser, A., and Villunger, A. (2008). Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J. Exp. Med. 205, 641–655.
Lai, Y., Chen, Y., Watkins, S. C., Nathaniel, P. D., Guo, F., Kochanek, P. M., Jenkins, L. W., Szabó, C., and Clark, R. S. (2008). Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 104, 1700–1711.
Lebart, M. C., and Benyamin, Y. (2006). Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J. 273, 3415–3426.
Lee, C. F., Qiao, M., Schröder, K., Zhao, Q., and Asmis, R. (2010). Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 106, 1489–1497.
Lehnert, M., Arteel, G. E., Smutney, O. M., Conzelmann, L. O., Zhong, Z., Thurman, R. G., and Lemasters, J. J. (2003). Dependence of liver injury after hemorrhage/resuscitation in mice on NADPH oxidase-derived superoxide. Shock 19, 345–351.
Li, X., Klaus, J. A., Zhang, J., Xu, Z., Kibler, K. K., Andrabi, S. A., Rao, K., Yang, Z. J., Dawson, T. M., Dawson, V. L., and Koehler, R. C. (2010). Contributions of poly (ADP-ribose) polymerase-1 and -2 to nuclear translocation of apoptosis-inducing factor and injury from focal cerebral ischemia. J. Neurochem. 113, 1012–1022.
Lin, Y., Choksi, S., Shen, H. M., Yang, Q. F., Hur, G. M., Kim, Y. S., Tran, J. H., Nedospasov, S. A., and Liu, Z. G. (2004). Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279, 10822–10828.
Masubuchi, Y., Suda, C., and Horie, T. (2005). Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol. 42, 110–116.
Millay, D. P., Sargent, M. A., Osinska, H., Baines, C. P., Barton, E. R., Vuagniaux, G., Sweeney, H. L., Robbins, J., and Molkentin, J. D. (2008). Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 14, 442–447.
Moquin, D., and Chan, F. K. (2010). The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 35, 434–441.
Moubarak, R. S., Yuste, V. J., Artus, C., Bouharrour, A., Greer, P. A., Menissier-de Murcia, J., and Susin, S. A. (2007). Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 27, 4844–4862.
Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H., Kubo, T., and Tsujimoto, Y. (2005). Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658.
Nakayama, H., Chen, X., Baines, C. P., Klevitsky, R., Zhang, X., Zhang, H., Jaleel, N., Chua, B. H., Hewett, T. E., Robbins, J., Houser, S. R., and Molkentin, J. D. (2007). Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Invest. 117, 2431–2444.
Pankotai, E., Lacza, Z., Murányi, M., and Szabó, C. (2009). Intra-mitochondrial poly(ADP-ribosyl)ation: potential role for alpha-ketoglutarate dehydrogenase. Mitochondrion 9, 159–164.
Rosenbaum, D. M., Degterev, A., David, J., Rosenbaum, P. S., Roth, S., Grotta, J. C., Cuny, G. D., Yuan, J., and Savitz, S. I. (2010). Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res. 88, 1569–1576.
Rossi, M. N., Carbone, M., Mostocotto, C., Mancone, C., Tripodi, M., Maione, R., and Amati, P. (2009). Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 284, 31616–31624.
Sancho, P., Fernández, C., Yuste, V. J., Amrán, D., Ramos, A. M., de Blas, E., Susin, S. A., and Aller, P. (2006). Regulation of apoptosis/necrosis execution in cadmium-treated human promonocytic cells under different forms of oxidative stress. Apoptosis 11, 673–686.
Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., Hetz, C., Danial, N. N., Moskowitz, M. A., and Korsmeyer, S. J. (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010.
Schulze-Osthoff, K., Bakker, A. C., Vanhaesebroeck, B., Beyaert, R., Jacob, W. A., and Fiers, W. (1992). Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 267, 5317–5323.
Smith, C. C., Davidson, S. M., Lim, S. Y., Simpkin, J. C., Hothersall, J. S., and Yellon, D. M. (2007). Necrostatin: a potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 21, 227–233.
Starkov, A. A., Fiskum, G., Chinopoulos, C., Lorenzo, B. J., Browne, S. E., Patel, M. S., and Beal, M. F. (2004). Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788.
Sun, X., Yin, J., Starovasnik, M. A., Fairbrother, W. J., and Dixit, V. M. (2002). Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 277, 9505–9511.
Szabó, I., De Pinto, V., and Zoratti, M. (1993). The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett. 330, 206–210.
Temkin, V., Huang, Q., Liu, H., Osada, H., Pope, R. M. (2006). Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol. Cell Biol. 26, 2215–2225.
Tiepolo, T., Angelin, A., Palma, E., Sabatelli, P., Merlini, L., Nicolosi, L., Finetti, F., Braghetta, P., Vuagniaux, G., Dumont, J. M., Baldari, C. T., Bonaldo, P., and Bernardi, P. (2009). The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1-/- myopathic mice. Br. J. Pharmacol. 157, 1045–1052.
Vahsen, N., Candé, C., Brière, J. J., Bénit, P., Joza, N., Larochette, N., Mastroberardino, P. G., Pequignot, M. O., Casares, N., Lazar, V., Feraud, O., Debili, N., Wissing, S., Engelhardt, S., Madeo, F., Piacentini, M., Penninger, J. M., Schägger, H., Rustin, P., and Kroemer, G. (2004). AIF deficiency compromises oxidative phosphorylation. EMBO J. 23, 4679–4689.
Vandenabeele, P., Vanden Berghe, T., and Festjens, N. (2006). Caspase inhibitors promote alternative cell death pathways. Sci. STKE 358, pe44.
Vande Velde, C., Cizeau, J., Dubik, D., Alimonti, J., Brown, T., Israels, S., Hakem, R., and Greenberg, A. H. (2000). BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 20, 5454–5468.
van Empel, V. P., Bertrand, A. T., van Oort, R. J., van der Nagel, R., Engelen, M., van Rijen, H. V., Doevendans, P. A., Crijns, H. J., Ackerman, S. L., Sluiter, W., and De Windt, L. J. (2006). EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J. Am. Coll. Cardiol. 48, 824–832.
van Wijk, S. J., and Hageman, G. J. (2005). Poly(ADP-ribose) polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic. Biol. Med. 39, 81–90.
Wang, Y., Kim, N. S., Li, X., Greer, P. A., Koehler, R. C., Dawson, V. L., and Dawson, T. M. (2009). Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J. Neurochem. 110, 687–696.
Xiao, C. Y., Chen, M., Zsengellér, Z., and Szabó, C. (2004). Poly(ADP-ribose) polymerase contributes to the development of myocardial infarction in diabetic rats and regulates the nuclear translocation of apoptosis-inducing factor. J. Pharmacol. Exp. Ther. 310, 498–504.
Xu, Y., Huang, S., Liu, Z. G., and Han, J. (2006). Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J. Biol. Chem. 281, 8788–8795.
Yamashima, T., and Oikawa, S. (2009). The role of lysosomal rupture in neuronal death. Prog. Neurobiol. 89, 343–358.
Yu, S. W., Andrabi, S. A., Wang, H., Kim, N. S., Poirier, G. G., Dawson, T. M., and Dawson, V. L. (2006). Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18314–18319.
Yu, S. W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., Poirier, G. G., Dawson, T. M., and Dawson, V. L. (2002). Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263.
Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., Dong, M. Q., and Han, J. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336.
Zheng, L., Bidere, N., Staudt, D., Cubre, A., Orenstein, J., Chan, F. K., and Lenardo, M. (2006). Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol. Cell. Biol. 26, 3505–3513.
Zhou, H. Z., Swanson, R. A., Simonis, U., Ma, X., Cecchini, G., and Gray, M. O. (2006). Poly(ADP-ribose) polymerase-1 hyperactivation and impairment of mitochondrial respiratory chain complex I function in reperfused mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 291, H714–H723.
Zhu, C., Wang, X., Huang, Z., Qiu, L., Xu, F., Vahsen, N., Nilsson, M., Eriksson, P. S., Hagberg, H., Culmsee, C., Plesnila, N., Kroemer, G., and Blomgren, K. (2007). Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 14, 775–784.
Keywords: necrosis, mitochondria, RIP kinases, PARP, reactive oxygen species
Citation: Baines CP (2010) Role of the mitochondrion in programmed necrosis. Front. Physio. 1:156. doi: 10.3389/fphys.2010.00156
Received: 22 August 2010;
Paper pending published: 16 September 2010;
Accepted: 16 November 2010;
Published online: 29 November 2010.
Edited by:
Miguel A. Aon, Johns Hopkins University School of Medicine, USAReviewed by:
Roberta A. Gottlieb, San Diego State University, USAPaolo Bernardi, University of Padova, Italy
Copyright: © 2010 Baines. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Christopher P. Baines, Department of Biomedical Sciences, Dalton Cardiovascular Research Center, University of Missouri-Columbia, 134 Research Park Drive, Columbia, MO 65211, USA. e-mail: bainesc@missouri.edu