
- Department of Medical Pharmacology and Physiology, Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, USA
The vascular myogenic response is characterized by arterial constriction in response to an increase in intraluminal pressure and dilatation to a decrease in pressure. This mechanism is important for the regulation of blood flow, capillary pressure, and arterial pressure. The identity of the mechanosensory mechanism(s) for this response is incompletely understood but has been shown to include the integrins as cell–extracellular matrix receptors. The possibility that a cell–cell adhesion receptor is involved has not been studied. Thus, we tested the hypothesis that N-cadherin, a cell–cell adhesion molecule in vascular smooth muscle cells (VSMCs), was important for myogenic responsiveness. The purpose of this study was to investigate: (1) whether cadherin inhibition blocks myogenic responses to increases in intraluminal pressure and (2) the effect of the cadherin or integrin blockade on pressure-induced changes in [Ca2+]i. Cadherin blockade was tested in isolated rat cremaster arterioles on myogenic responses to acute pressure steps from 60 to 100 mmHg and changes in VSMC Ca2+ were measured using fura-2. In the presence of a synthetic cadherin inhibitory peptide or a function-blocking antibody, myogenic responses were inhibited. In contrast, during N-cadherin blockade, pressure-induced changes in [Ca2+]i were not altered. Similarly, vessels treated with function-blocking β1- or β3-integrin antibodies maintained pressure-induced [Ca2+]i responses despite inhibition of myogenic constriction. Collectively, these data suggest that both cadherins and integrins play a fundamental role in mediating myogenic constriction but argue against their direct involvement in mediating pressure-induced [Ca2+]i increases.
Introduction
The myogenic response refers to the rapid vasomotor reaction of blood vessels to changes in intraluminal pressure. The vasomotor response is characterized by a blood vessel’s ability to constrict to an increase in intraluminal pressure and to dilate following a decrease in pressure. This mechanism, inherent to vascular smooth muscle, aids in the autoregulation of blood flow, arterial and capillary pressure, and maintenance of peripheral vascular resistance. Importantly, myogenic constriction provides a level of tone upon which vasodilators can act to lower peripheral resistance. In addition to playing a role in local control of microvascular hemodynamics, abnormalities in myogenic responsiveness have been associated with the vascular complications seen in hypertension and diabetes (Meininger and Trzeciakowski, 1988; Hill and Meininger, 1993; Schofield et al., 2002).
A number of cellular processes and signals, including activation/deactivation of ion channels and kinases/phosphatases and changes in [Ca2+]i, have been implicated in myogenic contraction (Jaggar et al., 1998; Zou et al., 2000; Wesselman et al., 2001; Cipolla et al., 2002; Bolz et al., 2003; Gokina et al., 2005).Specifically, with respect to [Ca2+]i, an increase in wall tension or cell stretch (i.e., resulting from an increase in pressure) leads to vascular smooth muscle cell (VSMC) membrane depolarization via activation of non-selective cation channels with subsequent voltage-gated Ca2+ entry leading to contraction of smooth muscle cells (Harder, 1984; Davis et al., 1992a,b; Zou et al., 1995).Thus, Ca2+ is generally perceived to play a pivotal and central role in the myogenic response. There are several reviews of this topic that provide a more detailed overview of possible cellular mechanisms contributing to the myogenic response (Johnson, 1977; Davis and Hill, 1999; Schubert and Mulvany, 1999; Hill et al., 2006).
In comparison to the information available on the intracellular signals involved in myogenic contraction, the identity of the sensory elements that can detect changes in intraluminal pressure remains incomplete. Specifically, less emphasis has been placed on the role of cell adhesion proteins, cytoskeletal elements, and extracellular matrix (ECM) proteins. Previous research in our laboratory has shown that inhibition of αvβ3 and α5β1-integrins prevents myogenic responsiveness in cremaster arterioles (Martinez-Lemus et al., 2005a) demonstrating the importance of a cell–ECM interaction. In addition to cell–matrix contacts the vascular wall contains a variety of cell–cell junctions (Hill et al., 2009) that have yet to be considered with respect to a role in myogenic responsiveness. Therefore, we considered the hypothesis that cell–cell adhesive interactions involving cadherins, a family of calcium-dependent transmembrane adhesion proteins, contribute to the mechanosensory pathway(s) that “sense” force and coordinate signaling between smooth muscle cells.
To test the hypothesis that cadherins are an important component in the myogenic response, intraluminal pressure was increased in cremaster arterioles in the absence and presence of an inhibitory antibody known to bind selectively N-cadherin or a cadherin inhibitory peptide (histidine–alanine–valine, HAV). N-cadherin was selected for investigation because previous reports have identified it as the prominent VSMC cadherin (Moiseeva, 2001). Additional studies, using the calcium-sensitive dye fura-2, examined whether cadherin and/or integrin signaling lie upstream of changes in [Ca2+]i.
Materials and Methods
Animal Handling
All experimental procedures were approved by the University of Missouri-Columbia Institutional Animal Care and Use Committee. Male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA) weighing 200–300 g were used in this study. The animals were anesthetized with an intraperitoneal injection of pentobarbital sodium (100 mg/Kg).
Vessel Isolation
After reaching a surgical level of anesthesia, the cremaster muscle was excised and pinned flat in a refrigerated (4°C) dissecting chamber containing a physiological saline solution (PSS) of the composition (in mM): 145.0 NaCl, 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4, 1.0 NaH2PO4, 5.0 D-glucose, 3.0 MOPS buffer, 2.0 pyruvate, 0.02 EDTA, and 0.15 BSA, pH 7.4. First-order feed arterioles were isolated from the muscle, cannulated in the absence of flow using two glass micropipettes, and pressurized to 60 mmHg as previously described (Falcone et al., 1991). Vessel preparations were superfused with a PSS (without albumin) as described in previous publications (Falcone et al., 1991; Meininger et al., 1991). The arteriole lumen diameter was recorded online using a video microscopy system and a calibrated video dimension analysis device.
Immunofluorescent Labeling of VSMCs
As an approach for immunohistochemical identification of N-cadherin, VSMCs were isolated from cremaster arterioles as previously described (Wu et al., 1998). Cells were allowed to grow until 50% confluent on glass-bottom tissue culture dishes, washed with DPBS, and fixed with 2% paraformaldehyde. After washing, glycine buffer (0.1 mM glycine) was added to quench any remaining paraformaldehyde. Subsequently, cells were incubated with a monoclonal N-cadherin primary antibody (clone GC-4; Sigma) at 4°C overnight. Cells were then washed six times with cold buffer (composed of 150 mM NaCl, 15 mM Na3C6H5O7, and 0.05% Triton X-100), followed by incubation with Alexa Fluor 488-conjugated secondary antibody (for 1 h at room temperature in a dark environment). Labeled cells were washed six times with cold buffer and imaged on a confocal microscope.
Antibody and Peptide Treatment
All antibodies and peptides were diluted in PSS and added abluminally to the cannulated arterioles in the superfusion solution. To block VSMC N-cadherin, arterioles were exposed to the HAV (476 μM) sequence-containing peptide LRAHAVDVNG (leucine–arginine–alanine–histidine–alanine–valine–aspartic acid–valine–asparagine–glycine-NH2) peptide (Bachem). The HGV-containing LRAHGVDVNG (leucine–arginine–alanine–histidine–glycine–valine–aspartic acid–valine–asparagine–glycine-NH2) peptide (476 μM; Bachem) was used similarly as an inactive control peptide. VSMC N-cadherin was also blocked with an inhibitory antibody, 200 μg/ml anti-N-cadherin clone GC-4 (Sigma). Two hundred microgram per milliliter of the anti-VE-cadherin (Sigma) was used as a control antibody.
Vascular smooth muscle cell β1- or β3-integrins were blocked with the following antibodies: 50 μg/ml anti-β3-integrin function-blocking antibody F11 or 50 μg/ml anti-β1-integrin antibody Ha2/5 (Pharmingen) as previously described in detail (Martinez-Lemus et al., 2005a).
[Ca2+]i and Vessel Diameter Measurements
Changes in [Ca2+]i were measured following incubation of vessels with 2.0 μM Fura-2-acetoxymethyl ester (Fura-2-AM; Molecular Probes) for 60–90 min at room temperature in PSS (without albumin) containing 0.5% dimethyl sulfoxide (DMSO) and 0.01% Pluronic F-127. The dye loading procedure was followed by a 30-min washout period with fresh, heated PSS (without albumin) solution. Details of this protocol have been previously published (Meininger et al., 1991).
Experimental Protocols
Effect of N-Cadherin Inhibition on Myogenic Responsiveness
Arterioles (60 mmHg intraluminal pressure) were warmed to 34.5°C for 1 h and allowed to develop spontaneous myogenic tone. A control series of step increments in intraluminal pressure (60–100, 20 mmHg increments) was performed at 5 min intervals. Diameter was recorded by a video microscopy system. After the initial series of pressure steps, the vessel was returned to 60 mmHg for 15 min. The arteriole was then incubated with either the N-cadherin antibody or peptide for 15 min. Next, a second series of pressure steps were performed in the continued presence of the antibody or peptide. The vessel was then returned to a pressure of 60 mmHg and washed with fresh PSS without the antibody or peptide for 15 min. At the completion of the blocking protocol, the arteriole was dilated with 100 μM adenosine and then followed by calcium-free PSS containing 100 μM adenosine to acquire the maximum passive diameter. Details of this protocol have been published previously (Martinez-Lemus et al., 2005a). Appropriate time controls were conducted to ensure reproducibility of the myogenic responses across the period of the study.
[Ca2+]i Measurements during Intraluminal Increases in Pressure
After vessels were incubated with the Ca2+ indicator, fura-2, the response to a step increment (60–100 mmHg) in intraluminal pressure was examined. The intraluminal pressure was returned to 60 mmHg for 10 min after which antibody or peptide was applied for 10 min. Intraluminal pressure was then increased from 60 to 100 mmHg for 5 min and changes in [Ca2+]i and diameter recorded. Intraluminal pressure was returned to 60 mmHg for 10 min followed by exposure of the vessel to 10 μM norepinephrine (in the presence of the antibody or peptide). Maximum diameter was measured following superfusion with calcium-free PSS containing 100 μM adenosine as above (Falcone et al., 1991; Meininger et al., 1991).
Data Analysis and Statistics
Diameter changes are presented as percentage of control, where control represents the pre-pressure step diameter value at 60 mmHg. The slope of the myogenic pressure–diameter relationship as determined by regression analysis was used as an index of myogenic gain. Thus, a negative slope of the myogenic pressure–diameter relationship represents myogenic vasoconstriction (i.e., the arterioles ability to reduce its diameter in response to an increase in intraluminal pressure). Analyses of slopes and calcium changes were performed using paired Student t-tests. All data are expressed as mean ± SEM. In all analyses, p < 0.05 represents the level of significance.
Results
Presence of N-Cadherin in VSMCs
To demonstrate the presence of N-cadherin in arteriolar VSMC, immunofluorescence microscopy was performed on cultured arteriolar VSMCs. As shown in Figure 1, N-cadherin was observed in individual VSMCs. N-cadherin was found localized to cell edges and particularly in areas of overlapping cell contact. This demonstrates the presence of N-cadherin and provides support for the hypothesis of possible involvement of this protein in myogenic behavior.
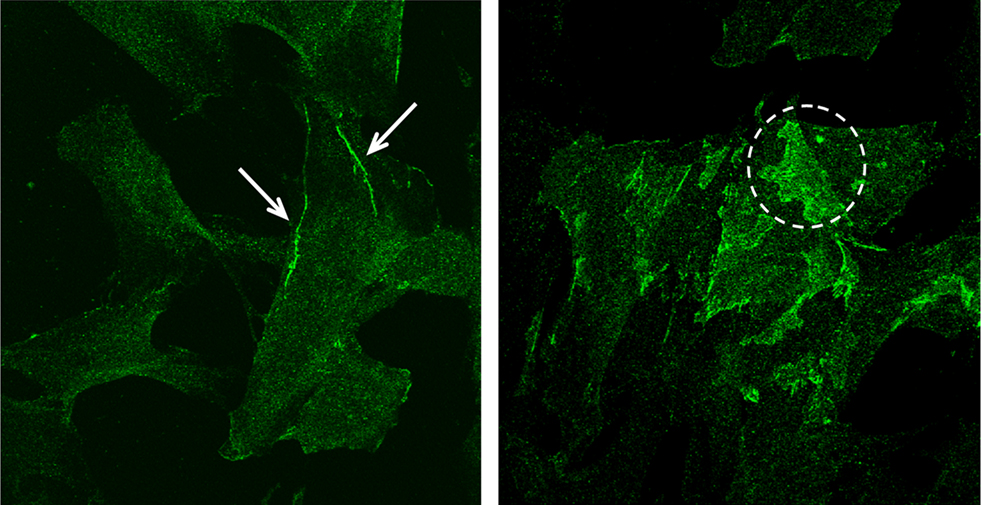
Figure 1. Fluorescent images of cultured VSMCs fluorescently immunolabeled with N-cadherin (green). N-cadherin was distributed along cell edges (arrows in left panel) and in areas of cell–cell overlap between adjacent cells (dashed circle in right panel).
Myogenic Response during N-Cadherin Inhibition is Abolished
Initial control experiments established that myogenic vasoconstrictor responses were reproducible over the time course required to conduct the protocols (Figures 2A,B). Group data showed that the slope of the pressure–diameter response (an index of myogenic reactivity) of an initial series of pressure steps (60 to 80 to 100 mmHg) to be −0.3 ± 0.1 and not statistically different compared to −0.3 ± 0.1 for a second control set of pressure steps performed under identical conditions and in the absence of peptide or antibody treatment (n = 5; Figure 2B).
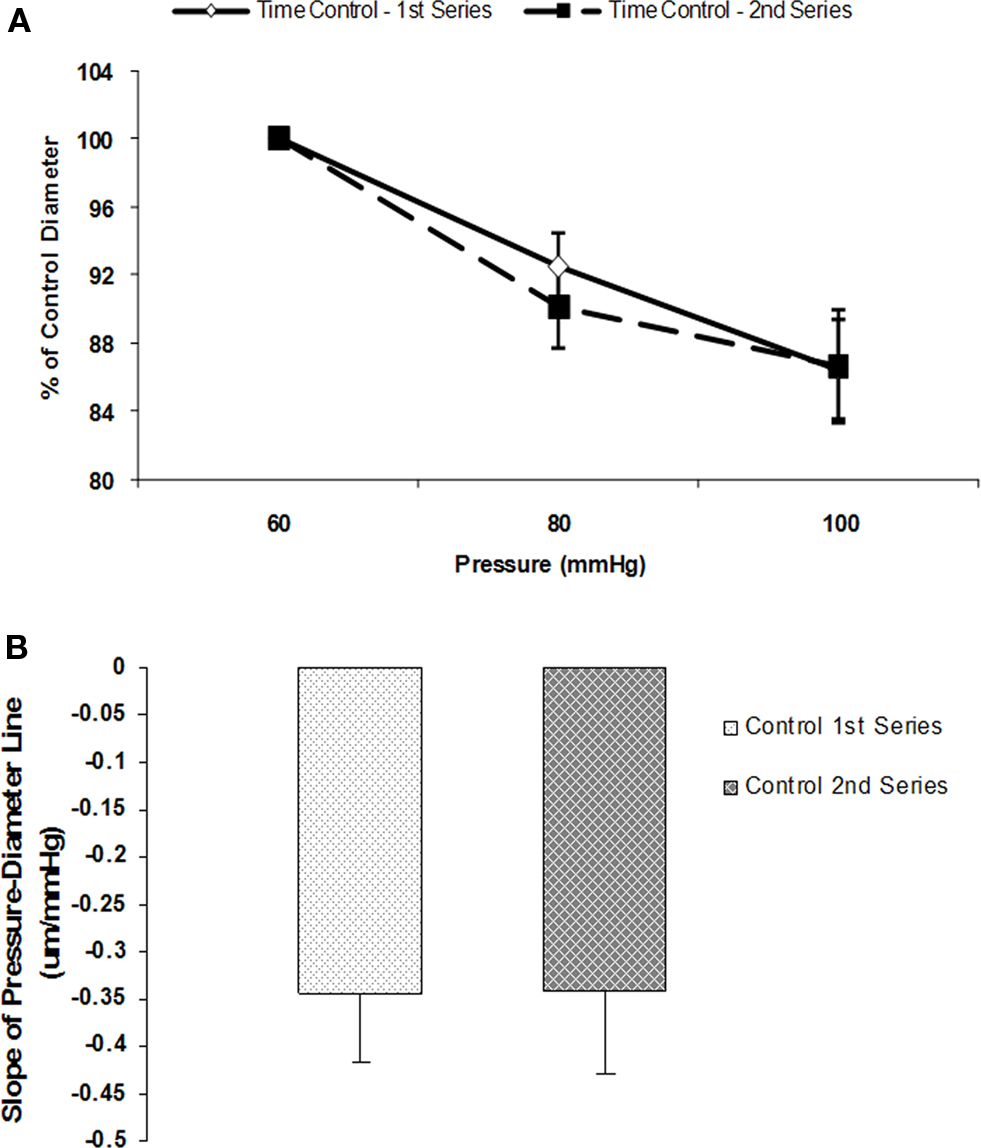
Figure 2. Myogenic responsiveness exhibited in isolated cremaster arterioles. Slope of myogenic pressure–diameter relationship as determined by regression analyses used as an index of myogenic gain. (A) Percentage control diameter of isolated arterioles exposed to increments in intraluminal pressure (time control-no drug treatment). (B) Slope before and after step increments in pressure for the two separate control series (n = 5). Data are presented as mean ± SEM.
To test whether N-cadherin inhibition would affect myogenic responsiveness, we performed two sets of experiments. Following measurement of a baseline response to pressure, inhibitory N-cadherin antibody (GC-4) was applied to the vessel and the effect of the treatment evaluated during a second series of pressure steps. In these experiments, the slope of the initial pressure–diameter response was −0.3 ± 0.04 which was significantly (p < 0.05) different to that after treatment with the antibody (slope not significantly different from 0) indicating inhibition of myogenic vasoconstriction (n = 6; Figures 3A,B). In contrast, treatment of the arterioles with an anti-VE-cadherin antibody, as a control, showed no significant change in myogenic responsiveness between the two series of pressure steps (−0.2 ± 0.05 compared to −0.2 ± 0.02, n = 4; Figures 3C,D).
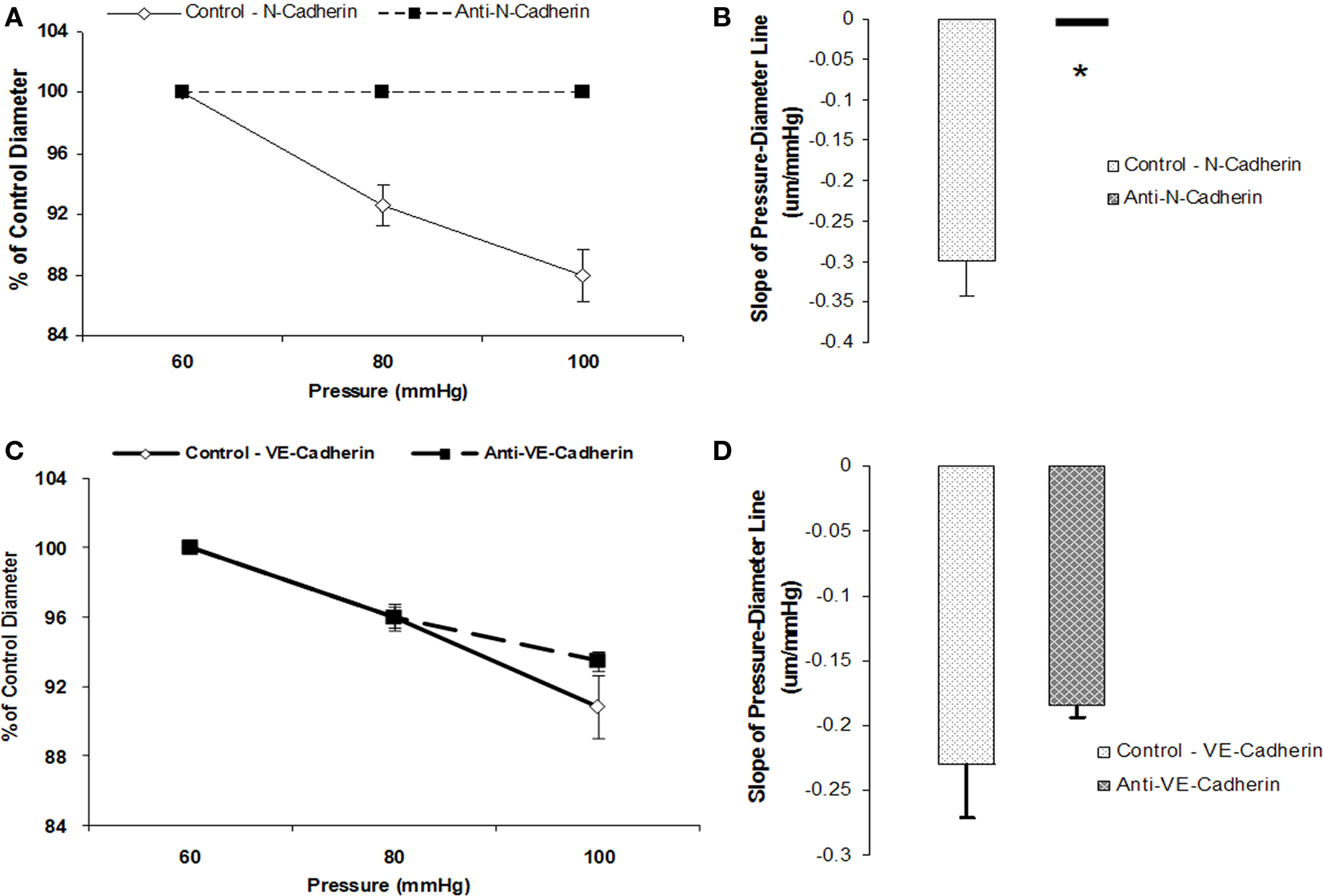
Figure 3. Anti-N-cadherin treatment inhibited pressure-induced myogenic vasoconstriction. The reduced slope of myogenic pressure–diameter relationship (i.e., index of myogenic gain) as determined by regression analyses demonstrates the ability of anti-N-Cadherin treatment to inhibit the myogenic response. (A) Percentage control diameter of isolated arterioles exposed to increments in intraluminal pressure before and after the anti-N-Cadherin antibody treatment. (B) Slope of the pressure–diameter regression line before the addition of the antibody and while in the presence of the inhibitory antibody (n = 6). (C) Percentage control diameter of isolated arterioles exposed to increments in intraluminal pressure before and after addition of the anti-VE-Cadherin antibody treatment as a control. (D) Before the addition of the antibody and while in the presence of the abluminal control antibody (n = 4). Data are presented as mean ± SEM. *Significantly different (p < 0.05) from control (first series of step increments in pressure in the same vessel).
In a second set of experiments, arterioles were treated with the synthetic inhibitory peptide, HAV, which inhibited the myogenic response. The slope of the response to the baseline pressure step was −0.2 ± 0.02 whereas the slope was not significantly different from 0 after treatment with the peptide (n = 6; Figures 4A,B). Vessels treated with a non-inhibitory control HGV-containing peptide did not show a significant change in myogenic responsiveness (slope −0.3 ± 0.03 compared to −0.2 ± 0.04, n = 6; Figures 4C,D).After exposure to either the anti-N-cadherin or HAV peptide, the arterioles continued to exhibit basal tone as resting diameters were not significantly altered, however the vessel myogenic response was abolished by both treatments (Figures 3A and 4A).
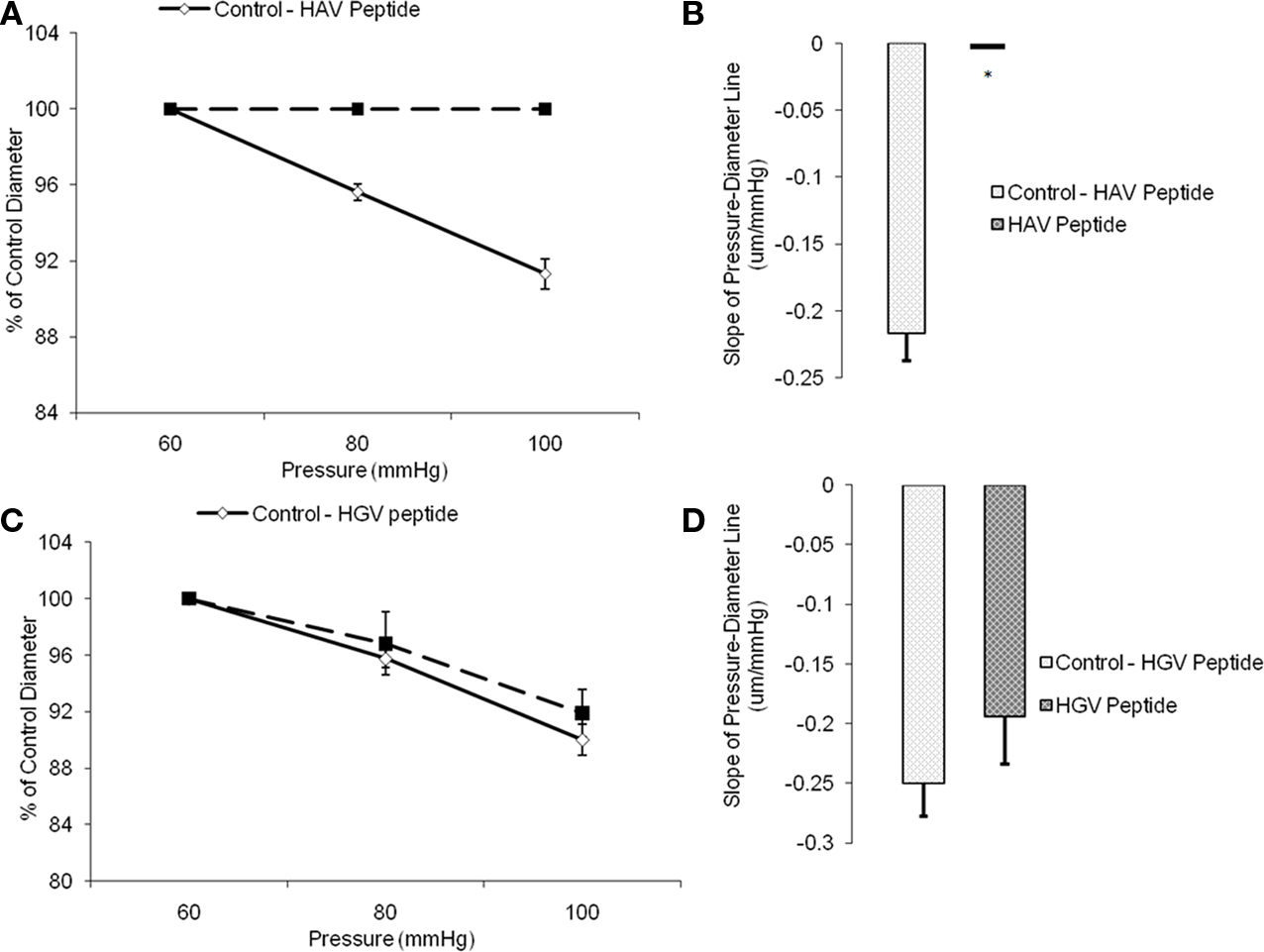
Figure 4. Histidine–alanine–valine peptide inhibited pressure-induced myogenic vasoconstriction. The reduced slope of myogenic pressure–diameter relationship (i.e., index of myogenic gain) as determined by regression analyses demonstrates the ability of HAV peptide to inhibit the myogenic response. (A) Percentage control diameter of isolated arterioles exposed to increments in intraluminal pressure before and after addition of the HAV peptide treatment. (B) Slope of the pressure–diameter regression line before the addition of the antibody and while in the presence of the abluminal inhibitory peptide (n = 6). (C) Percentage control diameter of isolated arterioles exposed to increments in intraluminal pressure before and after the control anti-VE-Cadherin antibody treatment. (D) Slope of the pressure–diameter regression line before the addition of the peptide and while in the presence of the abluminal control peptide (n = 6). Data are presented as mean ± SEM. *Significantly different from the first series of step increments in pressure in the same vessel.
Cadherin or Integrin Blockade Inhibits Myogenic Response while not Affecting [Ca2+]i
Changes in diameter and [Ca2+]i were simultaneously measured and compared before and after (following 5 min and reaching a steady state value) a step change in intravascular pressure (60–100 mmHg). Time controls showed no significant changes in [Ca2+]i or diameter (n = 6; Figure 5). During N-cadherin blockade, myogenic vasoconstriction was significantly reduced when treated with HAV peptide; however, changes in [Ca2+]i were not significantly altered. The first step increase in pressure caused a 11.7 ± 5.8% change in [Ca2+]i and −9.1 ± 2.6% in diameter while the second step increase in pressure exhibited a 6.8 ± 1.3% change in [Ca2+]i and 0.5 ± 0.6% in diameter (n = 9; Figures 6A,B). There were no significant changes in [Ca2+]i or diameter of the arterioles treated with the control HGV peptide (n = 7; Figures 6C,D). Arterioles treated with the inhibitory N-cadherin antibody showed a decrease in pressure-induced myogenic response (percentage of change in diameter −5.0 ± 1.8% compared to −0.9 ± 2.4%) but still showed an increase in [Ca2+]i (17.0 ± 6.7% compared to 11.3 ± 3.9%; n = 4; Figures 7A,B).
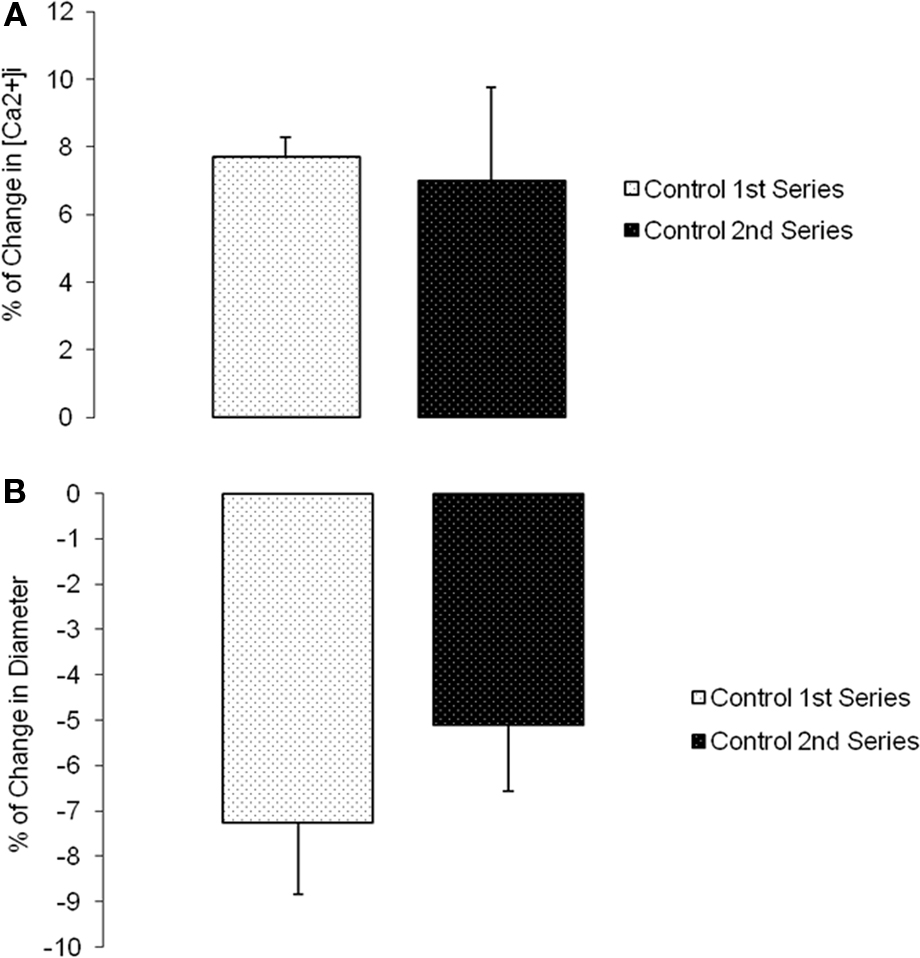
Figure 5. Myogenic response exhibited in following a step change in intravascular pressure from 60 to 100 mmHg in isolated cremaster arterioles. [Ca2+]i was measured in these arterioles using fura-2 to estimate VSMC Ca2+ changes in the vessel wall. (A) Percentage of [Ca2+]i change in arterioles exposed to increases in intraluminal pressure. (B) Percentage of change in diameter before and after repeated step increase in pressure as a time control (n = 6). Data are presented as mean ± SEM.
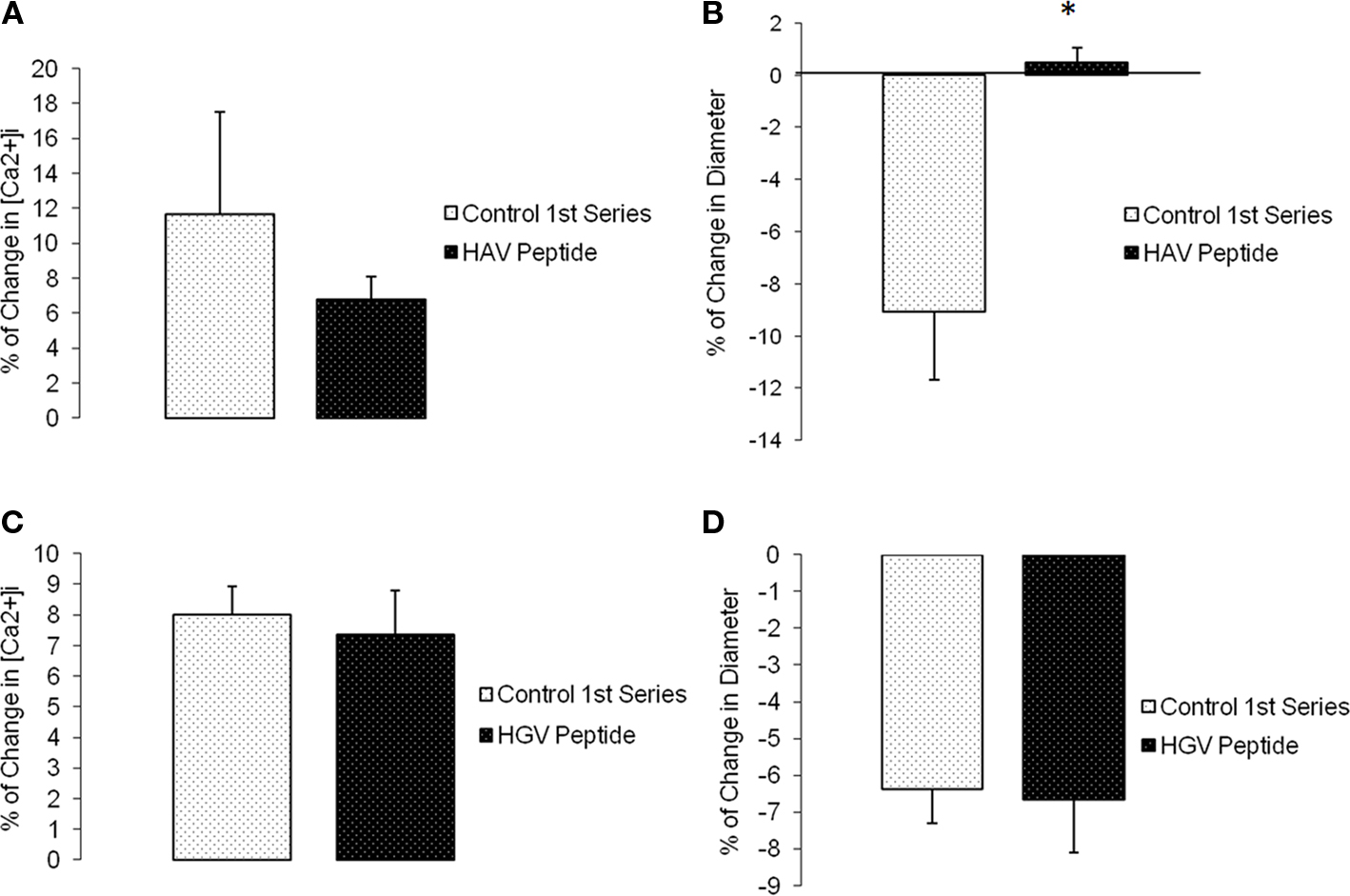
Figure 6. The N-cadherin binding inhibitory peptide, HAV, inhibited pressure-induced myogenic vasoconstriction but not [Ca2+]i changes in the vessel wall. (A) Percentage of [Ca2+]i change in arterioles exposed to an increase in intraluminal pressure before and after HAV peptide treatment. (B) Percentage of change in diameter before and after treatment with the HAV inhibitory peptide (n = 9). (C) Percentage of [Ca2+]i change in arterioles exposed to an increase in intraluminal pressure before and after HGV (control) peptide treatment. (D) Percentage of change in diameter before and after treatment with the HGV control peptide (n = 7). Data are presented as mean ± SEM. *Significantly different from before peptide treatment (first series of step increments in pressure in the same vessel).
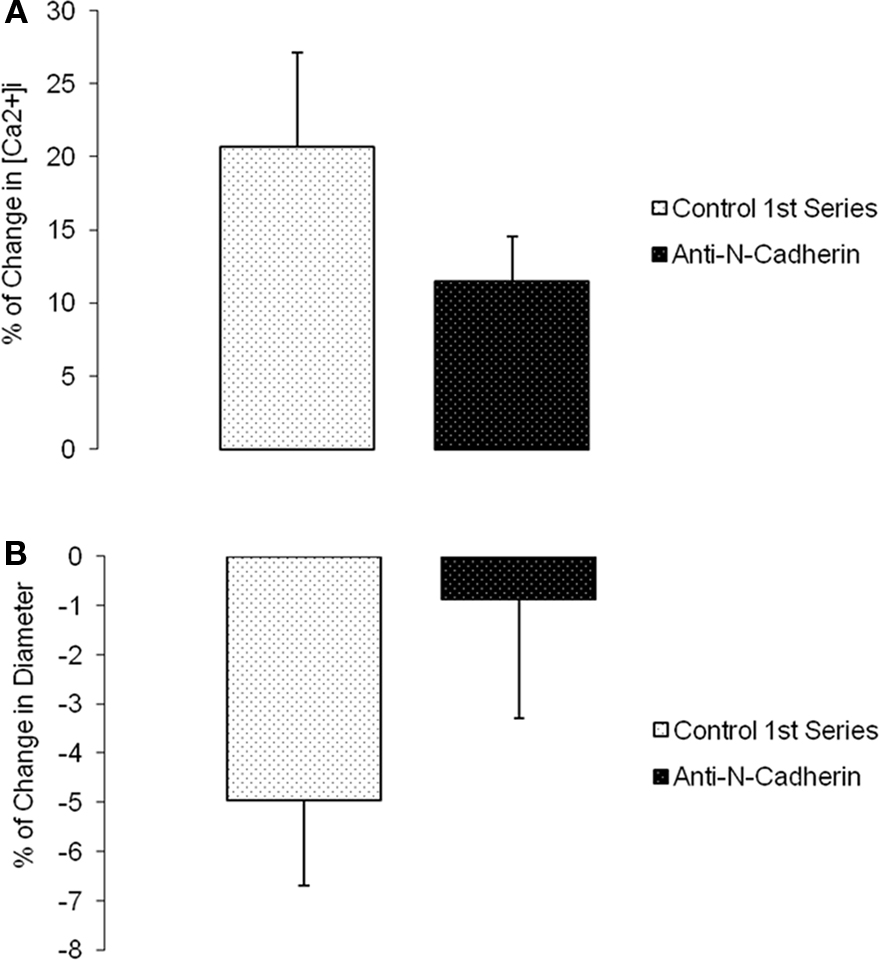
Figure 7. Anti-N-cadherin decreased pressure-induced myogenic vasoconstriction without a significant effect on [Ca2+]i increase in the vessel wall. (A) Percentage of [Ca2+]i change in arterioles exposed to an increase in intraluminal pressure before and after anti-N-cadherin treatment. (B) Percentage of change in diameter before and after treatment with the inhibitory antibody (n = 4). Data are presented as mean ± SEM.
For comparison, we investigated Ca2+ responses in arterioles in which the β1- or β3-integrins were inhibited. Previous published work from our laboratory has shown that inhibition of these integrins blocks myogenic responsiveness (Martinez-Lemus et al., 2005a). During blockade ofβ1-integrin with a function-blocking antibody, myogenic vasoconstriction was attenuated (percentage change in diameter −6.4 ± 1.3% compared to −1.2 ± 2.3%, p < 0.05) when intraluminal pressure was increased from 60 to 100 mmHg (n = 6; Figures 8A,B). However, the pressure-induced change in [Ca2+]i was unaffected; 5.6 ± 1.1% compared to 6.5 ± 1.2%. Similar observations were seen during exposure to the anti β3-integrin antibody (n = 5; Figures 8C,D). Thus, there was a significant inhibition in the diameter response (−8.2 ± 2.0% compared to −3.6 ± 1.2%, p < 0.05) while changes in [Ca2+]i responses were not significantly altered (5.8 ± 1.0% compared to 4.3 ± 0.5%).
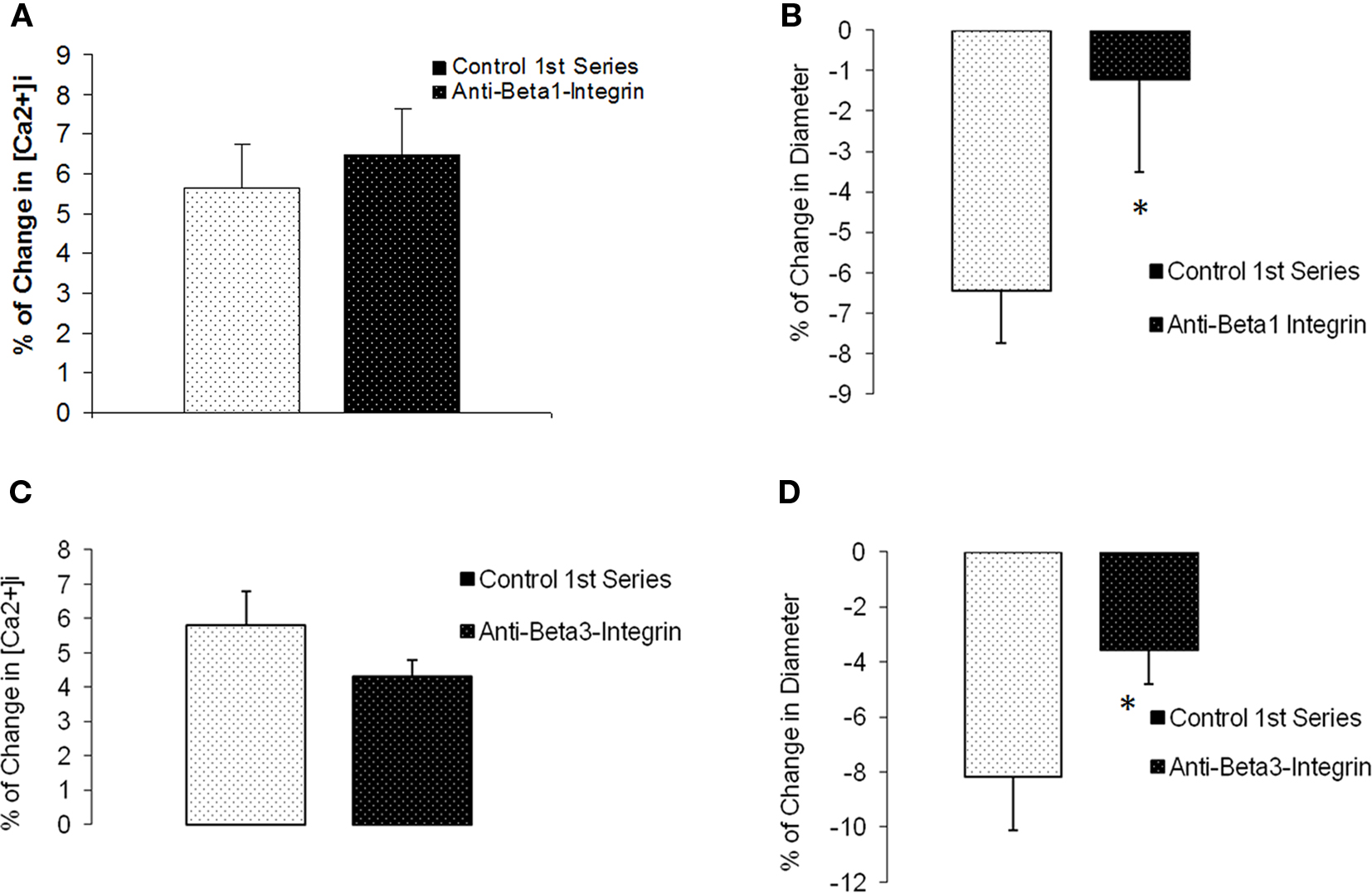
Figure 8. β1- and β3-integrin blockade diminished pressure-induced myogenic without affecting [Ca2+]i increases in response to pressure. (A) Percentage of [Ca2+]i change in arterioles exposed to an increase in intraluminal pressure before and after anti-β1-integrin. (B) Percentage of change in diameter before and after treatment with the inhibitory antibody (n = 6). (C) Percentage of [Ca2+]i change in arterioles exposed to an increase in intraluminal pressure before and after anti-β3-Integrin. (D) Percentage of change in diameter before and after treatment with the inhibitory antibody (n = 5). Data are presented as mean ± SEM.
Discussion
The signaling cascade underlying the arteriolar myogenic response is initiated when an intraluminal pressure stimulus activates VSMC. Key in this process is the ability of mechanosensors to detect changes in forces acting on the vessel wall leading to VSMC activation and subsequent vasomotor responses. Complete knowledge of the cellular events involved in this mechanism is unknown. We investigated the hypothesis that cadherins and downstream associated Ca2+ signaling events are involved in the myogenic response. Consistent with this hypothesis, the data from the present study show that inhibition of N-cadherin attenuates myogenic vasoconstriction however, inhibition occurs without altering pressure-induced increases in [Ca2+]i suggesting that it is not a downstream associated signal.
Cadherins are a superfamily of calcium-dependent, transmembrane cell–cell adhesion proteins that form adherens junctions. The cadherins have been reported to play a major role in embryogenesis, tissue morphogenesis, and other biological processes (Takeichi, 1991; George and Beeching, 2006). The cadherin superfamily includes classical cadherins, desmosomal cadherins, and proto cadherins (Leckband and Prakasam, 2006). The classical cadherins have been studied in more detail than the other subfamilies of cadherins. Classical cadherins are glycoproteins that are composed of an ectodomain containing calcium binding sites, transmembrane domain, and intercellular domain (Troyanovsky, 1999). It is widely recognized that cell–cell/cadherin to cadherin adhesion is mediated by a highly conserved tripeptide, HAV, located in the ectodomain (Noe et al., 1999). The intercellular domain binds to the cytoskeleton by interacting with scaffolding proteins known as catenins (Aberle et al., 1996). The cytoplasmic tail of the cadherin binds directly to β- or γ-catenin, which permits the binding of α-catenin to β- or γ-catenin. The cadherin–catenin complex therefore links the cadherin to the actin cytoskeleton. Thus, cadherins provide a potential pathway for connection of the intercellular junctions and intracellular environment that may contribute to mechanotransduction. Importantly, in regard to myogenic vasoconstriction, evidence has been presented previously for involvement of the actin cytoskeleton (Cipolla et al., 2002; Gokina and Osol, 2002). In the context of the present study it is important to consider that an acute action of pressure on cadherins is being considered. In situations of prolonged exposure to increased pressure (for example in hypertension and vascular remodeling) it is conceivable that regulation of cadherins may occur at the expression level.
Investigative studies focusing on the function of cadherins in vasomotor regulation are limited. Studies have shown that N-cadherin is normally present in the rat vasculature and provides cell–cell contact between VSMCs (Jones et al., 2002). Using immunofluorescence, our study also shows the presence of N-cadherin in cultured arteriolar VSMCs. Further, as most investigations of cadherins in VSMC have focused on migration, proliferation, apoptosis, and survival (for example as relates to restenosis) the current study is the first to show a direct role of N-cadherin in acute vasomotor responses.
Increasing attention has been focused on a mechanosensory mechanism to explain myogenic phenomena (Davis and Hill, 1999; Schubert and Mulvany, 1999; Hill et al., 2009). Recent evidence has supported the hypothesis that an extracellular matrix–integrin pathway is important (Martinez-Lemus et al., 2005b). Consistent with this the present study provides further support for the involvement of β1- and β3-integrins while extending this concept to include a role for an additional junctional complex, namely N-cadherin. Paradoxically, while blockade of both pathways inhibits myogenic responsiveness, pressure-induced increases in [Ca2+]i persisted. One possibility is that cadherins and integrins are more strongly linked to events controlling Ca2+ sensitivity rather than Ca2+ entry. In this regard, calcium sensitization has been suggested to be involved in the myogenic response (Schubert et al., 2008; Johnson et al., 2009). The activation of protein kinase C (PKC) and Rho kinase have been shown to regulate Ca2+ sensitization (Hill et al., 1990,1996; Meininger et al., 1991; Narayanan et al., 1994) and lead to an increase in phosphorylated myosin light chain (MLC), which results in VSMC contraction that is independent of [Ca2+]i concentrations (Hill et al., 1996). Actin polymerization (Gokina and Osol, 2002) has also been suggested to play a significant role in the arteriolar myogenic response, and lies downstream of cadherin activation. Further, earlier studies from our laboratories (Meininger et al., 1991; D’Angelo et al., 1997; Zou et al., 2000) and others (Schubert and Mulvany, 1999; Wesselman et al., 2001; Schubert et al., 2008) have shown that pressure induces relatively (as compared to an agonist) small changes in [Ca2+]i such that a change in sensitivity or actin polymerization could explain the paradoxical finding of inhibition of contraction despite a rise in [Ca2+]i. Due to the force and/or tension being placed on cadherins and integrins during pressure-induced stretch, these proteins may mechanically activate the mentioned protein-kinase pathways and actin remodeling processes resulting in vessel constriction.
Involvement of integrins and cadherins in myogenic vasoconstriction supports the notion that both of these proteins play an important role in the mechanosensory apparatus responsible for detecting forces associated with changes in intraluminal pressure. The fact that inhibition of either adhesion protein blocks myogenic vasoconstriction suggest that neither adhesion receptor is sufficient by itself to drive the vasomotor response. It has been suggested that both integrins and cadherins work together to maintain tissue homeostasis through a cooperative cytoskeleton-based mechanism (Brunton et al., 2004; Leckband and Prakasam, 2006; Mege et al., 2006). Cytoskeletal cooperation has been demonstrated through the interaction of actin filaments, intermediate filaments, and microtubules to provide integration of mechanical and cell signaling events (Noe et al., 1999; Ingber, 2006; Ren et al., 2009). In cancer models, for example, studies have suggested that there is a cadherin–integrin crosstalk that is important for tumor cell invasion and cell migration (Avizienyte et al., 2002; Zhang et al., 2006). Therefore, there may be a similar crosstalk between these two proteins in pressure-induced vasoconstriction.
In conclusion, these studies provide the first evidence for involvement of the cadherins in regulation of arteriolar contraction. These results further support the hypothesis that both integrins and cadherins play a role in myogenic vasoconstriction. The exact sensory mechanism of these proteins continues to require elucidation; however, the results suggest they may not function through a mechanism involving VSMC Ca2+ increase. Further investigation into the links between the Ca2+ sensitization and the actin polymerization pathway and integrin and cadherin signaling in the myogenic response is necessary. Delineation of these mechanisms may provide the rationale for the development of new therapeutic approaches in the treatment of vascular disease (Hill et al., 2009).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by NIH Grants to Gerald A. Meininger (HL095486-01), Luis A. Martinez-Lemus (HL088105-02), and Michael A. Hill (HL92241-02).
References
Aberle, H., Schwartz, H., and Kemler, R. (1996). Cadherin–catenin complex: protein interactions and their implications for cadherin function. J. Cell. Biochem. 61, 514–523.
Avizienyte, E., Wyke, A. W., Jones, R. J., McLean, G. W., Westhoff, M. A., Brunton, V. G., and Frame, M. C. (2002). Src-induced de-regulation of E-cadherin in colon cancer cell requires integrin signaling. Nat. Cell Biol. 4, 632–638.
Bolz, S. S., Vogel, L., Sollinger, D., Derwand, R., Boer, C., Pitson, S. M., Spiegel, S., and Pohl, U. (2003). Sphingosine kinase modulates microvascular tone and myogenic responses through activation of RhoA/Rho kinase. Circulation 108, 342–347.
Brunton, V. G., MacPherson, I. R., and Frame, M. C. (2004). Cell adhesion receptors, tyrosine kinases and actin modulators: a complex three-way circuitry. Biochim. Biophys. Acta 1692, 121–144.
Cipolla, M. J., Gokina, N. I., and Osol, G. (2002). Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 16, 72–76.
D’Angelo, G., Davis, M. J., and Meininger, G. A. (1997). Calcium and mechanotransduction of the myogenic response. Am. J. Physiol. Heart Circ. Physiol. 273, H175–H182.
Davis, M. J., Donovitz, J. A., and Hood, J. D. (1992a). Stretch-activated single-channel and whole currents in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 262, C1083–C1088.
Davis, M. J., Meininger, G. A., and Zawieja, D. C. (1992b). Stretch-induced increase in intracellular calcium of isolated vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 263, H1292–H1299.
Davis, M. J., and Hill, M. A. (1999). Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79, 387–423.
Falcone, J. C., Davis, M. J., and Meininger, G. A. (1991). Endothelial independence of myogenic response in isolated skeletal muscle arterioles. Am. J. Physiol. Heart Circ. Physiol. 260, H130–H135.
George, S. J., and Beeching, C. A. (2006). Cadherin: catenin complex: a novel regulator of vascular smooth muscle cell behavior. Atherosclerosis 188, 1–11.
Gokina, N. I., and Osol, G. (2002). Actin cytoskeletal modulation of pressure-induced depolarization and Ca(2+) influx in cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 282, H1410–H1420.
Gokina, N. I., Park, K. M., McElroy-Yaggy, K., and Osol, G. (2005). Effects of Rho kinase inhibition on cerebral artery myogenic tone and reactivity. J. Appl. Physiol. 98, 1940–1948.
Harder, D. R. (1984). Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ. Res. 55, 197–202.
Hill, M. A., Davis, M. J., Meininger, G. A., Potocnik, S. J., and Murphy, T. V. (2006). Arteriolar myogenic signaling mechanisms: implication for local vascular function. Clin. Hemorheol. Microcirc. 34, 67–79.
Hill, M. A., Davis, M. J., Song, J., and Zou, H. (1996). Calcium dependence of indolactam-mediated contractions in resistance vessels. J. Pharmacol. Exp. Ther. 276, 867–874.
Hill, M. A., Falcone, J. C., and Meininger, G. A. (1990). Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am. J. Physiol. Heart Circ. Physiol. 259, H1586–H1594.
Hill, M. A., and Meininger, G. A. (1993). Impaired arteriolar myogenic reactivity in early experimental diabetes. Diabetes 42, 1226–1232.
Hill, M. A., Meininger, G. A., Davis, M. J., and Laher, I. (2009). Therapeutic potential of pharmacologically targeting arteriolar myogenic tone. Trends Pharmacol. Sci. 30, 363–374.
Ingber, D. E. (2006). Cellular mechanotransduction: putting all the pieces together again. FASEB J. 20, 811–827.
Jaggar, J. H., Wellman, G. C., Heppner, T. J., Porter, V. A., Perez, G. J., Gollasch, M., Kleppisch, T., Rubart, M., Stevenson, A. S., Lederer, W. J., Knot, H. J., Bonev, A. D., and Nelson, M. T. (1998). Ca2+ channels, ryanodine receptors, and Ca2+–activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol. Scand. 164, 577–587.
Johnson, R. P., El-Yazbi, A. F., Takeya, K., Walsh, E. J., Walsh, M. P., and Cole, W. C. (2009). Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J. Physiol. 587, 2537–2553.
Jones, M., Sabatini, P. J., Lee, F. S., Bendeck, M. P., and Langille, B. L. (2002). N-cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler. Thromb. Vasc. Biol. 22, 1972–1977.
Leckband, D., and Prakasam, A. (2006). Mechanism and dynamics of cadherin adhesion. Annu. Rev. Biomed. Eng. 8, 259–287.
Martinez-Lemus, L. A., Crow, T., Davis, M. J., and Meininger, G. A. (2005a). Alphavbeta3- and alpha5beta1-integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am. J. Physiol. Heart Circ. Physiol. 289, H322–H329.
Martinez-Lemus, L. A., Sun, Z., Trache, A., Trzciakowski, J. P., and Meininger, G. A. (2005b). Integrins and regulation of the microcirculation: from arterioles to molecular studies using atomic force microscopy. Microcirculation 12, 99–112.
Mege, R. M., Gavard, J., and Lambert, M. (2006). Regulation of cell–cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18, 541–548.
Meininger, G. A., and Trzeciakowski, J. P. (1988). Vasoconstriction is amplified by autoregulation during vasoconstrictor-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 254, H709–H718.
Meininger, G. A., Zaweija, D. C., Falcone, J. C., Hill, M. A., and Davey, J. P. (1991). Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am. J. Physiol. Heart Circ. Physiol. 260, H950–H959.
Moiseeva, E. P. (2001). Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc. Res. 52, 372–386.
Narayanan, J., Imig, M., Roman, R. J., and Harder, D. R. (1994). Pressurization of isolated renal arteries increases inositol trisphosphate and diacylglycerol. Am. J. Physiol. Heart Circ. Physiol. 266, H1840–H1845.
Noe, V., Willems, J., Vandkerckhove, J., Roy, F. V., Bruyneel, E., and Mareel, M. (1999). Inhibition of adhesion and induction of epithelial cell invasion by HAV-containing E-cadherin-specific-peptides. J. Cell. Sci. 112, 127–135.
Ren, G., Crampton, M. S., and Yap, A. S. (2009). Cortactin: coordinating adhesion and the actin cytoskeleton at cellular protrusions. Cell Motil. Cytoskeleton 66, 865–873.
Schofield, I., Malik, R., Izzard, A., Austin, C., and Heagerty, A. (2002). Vascular structural and functional changes in type 2 diabetes mellitus: evidence for the roles of abnormal myogenic responsiveness and dyslipidemia. Circulation 106, 3037–3043.
Schubert, R., Lidington, D., and Bolz, S. S. (2008). The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc. Res. 77, 8–18.
Schubert, R., and Mulvany, M. J. (1999). The myogenic response: established facts and attractive hypotheses. Clin. Sci. 96, 331–326.
Takeichi, M. (1991). Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455.
Troyanovsky, S. M. (1999). Mechanism of cell–cell adhesion complex assembly. Curr. Opin. Cell Biol. 11, 561–566.
Wesselman, J. P., Spaan, J. A., van der Meulen, E. T., and VanBavel, E. (2001). Role of protein kinase C in myogenic calcium-contraction coupling of rat cannulated mesenteric small arteries. Clin. Exp. Pharmacol. Physiol. 28, 848–855.
Wu, X., Mogford, J. E., Platts, S. H., Davis, G. E., Meininger, G. A., and Davis, M. J. (1998). Modulations of calcium current in arteriolar smooth muscle, integrin receptor ligands. J. Cell Biol. 143, 241–252.
Zhang, W., Alt-Holland, A., Margulis, A., Shamis, Y., Fusenig, N. E., Rodeck, U., and Garlick, J. A. (2006). E-cadherin loss promotes the initiation of squamous cell carcinoma invasion through modulation of integrin-mediated adhesion. J. Cell. Sci. 119, 283–291.
Zou, H., Ratz, P. H., and Hill, M. A. (1995). Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am. J. Physiol. Heart Circ. Physiol. 269, H1590–H1596.
Keywords: vascular smooth muscle, myogenic response, mechanosensors, mechanotransduction, cell adhesion, integrins, cadherins, microcirculation
Citation: Jackson TY, Sun Z, Martinez-Lemus LA, Hill MA and Meininger GA (2010) N-cadherin and integrin blockade inhibit arteriolar myogenic reactivity but not pressure-induced increases in intracellular Ca2+. Front. Physio. 1:165. doi: 10.3389/fphys.2010.00165
Received: 26 July 2010;
Accepted: 09 December 2010;
Published online: 29 December 2010.
Edited by:
Daniel Henrion, Université d’Angers, FranceCopyright: © 2010 Jackson, Sun, Martinez-Lemus, Hill and Meininger. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Gerald A. Meininger, Dalton Cardiovascular Research Center, University of Missouri, 134 Research Park Drive, Columbia, MO 65211, USA. e-mail: meiningerg@missouri.edu