 Huaiyu Mi1,2* Paul D. Thomas2
Huaiyu Mi1,2* Paul D. Thomas2
- 1 SRI International, Menlo Park, CA, USA
- 2 Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
Ontologies and standards are very important parts of today’s bioscience research. With the rapid increase of biological knowledge, they provide mechanisms to better store and represent data in a controlled and structured way, so that scientists can share the data, and utilize a wide variety of software and tools to manage and analyze the data. Most of these standards are initially designed for computers to access large amounts of data that are difficult for human biologists to handle, and it is important to keep in mind that ultimately biologists are going to produce and interpret the data. While ontologies and standards must follow strict semantic rules that may not be familiar to biologists, effort must be spent to lower the learning barrier by involving biologists in the process of development, and by providing software and tool support. A standard will not succeed without support from the wider bioscience research community. Thus, it is crucial that these standards be designed not only for machines to read, but also to be scientifically accurate and intuitive to human biologists.
The Emergence of Ontologies and Standards in Bioscience Research
Ontologies and standards have emerged in bioscience research during the past decade in response to the fast accumulation of biology data, largely because of the advancement of bioscience research technologies. The main purposes of the ontologies and standards are to (1) organize the data in a controlled and structured format; (2) enable scientists from different research disciplines to share the data and collaborate; and (3) allow different computation tools and software to access and interpret the data in an unambiguous way. After 10 years of development, the concept of applying ontologies and standards in bioscience research has been well accepted, especially in the field of bioinformatics, systems biology, and computational biology. Although it has gained significant momentum in wet-lab research fields, especially those using high-throughput technologies, it still remains a challenge to be adopted by a wider research community.
The term ontology originated from ancient Greek philosophy. It is a study of properties, events, processes, and relations of existence (Smith, 2003). It is a formal structuring of knowledge. Contemporary ontology was first introduced in computer and information science. In these fields, the information representations built by different groups and communities often used a diverse variety of terms and concepts. The same concept could be represented with different nomenclatures by different groups, while identical labels might be used for different meanings. When the information needed to be shared and transferred among different groups or accessed by different platforms, a controlled and structured expression of the information became essential. As a result, ontology was used by computer and information scientists to describe these controlled terms, concepts, and descriptions of the information. This ontology differs from the classical philosophical ontology in that it is expressed in a machine-readable format, and that it assesses in terms of usefulness rather than truth.
The term ontology was introduced to biologists for very similar reasons. Gene Ontology (GO) was the first ontology designed as the formal representation of biological knowledge (Ashburner et al., 2000; Gene Ontology Consortium, 2010). Biology research used to be conducted within small research groups or communities that had common interests in a particular organism or a small group of molecules. Molecular biology required wider collaboration among researchers of diverse disciplines, such as geneticists, biochemists, and molecular biologists. A geneticist could name a gene differently from a molecular biologist, while scientists working on different organisms might use different naming conventions to name the same – or strictly speaking, evolutionarily related or orthologous – genes in the organism of their interest. For example, Drosophila geneticists first identified an allele that was important to potassium ion conductance on cell membrane. This allele was named Shaker based on the phenotype when it mutated (Salkoff and Wyman, 1981). Molecular biologists cloned the gene that was responsible for this phenotype at the allele. The gene was named after its original allele and is called shaker. Subsequently, a number of Drosophila shaker homologs and their orthologs in mammalians were cloned, and a variety of nomenclatures was used in naming these genes (e.g., shaw, shab, RCK1, drk1; Perney and Kaczmarek, 1991; Pongs, 1992). The urge to provide a more unified nomenclature system to facilitate communication and collaboration prompted the initial proposal of a naming standard for these potassium channels based on phylogenetic relationships (Chandy, 1991; Chandy and Gutman, 1993). The system was later refined and adopted by the community (Gutman et al., 2005). This case-by-case approach was quite common within various research communities until the genome-sequencing era. Near the end of the last century, a huge amount of molecular sequence data was produced because of the advancement of DNA sequencing technology. Sequences of the entire genome from various organisms are now available to the researchers. The conservation of sequences and functions of proteins across different organisms lets scientists believe that a unification of nomenclature should be given to characterize genes across all organisms. This led to the founding of the Gene Ontology Consortium and the creation of GO (Ashburner et al., 2000).
Gene Ontology covers three basic domains of biology knowledge: molecular function, biological process, and cellular component. Any given gene can be classified by one or more terms from any of the three domains. Significant progress has been made in GO annotation, mainly by major model organism databases (Hong et al., 2008; Tweedie et al., 2009; Bult et al., 2010; Harris et al., 2010) and Reference Genome Project (Reference Genome Group of the Gene Ontology Consortium, 2009). It is also supported by a large number of software tools1. Because of the success of this pioneer work, a number of ontologies were subsequently proposed and developed to cover other domains of biology, such as anatomy, structure, disease, phenotype, and pathways.
The rapid increase in the number of ontologies also created obstacles to integration. Thus, Open Biological and Biomedical Ontologies (OBO) Foundry was formed to provide a collaborative environment for coordinated expansion of ontologies that are interoperable and logically well formed (Smith et al., 2007). Most ontologies in OBO Foundry are driven by the biologists trying to understand the properties and functions of biological entities. Examples are, in addition to GO, ontology for chemical entities (CHEBI; Natale et al., 2007; Degtyarenko et al., 2008), protein ontology (PRO; Natale et al., 2007), ontology for phenotype quality (PATO; Sprague et al., 2008), and anatomy ontologies. The Foundry also includes a number of candidate ontologies that cover an even wider spectrum of biological knowledge.
Another intricate aspect of biology is that in the biological system, molecules often interact with each other. Dynamics and dependencies of various molecules through such interactions result in the formation of pathways and networks that regulate functions of individual molecules and, by doing so, impact the biological system. Scientists have been studying the interactions of biological entities and pathways in laboratories for decades. Most activities of this work, however, are isolated and are conducted in different conditions/cell types/organisms. It is often difficult to compose a comprehensive and intuitive overall pathway without great human labor and effort.
During the past decade, two new research approaches have emerged that revolutionized the paradigm of pathway research. Around 2000, systems biology emerged, spurred on by the completion of various genome-sequencing projects, together with the advancement in high-throughput experimental technology that resulted in a large increase in data from genomics and proteomics. Researchers in the fields have developed software and tools having the ability to integrate and analyze large amounts of complex data from various sources, and to build and model large pathway networks. Around the same time came the pathway databases, where large pathway maps have been created through careful curation by expert scientists. According to Pathguide2, a public pathway resource website, 325 different pathway databases are currently available. With the rapid growth of these fields, it is crucial for scientists to share the data and allow software and tools to access, interpret, and analyze the data.
When a controlled data representation of pathway knowledge became essential, several pathway standards emerged to address this need. In the field of systems biology – in order to better share, evaluate, and develop these models in a collaborative way – the community of systems biologists developed an information standard called the Systems Biology Markup Language (SBML; Hucka et al., 2003). SBML is a machine-readable format, written in Extensible Markup Language (XML) form, for representing pathway models. By supporting SBML as a format for reading and writing models, different software tools (including programs for building and editing models, simulation programs, databases, and other systems) can directly communicate and store the same computable representation of those models. Currently, more than 180 software packages support SBML. BioPAX is another standard through collaborative effort to create a data-exchange format for biological pathway data3. Its main purpose is to facilitate data access, sharing, and integration from multiple pathway databases. Pathway data that support BioPAX are stored in the web ontology language (OWL), and can be viewed using software that supports OWL, such as Protégé (Noy et al., 2003). While SBML is concentrated on mathematical modeling, BioPAX is more focused on qualitative pathway knowledge; therefore, these two standards complement each other.
Ontologies and Standards are for both Human and Computers
The development of ontologies and standards in bioscience is still in its infant stage. In order for them to succeed, it is crucial that the ontologies and standards are accepted and adopted by the entire bioscience research community. The ontologies and standards in bioscience should be designed to be not only readable by computers, but also scientifically accurate and intuitive to human biologists. The bioscience research workflow can be summarized as illustrated in Figure 1. It starts with hypotheses, followed by experimental design to test these hypotheses. Experiments are subsequently performed, and data are collected and analyzed by scientists, often with the assistance of tools and software. Finally, the conclusions are drawn, and new hypotheses are proposed to start a new round of research. Human involvement is essential at each of these steps, while computer software and tools will greatly facilitate the process by better storing, representing, and analyzing the data. At the current stage, the majority of the research data are generated in wet labs by bench scientists. If these scientists adopt the ontologies and standards, produce the data, and organize the data in compliance with the standards, it will enable computation biologists and systems biologists to directly analyze the data with computation tools and software. The results produced by the software and tools can be interpreted unambiguously by human scientists.
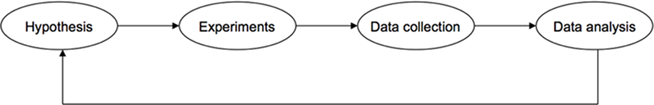
Figure 1. Biomedical research workflow. Human involvement is essential at each of the steps illustrated in this schematic diagram, while computer software and tools will greatly facilitate the process by better storing, representing, and analyzing the data. Thus, it is crucial to include ontologies and standards at each of these steps for unambiguous interpretation of data by both human and computers.
One fundamental element of an ontology is that it requires well-defined semantics. It is not just a list of controlled vocabulary, but rather a knowledge representation with well-defined structure and relationship. Therefore, it is usually expressed in terms that are not conventionally used by biologists. For example, homeobox protein is a common term used by biologists to describe a family of transcription factors that bind to a particular domain of DNA (homeobox) and usually are involved in the development (morphogenesis) of an organism. Because homeobox domains are found in genes involved in development, the term homeobox also has functional implications when used by biologists. In GO, homeobox is considered as a sequence structure, and therefore, it cannot be used to describe the molecular function of a protein. GO molecular function simply classify these proteins as sequence-specific DNA binding and transcription factory activity. In fact, the term homeobox cannot be found in any of the three ontologies in GO. This factor can deter biologists from learning and adopting it. Efforts must made to lower this learning barrier.
First, the ontologies and standards must be designed to accurately represent the biological knowledge. It is crucial to involve a large community in the process of the development. This community should include biologists, ontology experts, bioinformaticists, and software developers. Involvement of biologists and domain experts is necessary because they can ensure the accuracy and integrity of the knowledge, and ultimately they are the ones who will produce and interpret the data. Second, efforts must be spent to enable biologists to appreciate how biological knowledge is captured and represented by ontologies and standards. This would involve educating the biologists by providing documentation, hosting tutorials, including the biologists in the process of development, and encouraging them in the curation effort. Last, tools and software must be developed to facilitate the use of the ontology by biologists.
Gene Ontology is the best accepted and most widely used standard, but with a price. The project has been well funded for the past 10 years, and is supported by a number of well-funded model organism databases, including Saccharomyces Genome Database (SGD; Hong et al., 2008), FlyBase (Tweedie et al., 2009), WormBase (Harris et al., 2010), and Mouse Genome Database (MGD; Bult et al., 2010). From the very beginning of the GO development, scientists recognized the importance of reaching out to biologists. Various efforts have been focused on this goal, such as the involvement of biologists in the development, hosting GO-user meetings to discuss issues in the ontology, building tools to enable end users to utilize GO, and – most important – conducting large-scale genome curation using the ontology. Besides model organism databases that were actively participating in the genome curation effort using GO, other gene or protein databases also joined forces, such as UniProt (Camon et al., 2004) and PANTHER Classification System (Thomas et al., 2003b). This opened a window to the biologists for quick access to the ontologies. During this process, developers of the PANTHER Classification System, a protein evolution database, also recognized the gap between the conventional terms used by the biologists and the ontology terms. They built an index of controlled vocabulary, called PANTHER Index, to capture protein functions and biological processes in more conventional terms, but also mapped the Index to all GO terms (Thomas et al., 2003a). By doing so, users of the PANTHER system, mostly biologists, had a chance of getting used to the GO terms through the more familiar PANTHER Index terms, thus appreciating the rigorousness of the ontology. This became part of the education process to the biologists about the usefulness of the ontology. Because of the wide acceptance of GO, PANTHER retired its PANTHER Index in 2009 and developed GO-slim for its annotation (Mi et al., 2010).
In the field of pathway standards, things are a little different. Both SBML and BioPAX are designed more for data exchange and are in machine-readable formats. The purpose of SBML is to provide a standard data format for systems biologists (modelers) to exchange pathway models so that they can be accessed by different software and tools. BioPAX provides a more rigorous data format for pathway data exchange among different pathway databases. Neither standard is intuitive to biologists; therefore, specially trained experts (curators) are required to translate biological data into the standards using highly specialized tools. It is well known that pathways are always associated with diagrams. Pathway diagrams have been used for over 60 years. A diagram is an intuitive way to illustrate biological knowledge, and it is the most powerful method of communication among humans. To provide an unambiguous graphical representation of pathways that enables better pathway data sharing among scientists of different backgrounds and among different software programs and other tools, a consortium of molecular biologists, systems biologists, computational biologists, and software developers recently have proposed a graphical notation standard, called Systems Biology Graphical Notation (SBGN; Le Novère et al., 2009). By developing tools that support SBGN, biologists can easily draw pathway diagrams in a format with which they are familiar, and the tools can save those diagrams to either SBML or BioPAX. At the same time, the modelers can simulate pathway networks, and output the models in graphs that can be viewed and interpreted by a human. Database developers can easily illustrate pathway diagrams that accurately reflect the data stored in the database. Thus, the SBGN standard serves as an interface to the human biologists to read and write pathways that are compliant with community standards for modeling, exchanging, and storing.
Integration of Ontologies and Standards
One of the most intriguing aspects of biology is its complexity. It is not possible to capture and represent the entire biology with one standard or ontology. Each standard or ontology reflects only a particular domain of biology. Therefore, the development of standards and ontology should not be isolated. Collaboration and subsequent integration will be necessary for the understanding of biology as a whole. For example, the pathway exchange standard BioPAX captures pathway data where the entities are characterized by GO terms. Also, when analyzing data from high-throughput experiments – such as mass spectrometry, microarray, or genotyping analysis – it is often necessary to integrate the data from various sources. These sources may provide clinical, genomic, proteomic, and expression data. Therefore, integration of phenotype ontology, pathway standards, gene ontology, and protein ontology will be essential to facilitate such analysis. One such example is a recent study that utilizes an integrated ontology network by combining phenotype ontology, gene ontology, and pathway standards for a large genetic association study of nicotine addiction and treatment that includes epidemiological, clinical, and gene association data (Thomas et al., 2009). This is another reason why ontologies and standards should be intuitive to human biologists, so that scientists from different domains of expertise can understand them and utilize them for their research.
Summary
With the advance in technology, bioscience research is undergoing significant changes. Computer software and tools have been employed for better storage, representation, and analysis of ever-growing data knowledge. Ontologies and standards have been introduced to the field to facilitate data sharing among different platforms and software, and among scientists across different research fields. The fundamental research paradigm remains the same; it always starts with a hypothesis, followed by experiment design, data collection and careful analysis, and completed by drawing a conclusion. This paradigm relies heavily on human involvement to observe, interpret, and analyze the data. Therefore, when ontologies and standards are designed and developed, one must always keep in mind that ontologies and standards are not just for machine, but for human also.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29.
Bult, C. J., Kadin, J. A., Richardson, J. E., Blake, J. A., Eppig, J. T., and Mouse Genome Database Group. (2010). The Mouse Genome Database: enhancements and updates. Nucleic Acids Res. 38, D586–D592.
Camon, E., Magrane, M., Barrell, D., Lee, V., Dimmer, E., Maslen, J., Binns, D, Harte, N., Lopez, R., and Apweiler, R. (2004). The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res. 32, D262–D266.
Chandy, K. G., and Gutman, G. A. (1993). Nomenclature for mammalian potassium channel genes. Trends Pharmacol. Sci. 14, 434.
Degtyarenko, K., de Matos, P., Ennis, M., Hastings, J., Zbinden, M., McNaught, A., Alcántara, R., Darsow, M., Guedj, M., and Ashburner, M. (2008). ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 36, D344–D350.
Gene Ontology Consortium. (2010). The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 38, D331–D335.
Gutman, G. A., Chandy, K. G., Grissmer, S., Lazdunski, M., McKinnon, D., Pardo, L. A., Robertson, G. A., Rudy, B., Sanguinetti, M. C., Stühmer, W., and Wang, X. (2005). International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508.
Harris, T. W., Antoshechkin, I., Bieri, T., Blasiar, D., Chan, J., Chen, W. J., De La Cruz, N., Davis, P., Duesbury, M., Fang, R., Fernandes, J., Han, M., Kishore, R., Lee, R., Müller, H. M., Nakamura, C., Ozersky, P., Petcherski, A., Rangarajan, A., Rogers, A., Schindelman, G., Schwarz, E. M., Tuli ,M. A., Van Auken, K., Wang, D., Wang, X., Williams, G., Yook, K., Durbin, R., Stein, L. D., Spieth, J., and Sternberg, P. W. (2010). WormBase: a comprehensive resource for nematode research. Nucleic Acids Res. 38, D463–D467.
Hong, E. L., Balakrishnan, R., Dong, Q., Christie, K. R., Park, J., Binkley, G., Costanzo, M C., Dwight, S. S., Engel, S. R., Fisk, D. G., Hirschman, J. E., Hitz, B. C., Krieger, C. J., Livstone, M. S., Miyasato, S. R., Nash, R. S., Oughtred, R., Skrzypek, M. S., Weng, S., Wong, E. D., Zhu, K. K., Dolinski, K., Botstein, D., and Cherry, J. M. (2008). Gene Ontology annotations at SGD: new data sources and annotation methods. Nucleic Acids Res. 36, D577–D581.
Hucka, M., Finney, A., Sauro, H. M., Bolouri, H., Doyle, J. C., Kitano, H., Arkin, A. P., Bornstein, B. J., Bray, D., Cornish-Bowden, A., Cuellar, A. A., Dronov, S., Gilles, E. D., Ginkel, M., Gor, V., Goryanin, I. I., Hedley, W. J., Hodgman, T. C., Hofmeyr, J. H., Hunter, P. J., Juty, N. S., Kasberger, J. L., Kremling, A., Kummer, U., Le Novère, N., Loew, L. M., Lucio, D., Mendes, P., Minch, E., Mjolsness, E. D., Nakayama, Y., Nelson, M. R., Nielsen. P. F., Sakurada, T., Schaff, J. C., Shapiro, B. E., Shimizu, T. S., Spence, H. D., Stelling, J., Takahashi, K., Tomita, M., Wagner, J., and Wang, J. (2003). The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531.
Le Novère N., Hucka, M., Mi, H., Moodie, S., Schreiber, F., Sorokin, A., Demir, E., Wegner K, Aladjem MI, Wimalaratne S. M., Bergman, F. T., Gauges, R., Ghazal, P., Kawaji, H., Li, L., Matsuoka, Y., Villéger, A., Boyd, S. E., Calzone, L., Courtot, M., Dogrusoz, U., Freeman, T. C., Funahashi, A., Ghosh, S., Jouraku, A., Kim, S., Kolpakov, F., Luna, A., Sahle, S., Schmidt, E., Watterson, S., Wu, G., Goryanin, I., Kell, D. B., Sander, C., Sauro, H., Snoep, J. L., Kohn, K., and Kitano, H.(2009). The systems biology graphical notation. Nat. Biotechnol. 27, 735–741.
Mi, H., Dong, Q., Muruganujan, A., Gaudet, P., Lewis, S., and Thomas, P. D. (2010). PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the gene ontology consortium. Nucleic Acids Res. 38, D204–D210.
Natale, D. A., Arighi, C. N., Barker, W. C., Blake, J., Chang, T. C., Hu, Z., Liu, H., Smith, B., and Wu, C. H. (2007). Framework for a protein ontology. BMC Bioinformatics 8(Suppl. 9), S1. doi: 10.1186/1471-2105-8-S9-S1
Noy, N. F., Crubezy, M., Fergerson, R. W., Knublauch, H., Tu, S. W., Vendetti, J., and Musen, M. A. (2003). Protege-2000: an open-source ontology-development and knowledge-acquisition environment. AMIA Annu. Symp. Proc. 2003, 953.
Perney, T. M., and Kaczmarek, L. K. (1991). The molecular biology of K+ channels. Curr. Opin. Cell Biol. 3, 663–670.
Reference Genome Group of the Gene Ontology Consortium. (2009). The gene ontology’s reference genome project: a unified framework for functional annotation across species. PLoS Comput. Biol. 5, e1000431. doi: 10.1371/journal.pcbi.1000431
Salkoff, L., and Wyman, R. (1981). Genetic modification of potassium channels in Drosophila shaker mutants. Nature 293, 228–230.
Smith, B. (2003). “Ontology,” in Blackwell Guide to the Philosophy of Computing and Information, ed. L. Floridi (Oxford: Blackwell), 155–166.
Smith, B., Ashburner, M., Rosse, C., Bard, J., Bug, W., Ceusters, W., Goldberg, L. J., Eilbeck, K., Ireland, A., Mungall, C. J.; OBI Consortium, Leontis, N., Rocca-Serra, P., Ruttenberg, A., Sansone, S. A., Scheuermann, R. H., Shah, N., Whetzel, P. L., and Lewis, S. (2007). The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 25, 1251–1255.
Sprague, J., Bayraktaroglu, L., Bradford, Y., Conlin, T., Dunn, N., Fashena, D., Frazer, K., Haendel, M., Howe, D. G., Knight, J., Mani, P., Moxon, S. A., Pich, C., Ramachandran, S., Schaper, K., Segerdell, E., Shao, X., Singer, A., Song, P., Sprunger, B., Van Slyke, C. E., and Westerfield, M. (2008). The Zebrafish Information Network: the zebrafish model organism database provides expanded support for genotypes and phenotypes. Nucleic Acids Res. 36, D768–D772.
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., Diemer, K., Muruganujan, A., and Narechania, A. (2003a). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141.
Thomas, P. D., Kejariwal, A., Campbell, M. J., Mi, H., Diemer, K., Guo, N., Ladunga, I., Ulitsky-Lazareva, B., Muruganujan, A., Rabkin, S., Vandergriff, J. A., and Doremieux, O. (2003b). PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31, 334–341.
Thomas, P. D., Mi, H., Swan, G. E., Lerman, C., Benowitz, N., Tyndale, R. F., Bergen, A. W., Conti, D. V.; Pharmacogenetics of Nicotine Addiction and Treatment Consortium. (2009). A systems biology network model for genetic association studies of nicotine addiction and treatment. Pharmacogenet. Genomics 19, 538–551.
Keywords: ontology, standard, systems biology
Citation: Mi H and Thomas PD (2011) Ontologies and standards in bioscience research: for machine or for human. Front. Physio. 2:5. doi: 10.3389/fphys.2011.00005
Received: 24 September 2010;
Paper pending published: 16 November 2010;
Accepted: 07 February 2011;
Published online: 21 February 2011.
Edited by:
Hiroaki Kitano, The Systems Biology Institute, JapanReviewed by:
Hiroaki Kitano, The Systems Biology Institute, JapanSamik Ghosh, The Systems Biology Institute, Japan
Copyright: © 2011 Mi and Thomas. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Huaiyu Mi, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, 1450 Biggy Street, NRT2509F, Los Angeles, CA 90089, USA. e-mail: huaiyumi@usc.edu