
- 1 Department of Veterans Affairs, Southern California Research Center for Alcoholic Liver and Pancreatic Diseases, University of California, Los Angeles, CA, USA
- 2 Department of Medicine, Yale University and New England VA Health Care System, West Haven, CT, USA
- 3 Department of Cell Biology, Yale University and New England VA Health Care System, West Haven, CT, USA
The exocrine pancreas has the greatest protein synthetic capacity of any mammalian organ and is challenged with the synthesis, processing and transporting a large load of digestive enzymes. Based on recent findings we present a hypothesis proposing that mutations in the digestive enzymes and environmental risks impacting the pancreas (i.e., alcohol abuse, smoking, metabolic disorders, and drugs) cause endoplasmic reticulum (ER) stress. We review recent findings showing that in normal pancreas the ER stress resulting from alcohol abuse leads to an adaptive unfolded protein response (UPR) allowing for maintenance of protein synthesis, processing, and transport. However, when key pathways necessary for the adaptive UPR are altered, the exocrine cell of the pancreas is unable to maintain these processes and cellular pathology results. These findings may explain why some individuals with alcohol abuse disorders develop organ injury and disease while most do not. Further, the findings allow us to hypothesize that the UPR in the exocrine pancreas adapts the protein synthetic machinery of the ER stress resulting from mutational and environmental stressors. When the ability of the UPR to adapt to the stressors is exceeded, pathologic pathways and disease develop.
Cellular Functions of the Exocrine Pancreas
The exocrine pancreas functions are the synthesis, storage, and secretion of digestive enzymes into the lumen of the gastrointestinal tract where the enzymes are responsible for converting foodstuffs ingested into aqueous soluble molecules that can be absorbed by the intestinal epithelium. The two key epithelial cell types of the exocrine pancreas involved in digestive enzyme secretion are the acinar cell and the duct cell. The acinar cell is the site of digestive enzyme synthesis, storage, and regulated secretion while the duct cell provides ions and water necessary for transporting the acinar cell secreted products in the pancreatic ductal system to the intestinal lumen. Ductal cell ion secretions are rich in NaHCO3 which is involved in neutralization of gastric acid emptied into the duodenum. The neutralization of gastric acid is necessary because the pancreatic enzymes have optimal activity at neutral pH.
The acinar cell of the exocrine pancreas has the greatest rate of protein synthesis of any mammalian organ and thus is endowed by a highly developed endoplasmic reticulum (ER) system (Palade, 1975; Case, 1978). In addition to its functions in performing protein synthesis and processing, the ER is the major storage site for intracellular calcium which when released into the cytoplasm is the mediator of regulated secretion of stored digestive enzymes into the pancreatic ductal system (Petersen and Tepikin, 2008).
Each protein synthesized in the ER must undergo specific secondary modifications and folding before it can be transported to destination organelles such as Golgi, zymogen granule (storage for the digestive enzymes), and lysosome; or membrane sites. Further, because of variation in the demand for protein synthesis as a function of diet; and because protein processing in the ER could be adversely affected by environmental factors such as alcohol, smoking, altered metabolism, and xenobiotics, it is likely that there are regulatory systems within the ER that allow it to adapt its functions to such stressors.
Alcohol Abuse, Smoking, and the Diseases of the Exocrine Pancreas
The most common diseases of the exocrine pancreas are pancreatitis and pancreatic cancer. Alcohol abuse and smoking are key risk factors in the epidemiology of both diseases (Go et al., 2005; Pandol et al., 2009; Yadav and Whitcomb, 2010). In the case of alcohol abuse the increased risk for pancreatic cancer occurs largely through the effect of alcohol abuse causing chronic forms of pancreatitis (Lowenfels et al., 1993). Smoking also contributes to the development of pancreatitis and is a major risk factor for pancreatic cancer independent of pancreatitis (Lowenfels et al., 1993; Go et al., 2005; Pandol et al., 2009). Recent epidemiologic studies demonstrate that smoking accelerates the development of pancreatitis in alcoholic patients and may have an additive or multiplicative effect when combined with alcohol to cause pancreatitis (Maisonneuve et al., 2005; Yadav and Whitcomb, 2010). The mechanisms underlying the effects of alcohol and smoking on the development of pancreatic diseases are incompletely understood. An important and unexplained observation is that only a small proportion of heavy drinkers/smokers develop pancreatic diseases (Pandol et al., 2007). Although the reason for lack of development of pathology in the majority of those who drink and smoke is unknown, we hypothesize that an adaptive unfolded protein response (UPR) is sufficiently robust in most individuals to prevent pathology.
Overview of ER Stressors
Proteins enter the ER as unfolded polypeptides that require further processing for activation and targeting to the appropriate organelle or membrane site. Further, the ER is faced with several challenges in completing these functions with high fidelity. Figure 1 illustrates many of these challenges which are often referred to as ER stress that we have applied to our hypothesis of ER stress for the exocrine pancreas. At the bottom of the figure, we illustrate the UPR that is regulated by the stressors. A key implication from the figure is that UPR activation leads to an adaptive response in the ER. In this figure we hypothesize potential ER stressors in the pancreas from what is known about ER stressors in general (Ron and Walter, 2007). For example, it is likely that increases in protein synthesis rates that occur to replenish digestive enzyme stores after a meal will increase the demand for protein folding resulting in increased need for synthesis and functionality of chaperones and foldases in the ER. Because the rate of unfolded/misfolded protein formation would also increase with increased proteins synthesized, the ER quality control system to degrade these unusable proteins called ER-associated protein degradation (ERAD) is required to be present and functional to rid the cell from accumulation of permanently misfolded and unfolded proteins that could present toxicity to the cell.
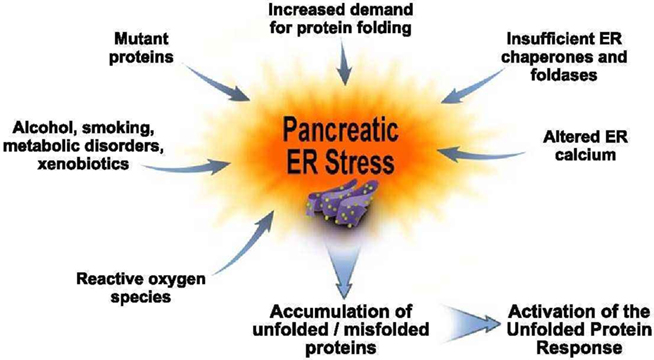
Figure 1. Pancreatic ER stress. This figure depicts factors we hypothesize cause pancreatic exocrine cell ER stress. For example, an increased folding demand will result when there is a need for increased digestive enzyme synthesis. Increased expression of chaperones and foldases are needed for processing these proteins. Altered ER calcium levels as occurs during forms of pancreatitis can also cause ER stress. On the left hand side of the figure are situations in which pancreatic ER stress is further amplified. These include genetic mutations in digestive enzymes; alcohol abuse; smoking; metabolic disorders such as diabetes and hyperlipidemia; xenobiotics such as drugs and intestinal bacterial metabolites; and reactive oxygen species that are generated in many of these situations as well as during acute and chronic pancreatitis. These pancreatic ER stressors lead to further accumulation of unfolded and misfolded proteins which, in turn, lead to further activation of the Unfolded Proteins Response in an attempt to adapt the pancreas to function in the face of the ER stressors.
There are several genetic, environmental, and disease-related stressors illustrated in Figure 1 that occur in the pancreas that are likely significantly increase ER stress requiring the acinar cell to activate its adaptive UPR or face the possibility of cellular pathologies. Examples include genetic mutations. Mutations in key protease digestive enzymes are known to lead to chronic forms of pancreatitis (hereditary pancreatitis) and increased rate of pancreatic cancer (Whitcomb, 2010). In fact, a recent report (Kereszturi et al., 2009) demonstrates that a mutation in human cationic trypsinogen causes ER stress in pancreatic cells. These results support a hypothesis that hereditary pancreatitis results from ER stress caused by a mutated protease.
Another example is altered ER calcium levels that occur in experimental models of pancreatitis (Sutton et al., 2008). Calcium is stored in the ER by its resident chaperones and foldases which act as low affinity, high capacity Ca2+-binding proteins (Gorlach et al., 2006). Importantly for ER stress as discussed below, their chaperone and foldase functions are markedly altered with perturbations in ER calcium homeostasis. Thus, alterations in ER calcium stores in cells can lead to pathologic consequences at least in part because of the ER stress induced. Further, as discussed in more detail below, manipulation of ER calcium stores in pancreatic acinar cells has been shown to activate ER stress signals.
Other potential stressors listed in Figure 1 include alcohol, smoking, metabolic disorders, and xenobiotics as well as reactive oxygen species (ROS). Except for information on alcohol abuse recently published (Lugea et al., 2010) and reviewed below, there is little information on whether these factors affecting the pancreas indeed cause ER stress and whether pathology results from an insufficiently robust UPR. However, alcohol, smoking, diabetes, obesity, hyperlipidemia, and drugs account for large percentage of the burden of pancreatic diseases (Pandol et al., 2007); and ROS participate in their pathogenesis. Further, ROS are generated in the inflammatory processes of pancreatic diseases themselves. Thus, the potential for roles of ER stress in pancreatic disease pathogenesis is great.
The Adaptive Unfolded Protein Response
The adaptive UPR has three major types of outputs (Marciniak et al., 2006; Ron and Walter, 2007; Rutkowski and Kaufman, 2007; Kaser et al., 2008; Kim et al., 2008). These include: (1) upregulation of the expression and function of chaperones and foldases to augment the folding and export capacity of the ER; (2) activation of the ER-associated protein degradation (ERAD) system to rid the ER of accumulated unfolded and misfolded proteins; and (3) a global reduction in translation of mRNA to decrease the processing demand for newly synthesized proteins.
The UPR also activates cell death programs under conditions of severe and/or prolonged ER stress when the adaptive UPR responses are exceeded or when a dysfunctional UPR is unable to correct the folding disorders presented to it.
As illustrated in Figure 2, the mammalian UPR is initiated mainly by three ER stress sensor–transducers located in the membrane of the ER (Ron and Walter, 2007; Rutkowski and Kaufman, 2007). These three are inositol-requiring protein-1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA (PKR)-like ER kinase (PERK). In each case the transmembrane sensor–transducer determines the folding status of proteins in the ER lumen and transmits this information across the ER membrane to the cell cytosol. In some cases, the transmembrane sensor–transducer is “silenced” by binding of an ER chaperone called immunoglobulin-binding protein (BiP) to its luminal domain. BiP is also known as also known as 78 kDa glucose-regulated protein (GRP78). ER stress unfolded and misfolded proteins compete for binding Bip resulting in removing its “silencing” effect resulting in activation of the sensor.
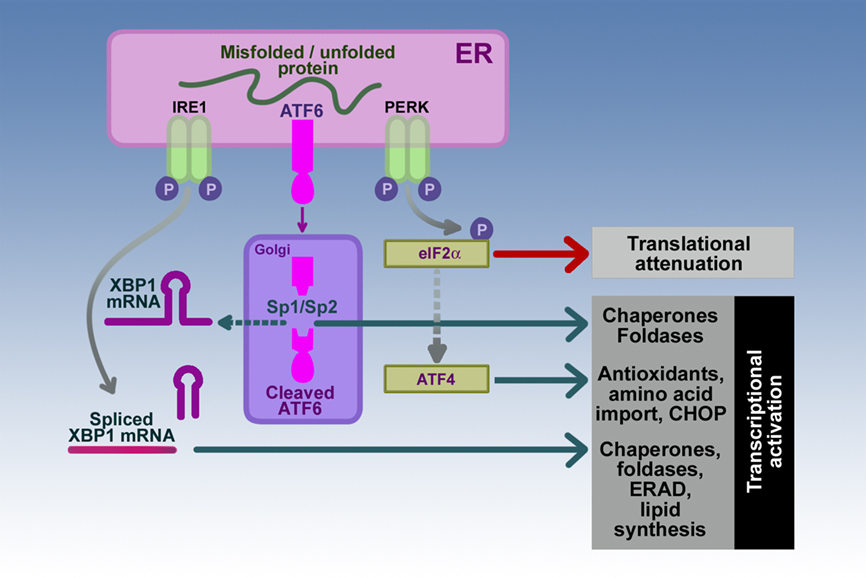
Figure 2. Sensor–transducers of the mammalian UPR. The three sensor–transducers of the UPR are inositol-requiring protein-1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA (PKR)-like ER kinase (PERK). These sensor–transducers determine the state of unfolded proteins in the ER lumen. Activation of each of the sensor–transducers is followed specific pathways resulting in transcriptional regulation of chaperones, foldases, and components of ER-associated protein degradation (ERAD) system and lipid synthesis for expansion of the ER mediated by the combined effects of IRE1 and ATF6; or translational attenuation and transcriptional upregulation pathways involved in antioxidant synthesis and cell death through the transcription factor C/EBP homologous protein (CHOP) as in the case of PERK. The participants in the pathways involved in the downstream effects of IRE1, ATF6, and PERK activation include X-box binding protein1 (XBP1), site-1 and site-2 proteases (Sp1/Sp2), and eukaryotic translation initiation factor-2a (eIF-2a), and activating transcription factor-4 (ATF4) as discussed in the text.
Activation of the ER transmembrane protein IRE1 initiates a response to increase the expression of ER chaperones and foldases in order to assist in protein folding and transport (Figure 2). ER stress induces IRE1 homodimerization and trans-autophosphorylation to activate its RNAse activity toward the mRNA for unspliced X-box binding protein1 (XBP1-U). Activated IRE1 removes a 26-nucleotide intron from XBP1-U resulting in a mRNA that translates into a potent transcription factor, spliced XBP1 (XBP1-S; Yoshida et al., 2001; Calfon et al., 2002; Lee et al., 2002). XBP1-S, in turn, binds to ER stress element (ERSE) and the UPR element (UPRE) DNA binding sites to upregulate many UPR target genes such as the chaperones BiP/GRP78 and GRP94 and the gene encoding XBP1-U (Yoshida et al., 2001, 2003; Calfon et al., 2002; Lee et al., 2002). This ability to increase transcription of XBP1-U leads to more substrate for expression of the XBP1-S transcription factor thus augmenting this protective response. The IRE1/XBP1 pathway also leads to increased expression of foldases such as protein disulfide isomerase (PDI), enzymes for lipid synthesis for expanding the ER membrane and ER capacity, components of the ER-associated degradation (ERAD), all protective mechanism to lessen ER stress (Yoshida et al., 2003).
The importance of XBP1 for the function of the pancreatic acinar cell has been demonstrated in XBP1 deficient mice with a transgene expressing XBP1 in hepatocytes to prevent embryonic lethality (Lee et al., 2005). These mice show abnormalities limited to the secretory organs, the exocrine pancreas, and salivary gland. The ER in the pancreatic acinar cells in these animals is poorly developed accompanied by a decreased in the expression of ER chaperones and marked apoptosis of the cells.
Activating transcription factor-6 represents a second ER transmembrane protein that is sensitive to ER stress (Figure 2). The N-terminal cytoplasmic domain contains a DNA transcription-activating domain while the C-terminal luminal domain is sensitive to ER stress. ATF6 release from BiP permits ATF6 transport to the Golgi compartment where it is cleaved by site-1 and site-2 proteases (Sp1/Sp2) to a 50- to 60-kDa fragment that migrates to the nucleus to activate transcription of XBP1-U and other UPR target genes (Shen et al., 2002). Thus, the IRE1 and ATF6 pathways work in concert to upregulate an ER protective response utilizing XBP1.
As indicated, XBP1-S is a potent transcription activator for many UPR target genes including the molecular chaperone BiP. The increased expression allows more BiP available to regulate the ER sensors. As discussed here, BiP has several functions including acting as a chaperone, a luminal sensor of unfolded proteins, a regulator of activation of UPR pathways UPR, and a regulator of its own expression. Recently, we found that genetic inhibition of BiP leads to augmented severity of experimental pancreatitis showing the importance of BiP in cellular protective responses (Ye et al., 2010).
The PERK plays a key role in adjusting the cell to ER stress by causing a significant attenuation of general protein synthesis (Figure 2). The activation of PERK by autophosphorylation (Thr980) leads to its phosphorylation of the alpha subunit of the eukaryotic translation initiation factor-2a (eIF-2a; Shen et al., 2002; Blais et al., 2010). The non-phosphorylated form of eIF-2a in its GTP-bound form is essential for translation initiation because it recruits the first tRNA (tRNAMet) to the ribosomal subunits to start translation of the attached mRNA. Phosphorylation of eIF2a at Ser51 by PERK blocks eIF2a-mediated initiation resulting in a general inhibition of protein synthesis. Cells with genetic deletion of PERK or cells containing eIF2a with position 51 containing alanine instead of serine to prevent phosphorylation do not attenuate protein synthesis with ER stress (Harding et al., 2000; Scheuner et al., 2001). As a consequence, cells are more sensitive to ER stress pointing out a potential protective role of the PERK signaling pathway in the ER stress response. Of note, previous studies in acinar cells have demonstrated that concentrations of cholecystokinin peptides which cause pancreatitis also inhibit translational initiation; and that this block in translation is mediated by phosphorylation of eIF2a (Perkins et al., 1997; Sans et al., 2002). Of interest, the phosphorylation of eIF2a is augmented by depletion of calcium from ER stores, showing that this manipulation of ER calcium homeostasis can initiate ER stress. Because cholecystokinin as well as acetylcholine can also deplete ER calcium stores, the findings suggest that changes in ER calcium provide an ER stress signal in the pancreatic acinar cell.
Persistent phosphorylation of eIF2a leads to specific translational upregulation of ATF4 that targets genes involved in antioxidant effects including synthesis of glutathione (Harding et al., 2003). ATF4 also upregulates the expression of the transcription factor C/EBP homologous protein (CHOP) which induces apoptosis (Oyadomari and Mori, 2004).
The mechanisms of the ERAD system to rid the ER of accumulated unfolded and misfolded proteins is complex, debated, and only partly understood in any cell (Brodsky and Wojcikiewicz, 2009; Yoshida and Tanaka, 2010). The degradation of ERAD substrates requires the multi-catalytic 26S protease called the proteasome. The proteasomes are not located in the ER but in the cytoplasm adjacent to the ER. Proteasomes act complementarily to the lysosome/vacuole to mediate the disassembly of proteins for recycling of their amino acids. The mechanisms of delivery of proteins from the ER to proteasomes are under active investigation and likely require a “proteinaceous channel” in the ER membrane needed to transfer proteins to the cytoplasmic located proteasomes. Further, most proteins submitted to the ERAD system are ubiquitinylated by ubiquitin ligases. The modification with ubiquitin both aids in transfer and targets a protein to the proteasome. Of importance, we recently reported (Lugea et al., 2010) that alcohol feeding in animals with genetic inhibition of XBP1 expression led to a marked decrease in expression of ER degradation-enhancing alpha-mannosidase-like 1 (EDEM1), a key participant involved in ERAD and targeting unfolded/misfolded proteins to the proteasome (Yoshida and Tanaka, 2010). These findings suggest a key role for ERAD in the adaptive response of the exocrine pancreas to environmental stressors.
Alcohol Abuse, ER Stress, and the UPR of the Exocrine Pancreas
Previous studies (Kubisch et al., 2006) demonstrated activation of all three ER stress sensor–transducers (i.e., IRE1, ATF6, and PERK) and their downstream pathways during the development of pancreatitis in experimental models pancreatitis suggesting involvement of ER stress responses in this disorder. However, there was no information on ER stress signals and the UPR in providing adaptation of environmental insults such as alcohol abuse.
Previous reports (Gukovsky et al., 2008) have demonstrated that long-term ethanol feeding in animal models causes little evidence of damage of the pancreatic parenchyma as determined by histology and blood concentrations of pancreatic digestive enzymes. For these reasons we designed studies to determine if alcohol abuse causes ER stress and activates the UPR in the exocrine pancreas; and studies to investigate the role of UPR signals in adapting the pancreas to the effects of alcohol abuse (Lugea et al., 2010).
For these studies rats and mice were fed control diets or ethanol containing diets for 4–6 weeks using the Tsukamoto–French intra-gastric model (Tsukamoto et al., 1985) which provides continuous feeding of the diets to the animals. As reportedly previously (Gukovsky et al., 2008), light microscopic examination of the exocrine pancreatic tissue demonstrated no obvious injury. However, careful structural examination by electron microscopy demonstrated extensive distention of the ER of acinar cells and altered distribution of zymogen granules. In addition, an investigation of the redox status of the ER demonstrated that there was a significant decrease in reduced and an increase in oxidized glutathione indicating that the alcohol feeding has a significant oxidative effect in the ER of the acinar cell. Taken together, these findings indicated that ethanol has the oxidative effect in the ER of acinar cells associated with morphologic signs of ER stress.
We then evaluated the effect of the alcohol treatment on the sensors and signals of the UPR. The major findings were significant effects of alcohol feeding on IRE1 and XBP1-S expression with smaller effects on PERK induction (Lugea et al., 2010). Also, we found that a key oxido-reductase, PDI, was significantly upregulated in the pancreas of the ethanol-fed animals. Further, we found that a much greater proportion of the PDI was in its oxidized state in the ethanol-fed animals compared to control fed. PDI is critical for protein folding by catalyzing disulfide bond formation, a key step for maturation of proteins in the secretory pathway (Ellgaard and Ruddock, 2005). The PDI protein folding function is often referred to as oxidative folding. That is, PDI requires reduced glutathione to sustain its catalytic redox cycling (Zhang and Kaufman, 2008). The combination of these findings suggest that PDI activity and thus appropriate folding of proteins in the secretory pathway of the acinar cell are impaired due to the oxidizing effects of alcohol feeding.
The findings that alcohol feeding increased IRE1 and XBP1-S expression suggested to us the possibility that these sensors and signals are important for adapting the ER of the acinar cell to the ER stress caused by the alcohol feeding. In order to test this possibility we obtained and tested animals with heterozygous deficiency in XBP1. Homozygous deletion of XBP1 is lethal and pancreatic tissue specific deletion leads to lack of development of the pancreas (Lee et al., 2005). We hypothesized that there would be an incomplete adaptive response to alcohol feeding in the heterozygous animals.
In contrast to wild-type animals, there were marked morphological and biochemical effects of alcohol feeding in animals with heterozygous deletion of XBP1. Of note, alcohol feeding increased levels of XBP1-S in the pancreas of heterozygous animals to the same level observed in wild-type animals receiving the control diet but not the augmented levels observed in the pancreas with alcohol feeding in the wild-type animals. Thus, the heterozygous deletion specifically prevented the ethanol feeding-induced augmentation in XBP1-S levels in the pancreas.
The predominant morphologic abnormalities in the XBP1 deficient animals receiving alcohol were a disorganized ultrastructure with decreased number of zymogen granules with the remaining ones inappropriately scattered throughout the cell and not localized to their normal apical position. There was extensive dilation of the ER with occasional dense luminal inclusions, a hallmark of ER stress, and a significant accumulation of autophagic vacuoles. There was also a marked decrease in levels of the digestive enzyme amylase corresponding to the decrease number of digestive enzyme storing zymogen granules. Finally, about 20% of the pancreas contained lesions representing severe injury. These areas showed acinar cell necrosis, apoptosis, and inflammation with replacement by stromal cells and regenerating ductular structures indicative of a response to severe injury.
An analysis of the UPR revealed that XBP1 deficiency prevented alcohol’s effect to increase PDI while it markedly promoted PERK and eIF2a phosphorylation and expression of ATF4, all features of prolonged and severe ER stress (Harding et al., 2003). Also of importance, XBP1 deficiency decreased levels of EDEM1, a key participant in the ERAD pathway for degradation of misfolded proteins. Of note, deficiencies in EDAM1 have been shown to result in increased degradation of misfolded proteins via autophagy (Hetz et al., 2009) which may explain the increased autophagy observed in our studies.
Conclusion
The results of the in vivo studies described above showing the role of the UPR in adapting the pancreas to alcohol treatments allow us to propose the model illustrated in Figure 3. We suggest that alcohol abuse usually does not cause pancreatic disease because of an adaptive UPR predominated by IRE1 and XBP1. The oxidative stress caused by alcohol metabolism in the ER activates IRE1 leading to splicing of XBP1 mRNA resulting in translation of the active transcription factor, XBP1-S. This transcription factor in turn mediates the expression of chaperones, PDI, ERAD proteins (for example EDEM1), and increased lipid synthesis to expand the capacity of the ER. These should all result in an attenuation of the ER stress. When there is an insufficient UPR response as shown in our experiments using animals with deficient XBP1, there is unresolved and augmented ER stress resulting in PERK activation causing global protein synthesis inhibition and even ATF4-mediated activation of cell death pathways via CHOP expression. These are only a few events that must be occurring in the cascade of pathobiologic events resulting in the significant tissue injury we observed.
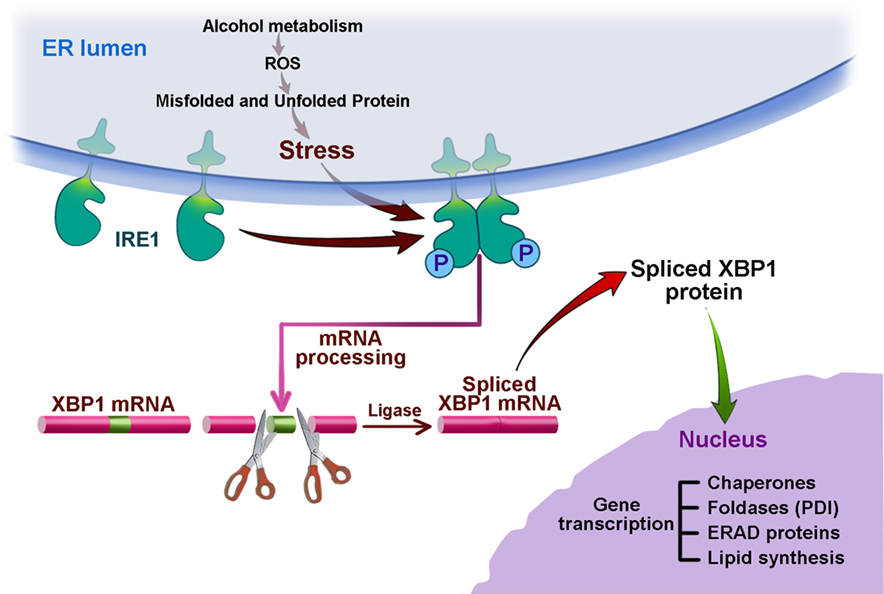
Figure 3. Alcohol, ER stress, and the IRE1 pathway of the UPR. This figure illustrates the results of our experiments in alcohol fed animals showing ER stress from ethanol and its metabolites, acetaldehyde, and fatty acid ethanol ester (FAEE), by inducing an oxidative state in the ER mediating misfolding and unfolding of proteins and oxidation of lipids. The ER stress leads to upregulation of the IRE1 pathway leading to splicing of XBP1 mRNA resulting in the translation of the transcription factor XBP1-S which is transported to the nucleus where it upregulates the expression of chaperones, oxido-reductases/foldases such as PDI, ERAD proteins, and lipid synthetic enzymes through DNA binding elements, ER stress element (ERSE) and the UPR element (UPRE). The activation of this pathway leads to adaptation of the pancreas to the stress of ethanol abuse and prevents pancreatic injury and disease.
The findings related to the oxidative state of glutathione and PDI provide important insights for ER stress and the UPR in the exocrine pancreas. Figure 4 illustrates the mechanism of oxidative folding of nascent proteins (Ellgaard and Ruddock, 2005; Hatahet and Ruddock, 2009; Shimizu and Hendershot, 2009). The ability of PDI to mediate folding of a nascent protein is a function of its ability to catalyze disulfide–dithiol exchange in the folding protein (substrate). PDI recognizes and binds to nascent unfolded proteins, semi-folded proteins and misfolded proteins but not correctly folded proteins through mechanisms that are not entirely known. Through electron transfer, oxidized PDI catalyzes disulfide bridge formation of the substrate protein as shown in the center of Figure 4. This action of PDI results in its own reduction. PDI needs then to be re-oxidized to carry out further disulfide catalysis.
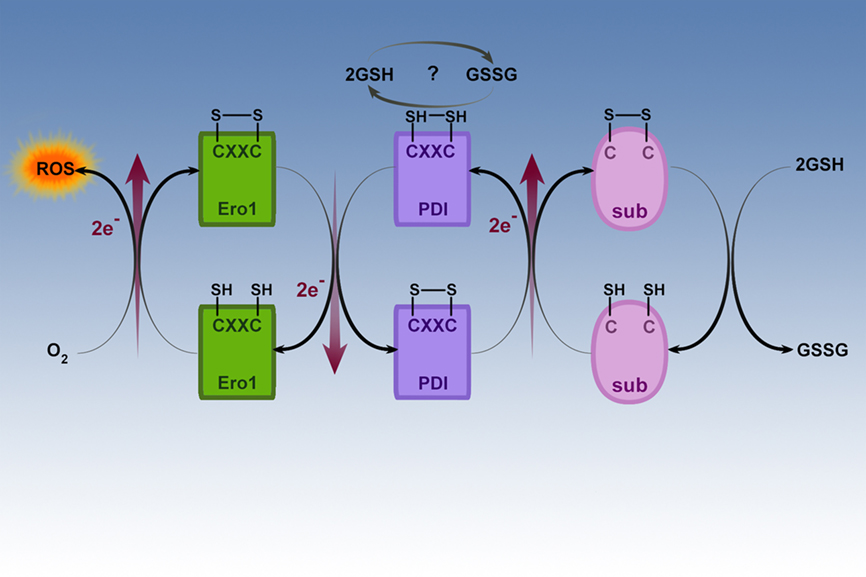
Figure 4. Oxidative folding in the ER. This figure illustrates the ER oxido-reductases involved in oxidative folding of nascent proteins in the ER. The oxido-reductases PDI and ERO1 are coupled and mediate the disulfide bridge formation and protein folding of proteins such as digestive enzymes synthesized in the ER. Another product of these coupled reactions is ROS. Reduced glutathione is necessary to reduce incorrectly placed disulfide bonds so that the protein can be recycled for correct disulfide bridge formation and folding.
The re-oxidation of PDI is catalyzed by another oxido-reductase called ER oxidase (ERO1) as shown on the left side of Figure 4. ERO1 is a flavo-enzyme that is induced by the UPR and it is directly oxidized by molecular oxygen in a FAD-dependent reaction (Shimizu and Hendershot, 2009; Tavender and Bulleid, 2010). FAD is synthesized in the cytoplasm and easily enters the ER. In the process ERO1 delivers the electrons that have been transferred from PDI to molecular oxygen with resultant ROS formation. It is known that PDI function and oxidative folding are dependent on ERO1 (Chakravarthi and Bulleid, 2004; Tavender and Bulleid, 2010). As illustrated on the right side of Figure 4, the reduction of disulfides is important for the reduction of incorrect disulfide bonds that can form during folding (Chakravarthi and Bulleid, 2004). Illustrated at the top of Figure 4 is the suggestion that glutathione may be involved in regeneration of the oxidized state of PDI in some systems.
The activities and direction of the reaction catalyzed by oxido-reductases such and PDI and ERO1 are dependent on the oxidative state of their catalytic sites as well as the local environments of the catalytic sites. The results from the experiments in the exocrine pancreas do not yet provide information on the detail of the oxidative state of the oxido-reductases or about their kinetics during the ER stress produced by alcohol treatments. However, the initial findings of a marked increase in the ratio of the oxidized to the reduced form of glutathione in the ER and the increase in the oxidized form of PDI suggest the likelihood of significant disturbances in oxido-reductase enzyme kinetics which would lead to the findings described.
In closing, we hypothesize that alcohol abuse and many other causes of pancreatitis including genetic mutations in digestive enzymes, smoking, metabolic disorders such as diabetes and hypertriglyceridemia, and drugs may cause the disease at least in part by causing ER stress. The key question related to why some individuals develop disease while others do not is not known. It is certainly possible that alterations in key components of the UPR through mutation (or environmental factors) are present in the pancreas of those individuals who develop tissue injury. In fact, genetic variants of XBP1 have been associated with the risk of development of inflammatory bowel disease (Kaser et al., 2008) supporting this possibility.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Department of Veterans Affairs; Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (P60 AA11999); UCLA Center for Excellence in Pancreatic Diseases (P01AT003960); and Alcohol and the Exocrine Pancreas ER Stress (R21 AA016010).
References
Blais, J. D., Chin, K. T., Zito, E., Zhang, Y., Heldman, N., Harding, H. P., Fass, D., Thorpe, C., and Ron, D. (2010). A small molecule inhibitor of endoplasmic reticulum oxidation 1 (ERO1) with selectively reversible thiol reactivity. J. Biol. Chem. 285, 20993–21003.
Brodsky, J. L., and Wojcikiewicz, R. J. (2009). Substrate-specific mediators of ER associated degradation (ERAD). Curr. Opin. Cell Biol. 21, 516–521.
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., Clark, S. G., and Ron, D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96.
Case, R. M. (1978). Synthesis, intracellular transport and discharge of exportable proteins in the pancreatic acinar cell and other cells. Biol. Rev. Camb. Philos. Soc. 53, 211–354.
Chakravarthi, S., and Bulleid, N. J. (2004). Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 279, 39872–39879.
Ellgaard, L., and Ruddock, L. W. (2005). The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32.
Go, V. L., Gukovskaya, A., and Pandol, S. J. (2005). Alcohol and pancreatic cancer. Alcohol 35, 205–211.
Gorlach, A., Klappa, P., and Kietzmann, T. (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391–1418.
Gukovsky, I., Lugea, A., Shahsahebi, M., Cheng, J. H., Hong, P. P., Jung, Y. J., Deng, Q. G., French, B. A., Lungo, W., French, S. W., Tsukamoto, H., and Pandol, S. J. (2008). A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G68–G79.
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., and Ron, D. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108.
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B., Paules, R., Stojdl, D. F., Bell, J. C., Hettmann, T., Leiden, J. M., and Ron, D. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633.
Hatahet, F., and Ruddock, L. W. (2009). Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850.
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., Martinez, G., Cuervo, A. M., Brown, R. H., and Glimcher, L. H. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306.
Kaser, A., Lee, A. H., Franke, A., Glickman, J. N., Zeissig, S., Tilg, H., Nieuwenhuis, E. E., Higgins, D. E., Schreiber, S., Glimcher, L. H., and Blumberg, R. S. (2008). XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756.
Kereszturi, E., Szmola, R., Kukor, Z., Simon, P., Weiss, F. U., Lerch, M. M., and Sahin- Toth, M. (2009). Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum. Mutat. 30, 575–582.
Kim, I., Xu, W., and Reed, J. C. (2008). Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7, 1013–1030.
Kubisch, C. H., Sans, M. D., Arumugam, T., Ernst, S. A, Williams, J. A., and Logsdon, C. D. (2006). Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G238–G245.
Lee, A. H., Chu, G. C., Iwakoshi, N. N., and Glimcher, L. H. (2005). XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 24, 4368–4380.
Lee, K., Tirasophon, W., Shen, X., Michalak, M., Prywes, R., Okada, T., Yoshida, H., Mori, K., and Kaufman, R. J. (2002). IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466.
Lowenfels, A. B., Maisonneuve, P., Cavallini, G., Ammann, R. W., Lankisch, P. G., Andersen, J. R., Dimagno, E. P., Andren- Sandberg, A., and Domellof, L. (1993). Pancreatitis and the risk of pancreatic cancer. International pancreatitis study group. N. Engl. J. Med. 328, 1433–1437.
Lugea, A., Tischler, D., Nguyen, J., Gong, J., Gukovsky, I., French, S. W., Gorelick, F. S., and Pandol, S. J. (2010). Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology. 140, 987–997.
Maisonneuve, P., Lowenfels, A. B., Mullhaupt, B., Cavallini, G., Lankisch, P. G., Andersen, J. R., Dimagno, E. P., Andren- Sandberg, A., Domellof, L., Frulloni, L., and Ammann, R. W. (2005). Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 54, 510–514.
Marciniak, S. J., Garcia- Bonilla, L., Hu, J., Harding, H. P., and Ron, D. (2006). Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 172, 201–209.
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389.
Pandol, S., Edderkaoui, M., Gukovsky, I., Lugea, A., and Gukovskaya, A. (2009). Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 7(Suppl. 11), S44–S47.
Pandol, S. J., Saluja, A. K., Imrie, C. W., and Banks, P. A. (2007). Acute pancreatitis: bench to the bedside. Gastroenterology 132, 1127–1151.
Perkins, P. S., Park, J. H., and Pandol, S. J. (1997). The role of calcium in the regulation of protein synthesis in the exocrine pancreas. Pancreas 14, 133–141.
Petersen, O. H., and Tepikin, A. V. (2008). Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 70, 273–299.
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529.
Rutkowski, D. T., and Kaufman, R. J. (2007). That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 32, 469–476.
Sans, M. D., Kimball, S. R., and Williams, J. A. (2002). Effect of CCK and intracellular calcium to regulate eIF2B and protein synthesis in rat pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G267–G276.
Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., Saunders, T., Bonner- Weir, S., and Kaufman, R. J. (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176.
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111.
Shimizu, Y., and Hendershot, L. M. (2009). Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal. 11, 2317–2331.
Sutton, R., Petersen, O. H., and Pandol, S. J. (2008). Pancreatitis and calcium signalling: report of an international workshop. Pancreas 36, e1–e14.
Tavender, T. J., and Bulleid, N. J. (2010). Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid. Redox Signal. 13, 1177–1187.
Tsukamoto, H., French, S. W., Benson, N., Delgado, G., Rao, G. A., Larkin, E. C., and Largman, C. (1985). Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology 5, 224–232.
Yadav, D., and Whitcomb, D. C. (2010). The role of alcohol and smoking in pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 7, 131–145.
Ye, R., Mareninova, O. A., Barron, E., Wang, M., Hinton, D. R., Pandol, S. J., and Lee, A. S. (2010). Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am. J. Pathol. 177, 2827–2836.
Yoshida, H., Matsui, T., Hosokawa, N., Kaufman, R. J., Nagata, K., and Mori, K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271.
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, p. 881–891.
Yoshida, Y., and Tanaka, K. (2010). Lectin-like ERAD players in ER and cytosol. Biochim. Biophys. Acta 1800, 172–180.
Keywords: UPR, pancreas, gastrointestinal, research, exocrine, pancreatic function
Citation: Pandol SJ, Gorelick FS and Lugea A (2011) Environmental and genetic stressors and the unfolded protein response in exocrine pancreatic function – a hypothesis. Front. Physio. 2:8. doi: 10.3389/fphys.2011.00008
Received: 09 November 2010;
Accepted: 24 February 2011;
Published online: 10 March 2011.
Edited by:
D. N. Granger, Louisiana State University Health Sciences Center, USAReviewed by:
Constanze Heike Kubisch, University of Munich, GermanyJohn A. Williams, University of Michigan, USA
Tak Yee Aw, Louisiana State University Health Sciences Center Shreveport, USA
Copyright: © 2011 Pandol, Gorelick and Lugea. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Stephen J. Pandol, Department of Veterans Affairs, University of California, Building 258, Room 340, 11310 Wilshire Boulevard, Los Angeles, CA 90073, USA.e-mail: stephen.pandol@va.gov