 Kun-Ze Lee1 Kai Qiu1 Milapjit S. Sandhu1 Mai K. Elmallah2 Darin J. Falk3,4 Michael A. Lane5 Paul J. Reier5 Barry J. Byrne3,4 David D. Fuller1*
Kun-Ze Lee1 Kai Qiu1 Milapjit S. Sandhu1 Mai K. Elmallah2 Darin J. Falk3,4 Michael A. Lane5 Paul J. Reier5 Barry J. Byrne3,4 David D. Fuller1*- 1 Department of Physical Therapy, McKnight Brain Institute, University of Florida, Gainesville, FL, USA
- 2 Pediatric Pulmonary Division, Department of Pediatrics, University of Florida, Gainesville, FL, USA
- 3 Division Cell and Molecular Therapy and Pediatric Cardiology, Department of Pediatrics, University of Florida, Gainesville, FL, USA
- 4 Powell Gene Therapy Center, University of Florida, Gainesville, FL, USA
- 5 Department of Neuroscience, McKnight Brain Institute, University of Florida, Gainesville, FL, USA
Pompe disease is a lysosomal storage disorder associated with systemic deficiency of acid α-glucosidase (GAA). Respiratory-related problems in Pompe disease include hypoventilation and upper airway dysfunction. Although these problems have generally been attributed to muscular pathology, recent work has highlighted the potential role of central nervous system (CNS) neuropathology in Pompe motor deficiencies. We used a murine model of Pompe disease to test the hypothesis that systemic GAA deficiency is associated with hypoglossal (XII) motoneuron pathology and altered XII motor output during breathing. Brainstem tissue was harvested from adult Gaa−/− mice and the periodic acid Schiff method was used to examine neuronal glycogen accumulation. Semi-thin (2 μm) plastic sections showed widespread medullary neuropathology with extensive cytoplasmic glycogen accumulation in XII motoneuron soma. We next recorded efferent XII bursting in anesthetized and ventilated Gaa−/− and B6/129 mice both before and after bilateral vagotomy. The coefficient of variation of respiratory cycle duration was greater in Gaa−/− compared to B6/129 mice (p < 0.01). Vagotomy caused a robust increase in XII inspiratory burst amplitude in B6/129 mice (239 ± 44% baseline; p < 0.01) but had little impact on burst amplitude in Gaa−/− mice (130 ± 23% baseline; p > 0.05). We conclude that CNS GAA deficiency results in substantial glycogen accumulation in XII motoneuron cell bodies and altered XII motor output. Therapeutic strategies targeting the CNS may be required to fully correct respiratory-related deficits in Pompe disease.
Introduction
Glycogen storage disease type II or Pompe disease is caused by a systemic lack of functional acid α-glucosidase (GAA). The prevalence of Pompe disease is approximately 1 per 40,000 births (Ausems et al., 1999; Hirschhorn and Reuser, 2001), and this condition is associated with extensive glycogen accumulation in skeletal muscle, visceral organs, and the central nervous system (CNS; Raben et al., 2002; Sidman et al., 2008; DeRuisseau et al., 2009). Clinically, Pompe syndromes can be classified as early (infantile) and late-onset (juvenile/adult). However, these classifications represent a disease continuum related to the extent of enzyme deficiency (Byrne et al., 2011a). Infantile Pompe disease is associated with complete or near complete GAA deficiency but late-onset patients maintain some residual GAA activity (Hirschhorn and Reuser, 2001; Raben et al., 2002). Respiratory insufficiency is a common symptom in both infantile and late-onset Pompe disease (Mellies et al., 2005; Pellegrini et al., 2005; Burghaus et al., 2006; Mellies and Lofaso, 2009). Pompe infants typically present at 4–6 months of age with “respiratory difficulty” being noted as the first symptom in 25% of cases (van den Hout et al., 2003). Respiratory failure is listed as the cause of death in 19% of documented cases (Byrne et al., 2011a). Late-onset patients usually show progressive respiratory weakness and eventual failure. Thus, approximately 75% of children and adolescents with Pompe disease eventually require mechanical ventilation (Haley et al., 2003; Marsden, 2005). In adult patients, vital capacity and peak inspiratory pressures are reduced, and arterial hypoxemia, and hypercapnia are common (Mellies et al., 2005). Approximately 33% of adult Pompe patients will eventually require mechanical ventilatory support and both pneumonia and bronchitis are prevalent (Hagemans et al., 2005).
Although motor problems in Pompe disease have historically been attributed to muscular pathology (Raben et al., 2002), glycogen accumulation in the CNS has been documented in Pompe tissues (Mancall et al., 1965; Gambetti et al., 1971; Martin et al., 1973; Martini et al., 2001; Teng et al., 2004; DeRuisseau et al., 2009), and in various Pompe animal models (Matsui et al., 1983; Sidman et al., 2008; DeRuisseau et al., 2009). Clinical case reports also highlight the involvement of the CNS in motor (Clement and Godman, 1950; Zellweger et al., 1955; Hogan et al., 1969; Willemsen et al., 1998; Teng et al., 2004) and possibly cognitive deficiencies (Rohrbach et al., 2010) in Pompe disease. We recently reported spinal (phrenic) respiratory motoneuron pathology and reduced minute ventilation in the adult Gaa−/− mouse model of Pompe disease (DeRuisseau et al., 2009; Mah et al., 2010). Similar neuropathology was noted in putative phrenic motoneurons in the cervical ventral horn of spinal tissue from a Pompe infant obtained at autopsy (DeRuisseau et al., 2009). We therefore have proposed that phrenic motoneuron pathology may contribute to respiratory insufficiency in Pompe disease (DeRuisseau et al., 2009). However, breathing also involves activation of pharyngeal and laryngeal muscles controlled by supraspinal motoneurons (Feldman and Del Negro, 2006). Hypoglossal (XII) motoneurons are of particular importance to upper airway patency as these cells regulate the shape, stiffness, and position of the tongue (Remmers et al., 1978; Fregosi and Fuller, 1997; Bailey and Fregosi, 2004; Gestreau et al., 2005). In particular, contraction of the extrinsic tongue muscles (e.g., the genioglossus muscle) can dilate and/or stiffen the pharyngeal lumen, thereby minimizing airway narrowing, and/or collapse in the face of negative inspiratory pressures (Fuller et al., 1999). Although there is clinical evidence of XII motor dysfunction in Pompe disease (Margolis et al., 1994; Muller et al., 2009; Jones et al., 2010; Byrne et al., 2011a), to our knowledge there have been no detailed studies of respiratory-related XII motor control in Pompe patients or relevant animal models. Sidman et al. (2008) recently described brainstem neuropathology in the 6neo/6neo mouse Pompe model but did not comment on the appearance of XII motoneurons. Accordingly, we used Gaa−/− mice (Raben et al., 1998) to test the hypothesis that systemic GAA deficiency is associated with XII motoneuron pathology and altered efferent XII motor output during breathing.
Materials and Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Florida. B6/129 mice (age 11 ± 1 month) and Gaa−/− mice of similar age (12 ± 1 month, p = 0.54) were used in the majority of this work. Histological studies were conducted using N = 7 B6/129 and N = 6 Gaa−/− mice. Neurophysiological studies (e.g., nerve recordings; see below) were completed in N = 10 B6/129 and N = 15 Gaa−/− mice. A few additional histological studies were conducted in 129 mice to test cholera toxin β-subunit (CT-β) labeling of the XII nucleus (N = 4, age 4–5 weeks). The Gaa−/− mice were previously described (Raben et al., 1998; DeRuisseau et al., 2009), and originally obtained from Taconic, Inc. Prior to initiating these experiments, a subset of Gaa−/− mice were genotyped using standard PCR methods to confirm the absence of the GAA gene (Raben et al., 1998; Mah et al., 2007). Briefly, DNA was isolated from tail snips and amplified using primers to exons five and seven of the mouse GAA gene. A 692-bp product indicated the wildtype allele and a 2-kb product confirmed the insertion of a neo cassette and was detected in Gaa−/− mice.
Brainstem Histology
Brainstem tissues were embedded in paraffin or in epoxy resin (Lane et al., 2008). Mice were anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and euthanized via systemic perfusion. The perfusate was either 4% paraformaldehyde (prior to paraffin tissue embedding) or 4% paraformaldehyde and 3.5% glutaraldehyde (prior to plastic embedding). Paraffin embedding of the tissue was done with standard procedures (Mah et al., 2007). The plastic embedding methods have also been described (Lane et al., 2007, 2008). Visual confirmation of glycogen accumulation was accomplished using the periodic acid Schiff (PAS) staining method (Guth and Watson, 1968; Sidman et al., 2008; DeRuisseau et al., 2009). Briefly, tissues were incubated in 0.5% periodic acid (Richard-Allan Scientific Inc., Kalamazoo, MI, USA) for 10 min at 60°C, rinsed with tap water and then stained with Schiff’s reagent (Richard-Allan Scientific Inc.) for 5 min, and then rinsed again in tap water. Plastic sections were then counterstained with toluidine blue (Lane et al., 2008) for 15–30 s. The XII motor nucleus was retrogradely labeled by injecting CT-β (0.1% in 0.1 M PBS) into the base of the tongue. Mice were anesthetized as described above and the tongue was gently protruded using fine forceps. One or two injections (2–5 μl/site) were then made lateral to the lingual frenulum. Animals were sacrificed 60 h post-injection and CT-β labeled neurons were detected as previously described (Lane et al., 2008).
XII Nerve Recordings
These procedures were adapted from our recent report (DeRuisseau et al., 2009). Mice were anesthetized with urethane (1.0–1.6 mg/kg, i.p., Sigma, St. Louis, MO, USA) and placed in a supine position. Body temperature was maintained at 37–38°C using a servo-controlled heating pad (model TC-1000, CWE, Ardmore, PA, USA). The trachea was cannulated below the larynx to enable mechanical ventilation (model SAR-830/AP; CWE) with a hyperoxic gas mixture (FIO2 = 0.50–0.60). The ventilator frequency was maintained at 150 cycles/min with inspiratory duration set at 0.33 s. The resulting minute ventilation values ranged from 15 to 18 ml/min/10 g body weight, and were based on prior descriptions of respiratory volumes in unanesthetized Gaa−/− and B6/129 mice (Mah et al., 2007; DeRuisseau et al., 2009). Pulse oximetry (MouseOx, STARR Life Science Corp., Oakmont, PA, USA) was used to measure the saturation of hemoglobin (SaO2,%) in five of nine B6/129 mice and 5 of 11 Gaa−/− mice. In each case the measured SaO2 exceeded 96% throughout the experiment. After initiating mechanical ventilation, mice were paralyzed by i.p. administration of pancuronium bromide (2.5 mg/kg, Hospira, Inc., Lake Forest, IL, USA). The phrenic and XII nerves were isolated unilaterally and the distal ends of each nerve were cut prior to initiating the recordings. Nerve activity was recorded using monopolar silver wire electrodes following amplification (1000×, Model 1700, A-M Systems, Carlsborg, WA, USA) and band-pass filtering (0.3–10 KHz). The raw neurograms were integrated using a 100-ms time constant (model MA-1000; CWE). Data were digitized (CED Power 1401) and recorded on a PC using Spike2 software (Cambridge Electronic Design Limited, Cambridge, England).
Data Analysis
Inspiratory time (TI), expiratory time (TE), and the overall respiratory cycle duration (TTOT) were calculated from the integrated phrenic neurogram as previously described (Lee et al., 2009). Efferent burst amplitude was assessed by measuring the peak height of integrated neurograms during each neural breath (Lee et al., 2009). Burst amplitude was first quantified using the “raw” output of the amplifier (arbitrary units, a.u.) and was also normalized to the baseline condition (% baseline). The “area under the curve” for each integrated neural breath was also calculated and expressed as a.u.*s or % baseline. However, all burst amplitude results were both qualitatively and quantitatively similar when analyzed as “peak height” or “area under the curve,” and accordingly we present only the peak height. The pre-inspiratory (Pre-I) component of the XII burst was calculated as previously described (Lee and Fuller, 2010a). Data from each individual mouse were averaged over 3 min periods, and the overall mean responses were calculated from these values. The variability of the respiratory pattern was quantified by calculating the coefficient of variation [i.e., 100 × (standard deviation/mean)] for all parameters. When all mice had measurements during both vagotomized and vagal-intact conditions (e.g., XII amplitude, a.u.), statistical comparisons between B6/129 and Gaa−/− mice (i.e., “strain effects”) were made using two way repeated measures (RM) analysis of variance (ANOVA). If data were not obtained in both conditions in all mice, a two way ANOVA was used to compare the strains. In cases in which only a single data point was compared (e.g., XII amplitude, % vagal-intact), a one-tailed t-test was used to compare the groups. All data are expressed as the mean ± standard error. Data were considered to be statistically different when the p-value was <0.05.
Results
Brainstem Histology
The XII nucleus was observed immediately caudal and lateral to the central canal in the medulla (Figures 1 and 2) as previously reported in mice (Millecamps et al., 2001). Our initial histochemistry experiments were performed using paraffin-embedded tissues, and PAS staining produced significant reaction product in neuronal cell bodies within the XII nucleus of Gaa−/− but not B6/129 mice (Figures 1A–D). The location of the XII nucleus was also confirmed using CT-β tongue injections (Figures 1E,F). Semi-thin (2 μm) plastic sections were also evaluated from Gaa−/− mice, and these revealed widespread medullary neuropathology (Figure 2). Deposits of PAS positive material were readily observed in XII motoneuron cytoplasm (Figures 2A–C) and nuclei were free of PAS positive material (Figure 2C). A gradient of cytoplasmic inclusions could be observed across cells with some neurons appearing to be virtually filled with glycogen droplets (e.g., Figure 2B). PAS positive inclusions were also noted in ependymal cells near the central canal (Figure 2A) as well as in astrocytic cells (Figure 2D). However, there was no indication of overt axonal pathology (Figures 2D,E). Although not a primary focus of this work, we also noted robust PAS staining in neuronal cytoplasm within the nucleus ambiguous (Figure 2F). The nucleus ambiguous contains pharyngeal and laryngeal motoneurons, and accordingly neuropathology in this region could contribute to upper airway dysfunction.
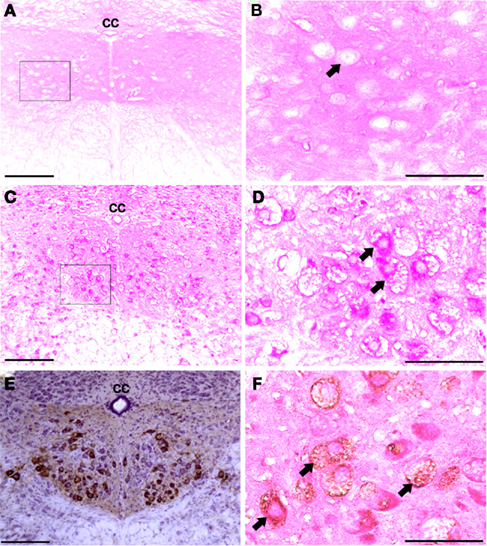
Figure 1. Medullary sections showing XII motor neurons. (A–D) Show paraffin-embedded tissues cut at 10 μm and stained with the PAS method. Positive PAS staining was not observed within neuronal cell bodies of B6/129 mice (A,B). In contrast, XII motoneurons and cells in the surrounding areas showed robust PAS staining in Gaa−/− mice (C,D). (B and D) Are higher magnification images of the areas indicated in (A and C); arrows indicate individual XII motoneurons. (E) Shows an example of CT-β labeling of the XII nuclei following tongue injection. (F) Shows a paraffin-embedded medullary section from a Gaa−/− mouse in which XII motoneurons were retrogradely labeled with CT-β. This section was also stained with the PAS method, and labeled XII motoneurons (arrows) show PAS staining consistent with what was presented in (C,D). cc, Central canal; scale bars: (A,C,E): 100 μm; (B,D,F): 50 μm.
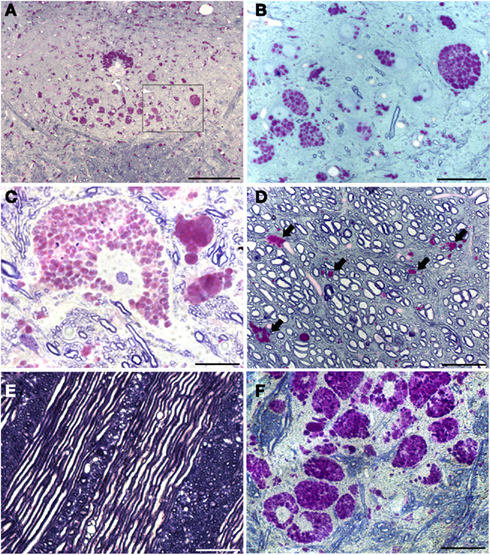
Figure 2. Plastic-embedded semi-thin medullary sections (2 μm) show neuronal glycogen accumulation in Gaa−/− mice. Sections were stained with PAS and toluidine blue. (A) Provides a low power view including both hypoglossal nuclei; (B) is a higher resolution image of the area indicated by the box in (A). These images demonstrate that PAS staining within the XII nuclei is restricted to neuronal cell bodies. Ependymal cells around the central canal (CC) also showed robust PAS staining (A). (C) Shows a high magnification of PAS staining within a XII motoneuron – note the extensive accumulation of glycogen droplets in the cytoplasm with no positive nuclear staining. The image in (D) is from the ventral medulla near the pyramidal decussation. PAS positive neuroglial cells were noted (indicated by the arrows) but axons appear to be healthy with no evidence of glycogen accumulation. Similarly, glycogen accumulation was not observed in medullary axons including cranial nerve fibers (E). Finally, (F) depicts the extensive neuronal glycogen accumulation within the nucleus ambiguous in the rostral medulla. Although not the primary focus of this work, this image is included to illustrate that neuropathology in upper airway motor systems in Gaa−/− mice is not limited to the XII motor nucleus. Scale bars: (A): 100 μm; (B,D–F): 50 μm; (C): 20 μm.
Pattern of Respiratory Output
Representative examples of XII neurograms recorded in anesthetized mice are provided in Figure 3. The “double bursting” pattern illustrated in Figure 3 for the Gaa−/−mouse occurred in 33% (5/15) of Gaa−/− mice and 10% (1/10) B6/129 mice.
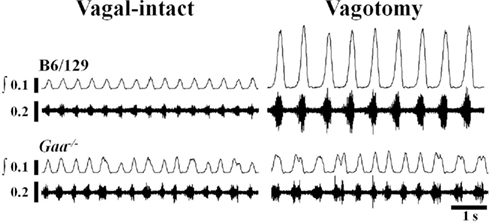
Figure 3. Examples of efferent XII nerve activity in B6/129 and Gaa−/− mice under vagal-intact and vagotomized conditions. Both “raw” and integrated (∫) neurograms are shown. In the vagal-intact condition, the amplitude of the XII signal is similar between the two mice, but the Gaa−/− mouse shows more variability in the timing of the respiratory cycle. Following bilateral cervical vagotomy there is a robust increase in XII burst amplitude in the B6/129 mouse whereas the Gaa−/− mouse shows little change.
In the vagal-intact condition, inspiratory burst frequency was similar between groups (B6/129: 123 ± 11 bursts/min; Gaa−/−: 126 ± 8 bursts/min). Following cervical vagotomy, inspiratory burst frequency was significantly reduced in both groups of mice (p < 0.05, Table 1). While overall burst frequency was not different, the respiratory cycle showed greater variability in Gaa−/− compared to B6/129 mice (Figure 4). On average, the coefficient of variation of TI, TE, and TTOT were all increased in Gaa−/− mice (two way ANOVA, strain effect, all p < 0.05; Figure 5).
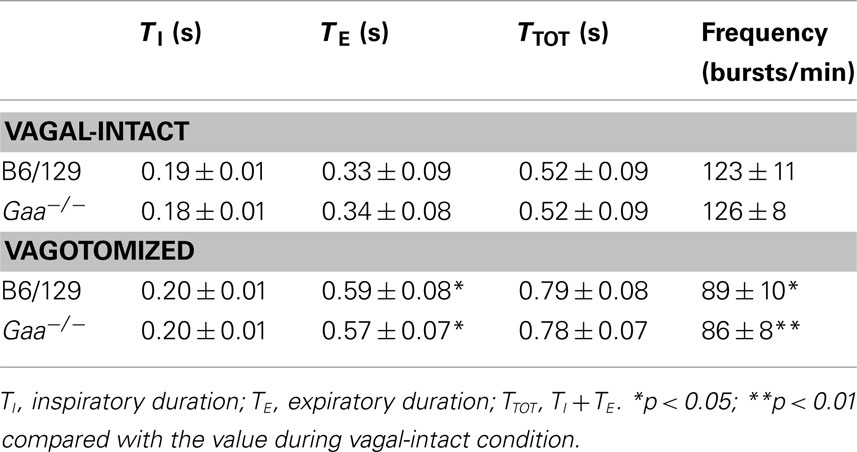
Table 1. Respiratory cycle duration and frequency in B6/129 and Gaa−/− mice.
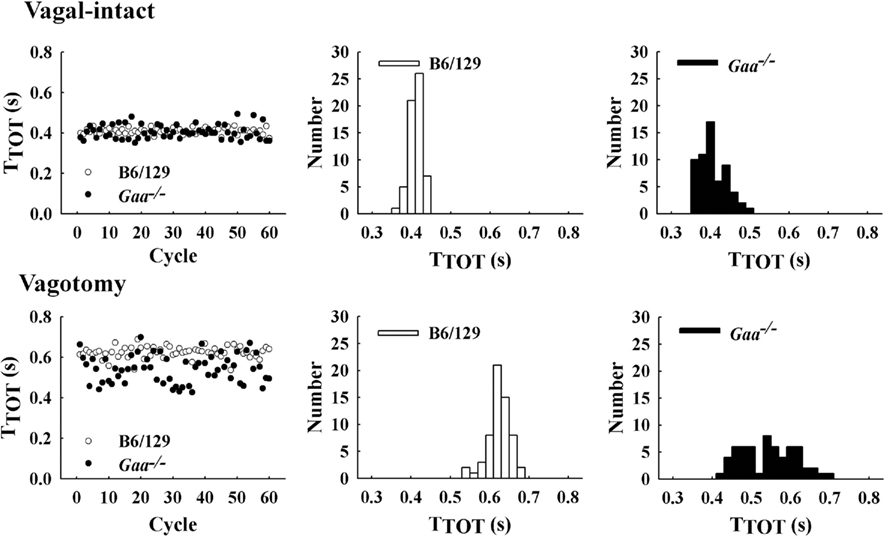
Figure 4. Examples of respiratory cycle variability in representative B6/129 and Gaa−/− mice. The left panel shows a sequential plot of the total duration of the respiratory cycle (TTOT) for 60 consecutive cycles. An increase in the breath-to-breath variability in TTOT can be seen in the Gaa−/− mouse under both vagal-intact and vagotomized conditions. The middle and right panels show histograms depicting the overall distribution of values for TTOT during the 60 respiratory cycles depicted in the left panel.
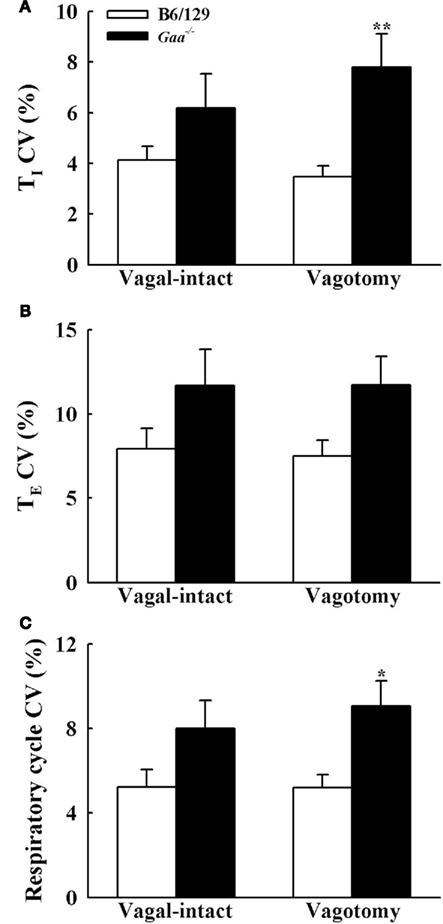
Figure 5. The coefficient of variation (CV) of inspiratory [TI, (A)], expiratory [TE, (B)], and total respiratory cycle [TTOT, (C)] duration during vagal-intact and vagotomized conditions. CV of TI and TTOT is significantly higher in vagotomized Gaa−/− mice than B6/129 mice. *p < 0.05; **p < 0.01 significant different from B6/129 mice. #p < 0.05 compared with the value during vagal-intact condition.
The inspiratory burst onset recorded in the phrenic and XII nerves was similar in the vagal-intact condition. In other words, Pre-I XII bursting (Lee et al., 2007b) could not be discerned in either experimental group with vagus nerves intact. However, following vagotomy clear Pre-I XII activity was observed in B6/129 but not Gaa−/− mice (Figure 6). The finding in B6/129 mice is consistent with our prior reports showing that Pre-I bursting is much more robust following vagotomy (Lee et al., 2007a, b). Pre-I bursting probably plays a role in stabilizing the upper airways before inspiration, and accordingly these data may indicate that Gaa−/− mice have reduced airway patency during conditions associated with increased respiratory drive.
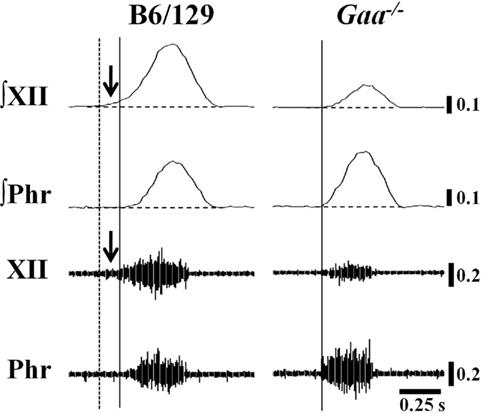
Figure 6. Examples of Pre-I XII bursting in B6/129 and Gaa−/− mice. These examples were taken from vagotomized, ventilated mice. We noted anecdotally that Pre-I XII bursting (i.e., bursting before the onset of the phrenic burst) was evident in B6/129 but not Gaa−/− mice. In the examples, the solid vertical line indicates the onset of inspiratory phrenic bursting and the vertical dashed line represents the onset of Pre-I XII activity. The horizontal line indicates the end-expiratory amplitude prior to inspiration. This particular Gaa−/− mouse example was chosen to illustrate that in cases where Gaa−/− mice showed more robust phrenic bursting, the same animal tended to show reduced efferent XII output (see text for further description).
Amplitude of XII and Phrenic Bursting
Assessment of inspiratory XII burst amplitude (a.u.) revealed an interaction (p = 0.045) between strain and condition with differences between B6/129 and Gaa−/− mice becoming evident after vagotomy. During the baseline vagal-intact condition, XII burst amplitude (a.u.) tended to be greater in B6/129 (0.18 ± 0.04) than Gaa−/− mice (0.11 ± 0.04), but this difference was not statistically significant (p > 0.05; Figure 7). Following vagotomy, however, XII burst amplitude (a.u.) was considerably greater in B6/129 (0.40 ± 0.13) than Gaa−/− mice (0.12 ± 0.03; p = 0.007; Figure 7). The normalized XII burst amplitudes confirmed this observation. Indeed, vagotomy triggered a robust increase in XII burst amplitude (239 ± 44% baseline) in B6/129 mice but did not consistently alter burst amplitude in Gaa−/− mice (130 ± 23% baseline; p = 0.02 vs. B6/129). The coefficient of variation of XII burst amplitude was not different between Gaa−/− and B6/129 mice, and vagotomy reduced amplitude variability in both groups (p < 0.05; Table 2).
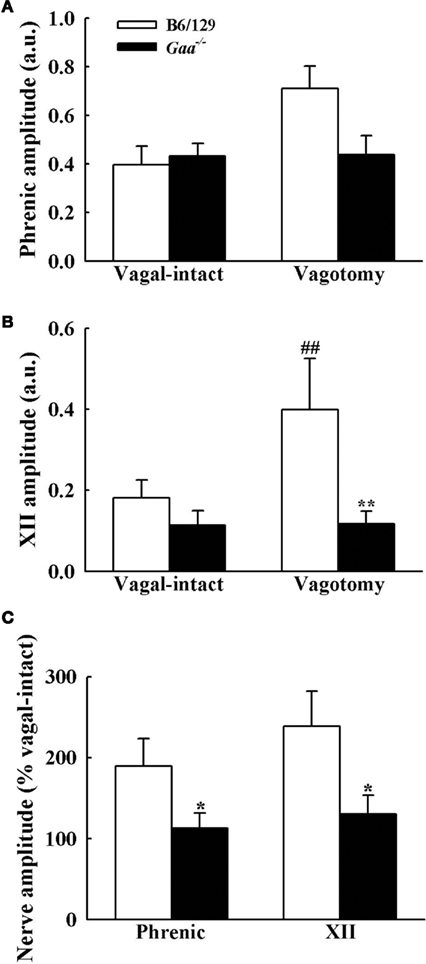
Figure 7. Inspiratory phrenic and XII burst amplitude B6/129 and Gaa−/− mice. The amplitude of phrenic (A) and XII (B) neurogram inspiratory burst amplitudes were expressed as arbitrary units (a.u.), and normalized to the activity recorded under vagal-intact conditions (C). Cervical vagotomy caused robust increases in XII burst amplitude in B6/129 but not Gaa−/− mice. See text for a commentary on the phrenic burst amplitude. *p < 0.05; **p < 0.01 significant different from B6/129 mice. #p < 0.05; ##p < 0.01 compared with the value during vagal-intact condition.
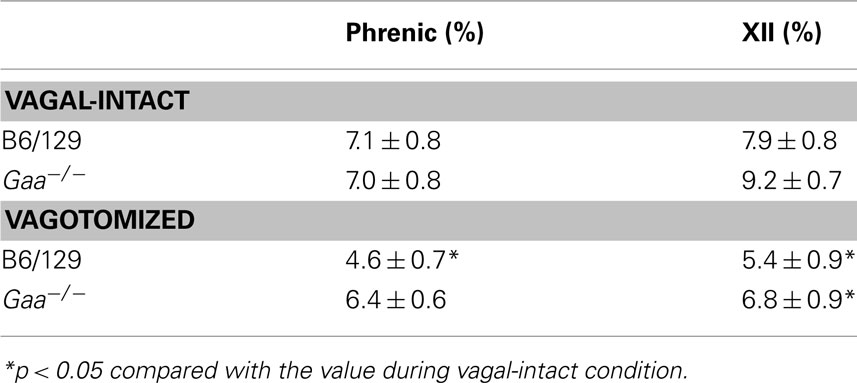
Table 2. The coefficient of variation of phrenic and XII nerve amplitude in B6/129 and Gaa−/− mice.
Phrenic burst amplitude (a.u.) was similar between B6/129 (0.40 ± 0.10) and Gaa−/− mice (0.45 ± 0.10) during the vagal-intact condition (Figure 7). However, vagotomy robustly increased phrenic burst amplitude in B6/129 (190 ± 32% baseline) but not Gaa−/− mice (113 ± 18% baseline; Figure 7). Thus, the normalized phrenic output (% baseline) was blunted in Gaa−/− mice (p = 0.03 vs. B6/129). The overall range of these values was also greater in B6/129 (from 96 to 339% baseline) compared to Gaa−/− mice (from 33 to 239% baseline). Following vagotomy, non-normalized (a.u.) Gaa−/− phrenic burst amplitude (0.43 ± 0.09) was approximately 60% of that of B6/129 mice (0.71 ± 0.08) but this did not reach statistical significance (p > 0.05). However, we noted that an outlier point skewed this result. Specifically, one Gaa−/− mouse demonstrated particularly robust phrenic bursting, and this data point met the criteria established by Dixon’s outlier test at the 95% confidence level (Verma and Quiroz-Ruiz, 2006). Removal of this point resulted in a statistically significant difference in raw phrenic bursting between Gaa−/− and B6/129 mice (p = 0.01). Nevertheless, this data point is included in the overall analyses because this may reflect the “clinical spectrum” of Pompe disease. Indeed the range of values observed for both raw and normalized neural output in this study may reflect variance in the relative rate of disease progression and associated neuropathology (Sidman et al., 2008; DeRuisseau et al., 2009). In this regard, it is interesting that a recent systematic review of a large sample of Pompe patients with symptom onset after 12 month of age found a wide range of pulmonary function. For example, forced vital capacity (FVC) ranged from as low as 3%, to as much as 123% of normal (Byrne et al., 2011a).
A few additional anecdotal observations are pertinent. First, we noted that those Gaa−/− mice with the most striking locomotor deficits tended to have the weakest phrenic output. Second, those Gaa−/− mice with more robust phrenic output tended to have blunted XII activity (e.g., Figure 6). Thus, the amplitude (a.u.) of the phrenic signal in Gaa−/− mice did not correlate with the XII burst amplitude (r2 = 0.048; p = 0.384). In contrast, the same two variables were significantly correlated in B6/129 mice (r2 = 0.272; p = 0.038). This observation could reflect a differential onset of neuropathology and associated symptoms between the XII and phrenic motor pools. This is potentially important since overt symptoms of altered tongue motor control can present before ventilatory failure in Pompe infants (Byrne, unpublished observations).
Discussion
This study provides the first evidence that medullary glycogen accumulation is associated with alterations in the neural control of XII motoneurons. PAS staining of brainstem tissues confirmed widespread neuropathology in Pompe mice (Sidman et al., 2008; DeRuisseau et al., 2009) and documented extensive somal glycogen accumulation in XII motoneurons. Neurophysiology experiments revealed altered XII nerve output and increased respiratory cycle variability in Gaa−/− mice. Thus, systemic GAA deficiency is associated with instability in the respiratory control circuits, and apparently reduced neural drive to the tongue. Altered control of the tongue and other pharyngeal muscles could contribute to upper airway instability as well as speech and/or swallow dysfunction in Pompe disease (Jones et al., 2010).
Respiratory Activity in Pompe Mice
Prior work has shown that Gaa−/− mice have respiratory-related functional impairments including reduced diaphragm muscle contractility and attenuated minute ventilation (Mah et al., 2007, 2010; DeRuisseau et al., 2009). The present observation of altered respiratory timing in Gaa−/− mice is consistent with the hypothesis that systemic GAA deficiency can alter the neural control of breathing (DeRuisseau et al., 2009). The mechanisms underlying the increased variability in the respiratory cycle are not clear, but previous studies may provide insight. For example, CNS deficiencies in brain-derived neurotrophic factor (Balkowiec and Katz, 1998), norepinephrine (Viemari et al., 2005) and serotonin (Erickson et al., 2007) are also associated with increased variability in the respiratory cycle. Thus, respiratory pattern irregularities can be triggered by various CNS pathologies, and we suggest that the present results may arise due to altered function in the neuronal circuitry controlling respiratory output in Gaa−/− mice.
The widespread neuropathology observed in Pompe animal models (current data; see also Mancall et al., 1965; Gambetti et al., 1971; Sidman et al., 2008) makes it hard to pinpoint a precise anatomical location to explain the enhanced variability in respiratory output. In preliminary experiments (unpublished observations) using the transynaptic tracer pseudo-rabies virus (PRV; see Lane et al., 2008), we noted that glycogen accumulates in Gaa−/− mice in the presumed location of the ventral respiratory group – a location that includes respiratory premotor neurons (Feldman et al., 2003). Facial motoneurons in Pompe mice also have enlarged lysosomes with positive PAS staining (Sidman et al., 2008). This is noteworthy since the facial nerve can show inspiratory bursting (Lee and Fuller, 2010b), and the facial motor nucleus is in close proximity to components of the respiratory rhythm circuitry (i.e., the parafacial respiratory group or pFRG). The pFRG may be essential to the production of the overall respiratory rhythm, or may function more exclusively as an expiratory rhythm generator (Feldman and Del Negro, 2006; Onimaru et al., 2006). Finally, the nucleus of the solitary tract (NTS) is a primary integrative site for peripheral afferents regulating cardiorespiratory function (Boscan et al., 2002; Kubin et al., 2006), and dysfunction within this neuronal population could contribute to the impaired vagal reflexes noted in this study (e.g., Figures 3 and 4). NTS dysfunction could also contribute to increased respiratory variability since impairment of chemoafferent input can destabilize respiration (Hofer, 1984). In the current study we noted glycogen accumulation in the putative region of the NTS, although the specific location was not confirmed with labeling techniques.
In addition to pattern abnormalities, analyses of XII bursting in Gaa−/− mice suggested alterations in efferent bursting. Specifically, we noted less robust bursting during baseline conditions and a blunted response to bilateral vagotomy (Figure 3). Vagotomy typically triggers substantial increases in efferent XII motor output (Fuller et al., 1999; Lee et al., 2007a), and accordingly our results suggest that either vagally mediated reflexes are impaired in Gaa−/− mice, or that there is a non-specific impairment in the ability to increase the baseline (i.e., “quiet breathing”) efferent respiratory output (DeRuisseau et al., 2009). The mechanisms associated with potentially blunted efferent output could be distinct from those associated with the altered respiratory timing. For example, glycogen accumulation in XII motoneuron cell bodies could directly lead to impaired motor output (DeRuisseau et al., 2009).
Another interesting observation was that Pre-I XII motor output was altered in Gaa−/− mice. The Pre-I XII burst may act to stabilize the pharyngeal airway prior to the onset of negative intraluminal pressure during inspiration (Lee et al., 2007b). Thus, the absence of Pre-I XII activity during elevated respiratory drive (e.g., post-vagotomy) could lead to an increase in airflow resistance during inspiration. Most work on the mechanisms of upper airway patency during breathing focused on the genioglossus which is an extrinsic tongue muscle that acts to protrude the tongue (Remmers et al., 1978; Fregosi and Fuller, 1997). However, the intrinsic muscles which comprise the body of the tongue, as well as the extrinsic muscles which serve to retract the tongue are also important to airway patency (Fuller et al., 1998, 1999; Bailey and Fregosi, 2004; Bailey et al., 2006). For example, the tongue retrusor muscles can reduce airway compliance (i.e., “stiffen” the airway; Fuller et al., 1999), and activation of the intrinsic tongue musculature facilitates airway dilation (Bailey et al., 2006). The motoneurons controlling the extrinsic tongue muscles are compartmentalized within the XII nucleus, whereas the intrinsic muscle motoneurons are distributed throughout the nucleus (Uemura-Sumi et al., 1988; Guo et al., 1996; McClung and Goldberg, 1999). In our study glycogen accumulation occurred in neuronal soma throughout the XII nucleus (Figures 1 and 2) suggesting that motoneurons projecting to both intrinsic and extrinsic tongue muscles were affected.
We noted similarities but also a discrepancy between the current data set and our prior report in Gaa−/− mice (DeRuisseau et al., 2009). However, the fundamental conclusions between the prior and current data sets are similar. Namely, both studies indicate that GAA deficiency is associated with pathology in and around respiratory motoneurons (see also Sidman et al., 2008). Indeed the neuronal medullary glycogen accumulation reported here (e.g., Figures 1 and 2) is similar to spinal cord histological and biochemical results from our prior report (DeRuisseau et al., 2009). Similarly, in our prior report we observed reduced phrenic efferent activity in vagotomized Gaa−/− vs. control (B6/129) mice. Here we found that normalized phrenic and XII bursting was less in Gaa−/− mice, and the raw phrenic burst amplitude was qualitatively consistent with our prior report (i.e., lower in Gaa−/− mice). However, we previously noted substantial reductions in burst frequency in Gaa−/− mice (DeRuisseau et al., 2009) whereas in the present study respiratory burst frequency was similar between groups. This discrepancy could reflect the overall health of the Gaa−/− mice, subtle differences in the experimental procedures, or possibly genetic factors. The Gaa−/− mice in our study were genotyped, but the current study was conducted several years after our initial report, and we cannot rule out possible differences within the Gaa−/− colony. For example, even within a particular rodent strain (e.g., Sprague-Dawley rats), differences in respiratory control mechanisms are present depending on the particular colony of animals (Fuller et al., 2001; Wilkerson and Mitchell, 2009; Baker-Herman et al., 2010).
Significance
Respiratory insufficiency in Pompe disease has historically been attributed to muscular failure (Pellegrini et al., 2005; Burghaus et al., 2006). While respiratory muscle pathology almost certainly makes a contribution to respiratory failure, the current data contribute to a growing consensus that motor failures in Pompe disease may also involve CNS pathology (Hogan et al., 1969; Martin et al., 1973; Matsui et al., 1983; Teng et al., 2004; Sidman et al., 2008; DeRuisseau et al., 2009; Burrow et al., 2010; Mah et al., 2010). Pompe respiratory deficits, in particular, may have both a spinal (DeRuisseau et al., 2009) and supraspinal component (current data). Consistent with the current mouse data, a few Pompe case reports have documented brainstem glycogen accumulation (Mancall et al., 1965; Teng et al., 2004). In addition, there are clinical reports of impaired upper airway function. For example, swallow dysfunction occurs in infantile Pompe disease (Jones et al., 2010), and speech problems are common, even after initiation of enzyme replacement therapy (Muller et al., 2009). Feeding difficulties are present in >70% of Pompe infants, and approximately 50% of patients are never fed by mouth (Byrne et al., 2011a). In addition, obstructive sleep apnea, which may reflect inadequate neural drive to the pharyngeal muscles during sleep (Horner, 2008), is prevalent in adult Pompe patients (Margolis et al., 1994). Each of these upper airway-related clinical problems could reflect, at least in part, impaired XII motoneuron function. Accordingly, the XII motoneuron pathology and altered output in Gaa−/− mice suggests that aspects of tongue dysfunction in Pompe patients may have a neural basis.
The current “best-practice” therapy for Pompe disease involves repeated intravenous infusion of recombinantly produced GAA enzyme (Beck, 2009; Byrne et al., 2011a). However, the recombinant GAA enzyme does not effectively cross the blood–brain-barrier where the protein may influence CNS GAA deficiency (Kikuchi et al., 1998; Raben et al., 2003). Accordingly, while intravenous GAA delivery can target cardiac and skeletal muscle (Levine et al., 2008; Sugai et al., 2010), it will not effectively mitigate CNS glycogen accumulation (Byrne et al., 2011b). This point is highlighted by our recent case study report in which a Pompe infant which had received enzyme replacement therapy showed considerable glycogen accumulation in the spinal cord on post-mortem examination (DeRuisseau et al., 2009). It should be emphasized however, that enzyme replacement therapy will prolong life in Pompe infants (Kishnani et al., 2009; Nicolino et al., 2009; Byrne et al., 2011a). Nevertheless, variability in the success of enzyme replacement (van den Hout et al., 2004) could reflect persistent CNS pathology. For example, Muller et al. (2009) reported that even with enzyme replacement therapy, children with Pompe disease are still at high risk for developing speech disorders. These authors speculated that this could reflect lower motoneuron involvement which would not be effectively targeted by the enzyme therapy. Based on the current data and both recent (Sidman et al., 2008; DeRuisseau et al., 2009; Muller et al., 2009) and historical reports (Mancall et al., 1965; Hogan et al., 1969; Gambetti et al., 1971), we hypothesize that therapies targeting both skeletal muscle and the CNS may be required to fully correct respiratory-related deficits, and in particular pharyngeal muscle control, in Pompe disease (Byrne et al., 2011b).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mr. Roland-Austin Federico for assistance with immunohistochemistry experiments. This work was supported by the National Institutes of Health Grants 1R01HD052682-01A1 (David D. Fuller), HL59412 (Barry J. Byrne) 5F32HL095282-02 (Darin J. Falk) NIDDK P01 DK58327-03 (Barry J. Byrne).
References
Ausems, M. G., Verbiest, J., Hermans, M. P., Kroos, M. A., Beemer, F. A., Wokke, J. H., Sandkuijl, L. A., Reuser, A. J., and van der Ploeg, A. T. (1999). Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counseling. Eur. J. Hum. Genet. 7, 713–716.
Bailey, E. F., and Fregosi, R. F. (2004). Coordination of intrinsic and extrinsic tongue muscles during spontaneous breathing in the rat. J. Appl. Physiol. 96, 440–449.
Bailey, E. F., Huang, Y. H., and Fregosi, R. F. (2006). Anatomic consequences of intrinsic tongue muscle activation. J. Appl. Physiol. 101, 1377–1385.
Baker-Herman, T. L., Bavis, R. W., Dahlberg, J. M., Mitchell, A. Z., Wilkerson, J. E., Golder, F. J., Macfarlane, P. M., Watters, J. J., Behan, M., and Mitchell, G. S. (2010). Differential expression of respiratory long-term facilitation among inbred rat strains. Respir. Physiol. Neurobiol. 170, 260–267.
Balkowiec, A., and Katz, D. M. (1998). Brain-derived neurotrophic factor is required for normal development of the central respiratory rhythm in mice. J. Physiol. (Lond.) 510(Pt 2), 527–533.
Beck, M. (2009). Alglucosidase alfa: long term use in the treatment of patients with Pompe disease. Ther. Clin. Risk Manag. 5, 767–772.
Boscan, P., Pickering, A. E., and Paton, J. F. (2002). The nucleus of the solitary tract: an integrating station for nociceptive and cardiorespiratory afferents. Exp. Physiol. 87, 259–266.
Burghaus, L., Liu, W., Neuen-Jacob, E., Gempel, K., and Haupt, W. F. (2006). Glycogenesis Type II (M. Pompe). Selective failure of the respiratory musculature – a rare first symptom. Nervenarzt 77, 181–182; 185–186.
Burrow, T. A., Bailey, L. A., Kinnett, D. G., and Hopkin, R. J. (2010). Acute progression of neuromuscular findings in infantile Pompe disease. Pediatr. Neurol. 42, 455–458.
Byrne, B. J., Kishnani, P. S., Case, L. E., Merlini, L., Muller-Felber, W., Prasad, S., and der Ploeg, A. (2011a). Pompe disease: design, methodology, and early findings from the Pompe registry. Mol. Genet. Metab. 103, 1–11.
Byrne, B. J., Falk, D. J., Pacak, C. A., Nayak, S., Herzog, R. W., Elder, M. E., Collins, S. W., Conlon, T. J., Clement, N., Cleaver, B. D., Cloutier, D. A., Porvasnik, S. L., Islam, S., Elmallah, M. K., Martin, A., Smith, B. K., Fuller, D. D., Lawson, L. A., and Mah, C. S. (2011b). Pompe disease gene therapy. Hum. Mol. Genet. 20, R61–R68.
Clement, D. H., and Godman, G. C. (1950). Glycogen disease resembling mongolism, cretinism, and amytonia congenita; case report and review of literature. J. Pediatr. 36, 11–30.
DeRuisseau, L. R., Fuller, D. D., Qiu, K., DeRuisseau, K. C., Donnelly, W. H. Jr., Mah, C., Reier, P. J., and Byrne, B. J. (2009). Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. U.S.A. 106, 9419–9424.
Erickson, J. T., Shafer, G., Rossetti, M. D., Wilson, C. G., and Deneris, E. S. (2007). Arrest of 5HT neuron differentiation delays respiratory maturation and impairs neonatal homeostatic responses to environmental challenges. Respir. Physiol. Neurobiol. 159, 85–101.
Feldman, J. L., and Del Negro, C. A. (2006). Looking for inspiration: new perspectives on respiratory rhythm. Nat. Rev. Neurosci. 7, 232–242.
Feldman, J. L., Mitchell, G. S., and Nattie, E. E. (2003). Breathing: rhythmicity, plasticity, chemosensitivity. Annu. Rev. Neurosci. 26, 239–266.
Fregosi, R. F., and Fuller, D. D. (1997). Respiratory-related control of extrinsic tongue muscle activity. Respir. Physiol. 110, 295–306.
Fuller, D. D., Baker, T. L., Behan, M., and Mitchell, G. S. (2001). Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol. Genomics 4, 175–181.
Fuller, D. D., Mateika, J. H., and Fregosi, R. F. (1998). Co-activation of tongue protrudor and retractor muscles during chemoreceptor stimulation in the rat. J. Physiol. (Lond.) 507(Pt 1), 265–276.
Fuller, D. D., Williams, J. S., Janssen, P. L., and Fregosi, R. F. (1999). Effect of co-activation of tongue protrudor and retractor muscles on tongue movements and pharyngeal airflow mechanics in the rat. J. Physiol. (Lond.) 519(Pt 2), 601–613.
Gambetti, P., DiMauro, S., and Baker, L. (1971). Nervous system in Pompe’s disease. Ultrastructure and biochemistry. J. Neuropathol. Exp. Neurol. 30, 412–430.
Gestreau, C., Dutschmann, M., Obled, S., and Bianchi, A. L. (2005). Activation of XII motoneurons and premotor neurons during various oropharyngeal behaviors. Respir. Physiol. Neurobiol. 147, 159–176.
Guo, Y., Goldberg, S. J., and McClung, J. R. (1996). Compartmental organization of styloglossus and hyoglossus motoneurons in the hypoglossal nucleus of the rat. Brain Res. 728, 277–280.
Guth, L., and Watson, P. K. (1968). A correlated histochemical and quantitative study on cerebral glycogen after brain injury in the rat. Exp. Neurol. 22, 590–602.
Hagemans, M. L., Winkel, L. P., Hop, W. C., Reuser, A. J., van Doorn, P. A., and van der Ploeg, A. T. (2005). Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology 64, 2139–2141.
Haley, S. M., Fragala, M. A., and Skrinar, A. M. (2003). Pompe disease and physical disability. Dev. Med. Child Neurol. 45, 618–623.
Hirschhorn, R. R., and Reuser, A. J. (2001). “Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency,” in Metabolic and Molecular Basis of Inherited Disease, eds C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York: McGraw Hill), 3389–3420.
Hofer, M. A. (1984). Lethal respiratory disturbance in neonatal rats after arterial chemoreceptor denervation. Life Sci. 34, 489–496.
Hogan, G. R., Gutmann, L., Schmidt, R., and Gilbert, E. (1969). Pompe’s disease. Neurology 19, 894–900.
Horner, R. L. (2008). Pathophysiology of obstructive sleep apnea. J. Cardiopulm. Rehabil. Prev. 28, 289–298.
Jones, H. N., Muller, C. W., Lin, M., Banugaria, S. G., Case, L. E., Li, J. S., O’Grady, G., Heller, J. H., and Kishnani, P. S. (2010). Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia 25, 277–283.
Kikuchi, T., Yang, H. W., Pennybacker, M., Ichihara, N., Mizutani, M., van Hove, J. L., and Chen, Y. T. (1998). Clinical and metabolic correction of Pompe disease by enzyme therapy in acid maltase-deficient quail. J. Clin. Invest. 101, 827–833.
Kishnani, P. S., Corzo, D., Leslie, N. D., Gruskin, D., van der Ploeg, A., Clancy, J. P., Parini, R., Morin, G., Beck, M., Bauer, M. S., Jokic, M., Tsai, C. E., Tsai, B. W., Morgan, C., O’Meara, T., Richards, S., Tsao, E. C., and Mandel, H. (2009). Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr. Res. 66, 329–335.
Kubin, L., Alheid, G. F., Zuperku, E. J., and McCrimmon, D. R. (2006). Central pathways of pulmonary and lower airway vagal afferents. J. Appl. Physiol. 101, 618–627.
Lane, M. A., Truettner, J. S., Brunschwig, J. P., Gomez, A., Bunge, M. B., Dietrich, W. D., Dziegielewska, K. M., Ek, C. J., Vandeberg, J. L., and Saunders, N. R. (2007). Age-related differences in the local cellular and molecular responses to injury in developing spinal cord of the opossum, Monodelphis domestica. Eur. J. Neurosci. 25, 1725–1742.
Lane, M. A., White, T. E., Coutts, M. A., Jones, A. L., Sandhu, M. S., Bloom, D. C., Bolser, D. C., Yates, B. J., Fuller, D. D., and Reier, P. J. (2008). Cervical prephrenic interneurons in the normal and lesioned spinal cord of the adult rat. J. Comp. Neurol. 511, 692–709.
Lee, K. Z., and Fuller, D. D. (2010a). Preinspiratory and inspiratory hypoglossal motor output during hypoxia-induced plasticity in the rat. J. Appl. Physiol. 108, 1187–1198.
Lee, K. Z., and Fuller, D. D. (2010b). Hypoxia-induced short-term potentiation of respiratory-modulated facial motor output in the rat. Respir. Physiol. Neurobiol. 173, 107–111.
Lee, K. Z., Fuller, D. D., Lu, I. J., Lin, J. T., and Hwang, J. C. (2007a). Neural drive to tongue protrudor and retractor muscles following pulmonary C-fiber activation. J. Appl. Physiol. 102, 434–444.
Lee, K. Z., Fuller, D. D., Tung, L. C., Lu, I. J., Ku, L. C., and Hwang, J. C. (2007b). Uncoupling of upper airway motor activity from phrenic bursting by positive end-expired pressure in the rat. J. Appl. Physiol. 102, 878–889.
Lee, K. Z., Reier, P. J., and Fuller, D. D. (2009). Phrenic motoneuron discharge patterns during hypoxia-induced short-term potentiation in rats. J. Neurophysiol. 102, 2184–2193.
Levine, J. C., Kishnani, P. S., Chen, Y. T., Herlong, J. R., and Li, J. S. (2008). Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease. Pediatr. Cardiol. 29, 1033–1042.
Mah, C., Pacak, C. A., Cresawn, K. O., Deruisseau, L. R., Germain, S., Lewis, M. A., Cloutier, D. A., Fuller, D. D., and Byrne, B. J. (2007). Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol. Ther. 15, 501–507.
Mah, C. S., Falk, D. J., Germain, S. A., Kelley, J. S., Lewis, M. A., Cloutier, D. A., DeRuisseau, L. R., Conlon, T. J., Cresawn, K. O., Fraites, T. J. Jr., Campbell-Thompson, M., Fuller, D. D., and Byrne, B. J. (2010). Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol. Ther. 18, 502–510.
Mancall, E. L., Aponte, G. E., and Berry, R. G. (1965). Pompe’s disease (diffuse glycogenosis) with neuronal storage. J. Neuropathol. Exp. Neurol. 24, 85–96.
Margolis, M. L., Howlett, P., Goldberg, R., Eftychiadis, A., and Levine, S. (1994). Obstructive sleep apnea syndrome in acid maltase deficiency. Chest 105, 947–949.
Marsden, D. (2005). Infantile onset Pompe disease: a report of physician narratives from an epidemiologic study. Genet. Med. 7, 147–150.
Martin, J. J., de Barsy, T., van Hoof, F., and Palladini, G. (1973). Pompe’s disease: an inborn lysosomal disorder with storage of glycogen. A study of brain and striated muscle. Acta Neuropathol. 23, 229–244.
Martini, C., Ciana, G., Benettoni, A., Katouzian, F., Severini, G. M., Bussani, R., and Bembi, B. (2001). Intractable fever and cortical neuronal glycogen storage in glycogenosis type 2. Neurology 57, 906–908.
Matsui, T., Kuroda, S., Mizutani, M., Kiuchi, Y., Suzuki, K., and Ono, T. (1983). Generalized glycogen storage disease in Japanese quail (Coturnix coturnix japonica). Vet. Pathol. 20, 312–321.
McClung, J. R., and Goldberg, S. J. (1999). Organization of motoneurons in the dorsal hypoglossal nucleus that innervate the retrusor muscles of the tongue in the rat. Anat. Rec. 254, 222–230.
Mellies, U., and Lofaso, F. (2009). Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respir. Med. 103, 477–484.
Mellies, U., Stehling, F., Dohna-Schwake, C., Ragette, R., Teschler, H., and Voit, T. (2005). Respiratory failure in Pompe disease: treatment with noninvasive ventilation. Neurology 64, 1465–1467.
Millecamps, S., Nicolle, D., Ceballos-Picot, I., Mallet, J., and Barkats, M. (2001). Synaptic sprouting increases the uptake capacities of motoneurons in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. U.S.A. 98, 7582–7587.
Muller, C. W., Jones, H. N., O’Grady, G., Suarez, A. H., Heller, J. H., and Kishnani, P. S. (2009). Language and speech function in children with infantile Pompe disease. J. Pediatr. Neurol. 7, 147–156.
Nicolino, M., Byrne, B., Wraith, J. E., Leslie, N., Mandel, H., Freyer, D. R., Arnold, G. L., Pivnick, E. K., Ottinger, C. J., Robinson, P. H., Loo, J. C., Smitka, M., Jardine, P., TatÓ, L., Chabrol, B., McCandless, S., Kimura, S., Mehta, L., Bali, D., Skrinar, A., Morgan, C., Rangachari, L., Corzo, D., and Kishnani, P. S. (2009). Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. 11, 210–219.
Onimaru, H., Kumagawa, Y., and Homma, I. (2006). Respiration-related rhythmic activity in the rostral medulla of newborn rats. J. Neurophysiol. 96, 55–61.
Pellegrini, N., Laforet, P., Orlikowski, D., Pellegrini, M., Caillaud, C., Eymard, B., Raphael, J. C., and Lofaso, F. (2005). Respiratory insufficiency and limb muscle weakness in adults with Pompe’s disease. Eur. Respir. J. 26, 1024–1031.
Raben, N., Danon, M., Gilbert, A. L., Dwivedi, S., Collins, B., Thurberg, B. L., Mattaliano, R. J., Nagaraju, K., and Plotz, P. H. (2003). Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 80, 159–169.
Raben, N., Nagaraju, K., Lee, E., Kessler, P., Byrne, B., Lee, L., LaMarca, M., King, C., Ward, J., Sauer, B., and Plotz, P. (1998). Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 273, 19086–19092.
Raben, N., Plotz, P., and Byrne, B. J. (2002). Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr. Mol. Med. 2, 145–166.
Remmers, J. E., deGroot, W. J., Sauerland, E. K., and Anch, A. M. (1978). Pathogenesis of upper airway occlusion during sleep. J. Appl. Physiol. 44, 931–938.
Rohrbach, M., Klein, A., Kohli-Wiesner, A., Veraguth, D., Scheer, I., Balmer, C., Lauener, R., and Baumgartner, M. R. (2010). CRIM-negative infantile Pompe disease: 42-month treatment outcome. J. Inherit. Metab. Dis. 33, 751–757.
Sidman, R. L., Taksir, T., Fidler, J., Zhao, M., Dodge, J. C., Passini, M. A., Raben, N., Thurberg, B. L., Cheng, S. H., and Shihabuddin, L. S. (2008). Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J. Neuropathol. Exp. Neurol. 67, 803–818.
Sugai, F., Kokunai, Y., Yamamoto, Y., Hashida, G., Shimazu, K., Mihara, M., Inoue, S., and Sakoda, S. (2010). Use of the muscle volume analyzer to evaluate enzyme replacement therapy in late-onset Pompe disease. J. Neurol. 257, 461–463.
Teng, Y. T., Su, W. J., Hou, J. W., and Huang, S. F. (2004). Infantile-onset glycogen storage disease type II (Pompe disease): report of a case with genetic diagnosis and pathological findings. Chang Gung Med. J. 27, 379–384.
Uemura-Sumi, M., Itoh, M., and Mizuno, N. (1988). The distribution of hypoglossal motoneurons in the dog, rabbit and rat. Anat. Embryol. 177, 389–394.
van den Hout, H. M., Hop, W., van Diggelen, O. P., Smeitink, J. A., Smit, G. P., Poll-The, B. T., Bakker, H. D., Loonen, M. C., de Klerk, J. B., Reuser, A. J., and van der Ploeg, A. T. (2003). The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 112, 332–340.
van den Hout, J. M., Kamphoven, J. H., Winkel, L. P., Arts, W. F., De Klerk, J. B., Loonen, M. C., Vulto, A. G., Cromme-Dijkhuis, A., Weisglas-Kuperus, N., Hop, W., van Hirtum, H., van Diggelen, O. P., Boer, M., Kroos, M. A., van Doorn, P. A., van der Voort, E., Sibbles, B., van Corven, E. J., Brakenhoff, J. P., van Hove, J., Smeitink, J. A., de Jong, G., Reuser, A. J., and van der Ploeg, A. T. (2004). Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 113, e448–e457.
Verma, S. P., and Quiroz-Ruiz, A. (2006). Critical values for six Dixon tests for outliers in normal samples up to sizes 100, and applications in science and engineering. Revista Mexicana de Ciencias Geológicas 23, 133–161.
Viemari, J. C., Roux, J. C., Tryba, A. K., Saywell, V., Burnet, H., Peña, F., Zanella, S., Bévengut, M., Barthelemy-Requin, M., Herzing, L. B., Moncla, A., Mancini, J., Ramirez, J. M., Villard, L., and Hilaire, G. (2005). Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J. Neurosci. 25, 11521–11530.
Wilkerson, J. E., and Mitchell, G. S. (2009). Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp. Neurol. 217, 116–123.
Keywords: acid α-glucosidase, Pompe, hypoglossal, brainstem, glycogen
Citation: Lee K-Z, Qiu K, Sandhu MS, Elmallah MK, Falk DJ, Lane MA, Reier PJ, Byrne BJ and Fuller DD (2011) Hypoglossal neuropathology and respiratory activity in Pompe mice. Front. Physio. 2:31. doi: 10.3389/fphys.2011.00031
Received: 11 March 2011; Accepted: 17 June 2011;
Published online: 30 June 2011.
Edited by:
Carlos Mantilla, Mayo Clinic, USAReviewed by:
Ken D. O’Halloran, University College Dublin, IrelandTracy L. Baker-Herman, University of Wisconsin, USA
Copyright: © 2011 Lee, Qiu, Sandhu, Elmallah, Falk, Lane, Lane, Byrne and Fuller. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: David D. Fuller, Department of Physical Therapy, McKnight Brain Institute, University of Florida, 100 S. Newell Dr., Gainesville, FL 32610, USA. e-mail: ddf@phhp.ufl.edu