
- 1 Hagey Laboratory for Regenerative Medicine, Department of Surgery, Division of Plastic and Reconstructive Surgery, Stanford University School of Medicine, Stanford, CA, USA
- 2 BG-Unfallklinik Ludwigshafen, Department of Plastic and Hand Surgery, University of Heidelberg, Heidelberg, Germany
- 3 Dipartimento die Scienze Chirurgiche, Anestesiologiche-Rianimatorie e dell’Emergenza “Giuseppe Zannini”, Università degli Studi di Napoli Federico II, Napoli, Italy
Craniosynostosis, the premature closure of cranial suture, is a pathologic condition that affects 1/2000 live births. Saethre-Chotzen syndrome is a genetic condition characterized by craniosynostosis. The Saethre-Chotzen syndrome, which is defined by loss-of-function mutations in the TWIST gene, is the second most prevalent craniosynostosis. Although much of the genetics and phenotypes in craniosynostosis syndromes is understood, less is known about the underlying ossification mechanism during suture closure. We have previously demonstrated that physiological closure of the posterior frontal suture occurs through endochondral ossification. Moreover, we revealed that antagonizing canonical Wnt-signaling in the sagittal suture leads to endochondral ossification of the suture mesenchyme and sagittal synostosis, presumably by inhibiting Twist1. Classic Saethre-Chotzen syndrome is characterized by coronal synostosis, and the haploinsufficient Twist1+/− mice represents a suitable model for studying this syndrome. Thus, we seeked to understand the underlying ossification process in coronal craniosynostosis in Twist1+/− mice. Our data indicate that coronal suture closure in Twist1+/− mice occurs between postnatal day 9 and 13 by endochondral ossification, as shown by histology, gene expression analysis, and immunohistochemistry. In conclusion, this study reveals that coronal craniosynostosis in Twist1+/− mice occurs through endochondral ossification. Moreover, it suggests that haploinsufficiency of Twist1 gene, a target of canonical Wnt-signaling, and inhibitor of chondrogenesis, mimics conditions of inactive canonical Wnt-signaling leading to craniosynostosis.
Introduction
The skull vault in mammalian organisms is predominantly formed by intramembranous ossification of membranes derived from the neural-crest or mesoderm tissues (Jiang et al., 2002). In this process, mesenchymal cells condense, form ossification centers and expand. Cranial sutures form as the margins of the developing bones approximate, and the mesenchymal tissue separating the bone fronts is recruited into the osteogenic fronts. Thus, a suture consists of the suture mesenchyme, flanked by osteogenic fronts on each side. Cranial sutures can either close, or remain patent and thereby serve as growth centers for the expansion of the skull vault at later stages of development. The architecture and arrangement of these sutures is surprisingly well conserved in mammals and other species (Opperman, 2000; Quarto and Longaker, 2005; Sahar et al., 2005). Two unpaired sutures, the posterior frontal (PF) between the frontal bones and the sagittal suture (SAG) between the parietal bones form the median axis of the skull vault. The paired coronal sutures (COR), located between the frontal and parietal bones as well as the lambdoid sutures (LAM) between the interparietal and parietal bones represent the coronal axis. The mesenchyme of PF and SAG suture is of neural-crest origin, whereas the COR suture mesenchyme is of mesoderm origin and a juxtaposition between the neural-crest derived frontal bone and the mesodermal parietal bone (Jiang et al., 2002). So far, the tissue origin of the LAM suture is undetermined. Among these four cranial sutures, only the PF (or metopic in humans) closes physiologically during the first months of life in humans and between day 13 and 15 postnatal in mice by endochondral ossification (Cohen, 1993; Sahar et al., 2005). Prolonged proliferation of suture mesenchyme or delayed differentiation of cranial osteoblasts results in pathological suture expansion, whereas impaired proliferation or accelerated differentiation causes premature fusion, or craniosynostosis, a common developmental disorder that can be either inherited or isolated (Wilkie, 1997). In the past decades, causative mutations in more than 10 genes have been identified. For example, mutations in genes encoding fibroblast growth factor receptors (FGFR) with craniosynostosis syndromes such as Pfeiffer, Crouzon, and Apert syndromes; mutations in TWIST, a basic helix-loop-helix transcription factor, cause Saethre-Chotzen syndrome (Jabs et al., 1993, 1994; Bellus et al., 1996; Howard et al., 1997; Wilkie, 1997; Twigg et al., 2004; Wieland et al., 2004). However, a specific genetic diagnosis can only be made in 28% of the cases (Morriss-Kay and Wilkie, 2005).
Craniosynostosis in the Saethre-Chotzen syndrome, which was first described 80 years ago (Saethre, 1931; Chotzen, 1932), involves the COR, PF, and LAM sutures. Since the Saethre-Chotzen syndrome is associated with heterozygous loss-of-function mutation in the TWIST1 gene (el Ghouzzi et al., 1997; Howard et al., 1997), Twist1+/− mice provide a useful genetic model to study this human genetic syndrome. Twist1+/− mice show COR fusion, thereby resembling the Saethre-Chotzen syndrome in humans (Bourgeois et al., 1998; Carver et al., 2002). However, it must be pointed out that over 100 mutations in the coding region of the TWIST1 gene have been found to cause Saethre-Chotzen syndrome (Cunningham et al., 2007).
In our recent study we have demonstrated, that active canonical Wnt-signaling is at least in part responsible for the patency of cranial sutures, and that low levels of this signaling are “permissive” for craniosynostosis (Behr et al., 2010). Furthermore, we have proposed a mechanism, in which decreased canonical Wnt-signaling leads to craniosynostosis by downregulating the expression of Twist1, a target gene of canonical Wnt-signaling (Howe et al., 2003). This would in turn allow the occurrence of chondrogenesis in the suture mesenchyme, a process which is otherwise inhibited by Twist1 (Reinhold et al., 2006). These findings were corroborated by the occurrence of partial synostosis in the SAG suture of Twist1+/− mice (Behr et al., 2010).
Based on our previous observations, in the current study we seeked to investigate in detail the timing and ossification mechanism of COR craniosynostosis in Twist1+/− mice. Moreover, using axin2-lacZ mice we have analyzed the activation of canonical Wnt-signaling in the four main sutures in order to determine, whether a correlation between suture patency and canonical Wnt-signaling exists.
Materials and methods
Mutant Animals
Animals were cared for in accordance to the Institutional Animal Care and the Use Committee of Stanford University. In order to study the activation of the canonical Wnt-signaling in cranial sutures, Axin2+/− mice were utilized. Axin2+/− mice, with a LacZ reporter gene were generated by breeding Axin2−/− males on a pure CD-1 background with wild-type CD-1 females. In addition, Wnt1Cre+/− and R26R+/− mice on a C57/BL6 background were mated to obtain double transgenic Wnt1Cre+/−/R26R+/− mice. Twist1+/− mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and previously described (Chen and Behringer, 1995). Genotyping was performed by PCR analysis of genomic DNA.
Tissues Harvesting and Processing
Animals were sacrificed on the exact postnatal day (p) based on birth date (day 0). Time points included: E18.5, p7, p9, p11, p13, p15, p25, and p180. For each time point at least three animals were sacrificed and processed for histology.
For tissues harvesting, animals were asphyxiated by CO2 and decapitated. The PF and the SAG suture including the adjacent osteogenic fronts of frontal and parietal bones were meticulously harvested using a stereomicroscope (LeicaMZ16). Accordingly, the COR and LAM sutures were harvested with the corresponding osteogenic fronts of frontal and parietal or parietal and interparietal bone. For RNA extraction, at least four suture complexes of littermates were harvested and homogenized with a Pellet Pestle Motor (Kontes) in 0.2 ml of Trizol (Invitrogen).
To obtain cryo-sections the suture complexes were briefly washed in cold PBS and then fixed with 0.4% PFA overnight at 4°C. After a second wash with cold PBS, tissue samples were decalcified in 19% EDTA at 4°C for the appropriate time (from 1 day up to 2 weeks), depending on the developmental stage of the skull. Specimens were then transferred in sucrose 10 and 30% (both incubations overnight) and cryo embedded in optimal cutting temperature compound (OCT-Tissue-Tek). To obtain paraffin sections, the suture complexes were washed in PBS and fixed in 10% neutral buffered formalin at room temperature overnight. After a second wash with PBS, tissue samples were decalcified as described above. Specimens then underwent dehydration in a graded series of alcohol and xylene and were paraffin embedded.
Histology
For both, cryo and paraffin sectioning the entire COR, LAM, PF, and SAG sutures were cut in 10 μm sections. For each suture, between 50 (E18.5) and 400 sections (p180) were obtained (three sections per slide). Every third slide of each cryo sectioned suture was stained with Xgal (Roche). For paraffin-sectioned sutures, each sixth slide was stained by pentachrome to determine the exact region within the suture. Sections were examined with a Zeiss Axioplan microscope. Images were captured by AxioVision (Zeiss) and processed with Adobe Photoshop (Adobe Systems). Xgal staining in the suture mesenchyme of Axin2+/− mice was quantified semi-automated by using the magic wand tool in Photoshop with the tolerance settings at 60. Data are presented in pixels. In order to calculate the fractions of Xgal staining in relation to the respective suture area, the suture area pixels were determined with the lasso tool in Photoshop. Fractions were generated by dividing Xgal positive pixels by the respective suture area.
Reverse Transcription-PCR
RNA isolation, reverse transcription (RT) quantitative real-time PCR (qPCR), and primers were described previously (Quarto et al., 2005, 2008; Sahar et al., 2005). Briefly, real-time RT-PCR was performed using the ABI Prism 7900 Sequence Detection System, TaqMan Gene Expression Master Mix, and TaqMan Gene Expression Assays (Applied Biosystems). Cycling conditions were initial denaturation at 95°C for 3 min, followed by 30 cycles consisting of a 15-s denaturation interval at 95°C and a 30-s interval for annealing and primer extension at 60°C. The relative mRNA levels in each sample were normalized to its Gapdh content. The results are presented as means ± SD of three independent experiments.
Immunohistochemistry
For immunohistochemistry of COR sutures, antigen retrieval was performed by incubating the slides with Proteinase K (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 10 min. Antibodies against Sox9 and Collagen II (ColII; SC-17340, SC-28887, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were used according to the manufacturer’s instruction. A goat or rabbit biotinylated secondary antibody followed by the AB reagent and NovaRed (Vector Laboratories, Burlingame, CA, USA) were used for detection. Irrelevant goat or rabbit IgG (Calbiochem) used as a negative control did not produce any staining (data not shown). Results were obtained from at least two animals and were carried out in duplicates.
Results
Different Occurrence of Craniosynostosis in Twist1+/− Cranial Sutures
Although, craniosynostosis is a well established phenotype in Twist1+/− mice (Bourgeois et al., 1998; Carver et al., 2002; Bialek et al., 2004), little is known about the detailed impact and ossification mechanism in the occurrence of craniosynostosis in these mice. For this reason, we performed coronal and sagittal sections on all four skull vault sutures of Twist1+/− mice at a mature time point for suture closure (p25; Figure 1). The PF suture, which physiologically closes during the second week of postnatal life through endochondral ossification (Sahar et al., 2005), showed a complete closure in Twist1+/− and wild-type mice. Interestingly, wild-type PF suture was larger and had a higher bone mass compared to Twist1+/− mice. As expected, the COR suture, which physiologically remains patent, was fused in 5/7 Twist1+/− mice. The SAG suture, which likewise stays patent, showed partial closure in Twist1+/− mice as previously reported (Behr et al., 2010). Finally, the LAM suture was patent in all examined Twist1+/− and wild-type mice.
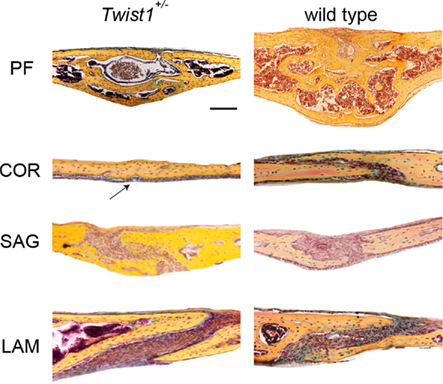
Figure 1. Pentachrome staining of the four representative cranial sutures: posterior frontal (PF), coronal (COR), sagittal (SAG), and lambdoid (LAM) at postnatal day (p) 25. A complete closure of COR suture is observed in Twist1+/− mice, while the suture remains patent in wild-type mice. The arrow indicates the fused coronal suture in Twist1+/− mice. Scale bar: 100 μm.
Craniosynostosis of Coronal Sutures in Twist1+/− Mice Occurs Through Endochondral Ossification
In Twist1+/− mice, which are viable, COR suture is known to be pathologically closed (Carver et al., 2002), mimicking craniosynostosis in Saethre-Chotzen syndrome in humans. Thus, it provides the opportunity to study suture closure in more detail. Since we previously uncovered, that physiological PF suture closure occurs through endochondral ossification (Sahar et al., 2005), which could also be observed in SAG suture synostosis elicited by antagonizing canonical Wnt-signaling (Behr et al., 2010), we addressed the question through which ossification mechanism craniosynostosis of COR suture in Twist1+/− mice occurs. For this purpose, COR sutures of Twist1+/− mice were examined at similar time points as predicted by PF suture closure (Sahar et al., 2005). Indeed, between p9 and p15, COR suture closure could be monitored in COR sutures of Twist1+/− mice. Pentachrome staining revealed chondrocytes in COR sutures of Twist1+/− mice at p9 and p11 in 4/7 animals (Figure 2). Of note, we did observe variations in the phenotype characteristics, which was most likely due to a varying penetrance of this mutation. To further prove that COR suture mesenchyme of Twist1+/− mice is undergoing endochondral ossification, we performed qPCR analysis for chondrogenic markers. As shown in Figure 3A, qPCR revealed a clear chondrogenic pattern with a temporal upregulation, followed by downregulation of Sox 9, the master regulator of chondrogenesis, and Collagen II (Coll II), component of chondrocytes extracellular matrix. At later time, upregulation of Collagen X (Coll X), a marker of hypertrophic chondrocyte, was observed. By postnatal day 15, high levels of Osteocalcin (Oc) were detected as result of bony bridge formation between the two osteogenic fronts of COR suture, and therefore its closure. Conversely, qPCR analysis of COR suture of wild-type mice did not reveal chondrogenic markers (data not shown). Immunohistochemistry analysis showed markedly elevated levels of Sox9 and Collagen II proteins in the suture mesenchyme of Twist1+/− as compared to wild type at p11 (Figure 3B).

Figure 2. Time course of COR suture closure in Twist1+/− mice. At day p11 pentachrome staining reveals the presence of cartilage (blue staining) between the bone plates of the COR suture of Twist1+/− mice, while no cartilage is present in wild-type mice. By day p13, pathological closure of COR suture is observed in Twist1+/− mice. Dashed lines indicate the bone plates. Scale bar: 50 μm.
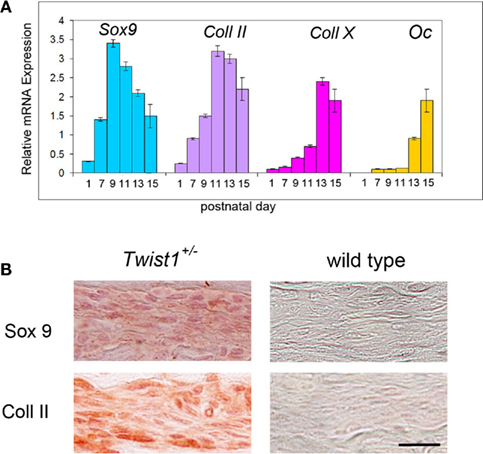
Figure 3. Presence of specific chondrogenic markers in the differentiating COR suture mesenchyme. (A) Expression analysis of chondrogenic markers by qPCR showing a chondrogenic pattern of the differentiating COR suture mesenchyme in Twist1+/− mice. By postnatal day 15 upregulation of osteocalcin (Oc) is observed, as result of the COR suture closure. (B) Immunohistochemistry for Sox9 and Col II confirm cartilage tissue formation in the COR suture of Twist1+/− at day p11. Scale bar: 10 μm Expression analysis of chondrogenic markers by qPCR showing a chondrogenic pattern of the differentiating COR suture mesenchyme.
Contribution of Neural-Crest Origin Cells to the Lambdoid Suture
We did establish that both, PF suture in wild-type mice and COR sutures in Twist1+/− mice close by endochondral ossification. These results were interesting, because these sutures do not share the same embryonic origin of other cranial sutures. While the PF and SAG suture mesenchymes are of neural-crest origin, the COR suture is derived from the mesoderm (Jiang et al., 2002). Since this study, which utilized Wnt1Cre/R26R mice, was carried out at embryonic stages, no information was available for LAM suture (Jiang et al., 2002). LAM sutures in Twist1+/− mice were unique in a way, that they did not show any signs of fusion, rather their osteogenic fronts were wide apart (Figure 1). Hence, we were interested in the embryonic origin and examined LAM sutures of p7 Wnt1Cre/R26R mice. Xgal staining revealed positive cells in the suture mesenchyme, indicating the contribution of neural-crest origin cells to the paired LAM suture (Figure 4A). In conclusion, among all four cranial skull vault sutures, only COR is of mesodermal origin. However, the tissue origin did not explain differences between a fusing COR suture and a patent LAM suture in Twist1+/− mice, since fusion likewise occurs in neural-crest derived sutures like the PF and SAG sutures.
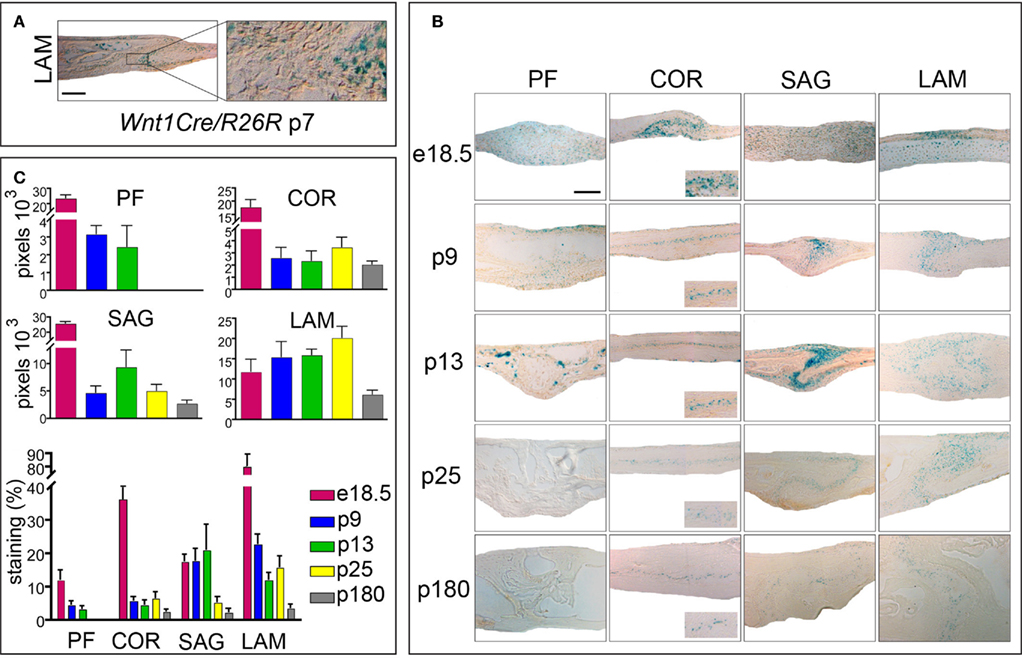
Figure 4. Activation of canonical Wnt-signaling in embryonic and postnatal cranial sutures. (A) Xgal staining of LAM suture harvested from Wnt1Cre/R26R mouse. Xgal staining indicates the neural-crest origin of the LAM suture mesenchyme. The boxed area is enlarged to the right. (B) Xgal staining of PF, COR SAG, and LAM suture mesenchymes in Axin2+/− mice from e18.5 to p180. Scale bar: 100 μm. Inserts represent zoomed areas of the coronal sutures. Dashed lines indicate the bone plates. (C) Quantification of Xgal staining of different cranial suture mesenchymes in Axin2+/− mice at day p25 and p180.
Canonical Wnt-Signaling Activity is Associated with Suture Patency and Closure
We have previously postulated that canonical Wnt-signaling tightly controls cranial suture closure or patency, with low canonical Wnt-signaling and finally its absence in fusing PF suture, which physiologically closes, whereas in SAG suture sustained canonical Wnt-signaling is associated with suture patency (Behr et al., 2010). We now seeked to investigate, whether this phenomena is likewise true for the mesoderm derived COR suture or the neural-crest derived LAM suture, which are both physiologically patent. For this purpose we used in Axin2+/− mice, in which Xgal staining monitors activation of canonical Wnt-signaling (Yu et al., 2005). Indeed, in reporter mice expressing Axin2-lacZ both the COR and LAM suture show a substantial activation of canonical Wnt-signaling (Figure 4B). After quantification of Xgal staining, which is indicative for canonical Wnt-signaling, it was evident, that canonical Wnt-signaling was most pronounced in LAM sutures followed by the SAG and finally the COR sutures at p25 and p180 (Figure 4C). As reported previously, absence of canonical Wnt-signaling was observed in PF sutures at p25 or later. By analyzing the anatomy of the LAM suture mesenchyme, an expansion, which could be linked to increased canonical Wnt-signaling, was evident. Taken together these results suggest that a strict correlation between suture patency and the presence of active canonical Wnt-signaling exists.
Discussion
In the current study, we demonstrated that craniosynostosis in COR sutures of Twist1+/− mice occurs by endochondral ossification between p9 and p15 as consequence of impaired expression of Twist1, a downstream target of canonical Wnt-signaling and inhibitor of chondrogenesis.
We have previously reported that physiological closure of the PF suture, which is of neural-crest origin, occurs by endochondral ossification (Sahar et al., 2005). Later, we found that this process is tightly regulated by canonical Wnt-signaling; i.e., high levels of canonical Wnt-signaling are associated with cranial suture patency, while low levels are associated with suture fusion (Behr et al., 2010). Endochondral ossification of suture mesenchyme in the cranial vault is somewhat surprising, since the default mode of osteogenesis in the skull vault is intramembranous ossification. However to date little is known about the ossification mechanism governing premature closure of sutures in pathologic conditions such as craniosynostosis.
The Twist1+/− mouse has been previously used to study the molecular pathogenesis of Saethre-Chotzen syndrome. Carver et al. (2002) reported fusions of the coronal suture in Twist1+/− mice at postnatal day 30, however this study was performed on whole-mount stained skulls, which does not allow a detailed anatomic analysis of the suture.
An independent study by Yoshida et al. (2005) showed that during establishment of the COR suture area Twist is required for the regulation of sutural cell proliferation and osteoblast differentiation. These authors demonstrated that inhibition of Twist synthesis, using morpholino-antisense oligonucleotides in calvarial organ culture resulted in a narrow COR sutural space and fusion of bone domains (Yoshida et al., 2005).
A hint that the Twist1+/− mouse model is relevant for understanding human Saethre-Chotzen syndrome was generated by a clinical case reporting, that suture fusion in a child also occurred at postnatal stages. While an X-ray performed at 4-month of age still showed patent COR sutures in a Saethre-Chotzen syndrome child, fusion was reported at 14-month of age (de Heer et al., 2004).
An interesting question is, whether endochondral ossification is a default mode for cranial suture mesenchyme undergoing craniosynostosis, or whether different mechanism(s) are also accountable. It has been proposed that COR suture fusion in Twist1+/− mice is caused by a boundary defect, induced by invading neural-crest cells into the otherwise undifferentiated mesodermal COR suture (Merrill et al., 2006). The authors further showed, that the migration of neural-crest cells is accompanied by an increase of Msx2 and reduction of ephrin-A4 expression (Merrill et al., 2006). A different mechanism for cranial suture closure in Twist1+/− mice was proposed with regards to Twist1 heterodimers and homodimers (Connerney et al., 2006, 2008). It was shown that a balance of Twist homodimers (location at the osteogenic fronts) and Twist1 heterodimers (location in the mid-suture) is tilted toward the homodimers in Twist1+/− mice (Connerney et al., 2008). In turn, Twist1 homodimers increase expression of Fgfr2, which then leads to craniosynostosis. Moreover, the authors demonstrated, that it was possible to rescue suture fusion in Twist1+/− mice by inhibiting FGF-signaling (Connerney et al., 2008). In line with this observation, Rice et al. (2000) reported alteration of Fgfr2 in the SAG suture mesenchyme of Twist1+/− mice. While these studies described alterations of other pathways in Twist1+/− mice such as Fgf or Msx, our study focused on the ossification process through which craniosynostosis occurs in Twist1+/− mice. Given that Twist1 inhibits chondrogenesis (Reinhold et al., 2006), reduced levels as found in Twist1+/− mice favor chondrogenesis, therefore paving the way for endochondral ossification in COR sutures.
Few studies investigated COR suture fusion in other models. A different hypothesis for the occurrence of COR craniosynostosis was generated with the first Apert syndrome mouse model Fgfr2250/+ (Chen et al., 2003). The authors proposed, that this mutation causes excessive apoptosis and therefore reduced thickness, a narrower suture mesenchyme and ultimately premature fusion of COR suture in Fgfr2250/+ mice (Chen et al., 2003). Later it was found that in Fgfr2S252W/+ skulls, which represent the most common mutation in Apert syndrome, COR craniosynostosis occurred during early stages of development (E16.5 to p1) starting from the base of the suture (Holmes et al., 2009), which is consistent with a report about COR suture fusion in Fgfr2-III+/Δ mice as early as E18 (Hajihosseini et al., 2001). In contrast to the first report about COR suture fusion in Fgfr2250/+ mice (Chen et al., 2003), the authors suggested that apoptosis is more a consequence rather than a cause of sutural fusion in Fgfr2S252W/+ mice (Holmes et al., 2009). To some extent COR suture fusion was also studied in a mouse Apert model with a P253R mutation of Fgfr2 (Yin et al., 2008). In this model, COR suture was fused at day p21. However, the fusion was subtler than in Twist1+/− mice, which showed a solid bone plate (Yin et al., 2008). Finally, Eswarakumar and colleagues reported complete fusion of the COR suture in Fgfr2cCLR/+-mice at p14, however they did not look at earlier stages (Eswarakumar et al., 2006). Indeed, it will be of interest to investigate whether COR suture closes through endochondral ossification also in these other mouse models.
Although in a different context, in earlier studies histological analysis revealed that PF suture “prematurely” fused in the Axin2-null mutants at 4 weeks postnatally, while only μCT analysis showed closure of COR suture (Liu et al., 2007). However, it must be pointed out that PF suture closes physiologically at 2 weeks postnatally. Moreover, in this Axin2-null mutant, only binding of β-catenin to the Axin-dependent degradation complex in the cytosol is disrupted. The interaction of β-catenin with the LEF/TCF transcription and the cadherin mediated adhesion complexes remains intact at the Axin2-null nucleus and plasma membrane, respectively (Liu et al., 2007). Further analysis of Axin2-null mice PF suture detected an expansion of the Sox9-expressing precursors only at postnatal days 0 and 8, earlier than chondrogenic differentiation of suture mesenchyme. However, there was no obvious difference in Sox9 expression during chondrogenesis of the Axin2+/+ and Axin2−/− nasal cartilages (Liu et al., 2007).
We have previously demonstrated that canonical Wnt-signaling is a crucial pathway, controlling sutures closure or patency (Behr et al., 2010). It is known that expression of Twist1 is regulated by canonical Wnt-signaling (Howe et al., 2003). Our earlier study indicated that in PF and SAG sutures Twist1 gene expression, as well Twist1 protein mirrored the different activation levels of canonical Wnt-signaling (Behr et al., 2010). Here we show that Twist1 haploinsufficiency mimicking a “context” of inactive canonical Wnt-signaling induces endochondral ossification of COR suture leading to craniosynostosis. Similarly, inhibition of canonical Wnt-signaling in the SAG suture leads to sagittal synostosis, a process occurring through endochondral ossification, paralleled by lower levels of Twist protein (Behr et al., 2010). As described previously for the SAG suture in Twist1+/− mice (Behr et al., 2010), reduced levels of Twist1 leads to the occurrence of endochondral ossification also in the COR suture mesenchyme. Thus, the current study further highlights the importance of canonical Wnt-signaling and Twist1 in determining and controlling cranial suture fate by regulating endochondral ossification.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Yang Chai for the Wnt1Cre/R26R mice and Dr. Roel Nusse for the Axin2lacZ/+ mice. This work was supported by the Hagey Family Program, The Oak Foundation, and NIH R21DE019274 to Michael T. Longaker and the German Research Foundation grant DFG BE 4169/1-1 to Björn Behr.
References
Behr, B., Longaker, M. T., and Quarto, N. (2010). Differential activation of canonical Wnt signaling determines cranial sutures fate: a novel mechanism for sagittal suture craniosynostosis. Dev. Biol. 344, 922–940.
Bellus, G. A., Gaudenz, K., Zackai, E. H., Clarke, L. A., Szabo, J., Francomano, C. A., and Muenke, M. (1996). Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat. Genet. 14, 174–176.
Bialek, P., Kern, B., Yang, X., Schrock, M., Sosic, D., Hong, N., Wu, H., Yu, K., Ornitz, D. M., Olson, E. N., Justice, M. J., and Karsenty, G. (2004). A twist code determines the onset of osteoblast differentiation. Dev. Cell 6, 423–435.
Bourgeois, P., Bolcato-Bellemin, A. L., Danse, J. M., Bloch-Zupan, A., Yoshiba, K., Stoetzel, C., and Perrin-Schmitt, F. (1998). The variable expressivity and incomplete penetrance of the twist-null heterozygous mouse phenotype resemble those of human Saethre-Chotzen syndrome. Hum. Mol. Genet. 7, 945–957.
Carver, E. A., Oram, K. F., and Gridley, T. (2002). Craniosynostosis in Twist heterozygous mice: a model for Saethre-Chotzen syndrome. Anat. Rec. 268, 90–92.
Chen, L., Li, D., Li, C., Engel, A., and Deng, C. X. (2003). A Ser250Trp substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 33, 169–178.
Chen, Z. F., and Behringer, R. R. (1995). Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 9, 686–699.
Chotzen, F. (1932). Eine eigenartige familiaere Entwicklungsstoer ung (Akrocephalosyndaktylie, Dysostosis craniofacialis und Hypertelorismus). Monatsschr. Kinderheilkd. 55, 97–122.
Cohen, M. M. Jr. (1993). Sutural biology and the correlates of craniosynostosis. Am. J. Med. Genet. 47, 581–616.
Connerney, J., Andreeva, V., Leshem, Y., Mercado, M. A., Dowell, K., Yang, X., Lindner, V., Friesel, R. E., and Spicer, D. B. (2008). Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev. Biol. 318, 323–334.
Connerney, J., Andreeva, V., Leshem, Y., Muentener, C., Mercado, M. A., and Spicer, D. B. (2006). Twist1 dimer selection regulates cranial suture patterning and fusion. Dev. Dyn. 235, 1345–1357.
Cunningham, M. L., Seto, M. L., Ratisoontorn, C., Heike, C. L., and Hing, A. V. (2007). Syndromic craniosynostosis: from history to hydrogen bonds. Orthod. Craniofac. Res. 10, 67–81.
de Heer, I. M., Hoogeboom, J., Vermeij-Keers, C., de Klein, A., and Vaandrager, J. M. (2004). Postnatal onset of craniosynostosis in a case of Saethre-Chotzen syndrome. J. Craniofac. Surg. 15, 1048–1052.
el Ghouzzi, V., Le Merrer, M., Perrin-Schmitt, F., Lajeunie, E., Benit, P., Renier, D., Bourgeois, P., Bolcato-Bellemin, A. L., Munnich, A., and Bonaventure, J. (1997). Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat. Genet. 15, 42–46.
Eswarakumar, V. P., Ozcan, F., Law, E. D., Bae, J. H., Tome, F., Booth, C. J., Adams, D. J., Lax, I., and Schlessinger, J. (2006). Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. PNAS 103, 18603–18608.
Hajihosseini, M. K., Wilson, S., De Moerlooze, L., and Dickson, C. (2001). A splicing switch and gain-of-function mutation in FgfR2-IIIc hemizygotes causes Apert/Pfeiffer-syndrome-like phenotypes. Proc. Natl. Acad. Sci. U.S.A. 98, 3855–3860.
Holmes, G., Rothschild, G., Roy, U. B., Deng, C. X., Mansukhani, A., and Basilico, C. (2009). Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev. Biol. 328, 273–284.
Howard, T. D., Paznekas, W. A., Green, E. D., Chiang, L. C., Ma, N., Ortiz de Luna, R. I., Garcia Delgado, C., Gonzalez-Ramos, M., Kline, A. D., and Jabs, E. W. (1997). Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15, 36–41.
Howe, L. R., Watanabe, O., Leonard, J., and Brown, A. M. (2003). Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 63, 1906–1913.
Jabs, E. W., Li, X., Scott, A. F., Meyers, G., Chen, W., Eccles, M., Mao, J. I., Charnas, L. R., Jackson, C. E., and Jaye, M. (1994). Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat. Genet. 8, 275–279.
Jabs, E. W., Muller, U., Li, X., Ma, L., Luo, W., Haworth, I. S., Klisak, I., Sparkes, R., Warman, M. L., and Mulliken, J. B. (1993). A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 75, 443–450.
Jiang, X., Iseki, S., Maxson, R. E., Sucov, H. M., and Morriss-Kay, G. M. (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106–116.
Liu, B., Yu, H. M., and Hsu, W. (2007). Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of beta-catenin in proliferation and differentiation. Dev. Biol. 301, 298–308.
Merrill, A. E., Bochukova, E. G., Brugger, S. M., Ishii, M., Pilz, D. T., Wall, S. A., Lyons, K. M., Wilkie, A. O., and Maxson, R. E. Jr. (2006). Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum. Mol. Genet. 15, 1319–1328.
Morriss-Kay, G. M., and Wilkie, A. O. (2005). Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J. Anat. 207, 637–653.
Opperman, L. A. (2000). Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 219, 472–485.
Quarto, N., Fong, K. D., and Longaker, M. T. (2005). Gene profiling of cells expressing different FGF-2 forms. Gene 356, 49–68.
Quarto, N., and Longaker, M. T. (2005). The zebrafish (Danio rerio): a model system for cranial suture patterning. Cells Tissues Organs (Print) 181, 109–118.
Quarto, N., Wan, D. C., and Longaker, M. T. (2008). Molecular mechanisms of FGF-2 inhibitory activity in the osteogenic context of mouse adipose – derived stem cells (mASCs). Bone 42, 1040–1052.
Reinhold, M. I., Kapadia, R. M., Liao, Z., and Naski, M. C. (2006). The Wnt-inducible transcription factor Twist1 inhibits chondrogenesis. J. Biol. Chem. 281, 1381–1388.
Rice, D. P., Aberg, T., Chan, Y., Tang, Z., Kettunen, P. J., Pakarinen, L., Maxson, R. E., and Thesleff, I. (2000). Integration of FGF and TWIST in calvarial bone and suture development. Development 127, 1845–1855.
Saethre, M. (1931). Ein Beitrag zum Turmschaedelproblem (Pathogenese, Erblichkeit und Symptomatologie). Dtsch. Z. Nervenheilkd. 119, 533–555.
Sahar, D. E., Longaker, M. T., and Quarto, N. (2005). Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Dev. Biol. 280, 344–361.
Twigg, S. R., Kan, R., Babbs, C., Bochukova, E. G., Robertson, S. P., Wall, S. A., Morriss-Kay, G. M., and Wilkie, A. O. (2004). Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc. Natl. Acad. Sci. U.S.A. 101, 8652–8657.
Wieland, I., Jakubiczka, S., Muschke, P., Cohen, M., Thiele, H., Gerlach, K. L., Adams, R. H., and Wieacker, P. (2004). Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am. J. Hum. Genet. 74, 1209–1215.
Yin, L., Du, X., Li, C., Xu, X., Chen, Z., Su, N., Zhao, L., Qi, H., Li, F., Xue, J., Yang, J., Jin, M., Deng, C., and Chen, L. (2008). A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone 42, 631–643.
Yoshida, T., Phylactou, L. A., Uney, J. B., Ishikawa, I., Eto, K., and Iseki, S. (2005). Twist is required for establishment of the mouse coronal suture. J. Anat. 206, 437–444.
Keywords: canonical Wnt-signaling, cranial suture, endochondral ossification, craniosynostosis, twist haploinsufficiency
Citation: Behr B, Longaker MT and Quarto N (2011) Craniosynostosis of coronal suture in Twist1+/− mice occurs through endochondral ossification recapitulating the physiological closure of posterior frontal suture. Front. Physio. 2:37. doi: 10.3389/fphys.2011.00037
Received: 18 May 2011;
Accepted: 28 June 2011;
Published online: 21 July 2011.
Edited by:
Anna Petryk, University of Minnesota, USAReviewed by:
Eric Everett, University of North Carolina at Chapel Hill, USADoug Spicer, Maine Medical Center Research Institute, USA
Raj Gopalakrishnan, University of Minnesota, USA
Copyright: © 2011 Behr, Longaker and Quarto. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Michael T. Longaker, Hagey Laboratory for Regenerative Medicine, Department of Surgery, Division of Plastic and Reconstructive Surgery, Stanford University School of Medicine, 257 Campus Drive, Stanford, CA 94305-5148, USA. e-mail: longaker@stanford.edu; Natalina Quarto, Department of Structural and Functional Biology, University of Naples Federico II, Complesso M. S. Angelo, Napoli, Italy. e-mail: quarto@unina.it