 Charles J. Billington Jr.1,2,3 Juliane E. Fiebig4 Cynthia L. Forsman1,2 Lan Pham5 Nathan Burbach1 Mu Sun6 Tina Jaskoll7 Kim Mansky8 Rajaram Gopalakrishnan5 Michael B. O’Connor2 Thomas D. Mueller4 Anna Petryk1,2*
Charles J. Billington Jr.1,2,3 Juliane E. Fiebig4 Cynthia L. Forsman1,2 Lan Pham5 Nathan Burbach1 Mu Sun6 Tina Jaskoll7 Kim Mansky8 Rajaram Gopalakrishnan5 Michael B. O’Connor2 Thomas D. Mueller4 Anna Petryk1,2*- 1 Department of Pediatrics, University of Minnesota, Minneapolis, MN, USA
- 2 Department of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN, USA
- 3 Medical Scientist Training Program, University of Minnesota, Minneapolis, MN, USA
- 4 Department for Molecular Plant Physiology and Biophysics, Julius-von-Sachs Institute, University of Würzburg, Würzburg, Germany
- 5 Diagnostic/Biological Sciences, School of Dentistry, University of Minnesota, Minneapolis, MN, USA
- 6 Neurodegeneration Discovery Performance Unit, GlaxoSmithKline Research & Development, Shanghai, China
- 7 Laboratory for Developmental Genetics, University of Southern California, Los Angeles, CA, USA
- 8 Department of Developmental and Surgical Sciences, School of Dentistry, University of Minnesota, Minneapolis, MN, USA
Twisted gastrulation (TWSG1) is a conserved, secreted glycoprotein that modulates signaling of bone morphogenetic proteins (BMPs) in the extracellular space. Deletion of exon 4 of mouse Twsg1 (mTwsg1) is associated with significant craniofacial defects. However, little is understood about the biochemical properties of the corresponding region of the protein. We have uncovered a significant role for exon 4 sequences as encoding the only two glycosylation sites of the mTWSG1 protein. Deletion of the entire exon 4 or mutation of both glycosylation sites within exon 4 abolishes glycosylation of mTWSG1. Importantly, we find that constructs with mutated glycosylation sites have significantly reduced BMP binding activity. We further show that glycosylation and activity of TWSG1 recombinant proteins vary markedly by cellular source. Non-glycosylated mTWSG1 made in E. coli has both reduced affinity for BMPs, as shown by surface plasmon resonance analysis, and reduced BMP inhibitory activity in a mandibular explant culture system compared to glycosylated proteins made in insect cells or murine myeloma cells. This study highlights an essential role for glycosylation in Twisted gastrulation action.
Introduction
Twisted gastrulation (TWSG1 in mammals, Tsg in non-mammals) is one of several key regulators of bone morphogenetic proteins (BMPs), including BMP2, BMP4, BMP7, in the extracellular space (Chang et al., 2001; Ross et al., 2001; Scott et al., 2001; Blitz et al., 2003; Zakin et al., 2005). TWSG1 is highly evolutionarily conserved and mutations in Drosophila, Xenopus, and Danio rerio Tsg disrupt normal dorsal–ventral patterning (Mason et al., 1994; Oelgeschlager et al., 2000; Ross et al., 2001; Scott et al., 2001). In mice, mutations in TWSG1 result in a range of craniofacial defects, including micrognathia, agnathia, cyclopia, and other midline facial defects due to abnormal development of the first branchial (mandibular) arch (Petryk et al., 2004; Zakin and De Robertis, 2004; Mackenzie et al., 2009; Billington et al., 2011).
The presumed biochemical basis of these craniofacial defects is impaired binding of TWSG1 to BMPs. Previous work in Xenopus has identified the importance of a set of conserved cysteines and a conserved tryptophan within the N-terminus of xTsg for binding (and inhibiting) BMPs. The C-terminus was shown to play a role in xTsg’s interaction with chordin and BMP-promoting activity (Oelgeschlager et al., 2000, 2003), most likely through facilitation of Chordin (CHRD) cleavage (Larrain et al., 2001). Deletion of exon 4 (coding exon 3), which removes neither the cysteine-rich domain nor the C-terminus, produces similar craniofacial and skeletal phenotypes to those resulting from targeting exons 2 and 3 (coding exons 1 and 2; Nosaka et al., 2003; Zakin and De Robertis, 2004; Gazzerro et al., 2006), raising the possibility that exon 4 encodes a region of the TWSG1 protein with important biochemical properties.
That Tsg is a glycoprotein has been known since the early stages of characterization of this protein (Mason et al., 1994). Despite this history, little research has actually investigated the role of glycosylation in TWSG1 function. There are two common types of glycosylation, N- and O-linked. In N-linked glycosylation, sugar chains are linked to asparagine residues and are generally seen in an Asn-X-Ser/Thr triplet (where X is not proline; Neuberger and Marshall, 1968; Gavel and Von Heijne, 1990; Spiro, 2002). In O-glycosylation sugar modifications occur on serines or threonines (Spiro, 2002). In this report we demonstrate a role for exon 4-encoded sequences as the exclusive site of N-glycosylation of mTWSG1 and show that the glycosylation of mTWSG1 is critical for mediating BMP binding and activity of mTWSG1.
Materials and Methods
Animal Care
Use and care of the mice in this study was approved by the University of Minnesota Institutional Animal Care and Use Committee. Wild type mice were in the C57BL/6 background.
Protein Sequence Analysis
Twisted gastrulation homolog protein sequences were downloaded from NCBI for Mus musculus, Rattus norvegicus, Homo sapiens, Bos taurus, Canis lupus familiaris, Monodelphis domestica, Gallus gallus, and Xenopus laevis. Protein sequences were aligned using ClustalW in the MacVector (MacVector, Cary, NC, USA) program. Protein phylogeny was calculated using MacVector’s phylogeny tool. Glycosylation sites were predicted in each protein using EnsemblGly1 (Caragea et al., 2007) and confirmed using NetNGlyc 1.02
Plasmids
All plasmids for expression of mTWSG1 in HEK 293 cells were constructed in the C-terminal FLAG-tagged expression vector pCMVTag4c (Stratagene, Santa Clara, CA, USA). A full-length wild type Twsg1 ORF and a Twsg1 ORF lacking exon 4 were each cloned into this vector using EcoRI and XhoI to create pCMVTag-Twsg1WT and pCMVTag-Twsg1Δex4. Constructs with glycosylation site mutations (pCMVTag-Twsg1N80Q, pCMVTag-Twsg1N146Q) were generated from the wild type construct using the QuickChange site directed mutagenesis kit (Stratagene) employing the following primers N80Q F: 5′-atgtgcaaccctcggcagt acagcgacaccccg, N80Q R: 5′-cggggtgtcgctgtactgccgagggttgcacat, N146Q F: 5′-ccagctgcaccaccaacaggtgtctgttcccagc, N146Q R: 5′-gctgggaacagacacctgttggtggtgcagctgg. The double glycosylation site mutant was generated from the N80Q plasmid, adding the N146Q mutation to generate pCMVTag-Twsg1QQ. A glycosylation site at Asn51 was restored into mouse TWSG1 using the primers P53S F: 5′-tggagaagggaactgcagctgctgtaaggagtgc and P53S R: 5′-gcactccttacagcagctgcagttcccttctcca. Following mutation, plasmids were assessed by restriction digests to confirm mutation (RsaI for N80Q, AflIII for N146Q, PstI for P53S) and sequenced to confirm no other changes had been introduced.
For production of recombinant proteins in E. coli the cDNA mature parts of mTWSG1 and xTsg were cloned into the expression vector pET28b(+) (Novagen, Darmstadt) via the restriction sites NdeI and BamHI, resulting in proteins carrying an N-terminal His6-Tag, which can be proteolytically removed using the protease thrombin. The expression construct for recombinant xTsg from insect cell expression was derived by cloning the mature part of xTsg into a modified version of the transfer vector pBAC3 (Novagen, Darmstadt) using the restriction sites BamHI and XhoI. The resulting construct for xTsg harbors a gp64 signal peptide for secretion into the medium and an N-terminal His6-Tag, which can be removed by thrombin proteolysis. The cDNAs encoding for mTWSG1 and xTsg were obtained from imaGenes (Berlin) and the correctness of all cloned cDNAs was verified by sequencing.
Recombinant Protein Preparations
The E. coli strain BL21(DE3) was used for bacterial protein expression. Transformed cells were grown in shaking flasks in LB-Medium (Melford, Chelsworth) at 37°C and 130 rpm, selection of transformed cells was achieved using 30 μg/ml kanamycin. Gene expression was induced at an optical density of OD600 = 0.6 by adding isopropyl-1-thio-β-D-galactopyranoside (IPTG) to a final concentration of 1 mM and protein expression was continued for 4 h and 37°C. Cells were harvested by centrifugation (6000 × g, 15 min, 4°C) and the pellet was resuspended in TBSE buffer (10 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA). The recombinant proteins mTWSG1 and xTsg were expressed in insoluble form as so-called inclusion bodies. To isolate the proteins, 10 g bacterial cells were lysed on ice by sonication (Bandelin Sonopuls HD3200, 10 min, 150 W). The suspension was centrifuged to harvest the insoluble protein fraction. Inclusion bodies were purified by mechanical washing and centrifugation using TBS buffer (10 mM Tris–HCl pH 8.0, 150 mM NaCl) with and without 1% Triton X-100. To extract the recombinant Tsg proteins the inclusion bodies were dissolved in 20 volumes (v/w) 100 mM Na-Phosphate, 10 mM Tris–HCl pH 8.0, 6 M Guanidinium chloride (GuCl) and the mixture was stirred for 12 h at 20°C. After centrifugation to remove insoluble impurities the supernatant was subjected to metal affinity chromatography (IMAC) using Ni2+-NTA resin (Amocol Bioprocedures Ltd., Teltow). Tsg protein was eluted employing a step gradient and 100 mM Na-Phosphate, 10 mM Tris pH 8.0, 6 M GuCl, 300 mM imidazole. Tsg-containing fractions were pooled, dialyzed against 50 mM Tris–HCl pH 8.0, 6 M GuCl, 5 mM EDTA and concentrated to a protein concentration of 5 mg/ml. Refolding of Tsg was carried out by rapid dilution using the conditions reported for preparation of BMP2 from E. coli (Ruppert et al., 1996). The solution containing refolded Tsg was dialyzed against 50 volumes 20 mM HEPES pH 8.0, 300 mM NaCl and Tsg was purified first by an IMAC chromatography step under non-denaturing conditions. Tsg-containing fractions were pooled and dialyzed against 50 volumes 20 mM HEPES pH 7.4, 500 mM NaCl. As a final purification step, size exclusion chromatography was performed to separate monomeric Tsg protein from accompanying dimeric and oligomeric species (Superdex 75 16/60 prep grade, GE Healthcare, Freiburg).
Recombinant xTsg protein from a eukaryotic source was generated by transient expression in baculovirus-transfected insect cells. The virus was prepared by co-transfection of the transfer vector pBAC3 containing the mature part of xTsg along with linearized virus DNA, using the BacVector-3000 DNA Kit (Merck KGaA, Darmstadt) into Sf9 insect cells according to the manufacturer’s instructions. Recombinant virus was identified and picked by a plaque-assay and protein expression was tested from six clones during virus amplification. A high-expression virus clone was then amplified in Sf21 insect cells to a titer of ≥107–108 pfu/ml.
For protein production, HighFive insect cells (BTI-TN-5B1-4, gift of R. Grabherr, VIBT, Vienna, Austria) were used, which were grown as adherent cell cultures in IPL-41 medium supplemented with 1% Penicillin/Streptomycin (LONZA, Köln) and 1% Lipid Medium Supplement (Sigma-Aldrich, München) at 27°C. For large-scale expression HighFive cells were transferred to RollerBottles (Greiner Bio-One, Frickenhausen) and adapted to suspension culture. 8 × 105 cells/ml were infected with recombinant baculovirus at a multiplicity of infection of 5 and protein expression was continued for additional 4–5 days. The cell suspension was clarified by centrifugation (6000 g, 20 min, 4°C) and the protein-containing supernatant was dialyzed against 20 mM Tris–HCl pH 7.9, 500 mM NaCl, 20 mM imidazole for IMAC purification. Pure xTsg was eluted in a step gradient using 20 mM Tris–HCl pH 7.9, 500 mM NaCl, 300 mM imidazole. Tsg-containing fractions were pooled, dialyzed twice against 50 volumes 20 mM HEPES pH 7.4, 500 mM NaCl and frozen at −20°C until further use.
Cell Culture, Protein Isolation, and Deglycosylation
HEK293 cells were cultured in Dulbecco’s minimum essential media supplemented with 10% fetal bovine serum and 1% 100× Anti-Anti antibiotic–antimycotic (GIBCO, Carlsbad, CA, USA). Cells were transfected using Lipofectamine with PLUS reagent (Invitrogen, Carlsbad, CA, USA) in media without serum or antibiotics and lysed to isolate proteins after 16 h. Proteins from transfected cells were isolated in lysis buffer (50 mM Tris–HCl, 125 mM NaCl, 1% NP40 Substitute (BioChemika #74385), and 0.5% sodium deoxycholate) with the addition of Complete protease inhibitor (Roche, Indianapolis, IN, USA). Lysates were spun for 10 min at 10000 g to pellet cell debris. Supernatant fractions were analyzed using SDS-PAGE with and without deglycosylation. Transfection product proteins, insect cell xTsg, E. coli mTWSG1 and were deglycosylated using PNGase F (New England Biolabs, Ipswich, MA, USA) according to manufacturer’s instructions. Recombinant mouse TWSG1 from murine myeloma cells (R&D systems, Minneapolis, MN, USA) was deglycosylated using PNGase F or protein deglycosylation mix (NEB) according to manufacturer’s instructions. Blots of FLAG-tagged proteins were probed with 1:1000 Rabbit Anti-DYKDDDDK in 0.1% casein/PBS (Cell Signaling, Danvers, MA, USA). Untagged recombinant proteins were visualized using a rabbit antibody custom generated (Open Biosystems, Thermo Fisher Scientific) against the peptide EGDTQLNWNIVSFPVAEE (mTWSG1 aa.108–125) at 1:250 in 0.1% casein/PBS. All blots were pre-blocked in 0.1% casein/PBS.
Immunoprecipitation
HEK293 cells were transfected with FLAG-tagged WT, QQ or Δex4 constructs while one plate was left untransfected. Cells were lysed after 16 h in 250 μl of 140 mM NaCl, 0.4 mM Tris–HCl pH 8.0, 1% Glycerol, 1% NP40 Substitute, 2% BSA with protease inhibitor cocktail. Lysates were spun at 10000 g for 10 min and supernatant was combined with EZview anti-FLAG M2 affinity beads (Sigma, St. Louis, MO, USA) as well as 100 ng recombinant BMP2 (R&D). IP beads, BMP ligand, and lysate were allowed to interact for 6 h at 4°C in a rotary shaker. IP beads were washed four times in 280 mM NaCl, 0.4 mM Tris–HCl pH 8.0, 1% Glycerol, 1% NP40 Substitute. Samples were eluted by boiling 5 min in sample loading buffer (62.5 mM Tris–HCl pH6.8, 2% SDS, 10% glycerol 0.002% bromophenol blue) and analyzed by SDS-PAGE immunoblotting. Duplicate blots of immunoprecipitated samples were probed with Rabbit Anti-DYKDDDDK (Cell Signaling; 1:5000) or with anti BMP2 (R&D; 1:5000). Blots were blocked in 5% BSA and primary antibodies were probed in 5% dry milk.
Surface Plasmon Resonance Analysis
In vitro interaction analysis employing surface plasmon resonance (SPR) was performed using a ProteON XPR36 system (Bio-Rad Laboratories Inc., Technion, Haifa, Israel). Recombinant BMP2 derived from E. coli expression was immobilized onto a ProteOn GLC-chip (Bio-Rad Laboratories Inc., Technion, Haifa, Israel) via amino coupling. The alginate polymer surface was first activated with a freshly prepared mixture of 100 mM 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) and 25 mM N-hydroxysulfosuccinimide (Sulfo-NHS). Then recombinant human BMP2 was immobilized on flow cells 1 and 2 by perfusing a solution of 200 nM BMP2 in 10 mM sodium acetate pH 4.0 over the activated alginate surface until protein densities of 100–200 (low density coating) and 400–500 RU (medium density coating) on the biosensor surface were obtained. Non-glycosylated xTsg and mTsg from E. coli as well as glycosylated xTsg from insect cells were perfused as analytes over the biosensor surface. Six different analyte concentrations were used in a one-shot kinetic measurement setup. All measurements were performed at 25°C in HBST buffer (10 mM HEPES pH 7.4, 500 mM NaCl, 0.005% Tween20, Sigma) using a flow rate of 100 μl/min. The association phase was set to 240 s, the dissociation phase was measured for 660 s. The BMP2 biosensor surface was regenerated using a short pulse 30 s of perfusion with 10 mM glycine pH 1.5. The correct function of the BMP2 biosensor was tested using perfusion with the BMP type I receptor BMPRIA and the values obtained were compared to reported binding affinities in the literature (Heinecke et al., 2009).
To account for bulk face effects a control flow cell (without BMP2 coating) was subtracted from all measurement flow cells. Interaction data were evaluated using the software ProteON Manager 2.1.2 Version 2.1.2.05. Apparent equilibrium binding constants KD were derived from the kinetic rate constants for association ka and dissociation kd by the equation KD = kd/ka. All measurements were performed in duplicate.
Mandibular Explants
Embryos were collected at embryonic day 10.5 (E10.5). Organ culture of mandibular processes was performed according to previously reported protocols (Jaskoll et al., 2005; Melnick et al., 2005). Briefly, the mandibular component of the first branchial arch was isolated by microdissection and explanted into a culture system using serum-free chemically defined BGJb medium supplemented with 0.1 mg/ml ascorbic acid (Sigma) and 1% penicillin/streptomycin (Cellgro, Manassa, VA, USA). Explants were allowed to develop at 37°C and 5% CO2 for 1 h before bead placement. Beads were soaked in either 1% BSA/PBS, 100 ng/μl BMP4 (R&D systems), 50 ng/μl mTWSG1 (R&D Systems), 50 ng/μl xTsg derived from insect cell expression or 50 ng/μl mTWSG1 produced by bacterial expression in E. coli and placed in the distal region of the explant with microforceps. Explants were cultured for additional 24 h following bead placement. In situ hybridization for Msx2 was performed as previously published (Mackenzie et al., 1992; Sasaki and Hogan, 1993).
Results
Twisted Gastrulation Proteins Have Evolutionarily Conserved Glycosylation Sites, Mapping in Rodents to Exon 4
Analysis of the mouse TWSG1 protein sequence using the EnsemblGly software predicts two putative N-linked glycosylation sites, at asparagines Asn80 and Asn146. No O-linked glycosylation sites were predicted (Caragea et al., 2007). Other vertebrate TWSG1/Tsg protein sequences were also analyzed. All other tetrapods also have the two putative glycosylation sites corresponding to those predicted in mice (Figure 1). A third putative glycosylation site at Asn51 is widely conserved in other organisms (Vilmos et al., 2001; Oelgeschlager et al., 2004) and has been shown in Xenopus to be an active site of in vivo glycosylation (Oelgeschlager et al., 2003). This site is not present in either mice or rats, where a consensus serine is substituted by proline, altering the N-X-S/T glycosylation sequon. Given that this site is widely conserved throughout other tetrapods, we infer that a mutation specifically in the rodent lineage led to a loss of this site. Interestingly, phylogenetic analysis of the protein sequences for these selected tetrapods recapitulated the expected species phylogeny with the rodents grouping together, the marsupial as an outgroup to the other mammals and avian and amphibian representatives on a more distant branch.
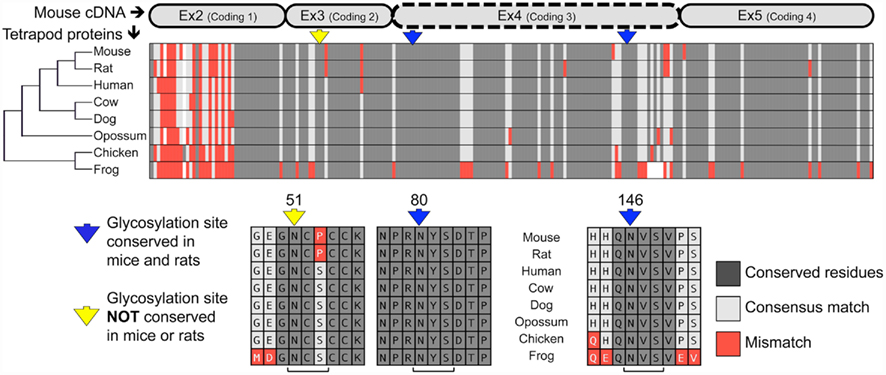
Figure 1. Twisted gastrulation is a conserved glycoprotein with predicted conserved and non-conserved glycosylation sites. ClustalW aligned proteins from various tetrapods are shown to highlight the high degree of conservation of the Twisted gastrulation protein and to show the locations of conserved and non-conserved glycosylation sites. A protein phylogenetic tree is shown at left, which also correlates with species phylogeny. The alignment of the protein sequences to the mouse is shown at the top. The forth exon deleted in a mouse knockout with craniofacial defects (Petryk et al., 2004) is indicated by a dashed line. The N-glycosylation sequons are indicated in context below with brackets at the bottom indicating the N-X-S sequons, while arrows in yellow or blue at the top indicate the asparagines to which sugars can be linked.
Most notably, both predicted glycosylation sites in mouse TWSG1 (Asn80 and Asn146) are in the region encoded by exon 4, suggesting that deletion of this exon in rodents would completely ablate the glycosylation of any truncated protein product.
Mutational Analysis Confirms Putative Glycosylation Sites
Our analysis of mTWSG1 sequences suggested that mTWSG1 is exclusively N-glycosylated. We tested this by first deglycosylating the protein using a cocktail of endoglycosidases that is able to remove both N- and O-linked glycosylation from recombinant mTWSG1 (data not shown). We also deglycosylated the protein using PNGase F, which specifically removes N-glycosylation directly at the asparagine–carbohydrate linkage, and observed an equivalent increase in electrophoretic mobility (Figure 2A). This indicated that, as predicted, mTWSG1 is solely N-glycosylated.
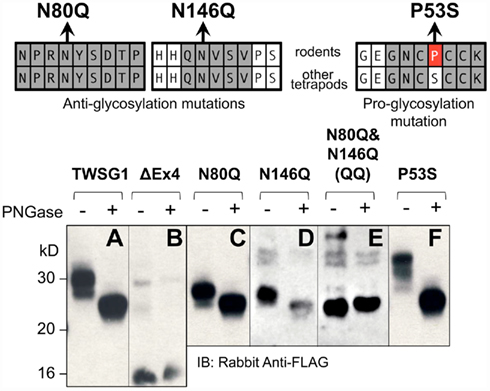
Figure 2. Twisted gastrulation in rodents is N-glycosylated at two residues, both in exon 4. (Top) Locations of mutations that remove or add glycosylation at conserved sites are shown with reference to the sequences in rodents and other tetrapods. (A) Wild type mTWSG1 is glycosylated and runs on SDS-PAGE as a doublet of about 27 kD and 30 kD. (B) Exon 4-deleted mTWSG1 is not glycosylated. (C) The N80Q mutation removes one glycosylation site but is still glycosylated. (D) The N146Q mutation removes one glycosylation site but the protein is still glycosylated. (E) When the N80Q and N146Q mutations are combined the resulting QQ-TWSG1 is not glycosylated. (F) Mutation of a proline in mTWSG1 to the consensus serine restores a third glycosylation site.
Although two N-glycosylation sites are predicted in mTWSG1, they have not been previously confirmed experimentally. The only mutational analysis performed to confirm a glycosylation site on a Tsg molecule had been done in xTsg at the site not conserved in rodents. Therefore, we mutated each of the putative rodent glycosylation sites, replacing asparagine with glutamine as had been done on other glycoproteins (Barbosa et al., 1987; Yamaguchi et al., 1991; Buller et al., 1994), reasoning that this would prevent glycosylation without disrupting the overall protein structure (Figure 2).
As expected, expression of an exon 4 deletion mutant construct (TWSG1Δex4) showed no change in protein mobility with PNGase F treatment, indicating the expected lack of glycosylation (Figure 2B). Mutating either Asn80 or Asn146 alone to glutamine increased electrophoretic mobility compared to the wild type protein (Figures 2C,D). Notably, whereas the wild type protein runs on SDS-PAGE as a doublet of roughly 27 and 30 kD, the mutant forms no longer show the 30-kD band and instead run as doublets of ~25 and 27 kD. These single mutant proteins showed further increased mobility, to a single band at ~25 kD, the molecular weight predicted for the unmodified amino acid sequence, when treated with PNGase F, suggesting that they were still glycosylated, although to a lesser degree than the wild type protein. When both mutations were combined, no change in mobility with PNGase treatment was observed and a single band was observed at ~25 kD, indicating a complete lack of glycosylation in the double mutant (Figure 2E).
Based on data for xTsg in which glycosylation at Asn52 has been shown to occur (Oelgeschlager et al., 2003), we hypothesized that if the consensus serine at position 53 were restored in rodent TWSG1, Asn51 of mTWSG1 would also be a glycosylation site. As predicted, when Pro53 was mutated in the mouse sequence to serine, a decrease in electrophoretic mobility was observed, and a triplet of bands appeared on SDS-PAGE analysis at 27, 30, and 33 kD (Figure 2F). The glycosylation ability of this restored site strongly suggests that other tetrapods, including humans, have three active glycosylation sites on TWSG1.
The observation that the number of bands in SDS-PAGE analysis roughly correlates with the number of glycosylation sites suggests that, at least in this in vitro system, there can be an equilibrium between fully and partially or non-glycosylated forms of the protein.
Lack of Glycosylation Limits BMP Binding
Since TWSG1’s primary known function is to modulate BMP activity in the extracellular space, we tested if glycosylation affected the binding of TWSG1 to BMPs. We also wanted to confirm that the exon 4 deletion, presumed to be defective for BMP binding, actually did prevent binding of TWSG1 to BMPs.
Immunoprecipitation of BMP2 by the wild type TWSG1, but not by the Δex4 mutant protein nor the QQ double glycosylation mutant, indicates that neither of these mutant proteins is able to bind BMP2 (Figure 3). Immunoprecipitation experiments were also performed using BMP4 protein and showed similar results (data not shown).
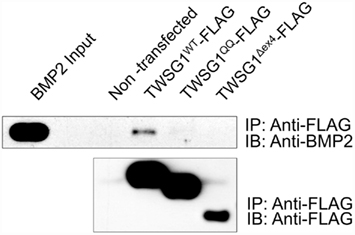
Figure 3. Immunoprecipitation indicates that glycosylation and exon 4 are necessary for BMP binding. Immunoprecipitation (IP) was performed with anti-FLAG and immunoblots (IB) were probed with either anti BMP2 (top) or anti-FLAG (bottom); BMP2 immunoprecipitates with wild type but not glycosylation defective TWSG1QQ or exon 4-deleted TWSG1Δex4.
Glycosylation of TWSG1 Recombinant Proteins Varies Markedly by Cellular Source
Cells derived from different species have variable abilities to glycosylate proteins. For example, in Gram-negative bacteria glycosylation is an extremely rare event, with E. coli only having two glycoproteins, AIDA-I, and the TibA adhesin, both of which are potential virulence factors (Lindenthal and Elsinghorst, 1999; Benz and Schmidt, 2001). To allow for the heterologous expression of O- or N-glycosylated proteins from bacteria, a very complex setup is required using either coexpression of certain UDP-GlcNac epimerase and transferase enzymes (Henderson et al., 2011) or the transfer of the glycosylation machinery of another bacterium, i.e., from the ε-proteobacterium Campylobacter jejuni into E. coli (Wacker et al., 2002). Thus mammalian glycoproteins are usually obtained in unglycosylated form when expressed in E. coli. On the other hand, insect cells are able to glycosylate these proteins although they may do so with slightly different sugar moieties and generally reduced saccharide chain complexity (Jarvis et al., 1998; Tomiya et al., 2004). Here N-glycosylation usually displays a high-mannose type unless engineered insect cells are used, which can mimic mammalian-type glycosylation. Following a common approach for investigating the role of sugar modifications on proteins (Walsh and Jefferis, 2006), we exploited this naturally occurring variation in glycosylation modification to test the biochemical and biological properties of mTWSG1.
We expressed TWSG1/Tsg in E. coli, which do not glycosylate the protein, as well as in HighFive insect cells, which should glycosylate the protein, and also used a commercially available preparation of mTWSG1 produced in murine myeloma cells for comparison. When we treated TWSG1/Tsg proteins with PNGase F and analyzed them by electrophoresis, we were able to confirm differences in glycosylation (Figure 4). Murine TWSG1 made in murine cells was fully glycosylated. Xenopus Tsg made in the HighFive insect cell line was also N-glycosylated, although this glycosylation may differ in the kinds or size of sugar moieties compared to mammalian glycosylation, since the shift in electrophoretic mobility was less than the apparent mobility shift observed upon deglycosylation of mTWSG1 from murine cells. Murine TWSG1 made in E. coli was not glycosylated and hence showed no change in mobility with PNGase F treatment.
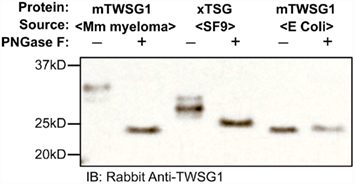
Figure 4. TWSG1 glycosylation varies depending on host source. mTWSG1 made in murine myeloma cells is markedly glycosylated as indicated by the increase in mobility with PNGase F treatment. Xenopus Tsg made in insect cells is also glycosylated but shows a smaller mobility shift. Murine TWSG1 made in E. coli is not glycosylated and shows no shift in mobility after treatment with PNGase F.
Lack of Glycosylation Decreases the Affinity of the BMP–TWSG1 Interaction
Our immunoprecipitation data suggest that TWSG1 can bind BMP2/4 only when glycosylated. However, SPR analysis allows for a more precise quantification of this change in affinity. To measure the affinity of the Tsg proteins for BMP2, the ligand BMP2 was immobilized on a biosensor chip surface by amino coupling. Varying concentrations of xTsg or mTWSG1 proteins made in insect cells or E. coli, respectively, were allowed to perfuse over the BMP2 biosensor surface and association and dissociation kinetics were measured (Figure 5). Quantitative analysis of the sensorgram reveals that glycosylated xTsg has a three to four-fold slower dissociation rate compared to non-glycosylated xTsg derived from E. coli (Table 1; Figures 5A,B) and also a three-fold faster association. The change in association and dissociation kinetics results in an approximately 10-fold increased affinity of N-glycosylated xTsg compared to the non-glycosylated form for BMP2. The difference in affinity and binding kinetics becomes most apparent by a overlay of two SPR sensorgrams of insect cell derived xTsg and E. coli derived xTsg at identical protein concentrations (Figure 5C).
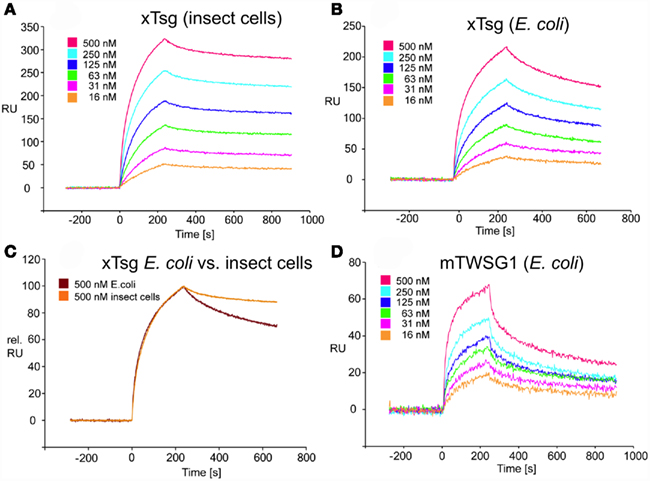
Figure 5. SPR interaction analysis of BMP2 binding to recombinant Tsg proteins derived from different sources. Binding of recombinant Tsg proteins to a BMP2 biosensor was analyzed using SPR. BMP2 was immobilized on a GLC-chip via amino coupling. Indicated concentrations of Tsg proteins were perfused over the biosensor surface, injection started at time point 0 with a duration of 240 s (association phase) after which only buffer was again perfused for 660 s to record the dissociation of Tsg from BMP2. (A,B). The BMP2 biosensor was perfused with varying concentrations of (A) xTsg derived from expression in insect cells or (B) xTsg produced in E. coli. (C) The different dissociation rate constants between xTsg protein derived from either a eukaryotic or a prokaryotic host is apparent from an overlay of two normalized SPR sensorgrams recorded for the interaction of different xTsg proteins (500 nM concentration) with BMP2. (D) The interaction of mTWSG1 derived from E. coli expression reveals a biphasic interaction with BMP2. The binding kinetics following a slow association and a slow dissociation yields an equilibrium binding constant very similar to that of xTsg produced in bacteria.
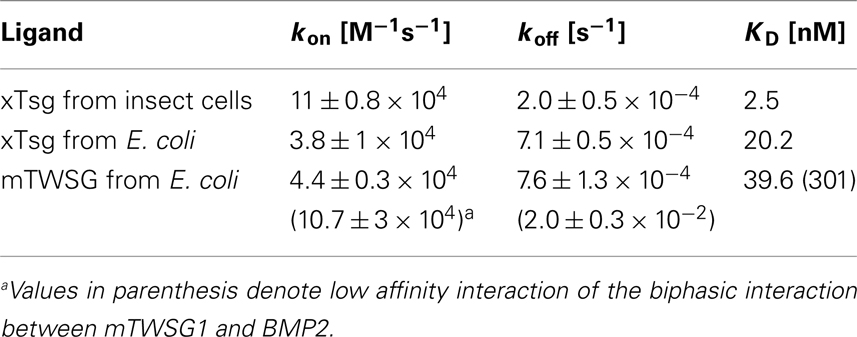
Table 1. Tsg affinity for BMP2 varies depending on host source.
Despite large efforts mTWSG1 could not be obtained from insect cell expression, however mTWSG1 could be produced in sufficient quantities in E. coli using the same expression and purification scheme as used for the production of xTsg. Measuring the interaction of BMP2 and mTWSG1 from E. coli via SPR also revealed a fast dissociation rate constant. In contrast to the interaction of xTsg and BMP2 the binding of mTWSG1 to BMP2 exhibited a biphasic binding kinetics, which could not be analyzed by a simple 1:1 Langmuir type interaction model (Figure 5D). Instead one interaction showed a fast association (on-rate) and a fast dissociation (off-rate) whereas the other interaction exhibited a slow on- and a slow off-rate. From the latter kinetics binding parameters could be deduced that were very similar to the ones determined for xTsg derived from E. coli (Table 1). Although we have no explanation why mTWSG1 shows two different binding kinetics for binding to BMP2 the comparison of mTWSG1 and xTsg produced in E. coli clearly suggests that the presence of the carbohydrate leads to a significant increase in binding affinity for BMP2.
TWSG1 Proteins have Different Biological Activity Depending on Expression System and Glycosylation
To demonstrate the biological effect of glycosylation loss, we chose a mandibular explant system, which has been used previously to examine the activity of BMPs and BMP binding proteins (Ekanayake and Hall, 1997; Tucker et al., 1998; Mina et al., 2002). Expression of the gene Msx2 was used as a reporter to assay of the effect of TWSG1/Tsg on BMP signaling in these explants.
As previously shown (Semba et al., 2000), treatment with BMP4 strongly induced Msx2 expression (Figure 6A). When a bead soaked with glycosylated mTWSG1 (expressed in murine cells) was placed in the Msx2 expression zone the TWSG1 was able to suppress Msx2 expression indicating local inhibition of BMP signaling (Figure 6B). Glycosylated xTsg from insect cells was similarly able to suppress Msx2 expression, indicating the competency of N-glycosylated protein from a different species and a heterologous expression system to inhibit BMP signaling (Figure 6C). On the other hand, mTWSG1 derived from E. coli expression, which is not glycosylated, showed no repression of Msx2, suggesting that it is unable to inhibit BMP signaling (Figure 6D). Thus, glycosylation appears to be essential for TWSG1’s BMP binding and BMP inhibitory functions in this tissue assay.
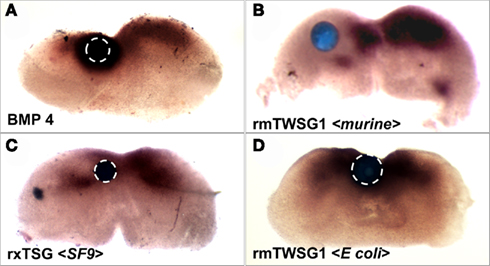
Figure 6. TWSG1 biological activity varies depending on host source. Mandibular explants were treated with beads soaked in ligand solutions for 24 h. In situ hybridization was performed to examine the expression of Msx2. Bead placements are indicated with dashed white lines where less visible due to dark Msx2 staining. Bead treatments were: (A) BMP4, showing induction of Msx2, (B) mTWSG, expressed in murine myeloma cells, showing suppression of Msx2 and indicating inhibition of BMP signaling, (C) Xenopus derived from insect cell expression, with the same result, and (D) mTWSG1 expressed in E. coli, which shows no effect on Msx2 expression, indicating a lack of impact on BMP signaling.
Discussion
Although mTWSG1 protein has been presumed to be glycosylated since its initial characterization, the role of glycosylation in mTWSG1 activity has not been studied in much detail. We report here, for the first time, that TWSG1 binding to BMPs is dependent on N-glycosylation. We also show that reduced BMP binding correlates with diminished TWSG1 BMP inhibitory activity.
Our data complement those previously published (Oelgeschlager et al., 2003) in which mutation of a glycosylation site in xTsg led to a stronger biological effect than that of the wild type protein. When serine 54 of xTsg (corresponding to proline 53 in mice) was mutated to alanine, the electrophoretic mobility of the protein was increased. In addition, this S54A mutant showed enhanced ventralizing activity compared to wild type xTsg, which the authors attributed to increased diffusibility of the protein in the tissue. Based on our results, it is also possible that BMP binding may have been compromised by reduced glycosylation. We predict that this would limit BMP binding but may not interfere with the ability of Tsg to stimulate CHRD cleavage. This model is consistent with other results reported by Oelgeschlager et al. (2003), in which they showed that N-terminal mutations of xTsg that were unable to bind BMPs still retained BMP-promoting activity via the C-terminus of the protein, which resulted in enhanced ventralization. These explanations are not mutually exclusive and diffusibility differences of variable TWSG1 glycoforms could represent an exciting new line of investigation.
Our findings raise the possibility that TWSG1’s BMP modulatory activity may be dependent on, or regulated by, glycosylation and highlight the importance of sugar modifications in extracellular signaling interactions. Given that several other extracellular BMP modulating proteins are also predicted glycoproteins, for example CHRD (Gumienny and Padgett, 2002), CV2/BMPER (Kamimura et al., 2004), Noggin (Groppe et al., 2002), and the DAN family of BMP binding proteins (Stanley et al., 1998; Avsian-Kretchmer and Hsueh, 2004), glycosylation may play a wider role in extracellular BMP regulation than presently recognized. The role of glycosylation in interactions of other proteins will have to be verified however. For example, Xenopus Lefty, which plays a role in Nodal signaling analogous to the role of TWSG1 in BMP signaling, is also a glycoprotein but does not require glycosylation for normal function (Westmoreland et al., 2007). In addition to the BMP binding proteins, glycosylation of the ligand itself is also likely to play a role in regulation of extracellular signaling as illustrated by the absolute requirement of N-glycosylation for the specific interaction of BMP6 with its BMP type I receptor Alk2 (Saremba et al., 2008). Likewise, given the important role of the metalloproteinase Tolloid/Xolloid/BMP1 in regulating BMP action in combination with TWSG/Tsg and CHRD (Larrain et al., 2001), it is interesting that glycosylation has been identified as regulating the activity of the metalloproteinase MT1-MMP (Wu et al., 2004).
Twisted gastrulation made in cells of various species may have differential glycosylation or complete lack of glycosylation. Given the correlation reported here between protein source, glycosylation, BMP binding activity, and biological activity, we suggest that experiments using recombinant BMP modulating proteins should carefully consider the source and glycosylation status of these proteins. It is also interesting to speculate whether glycosylation of TWSG1 or other proteins may be regulated at a tissue specific level, allowing fine-tuning of protein activity in various contexts.
The exon 4 deletion mutant, which led us to this line of inquiry, is deficient for normal TWSG1 activity and a lack of glycosylation may contribute to this deficiency and the ensuing craniofacial phenotypes. In the mandibular explant system, application of exogenous glycosylated mTWSG1 resulted in a reduction of Msx2 expression, consistent with TWSG1’s role as a BMP antagonist. The interactions between TWSG1 and BMPs in vivo, however, are more complex. Functional TWSG1 serves to establish a BMP signaling gradient in the developing mandibular arch. Deletion of exon 4 disrupts the interactions between TWSG1 and BMPs, which, in affected embryos, results in loss of the BMP gradient, more diffuse BMP signaling, and ectopic apoptosis (Mackenzie et al., 2009). Correspondingly, the expression of BMP target gene Msx2 is shifted ventrally at E9.5 and then mostly lost by E10.5 (Mackenzie et al., 2009; Billington et al., 2011). We believe that loss of expression of Msx2 as well as other BMP target genes by E10.5 is due to the loss of a proper BMP gradient and the resultant apoptosis of the distal tissues of the mandibular arch. While TWSG1 antagonizes BMP signaling within the mandibular arch in the explant culture system, we suggest that in vivo the BMP limiting action of TWSG1 can act to actually maintain proper BMP signaling levels in mandibular development and that this action is dependent on glycosylation of TWSG1.
Our results highlight the importance of a better understanding of both the various signaling molecules in craniofacial development as well as their potential post-translational modifications. The complex extracellular regulation of BMPs has been compared to a baroque “court of intrigue” (Bier, 2008) based on all the surprising and nuanced interactions of the many players in BMP signaling. Our study adds to this complexity by highlighting the role of post-translational modifications.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge BreAnne MacKenzie for assistance with mandibular explants, Brian Schmidt and Taylor Sulerud for technical assistance as well as Dr. Eric Jensen and Dr. Hiroshi Nakato for helpful advice and stimulating discussion. We would also like to thank Dr. Reingard Grabherr from the Vienna Institute for Biotechnology, Austria for the kind gift of suspension-adapted HighFive insect cells and his support. This work was supported by the NIH grant R01 DE016601 to Anna Petryk, R01 GM095746 to Michael B. O’Connor, R01 AR056642 to Rajaram Gopalakrishnan, and Deutsche Forschungsgemeinschaft grant MU1095/3-2 to Thomas D. Mueller. Charles J. Billington was supported by the University of Minnesota Musculoskeletal Research Training Program T32 AR050938 and the Medical Scientist Training Program T32 GM008244.
Footnotes
References
Avsian-Kretchmer, O., and Hsueh, A. J. (2004). Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol. Endocrinol. 18, 1–12.
Barbosa, J. A., Santos-Aguado, J., Mentzer, S. J., Strominger, J. L., Burakoff, S. J., and Biro, P. A. (1987). Site-directed mutagenesis of class I HLA genes. Role of glycosylation in surface expression and functional recognition. J. Exp. Med. 166, 1329–1350.
Benz, I., and Schmidt, M. A. (2001). Glycosylation with heptose residues mediated by the aah gene product is essential for adherence of the AIDA-I adhesin. Mol. Microbiol. 40, 1403–1413.
Billington, C. J. Jr., Ng, B., Forsman, C., Schmidt, B., Bagchi, A., Symer, D. E., Schotta, G., Gopalakrishnan, R., Sarver, A. L., and Petryk, A. (2011). The molecular and cellular basis of variable craniofacial phenotypes and their genetic rescue in Twisted gastrulation mutant mice. Dev. Biol. 355, 21–31.
Blitz, I. L., Cho, K. W., and Chang, C. (2003). Twisted gastrulation loss-of-function analyses support its role as a BMP inhibitor during early Xenopus embryogenesis. Development 130, 4975–4988.
Buller, A. L., Hastings, G. A., Kirkness, E. F., and Fraser, C. M. (1994). Site-directed mutagenesis of N-linked glycosylation sites on the gamma-aminobutyric acid type A receptor alpha 1 subunit. Mol. Pharmacol. 46, 858–865.
Caragea, C., Sinapov, J., Silvescu, A., Dobbs, D., and Honavar, V. (2007). Glycosylation site prediction using ensembles of support vector machine classifiers. BMC Bioinformatics 8, 438.
Chang, C., Holtzman, D. A., Chau, S., Chickering, T., Woolf, E. A., Holmgren, L. M., Bodorova, J., Gearing, D. P., Holmes, W. E., and Brivanlou, A. H. (2001). Twisted gastrulation can function as a BMP antagonist. Nature 410, 483–487.
Ekanayake, S., and Hall, B. K. (1997). The in vivo and in vitro effects of bone morphogenetic protein-2 on the development of the chick mandible. Int. J. Dev. Biol. 41, 67–81.
Gavel, Y., and Von Heijne, G. (1990). Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3, 433–442.
Gazzerro, E., Deregowski, V., Stadmeyer, L., Gale, N. W., Economides, A. N., and Canalis, E. (2006). Twisted gastrulation, a bone morphogenetic protein agonist/antagonist, is not required for post-natal skeletal function. Bone 39, 1252–1260.
Groppe, J., Greenwald, J., Wiater, E., Rodriguez-Leon, J., Economides, A. N., Kwiatkowski, W., Affolter, M., Vale, W. W., Belmonte, J. C., and Choe, S. (2002). Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 420, 636–642.
Gumienny, T. L., and Padgett, R. W. (2002). The other side of TGF-beta superfamily signal regulation: thinking outside the cell. Trends Endocrinol. Metab. 13, 295–299.
Heinecke, K., Seher, A., Schmitz, W., Mueller, T. D., Sebald, W., and Nickel, J. (2009). Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC Biol. 7, 59.
Henderson, G. E., Isett, K. D., and Gerngross, T. U. (2011). Site-specific modification of recombinant proteins: a novel platform for modifying glycoproteins expressed in E. coli. Bioconjug. Chem. 22, 903–912.
Jarvis, D. L., Kawar, Z. S., and Hollister, J. R. (1998). Engineering N-glycosylation pathways in the baculovirus-insect cell system. Curr. Opin. Biotechnol. 9, 528–533.
Jaskoll, T., Abichaker, G., Witcher, D., Sala, F. G., Bellusci, S., Hajihosseini, M. K., and Melnick, M. (2005). FGF10/FGFR2b signaling plays essential roles during in vivo embryonic submandibular salivary gland morphogenesis. BMC Dev. Biol. 5, 11.
Kamimura, M., Matsumoto, K., Koshiba-Takeuchi, K., and Ogura, T. (2004). Vertebrate crossveinless 2 is secreted and acts as an extracellular modulator of the BMP signaling cascade. Dev. Dyn. 230, 434–445.
Larrain, J., Oelgeschlager, M., Ketpura, N. I., Reversade, B., Zakin, L., and De Robertis, E. M. (2001). Proteolytic cleavage of chordin as a switch for the dual activities of Twisted gastrulation in BMP signaling. Development 128, 4439–4447.
Lindenthal, C., and Elsinghorst, E. A. (1999). Identification of a glycoprotein produced by enterotoxigenic Escherichia coli. Infect. Immun. 67, 4084–4091.
Mackenzie, A., Ferguson, M. W., and Sharpe, P. T. (1992). Expression patterns of the homeobox gene, Hox-8, in the mouse embryo suggest a role in specifying tooth initiation and shape. Development 115, 403–420.
Mackenzie, B., Wolff, R., Lowe, N., Billington, C. J., Peterson, A., Schmidt, B., Graf, D., Mina, M., Gopalakrishnan, R., and Petryk, A. (2009). Twisted gastrulation limits apoptosis in the distal region of the mandibular arch in mice. Dev. Biol. 328, 13–23.
Mason, E. D., Konrad, K. D., Webb, C. D., and Marsh, J. L. (1994). Dorsal midline fate in Drosophila embryos requires Twisted gastrulation, a gene encoding a secreted protein related to human connective tissue growth factor. Genes Dev. 8, 1489–1501.
Melnick, M., Witcher, D., Bringas, P. Jr., Carlsson, P., and Jaskoll, T. (2005). Meckel’s cartilage differentiation is dependent on hedgehog signaling. Cells Tissues Organs 179, 146–157.
Mina, M., Wang, Y. H., Ivanisevic, A. M., Upholt, W. B., and Rodgers, B. (2002). Region- and stage-specific effects of FGFs and BMPs in chick mandibular morphogenesis. Dev. Dyn. 223, 333–352.
Neuberger, A., and Marshall, R. D. (1968). “Aspects of the structure of glycoproteins,” in Symposium on Foods – Carbohydrates and their Roles, eds H. W. Schultz, R. F. Cain, and R. W. Wrolstad (Westport, CT: Avi Publishing Co.), 115–132.
Nosaka, T., Morita, S., Kitamura, H., Nakajima, H., Shibata, F., Morikawa, Y., Kataoka, Y., Ebihara, Y., Kawashima, T., Itoh, T., Ozaki, K., Senba, E., Tsuji, K., Makishima, F., Yoshida, N., and Kitamura, T. (2003). Mammalian Twisted gastrulation is essential for skeleto-lymphogenesis. Mol. Cell. Biol. 23, 2969–2980.
Oelgeschlager, M., Larrain, J., Geissert, D., and De Robertis, E. M. (2000). The evolutionarily conserved BMP-binding protein Twisted gastrulation promotes BMP signalling. Nature 405, 757–763.
Oelgeschlager, M., Reversade, B., Larrain, J., Little, S., Mullins, M. C., and De Robertis, E. M. (2003). The pro-BMP activity of Twisted gastrulation is independent of BMP binding. Development 130, 4047–4056.
Oelgeschlager, M., Tran, U., Grubisic, K., and De Robertis, E. M. (2004). Identification of a second Xenopus Twisted gastrulation gene. Int. J. Dev. Biol. 48, 57–61.
Petryk, A., Anderson, R., Jarcho, M. P., Leaf, H., Carlson, C. S., Klingensmith, J., Shawlot, W., and O’connor, M. B. (2004). The mammalian Twisted gastrulation gene functions in foregut and craniofacial development. Dev. Biol. 267, 374–386.
Ross, J. J., Shimmi, O., Vilmos, P., Petryk, A., Kim, H., Gaudenz, K., Hermanson, S., Ekker, S. C., O’connor, M. B., and Marsh, J. L. (2001). Twisted gastrulation is a conserved extracellular BMP antagonist. Nature 410, 479–483.
Ruppert, R., Hoffmann, E., and Sebald, W. (1996). Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur. J. Biochem. 237, 295–302.
Saremba, S., Nickel, J., Seher, A., Kotzsch, A., Sebald, W., and Mueller, T. D. (2008). Type I receptor binding of bone morphogenetic protein 6 is dependent on N-glycosylation of the ligand. FEBS J. 275, 172–183.
Sasaki, H., and Hogan, B. L. (1993). Differential expression of multiple fork head related genes during gastrulation and axial pattern formation in the mouse embryo. Development 118, 47–59.
Scott, I. C., Blitz, I. L., Pappano, W. N., Maas, S. A., Cho, K. W., and Greenspan, D. S. (2001). Homologues of Twisted gastrulation are extracellular cofactors in antagonism of BMP signalling. Nature 410, 475–478.
Semba, I., Nonaka, K., Takahashi, I., Takahashi, K., Dashner, R., Shum, L., Nuckolls, G. H., and Slavkin, H. C. (2000). Positionally-dependent chondrogenesis induced by BMP4 is co-regulated by Sox9 and Msx2. Dev. Dyn. 217, 401–414.
Spiro, R. G. (2002). Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R–56R.
Stanley, E., Biben, C., Kotecha, S., Fabri, L., Tajbakhsh, S., Wang, C. C., Hatzistavrou, T., Roberts, B., Drinkwater, C., Lah, M., Buckingham, M., Hilton, D., Nash, A., Mohun, T., and Harvey, R. P. (1998). DAN is a secreted glycoprotein related to Xenopus cerberus. Mech. Dev. 77, 173–184.
Tomiya, N., Narang, S., Lee, Y. C., and Betenbaugh, M. J. (2004). Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines. Glycoconj. J. 21, 343–360.
Tucker, A. S., Matthews, K. L., and Sharpe, P. T. (1998). Transformation of tooth type induced by inhibition of BMP signaling. Science 282, 1136–1138.
Vilmos, P., Gaudenz, K., Hegedus, Z., and Marsh, J. L. (2001). The Twisted gastrulation family of proteins, together with the IGFBP and CCN families, comprise the TIC superfamily of cysteine rich secreted factors. Mol. Pathol. 54, 317–323.
Wacker, M., Linton, D., Hitchen, P. G., Nita-Lazar, M., Haslam, S. M., North, S. J., Panico, M., Morris, H. R., Dell, A., Wren, B. W., and Aebi, M. (2002). N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793.
Walsh, G., and Jefferis, R. (2006). Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 24, 1241–1252.
Westmoreland, J. J., Takahashi, S., and Wright, C. V. (2007). Xenopus lefty requires proprotein cleavage but not N-linked glycosylation to inhibit nodal signaling. Dev. Dyn. 236, 2050–2061.
Wu, Y. I., Munshi, H. G., Sen, R., Snipas, S. J., Salvesen, G. S., Fridman, R., and Stack, M. S. (2004). Glycosylation broadens the substrate profile of membrane type 1 matrix metalloproteinase. J. Biol. Chem. 279, 8278–8289.
Yamaguchi, K., Akai, K., Kawanishi, G., Ueda, M., Masuda, S., and Sasaki, R. (1991). Effects of site-directed removal of N-glycosylation sites in human erythropoietin on its production and biological properties. J. Biol. Chem. 266, 20434–20439.
Zakin, L., and De Robertis, E. M. (2004). Inactivation of mouse Twisted gastrulation reveals its role in promoting Bmp4 activity during forebrain development. Development 131, 413–424.
Keywords: Twisted gastrulation, BMP, glycosylation, mandibular explants, Msx2, surface plasmon resonance analysis
Citation: Billington CJ Jr., Fiebig JE, Forsman CL, Pham L, Burbach N, Sun M, Jaskoll T, Mansky K, Gopalakrishnan R, O’Connor MB, Mueller TD and Petryk A (2011) Glycosylation of Twisted gastrulation is required for BMP binding and activity during craniofacial development. Front. Physio. 2:59. doi: 10.3389/fphys.2011.00059
Received: 02 July 2011; Paper pending published: 31 July 2011;
Accepted: 23 August 2011; Published online: 12 September 2011.
Edited by:
Christopher Evans, Harvard Medical School, USAReviewed by:
Ugo Ripamonti, University of the Witwatersrand, South AfricaJavier Catón, Universidad Complutense de Madrid, Spain
Copyright: © 2011 Jr., Fiebig, Forsman, Pham, Burbach, Sun, Jaskoll, Mansky, Gopalakrishnan, O’Connor, Mueller and Petryk. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Anna Petryk, Pediatric Endocrinology, University of Minnesota, East Building Room MB671, 2450 Riverside Avenue, Minneapolis, MN 55454, USA. e-mail: petry005@umn.edu