 Jianchao Zhang Vikram Shettigar George C. Zhang Daniel G. Kindell Xiaotong Liu Joseph J. López Vinatham Yerrimuni Grace A. Davis Jonathan P. Davis*
Jianchao Zhang Vikram Shettigar George C. Zhang Daniel G. Kindell Xiaotong Liu Joseph J. López Vinatham Yerrimuni Grace A. Davis Jonathan P. Davis*- Department of Physiology and Cell Biology, The Ohio State University, Columbus, OH, USA
Parvalbumin (PV), an EF-hand protein family member, is a delayed calcium buffer that exchanges magnesium for calcium to facilitate fast skeletal muscle relaxation. Genetic approaches that express parvalbumin in the heart also enhance relaxation and show promise of being therapeutic against various cardiac diseases where relaxation is compromised. Unfortunately, skeletal muscle PVs have very slow rates of Ca2+ dissociation and are prone to becoming saturated with Ca2+, eventually losing their buffering capability within the constantly beating heart. In order for PV to have a more therapeutic potential in the heart, a PV with faster rates of calcium dissociation and high Mg2+ affinity is needed. We demonstrate that at 35°C, rat β-PV has an ~30-fold faster rate of Ca2+ dissociation compared to rat skeletal muscle α-PV, and still possesses a physiologically relevant Ca2+ affinity (~100 nM). However, rat β-PV will not be a delayed Ca2+ buffer since its Mg2+ affinity is too low (~1 mM). We have engineered two mutations into rat β-PV, S55D and E62D, when observed alone increase Mg2+ affinity up to fivefold, but when combined increase Mg2+ affinity ~13-fold, well within a physiologically relevant affinity. Furthermore, the Mg2+ dissociation rate (172/s) from the engineered S55D, E62D PV is slow enough for delayed Ca2+ buffering. Additionally, the engineered PV retains a high Ca2+ affinity (132 nM) and fast rate of Ca2+ dissociation (64/s). These PV design strategies hold promise for the development of new therapies to remediate relaxation abnormalities in different heart diseases and heart failure.
Introduction
Diastolic dysfunction, the inability of the heart to properly relax, is a hallmark of many heart diseases and heart failure (Periasamy and Janssen, 2008). During this condition, it is generally thought that the cardiac myocyte loses the ability to efficiently and effectively manage intracellular Ca2+, prolonging relaxation (Bers, 2006; van der Velden, 2011). Attempts to restore the Ca2+ balance show promise of alleviating the symptoms of this debilitating cardiac condition (Wang et al., 2009; Gwathmey et al., 2011; McCauley and Wehrens, 2011; Rohde et al., 2011).
One novel approach to counter the Ca2+ imbalance has been borrowed from a specialized mechanism in fast twitch skeletal muscle that aids in relaxation. This mechanism utilizes a protein called parvalbumin (PV) to achieve faster relaxation in combination with the sarcoplasmic reticulum Ca2+ ATPase (Hou et al., 1993). PV is a small, cytosolic Ca2+ buffering protein found in high concentrations within fast-relaxing muscle ranging in species from fish to humans (Heizmann et al., 1982; Wilwert et al., 2006). Skeletal muscle PV binds both Ca2+ and Mg2+ competitively, typically with a Ca2+ affinity three to four orders of magnitude greater than  ;
;  ; Pauls et al., 1993; Eberhard and Erne, 1994). In a resting muscle, the free concentration of Ca2+ is very low (~100 nM), while the free Mg2+ concentration is very high (~1 mM; Williams, 1993). Thus, in a relaxed muscle, PV is bound with Mg2+ and cannot bind Ca2+ until Mg2+ dissociates (Hou et al., 1991). The Mg2+ dissociation rate from PV is quite slow (less than 10/s) making PV a delayed Ca2+ buffer, allowing troponin C to bind Ca2+ and initiate contraction before PV exchanges Mg2+ for Ca2+ to facilitate relaxation (Hou et al., 1992). The inferred physiological Ca2+ and Mg2+ exchange rates are nearly identical to those measured biochemically (Hou et al., 1993; Jiang et al., 1996; Lee et al., 2000).
; Pauls et al., 1993; Eberhard and Erne, 1994). In a resting muscle, the free concentration of Ca2+ is very low (~100 nM), while the free Mg2+ concentration is very high (~1 mM; Williams, 1993). Thus, in a relaxed muscle, PV is bound with Mg2+ and cannot bind Ca2+ until Mg2+ dissociates (Hou et al., 1991). The Mg2+ dissociation rate from PV is quite slow (less than 10/s) making PV a delayed Ca2+ buffer, allowing troponin C to bind Ca2+ and initiate contraction before PV exchanges Mg2+ for Ca2+ to facilitate relaxation (Hou et al., 1992). The inferred physiological Ca2+ and Mg2+ exchange rates are nearly identical to those measured biochemically (Hou et al., 1993; Jiang et al., 1996; Lee et al., 2000).
Naturally, PV is not typically expressed in the heart (Szatkowski et al., 2001). Due to its delayed Ca2+ buffer capability and ATP-independent mechanism, PV has a potential to be used therapeutically in the heart (Raake et al., 2011). In fact, in vitro and in vivo gene transfer of PV into the cardiac myocyte has been shown to increase the rate and extent of relaxation in normal and diseased states (Wang et al., 2009). However, with increasing frequency of contraction, PV loses its Ca2+ buffering potential (therefore losing its enhanced relaxing properties) due to its slow rate of Ca2+ dissociation (less than 3/s; Hou et al., 1992; Day et al., 2008). In order to recharge PV’s relaxing capability, the muscle must rest to give time for PV to re-exchange Ca2+ for Mg2+. Unlike skeletal muscle, cardiac muscle does not have the liberty to rest for prolonged periods of time.
One potential way to overcome this inherent problem is to use a PV that has a faster rate of Ca2+ dissociation with a high affinity for Mg2+. There are two isoforms of PV found in nature, α-PV and β-PV (Arif, 2009). Mammals utilize α-PV, while fish utilize β-PV in their skeletal muscle. The PVs from both mammalian and fish skeletal muscle have relatively similar cation binding properties, especially with regard to possessing slow rates of Ca2+ dissociation (White, 1988; Eberhard and Erne, 1994; Lee et al., 2000; Erickson and Moerland, 2006). On the other hand, mammalian β-PV, which is found in the brain, ear, placenta, and macrophages [not normally found in muscle (Belkacemi et al., 2002; Yin et al., 2006; Csillik et al., 2010)] has drastically lower affinity for both Ca2+ and Mg2+ compared to mammalian α-PV (Hapak et al., 1989). The rates of Ca2+ and Mg2+ exchange from mammalian β-PV are currently unknown and will be addressed in this manuscript. In any regard, mammalian β-PV still possesses a high enough Ca2+ affinity (~100 nM) to buffer Ca2+ in the heart. However, due to its very low Mg2+ affinity (greater than 1 mM), much of the PV will not be bound by Mg2+. In this case, mammalian β-PV will not actually be a delayed Ca2+ buffer and will compromise force production in the heart. Thus, we set out to engineer a higher Mg2+ affinity β-PV (while maintaining its Ca2+ affinity) that should function properly in the heart.
A great deal of work has been put into understanding the molecular mechanisms that control Ca2+ and Mg2+ binding to PV. For instance, Henzl et al. (1996) have previously shown that replacing one of the Ca2+ chelating residues in rat β-PV, Ser 55 with Asp, greatly increased Mg2+ affinity at room temperature using the flow dialysis method. In this manuscript, we have utilized this modification and a novel mutation, Glu 62 Asp, to engineer a mammalian β-PV that has appropriate affinities for both Ca2+ and Mg2+, as well as fast enough exchange kinetics, to potentially be a beneficial Ca2+ buffer in the constantly beating heart. This work represents the first step in designing a PV for the heart.
Materials and Methods
Materials
DEAE Sepharose™ Fast Flow was purchased from GE Healthcare (Piscataway, NJ, USA). Quin-2 was purchased from Molecular Probes (Eugene, OR, USA). MOPS, Ethidium Bromide, and EGTA were purchased from Sigma Chemical Company (St. Louis, MO, USA). All other chemicals were of analytical grade.
Protein Over-Expression, Purification, and Mutagenesis
All DNA manipulations were performed using standard molecular biology techniques (Sambrook and Rusell, 1989). Plasmids containing rat α- and β-PV were generous gifts from Dr. Michael Henzl (University of Missouri). The two rat PV coding sequences were individually sub-cloned into the over-expression vector Pet3b (kindly provided by Dr. Brandon Biesiadecki, The Ohio State University) by PCR to produce Pet3b/α-PV and Pet3b/β-PV.
Conditions for purification of the two proteins were optimized based on an existing protocol with the following modifications (Hapak et al., 1989). For α-PV, the Pet3b/α-PV plasmid was transformed into BL21(DE3) bacteria (Novagen). A single colony was grown to inoculate 1 l LB/Ampicillin media and 1 mM IPTG was added after the OD600 of the culture was greater than 1.0. After 4 h, the cells were harvested by centrifugation. The cell pellet was resuspended in 25 ml of resuspension buffer (20 mM MOPS, 240 mM KCl, 2 mM EDTA, 1 mM DTT, pH 7.4) containing 1 mM PMSF and the cells were broken by sonication. The cell lysate was clarified by centrifugation. Mg2+ and Ca2+ were then added to the lysate to a final concentration of 10 and 1 mM, respectively. Ammonium sulfate (AMS) fractionation to 100% was performed on the cell lysate. The supernatant was dialyzed against three, 4 l of buffer A (1 mM MOPS, 1 mM EDTA, pH 7.4). After dialysis, the supernatant was loaded onto a DEAE column equilibrated with buffer A and then washed with buffer A for at least 30 min at 1.5 ml/min. The protein was eluted with a linear Ca2+ gradient from 0 to 50 mM Ca2+ prepared in buffer A using a Bio-Rad Econo Gradient Pump running at 0.75 ml/min for a total 260 min. Fractions of the flow through were collected. OD280 of each fraction was measured and SDS-PAGE was run for the fractions that had OD280 greater than 0.3. In order to ensure nucleic acid contamination was separate from the protein fractions, the stained gel was soaked in TAE buffer (40 mM Tris–Acetate, 1 mM EDTA) and re-stained with Ethidium Bromide (~0.25 μg/ml). After 1 h staining, the gel was washed with the TAE buffer and pictures of the gel were taken under UV light. Protein samples from fractions containing the correct molecular weight band, with negligible protein or nucleic acid contamination, were pooled. The pooled protein fractions were dialyzed against three, 4 l experimental buffer (10 mM MOPS, 150 mM KCl, 1 mM DTT, pH 7.0). Protein stock concentration was calculated by OD280 using an extinction coefficient predicted by the web based program, Protein Calculator v3.3, according to its protein sequence (http://www.scripps.edu/~cdputnam/protcalc.html). Typically, 80–120 mg of PV per liter was purified.
For β-PV, a similar procedure was performed with the following changes. The supernatant after 100% AMS treatment was dialyzed against buffer B (20 mM MOPS, 2 mM DTT, pH 7.4). After dialysis, the supernatant was loaded onto a DEAE column equilibrated with buffer B. The β-PV protein was eluted with a linear salt gradient from 0 to 0.6 M NaCl prepared in buffer B.
All site-directed mutagenesis was performed using Stratagene’s Quick-Change Site-Directed Mutagenesis Kit (La Jolla, CA, USA). β-PV has one solvent-exposed Cys residue at position 18. To avoid possible oxidation and dimerization of β-PV during purification and storage, a C18S mutation was introduced. All the F102W β-PV constructs contained C18S and is not included in the nomenclature throughout the manuscript. As has been previously used to facilitate the steady-state and kinetic studies of Ca2+ and Mg2+ binding, Phe 102 was replaced by Trp (F102W) in both α-PV and β-PV (Hutnik et al., 1990; Pauls et al., 1993). In order to increase the Mg2+ affinity of β-PV, the single mutations S55D, E62D, and double mutations (S55D, E62D) were made. All mutations were confirmed by DNA sequence analysis and were expressed and purified the same way as described above. The numbering scheme for the rat PVs utilized in this manuscript does not include the initial Met residue.
Determination of Ca2+ and Mg2+ Affinities
All steady-state fluorescence measurements were performed using a Perkin-Elmer LS55B Spectrofluorimeter at 35°C. This temperature was utilized for direct comparison to previous studies and is very close to that of mammalian body temperature (Eberhard and Erne, 1994). Ca2+ and Mg2+ titrations were performed by adding microliter amounts of CaCl2 or MgCl2 to 2 ml of the proteins (1 μM) in 200 mM MOPS, 150 mM KCl, 4 mM EGTA, 1 mM DTT, pH 7.0 with constant stirring. The [Ca2+]free and [Mg2+]free at 35°C were calculated using the computer program EGCA02 developed by Robertson and Potter (1984). Trp fluorescence was excited at 295 nm and measured at 330 nm. The Ca2+ and Mg2+ affinities are reported as dissociation constants [Kd(Ca)] and [Kd(Mg)], respectively. Each [Kd(Ca)] or [Kd(Mg)] represents a mean of at least three titrations fit with a logistic sigmoid function mathematically equivalent to the Hill equation, as previously described (Tikunova et al., 2002).
Determination of Ca2+ and Mg2+ Dissociation Kinetics
Ca2+ and Mg2+ dissociation rates were measured using an Applied Photophysics Ltd. (Leatherhead, UK) model SX.18 MV stopped-flow instrument at 35°C. Trp fluorescence was excited using a 150-W xenon arc source excited at 295 nm with emission monitored through a narrow band-pass filter centered at 334 nm (Oriel, Stratford, CT, USA). Direct Ca2+ dissociation rates were also measured using the fluorescent Ca2+ chelator Quin-2 (Tikunova et al., 2002; Davis et al., 2004). Quin-2 was excited at 330 nm with its emission monitored through a 510-nm broad band-pass interference filter (Oriel, Stratford, CT, USA). The buffer used for the stopped-flow experiments was 10 mM MOPS, 150 mM KCl, 1 mM DTT, at pH 7.0. To measure the kinetics of Ca2+ dissociation from PV, 10 μM Ca2+ was equilibrated with 5 μM protein and rapidly mixed with buffer containing 30 mM EDTA. To measure the kinetics of Mg2+ dissociation from PV, 500 μM Mg2+ and 5 mM EGTA (to remove contaminating Ca2+) were equilibrated with 5 μM protein and rapidly mixed with buffer contain 30 mM EDTA. For the Quin-2 studies, there was enough contaminating Ca2+ in the buffer to observe the Ca2+ dissociation rates when 6 μM protein was rapidly mixed with the buffer containing 150 μM Quin-2.
Data Analysis and Statistics
Statistical significance was determined by ANOVA followed by a Dunnett’s post hoc t-test, using the statistical analysis software Minitab (State College, PA, USA). Two means were considered to be significantly different when the P value was <0.05. The data is shown as a mean value ± SEM.
Results
PV is an unusually stable protein, especially in the presence of Ca2+ and/or Mg2+ (Filimonov et al., 1978). In order to simplify the purification protocol for PV, we speculated that unlike other proteins, it would not denature and precipitate in 100% saturating AMS when in the presence of Ca2+ and Mg2+. Consistent with this idea, 100% AMS saturation precipitated nearly all the bacterial proteins, leaving PV and nucleic acids in the supernatant as judged by Coomassie Brilliant Blue and Ethidium Bromide staining as can be seen in Figures 1A,B (lane 1, data shown for E62D F102W β-PV). The contaminating nucleic acids (lanes 8 through 15) were then easily removed from the PV (lanes 2 through 7) by a single DEAE chromatography step (Figures 1A,B). Similar results were obtained with all the PVs used in this study (data not shown). Thus, we were able to quickly and efficiently purify an abundance of the wild-type and mutant PVs with very high purity for the following studies.
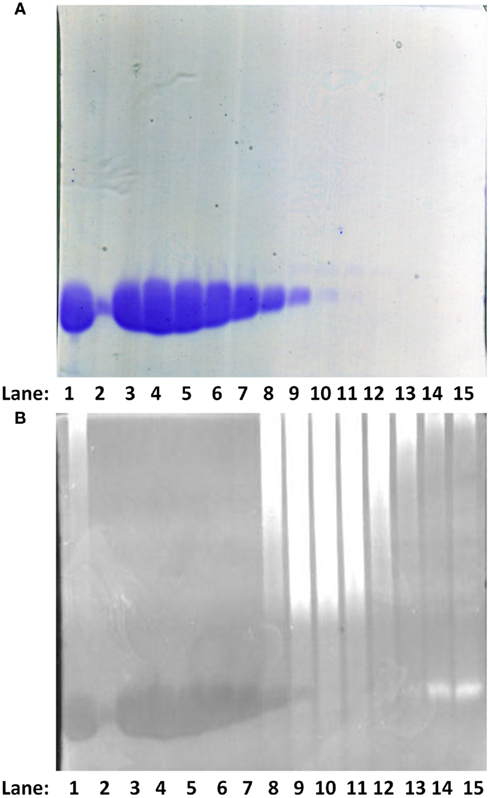
Figure 1. Purification of PV. (A) SDS-PAGE analysis of the DEAE column purification profile for the E62D, F102W β-PV. The gel was stained with Coomassie Brilliant Blue. Lane 1: a positive control sample from rat F102W β-PV, which was characterized before the nucleic acid contamination issue was resolved. Lanes 2–15 correspond to elution fractions from 21 to 35 after the DEAE column (refer to Materials and Method). (B) the same SDS-PAGE from (A), but was re-stained with Ethidium Bromide and viewed with UV light (refer to Materials and Methods).
There is ample evidence that PV increases the relaxation rate of skeletal muscle (Hou et al., 1991, 1993). Gene transfer of both skeletal muscle α-PV and β-PV has been shown to do the same in cardiac myocytes (Rodenbaugh et al., 2007). However, the skeletal muscle PVs are not designed to work in a muscle that constantly contracts and relaxes and will eventually saturate with Ca2+ (Hou et al., 1993; Szatkowski et al., 2001). Similar to previous studies (Eberhard and Erne, 1994), Figures 2A,B demonstrate that rat skeletal muscle F102W α-PV binds Ca2+ with a Kd(Ca) of 1.9 ± 0.4 nM and Mg2+ with a Kd(Mg) of 26 ± 2 μM at 35°C (Table 1). Also similar to previous studies (Hapak et al., 1989), Figures 2A,B show that F102W β-PV binds Ca2+ ~49-fold weaker and Mg2+ ~35-fold weaker than F102W α-PV (Table 1). Although the Ca2+ and Mg2+ affinities of F102W β-PV are within the physiological range for these cations, the Mg2+ affinity of F102W β-PV is too weak to be a useful delayed Ca2+ buffer in the heart.
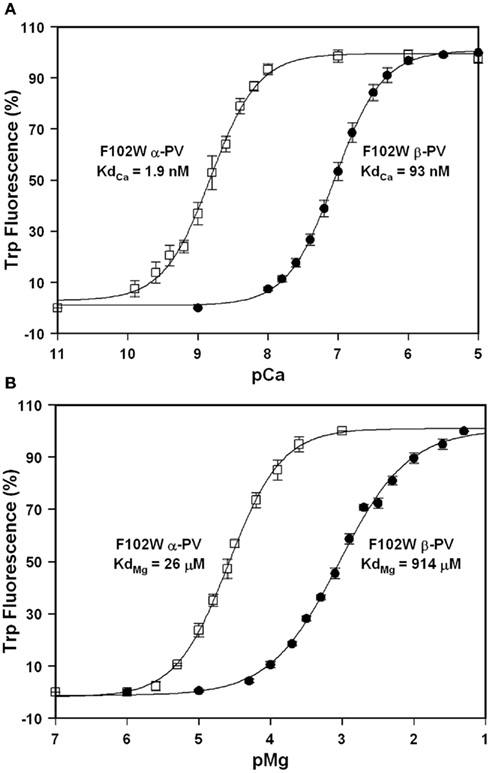
Figure 2. Ca2+ and Mg2+ binding to the F102W α-PV and F102W β-PV. (A) The Ca2+ dependent increase in Trp fluorescence is shown as a function of −Log[Ca2+] (pCa) for F102W α-PV (□) and F102W β-PV (•). Increasing concentrations of Ca2+ were added to 1 μM protein in 2 ml of 200 mM MOPS, 150 mM KCl, 4 mM EGTA, pH 7.0 at 35°C. Trp fluorescence was monitored at 330 nm with excitation at 295 nm. Each data point represents the mean ± SE of at least three titrations. (B) The Mg2+ dependent increase in Trp fluorescence is shown as a function of −Log[Mg2+] (pMg) for F102W α-PV (□) and F102W β-PV (•). The experimental conditions were the same as described for (A).
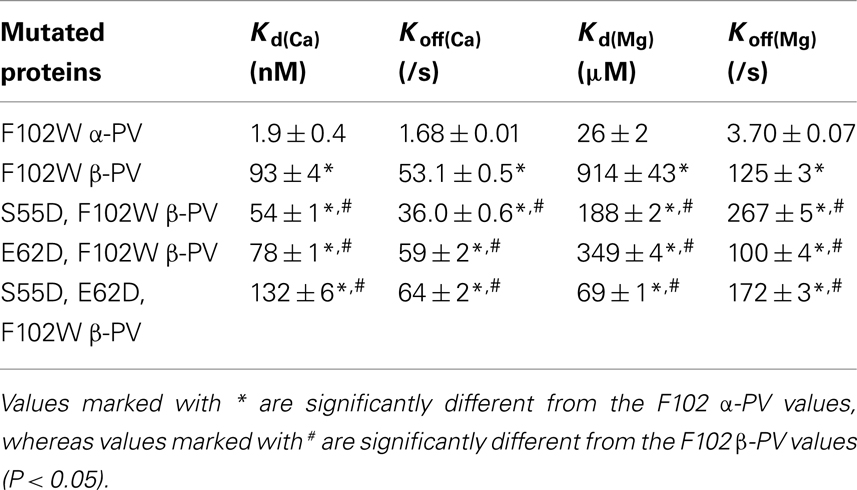
Table 1. Summary of the Ca2 + and Mg2 + binding properties of the modified PVs.
In addition to their extremely high Ca2+ affinity, another reason why the skeletal muscle PVs saturate with Ca2+ upon repeated or prolonged contraction is their slow rates of Ca2+ dissociation (Hou et al., 1992; Day et al., 2008). Consistent with previous studies (Lee et al., 2000), Figure 3A shows that the rate of Ca2+ dissociation from F102W α-PV is 1.68 ± 0.01/s at 35°C. Consistent with its weaker Ca2+ affinity, Figure 3A shows that F102W β-PV has an ~32-fold faster rate of Ca2+ dissociation compared to F102W α-PV. The Ca2+ dissociation rates reported by Trp were nearly identical to those measured with Quin-2 (data not shown), suggesting that the change in F102W fluorescence follows cation binding. Neither the F102W nor C18S mutations affected the rates of Ca2+ dissociation from the PVs as measured by Quin-2 (data not shown).
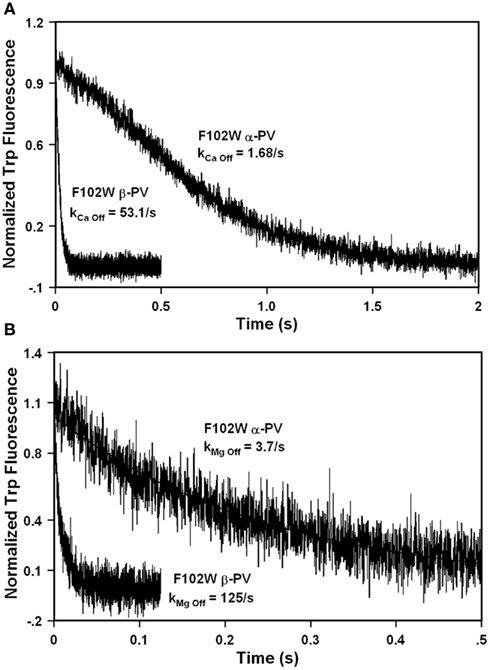
Figure 3. Rates of Ca2+ and Mg2+ dissociation from F102W α-P and F102W β-PV. (A) The time course of Trp fluorescence is shown as EDTA rapidly chelates Ca2+ causing dissociation of Ca2+ from F102W α-PV and F102W β-PV. Each protein (5 μM) in 10 mM MOPS, 150 mM KCl, 10 μM Ca2+, pH 7.0 at 35°C was rapidly mixed with equal volume of 30 mM EDTA in 10 mM MOPS, 150 mM KCl, pH 7.0. (B) The time course of Trp fluorescence is shown as EDTA rapidly chelates Mg2+ causing dissociation of Mg2+ from F102W α-PV and F102W β-PV. Each protein (5 μM) in 10 mM MOPS, 150 mM KCl, 5 mM EGTA, 500 μM Mg2+, pH 7.0 at 35°C was rapidly mixed with equal volume of 30 mM EDTA in 10 mM MOPS, 150 mM KCl, pH 7.0. Trp fluorescence was monitored through a narrow band-pass filter centered at 334 nm with an excitation wavelength of 295 nm. Each trace is an average of at least five traces fit with a single exponential equation. All kinetic traces were triggered at time zero.
One reason why skeletal muscle PV does not interfere with the initial, nearly diffusion controlled, binding of Ca2+ to TnC is its delayed Ca2+ binding due to its slow rate of Mg2+ dissociation (Hou et al., 1992). Similar to these findings, Figure 3B shows that Mg2+ dissociates from F102W α-PV at 3.70 ± 0.07/s (Table 1). Consistent with its weaker Mg2+ affinity, Figure 3B shows that F102W β-PV has an ~34-fold faster rate of Mg2+ dissociation compared to F102W α-PV (Table 1). Thus, if F102W β-PV is bound by Mg2+, it will still have a relatively slow rate of Ca2+ association compared to the nearly diffusion controlled rate of Ca2+ binding to TnC. However, due to its low Mg2+ affinity, much of F102W β-PV would not be bound by Mg2+ and thus would actually not be a delayed Ca2+ buffer.
Previously, Henzl et al. (1996) demonstrated that the S55D mutation in rat β-PV modestly increased the Ca2+ affinity, but drastically increased the Mg2+ affinity of the protein. This type of EF-hand chelating residue modification is thought to make the cation binding pocket smaller and bring a negatively charged ligand closer to the bound cation (Davis et al., 2002). Consistent with the previous findings and theory, Figure 4A shows that the S55D mutation in F102W β-PV increased the Ca2+ affinity ~1.7-fold (Table 1). Figure 4B demonstrates that the S55D mutation in F102W β-PV also slowed the rate of Ca2+ dissociation ~1.5-fold (Table 1). Similarly, the S55D mutation increased the Mg2+ affinity of F102W β-PV ~fivefold (Figure 4C; Table 1), but also increased the rate of Mg2+ dissociation ~twofold (Figure 4D; Table 1). Thus, the S55D mutation begins to bring the Mg2+ affinity of rat β-PV within a physiological range to make it a delayed Ca2+ buffer.
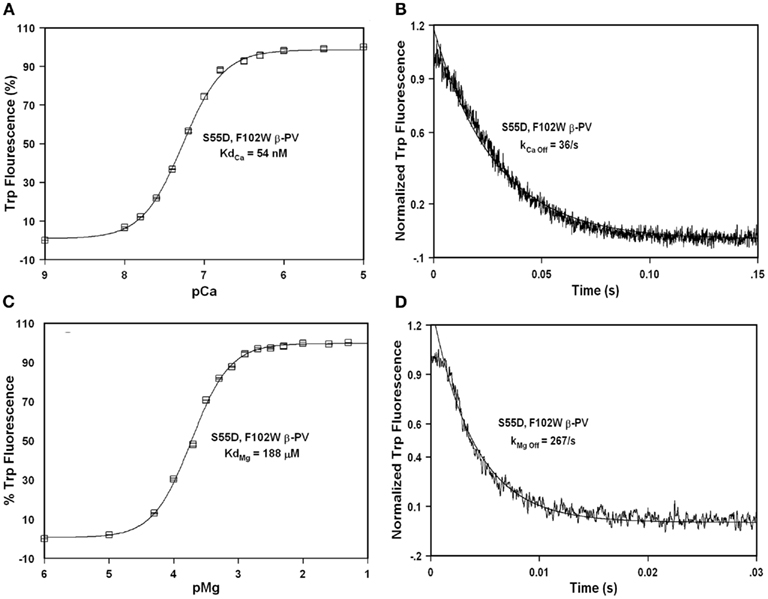
Figure 4. Ca2+ and Mg2+ binding and dissociation from S55D, F102W β-PV. (A) The Ca2+ dependent increase in Trp fluorescence is shown as a function of −Log[Ca2+] (pCa) for S55D, F102W β-PV. (B) The time course of Trp fluorescence is shown as EDTA rapidly chelates Ca2+ causing dissociation of Ca2+ from S55D, F102W β-PV. (C) The Mg2+ dependent increase in Trp fluorescence is shown as a function of −Log[Mg2+] (pMg) for S55D, F102W β-PV. (D) The time course of Trp fluorescence is shown as EDTA rapidly chelates Mg2+ causing dissociation of Mg2+ from S55D, F102W β-PV. All the measurements were performed as previously mentioned in the legends of Figures 2 and 3.
The EF-hand −Z chelating residue is primarily Glu, but is Asp in the sarcoplasmic calcium-binding protein from Nereis diversicolor (Vijay-Kumar and Cook, 1992). This particular EF-hand has a smaller cation binding pocket more preferable for Mg2+ binding. Consistent with this idea substitution of the −Z chelating ligand from Glu to Asp in carp β-PV increased Mg2+ affinity ~10-fold, but also decreased the Ca2+ affinity ~100-fold (Cates et al., 1999). We speculated that if we mutated the −Z Glu at position 62 with Asp we might increase the Mg2+ affinity of rat β-PV without influencing Ca2+ binding since the rat β-PV already has a weaker Ca2+ affinity. Consistent with this idea, the E62D mutation in F102W β-PV actually modestly increased the Ca2+ affinity ~1.2-fold (Figure 5A; Table 1) and slightly increased the Ca2+ dissociation rate ~1.1-fold (Figure 5B; Table 1). Significantly, the E62D mutation increased the Mg2+ affinity of F102W β-PV ~threefold (Figure 5C; Table 1), and decreased the rate of Mg2+ dissociation ~1.3-fold (Figure 5D; Table 1). Thus, the E62D mutation increases the Mg2+ affinity of β-PV (without drastically altering the Ca2+ binding properties) and maintains a relatively slow rate of Mg2+ dissociation.
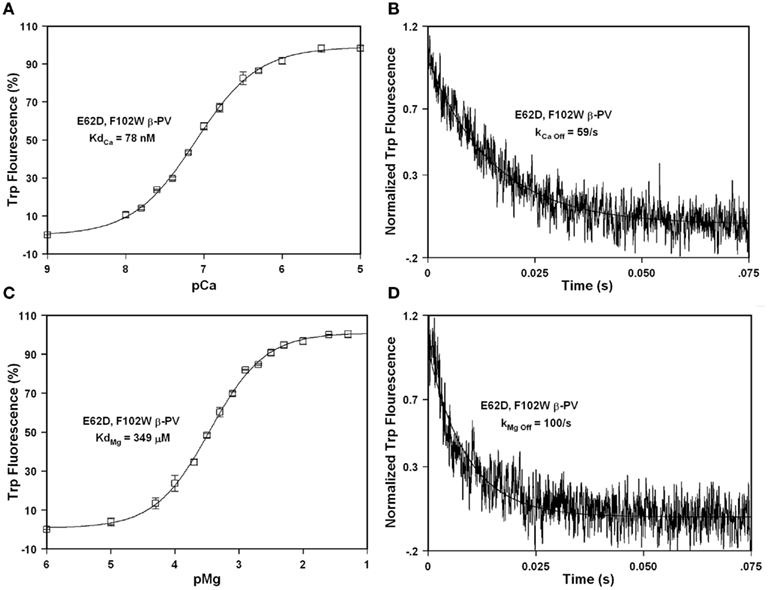
Figure 5. Ca2+ and Mg2+ binding and dissociation from E62D, F102W β-PV. (A) The Ca2+ dependent increase in Trp fluorescence is shown as a function of −Log[Ca2+] (pCa) for E62D, F102W β-PV. (B) The time course of Trp fluorescence is shown as EDTA rapidly chelates Ca2+ causing dissociation of Ca2+ from E62D, F102W β-PV. (C) The Mg2+ dependent increase in Trp fluorescence is shown as a function of −Log[Mg2+] (pMg) for E62D, F102W β-PV. (D) The time course of Trp fluorescence is shown as EDTA rapidly chelates Mg2+ causing dissociation of Mg2+ from E62D, F102W β-PV. All the measurements were performed as previously mentioned in the legends of Figures 2 and 3.
Since the S55D and E62D mutations are thought to increase Mg2+ affinity through different mechanisms (and have little impact on Ca2+ binding), we speculated that the combination of these two mutations might be additive on Mg2+ affinity. Consistent with this idea, Figures 6A,B show that the double mutation S55D, E62D had a minor effect on the Ca2+ affinity or dissociation rate compared to F102W β-PV (~1.4-fold, Table 1). Furthermore, the double mutation increased the Mg2+ affinity ~13-fold with a minor effect on the rate of Mg2+ dissociation (~1.4-fold increase) as compared to F102W β-PV (Figures 6C,D; Table 1). Thus, the double mutation actually had a multiplicative effect of the two single mutations on the Mg2+ binding properties of F102W β-PV. Thus, the S55D, E62D, F102W β-PV now has Ca2+ and Mg2+ sensitivities and kinetics that should make it an ideal Ca2+ buffering protein for the heart.
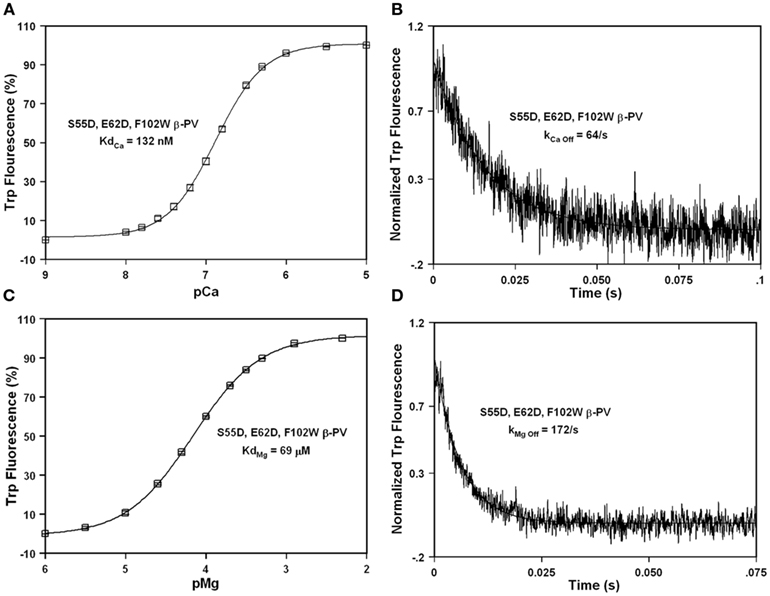
Figure 6. Ca2+ and Mg2+ binding and dissociation from S55D, E62D, F102W β-PV. (A) The Ca2+ dependent increase in Trp fluorescence is shown as a function of −Log[Ca2+] (pCa) for S55D, E62D, F102W β-PV. (B) The time course of Trp fluorescence is shown as EDTA rapidly chelates Ca2+ causing dissociation of Ca2+ from S55D, E62D, F102W β-PV. (C) The Mg2+ dependent increase in Trp fluorescence is shown as a function of −Log[Mg2+] (pMg) for S55D, E62D, F102W β-PV. (D) The time course of Trp fluorescence is shown as EDTA rapidly chelates Mg2+ causing dissociation of Mg2+ from S55D, E62D, F102W β-PV. All the measurements were performed as previously mentioned in the legends of Figures 2 and 3.
Discussion
It is clear that PV functions in skeletal muscle as a delayed Ca2+ buffer to temporarily aid relaxation (Hou et al., 1991). Metzger and co-workers have been the pioneers in studying the potential therapeutic value of using native PVs to help relax cardiac muscle (Rodenbaugh et al., 2007). They have clearly shown the proof of principle that PV can increase the rate and extent of relaxation in healthy and diseased cardiac myocytes in vitro and in vivo, as well as in small and large animal models (Wang et al., 2009). However, the PVs used to date (skeletal muscle rat α-PV and carp β-PV) have similarly slow rates of Ca2+ dissociation and are prone to Ca2+ saturation with repeated contractions, especially at high frequencies of contraction (Szatkowski et al., 2001). These studies strongly suggested that a different PV with modified Mg2+ and/or Ca2+ affinities would be needed to work in the heart. To achieve this goal, we could either explore additional existing PVs with more appropriate cation binding properties or re-engineer an existing well-studied PV.
There are four intrinsic factors of PV that must be considered in order for PV to work in the heart, these include the: Mg2+ affinity, Mg2+ dissociation rate, Ca2+ affinity, and Ca2+ dissociation rate. It is not entirely clear what properties an ideal PV for the heart should possess. Potentially, one could theoretically determine an ideal PV for the heart if there were a reliable mathematical model for cardiac muscle contraction and relaxation (Trayanova and Rice, 2011). In any regard, it should be a delayed Ca2+ buffer, in that it binds Mg2+ with an affinity at least three times lower than its physiological concentration, to ensure there is little to no unbound PV available to rapidly chelate Ca2+. The Mg2+ dissociation rate must also be substantially slower than the rate of Ca2+ binding to TnC, so that the PV is a delayed Ca2+ buffer and will not interfere with force production. The Ca2+ affinity should be high enough so that it is able to out-compete Mg2+ binding during the relaxation phase of the muscle, but not so high that Mg2+ cannot out-compete Ca2+ binding during the resting periods between beats. Finally, the Ca2+ dissociation rate must be fast enough to allow the PV to continuously buffer Ca2+ effectively on a beat-to-beat basis, without becoming saturated with Ca2+. To the best of our knowledge, there is no naturally occurring PV that meets these requirements, but this does not mean one does not exist.
There are hundreds of unique PV sequences in the protein databases that are found in species that live in very diverse climates and environments. Unfortunately, there is no algorithm that can predict the Ca2+ or Mg2+ binding properties of an EF-hand protein based on its protein sequence. Additionally, there are extremely diverse skeletal muscles in these various species that utilize PV to help aid relaxation, some of which can contract and relax over 100 Hz, such as the toadfish swim bladder (yet, for only brief periods of time; Tikunov and Rome, 2009). It is clear from the steady-state sensitivities of temperate and cold-adapted fish, that their PV isoforms are also adapted to function similarly only at their native temperature (Erickson and Moerland, 2006). One of these PVs might have properties that will work in the heart. However, only a small subset of PVs have been characterized for their steady-state Ca2+ and Mg2+ binding properties, and only a handful of these have had their Ca2+ and Mg2+ kinetics measured. So far, all of the characterized PVs have very slow Ca2+ dissociation rates (Ogawa and Tanokura, 1986; Permyakov et al., 1987; White, 1988; Hou et al., 1992; Lee et al., 2000). Therefore, it may take a long time and great effort to find an appropriate natural PV that would work in the heart.
Another way to obtain a PV that might function appropriately in the heart is to re-engineer an existing PV. We have a great deal of experience designing mutations in other EF-hand Ca2+ binding proteins, including calmodulin, cardiac TnC and skeletal TnC, that alter both the steady-state and kinetics of Ca2+ and Mg2+ binding (Tikunova et al., 2001, 2002; Davis et al., 2002, 2004; Tikunova and Davis, 2004). In this manuscript we chose to re-engineer rat β-PV. The reasons for this choice are that rat β-PV has: (1) an intrinsically lower Ca2+ affinity than the skeletal muscle PVs (Hapak et al., 1989), but still within the physiological range of the heart; (2) a more rapid rate of Ca2+ dissociation than the skeletal muscle PVs (shown in this manuscript); (3) a relatively slow rate of Mg2+ dissociation (shown in this manuscript) so that it will be a delayed Ca2+ buffer; and (4) a great deal of work has previously been performed on understanding its Ca2+ and Mg2+ sensitivities (Hapak et al., 1989; Henzl et al., 1996). As we mentioned above, the down side to using rat β-PV directly in the heart is its extremely low Mg2+ affinity (Hapak et al., 1989). However, we have shown in this manuscript that this problem can be overcome by rationally designed mutagenesis of rat β-PV.
Although this work represents the first step in designing a PV for the heart, the only way to know for certain that we have designed an appropriate PV for the heart will be to use gene transfer techniques to express this engineered PV in the heart. Studies are currently being designed to approach this goal. For these studies to be successful, not only the cation binding properties, but also the concentration of the PV must be considered (Day et al., 2008). Additional approaches are also underway in our lab to further refine the engineered rat β-PV (further slowing the Mg2+ dissociation rate), and design a synthetic PV based on TnC. These engineered PVs hold promise for the development of new therapies to remediate relaxation abnormalities in different heart diseases and heart failure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was funded, in part, by NIH grant HL091986 to Jonathan P. Davis.
References
Arif, S. H. (2009). A Ca(2+)-binding protein with numerous roles and uses: parvalbumin in molecular biology and physiology. Bioessays 31, 410–421.
Belkacemi, L., Simoneau, L., and Lafond, J. (2002). Calcium-binding proteins: distribution and implication in mammalian placenta. Endocrine 19, 57–64.
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21, 380–387.
Cates, M. S., Berry, M. B., Ho, E. L., Li, Q., Potter, J. D., and Phillips, G. N. Jr. (1999). Metal-ion affinity and specificity in EF-hand proteins: coordination geometry and domain plasticity in parvalbumin. Structure 7, 1269–1278.
Csillik, B., Schwaller, B., Mihaly, A., Henzi, T., Losonczi, E., and Knyihar-Csillik, E. (2010). Upregulated expression of oncomodulin, the beta isoform of parvalbumin, in perikarya and axons in the diencephalon of parvalbumin knockout mice. Neuroscience 165, 749–757.
Davis, J. P., Rall, J. A., Alionte, C., and Tikunova, S. B. (2004). Mutations of hydrophobic residues in the N-terminal domain of troponin C affect calcium binding and exchange with the troponin C-troponin I96-148 complex and muscle force production. J. Biol. Chem. 279, 17348–17360.
Davis, J. P., Rall, J. A., Reiser, P. J., Smillie, L. B., and Tikunova, S. B. (2002). Engineering competitive magnesium binding into the first EF-hand of skeletal troponin C. J. Biol. Chem. 277, 49716–49726.
Day, S. M., Coutu, P., Wang, W., Herron, T., Turner, I., Shillingford, M., Lacross, N. C., Converso, K. L., Piao, L., Li, J., Lopatin, A. N., and Metzger, J. M. (2008). Cardiac-directed parvalbumin transgene expression in mice shows marked heart rate dependence of delayed Ca2+ buffering action. Physiol. Genomics 33, 312–322.
Eberhard, M., and Erne, P. (1994). Calcium and magnesium binding to rat parvalbumin. Eur. J. Biochem. 222, 21–26.
Erickson, J. R., and Moerland, T. S. (2006). Functional characterization of parvalbumin from the Arctic cod (Boreogadus saida): similarity in calcium affinity among parvalbumins from polar teleosts. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 143, 228–233.
Filimonov, W., Pfeil, W., Tsalkova, T. N., and Privalov, P. L. (1978). Thermodynamic investigations of proteins. IV. Calcium binding protein parvalbumin. Biophys. Chem. 8, 117–122.
Gwathmey, J. K., Yerevanian, A. I., and Hajjar, R. J. (2011). Cardiac gene therapy with SERCA2a: from bench to bedside. J. Mol. Cell. Cardiol. 50, 803–812.
Hapak, R. C., Lammers, P. J., Palmisano, W. A., Birnbaum, E. R., and Henzl, M. T. (1989). Site-specific substitution of glutamate for aspartate at position 59 of rat oncomodulin. J. Biol. Chem. 264, 18751–18760.
Heizmann, C. W., Berchtold, M. W., and Rowlerson, A. M. (1982). Correlation of parvalbumin concentration with relaxation speed in mammalian muscles. Proc. Natl. Acad. Sci. U.S.A. 79, 7243–7247.
Henzl, M. T., Hapak, R. C., and Goodpasture, E. A. (1996). Introduction of a fifth carboxylate ligand heightens the affinity of the oncomodulin CD and EF sites for Ca2+. Biochemistry 35, 5856–5869.
Hou, T. T., Johnson, J. D., and Rall, J. A. (1991). Parvalbumin content and Ca2+ and Mg2+ dissociation rates correlated with changes in relaxation rate of frog muscle fibres. J. Physiol. (Lond.) 441, 285–304.
Hou, T. T., Johnson, J. D., and Rall, J. A. (1992). Effect of temperature on relaxation rate and Ca2+, Mg2+ dissociation rates from parvalbumin of frog muscle fibres. J. Physiol. (Lond.) 449, 399–410.
Hou, T. T., Johnson, J. D., and Rall, J. A. (1993). Role of parvalbumin in relaxation of frog skeletal muscle. Adv. Exp. Med. Biol. 332, 141–151; discussion 151–143.
Hutnik, C. M., MacManus, J. P., Banville, D., and Szabo, A. G. (1990). Comparison of metal ion-induced conformational changes in parvalbumin and oncomodulin as probed by the intrinsic fluorescence of tryptophan 102. J. Biol. Chem. 265, 11456–11464.
Jiang, Y., Johnson, J. D., and Rall, J. A. (1996). Parvalbumin relaxes frog skeletal muscle when sarcoplasmic reticulum-ATPase is inhibited. 270, C411–C417.
Lee, S. H., Schwaller, B., and Neher, E. (2000). Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J. Physiol. (Lond.) 525(Pt 2), 419–432.
McCauley, M. D., and Wehrens, X. H. (2011). Targeting ryanodine receptors for anti-arrhythmic therapy. Acta Pharmacol. Sin. 32, 749–757.
Ogawa, Y., and Tanokura, M. (1986). Kinetic studies of calcium binding to parvalbumins from bullfrog skeletal muscle. J. Biochem. 99, 81–89.
Pauls, T. L., Durussel, I., Cox, J. A., Clark, I. D., Szabo, A. G., Gagne, S. M., Sykes, B. D., and Berchtold, M. W. (1993). Metal binding properties of recombinant rat parvalbumin wild-type and F102W mutant. J. Biol. Chem. 268, 20897–20903.
Periasamy, M., and Janssen, P. M. (2008). Molecular basis of diastolic dysfunction. Heart Fail. Clin. 4, 13–21.
Permyakov, E. A., Ostrovsky, A. V., and Kalinichenko, L. P. (1987). Stopped-flow kinetic studies of Ca(II) and Mg(II) dissociation in cod parvalbumin and bovine alpha-lactalbumin. Biophys. Chem. 28, 225–233.
Raake, P. W., Tscheschner, H., Reinkober, J., Ritterhoff, J., Katus, H. A., Koch, W. J., and Most, P. (2011). Gene therapy targets in heart failure: the path to translation. Clin. Pharmacol. Ther. 90, 542–553.
Robertson, S. P., and Potter, J. D. (1984). The regulation of free calcium ion concentration by metal chelators. Methods Pharmacol. 5, 63–75.
Rodenbaugh, D. W., Wang, W., Davis, J., Edwards, T., Potter, J. D., and Metzger, J. M. (2007). Parvalbumin isoforms differentially accelerate cardiac myocyte relaxation kinetics in an animal model of diastolic dysfunction. Am. J. Physiol. Heart Circ. Physiol. 293, H1705–H1713.
Rohde, D., Brinks, H., Ritterhoff, J., Qui, G., Ren, S., and Most, P. (2011). S100A1 gene therapy for heart failure: a novel strategy on the verge of clinical trials. J. Mol. Cell. Cardiol. 50, 777–784.
Sambrook, J., and Rusell, D. W. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Szatkowski, M. L., Westfall, M. V., Gomez, C. A., Wahr, P. A., Michele, D. E., DelloRusso, C., Turner, II Hong, K. E., Albayya, F. P., and Metzger, J. M. (2001). In vivo acceleration of heart relaxation performance by parvalbumin gene delivery. J. Clin. Invest. 107, 191–198.
Tikunov, B. A., and Rome, L. C. (2009). Is high concentration of parvalbumin a requirement for superfast relaxation? J. Muscle Res. Cell Motil. 30, 57–65.
Tikunova, S. B., Black, D. J., Johnson, J. D., and Davis, J. P. (2001). Modifying Mg2+ binding and exchange with the N-terminal of calmodulin. Biochemistry 40, 3348–3353.
Tikunova, S. B., and Davis, J. P. (2004). Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J. Biol. Chem. 279, 35341–35352.
Tikunova, S. B., Rall, J. A., and Davis, J. P. (2002). Effect of hydrophobic residue substitutions with glutamine on Ca(2+) binding and exchange with the N-domain of troponin C. Biochemistry 41, 6697–6705.
Trayanova, N. A., and Rice, J. J. (2011). Cardiac electromechanical models: from cell to organ. Front. Physiol. 2:43. doi:10.3389/fphys.2011.00043
van der Velden, J. (2011). Diastolic myofilament dysfunction in the failing human heart. Pflugers Arch. 462, 155–163.
Vijay-Kumar, S., and Cook, W. J. (1992). Structure of a sarcoplasmic calcium-binding protein from Nereis diversicolor refined at 2.0 A resolution. J. Mol. Biol. 224, 413–426.
Wang, W., Martindale, J., and Metzger, J. M. (2009). Parvalbumin: targeting calcium handling in cardiac diastolic dysfunction. Gen. Physiol. Biophys. 28, F3–F6.
White, H. D. (1988). Kinetic mechanism of calcium binding to whiting parvalbumin. Biochemistry 27, 3357–3365.
Wilwert, J. L., Madhoun, N. M., and Coughlin, D. J. (2006). Parvalbumin correlates with relaxation rate in the swimming muscle of sheepshead and kingfish. J. Exp. Biol. 209, 227–237.
Keywords: parvalbumin, relaxation, calcium, magnesium
Citation: Zhang J, Shettigar V, Zhang GC, Kindell DG, Liu X, López JJ, Yerrimuni V, Davis GA and Davis JP (2011) Engineering parvalbumin for the heart: optimizing the Mg2+ binding properties of rat β-parvalbumin. Front. Physio. 2:77. doi: 10.3389/fphys.2011.00077
Received: 11 September 2011;
Paper pending published: 27 September 2011;
Accepted: 10 October 2011;
Published online: 31 October 2011.
Edited by:
Jolanda van der Velden, VU University Medical Center, NetherlandsReviewed by:
Regis Lamberts, University of Otago, New ZealandJon Kentish, King’s College London, UK
Copyright: © 2011 Zhang, Shettigar, Zhang, Kindell, Liu, López, Yerrimuni, Davis and Davis. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jonathan P. Davis, Department of Physiology and Cell Biology, The Ohio State University, 1645 Neil Avenue, 209 Hamilton Hall, Columbus, OH 43210, USA. e-mail: davis.812@osu.edu