
- 1 Artialis S.A., Liège, Belgium
- 2 Bone and Cartilage Research Unit, University of Liege, Liege, Belgium
After the genomic era, proteomic corresponds to a wide variety of techniques that study the protein content of cells, tissue, or organism and that allow the isolation of protein of interest. It offers the choice between gel-based and gel-free methods or shotgun proteomics. Applications of proteomic technology may concern three principal objectives in several biomedical or clinical domains of research as in osteoarthritis: (i) to understand the physiopathology or underlying mechanisms leading to a disease or associated with a particular model, (ii), to find disease-specific biomarker, and (iii) to identify new therapeutic targets. This review aimed at gathering most of the data regarding the proteomic techniques and their applications to osteoarthritis research. It also reported technical limitations and solutions, as for example for sample preparation. Proteomics open wide perspectives in biochemical research but many technical matters still remain to be solved.
Introduction
Osteoarthritis
Osteoarthritis (OA) is a progressive disorder characterized by the destruction of articular cartilage accompanied by subchondral bone sclerosis and synovial inflammation. Pain is the main symptom of OA. In the early stages of disease, pain occurs only when the joint is required. In the later stages of disease, pain is felt not only at every movement and but also at rest. The movements of the involved joints are limited. It installs a very debilitating functional impairment for the patient. Physical inactivity is settled down with its own set of physiological and psychological impacts. OA is the major cause of physical disability in people over 50 years of age, excluding traumatic causes. After 65 years, 70–80% of individuals show radiological signs of OA in at least one joint.
Multiple causes lead to OA development. Genetic predispositions were identified in specific hand or hip localization of OA (Hunter et al., 2004; MacGregor et al., 2009) or in generalized OA (Miyamoto et al., 2007; Evangelou et al., 2009) and could be implicated in 15% of OA (Felson, 2010). Aging, obesity, and being a female are well-known risk factors of OA but others parameters implicated in the physiopathology of the disease remain to be detailed. In recent years, an association between diabetes mellitus, metabolic syndrome, and other conditions characterized by impaired glucose metabolism have also been postulated to be associated with OA (Burner and Rosenthal, 2009; Rosa et al., 2009, 2011; Berenbaum, 2011).
Currently, the diagnosis of OA is based on symptoms and radiological signs which occur late during disease progression. More specifically, diagnosis is based on cartilage integrity. However, articular cartilage is invisible on radiographs and must be assessed indirectly by the joint space width corresponding to the spacing between subchondral bone ends in a joint. This method does not allow detection of early structural damage, and its use in follow-up of the disease is not recommended. Moreover, the diagnosis of OA occurs with the appearance of pain, when cartilage degradation is often already advanced. Patients are therefore treated for symptoms with anti-inflammatory drugs and analgesics because no disease-modifying drugs (DMOAD) are currently available.
Proteomics
Proteome was originally defined as the complete protein content of a cell, a tissue or an organism. The term was proposed by Wilkins in 1994 (Wilkins et al., 1996). Because protein expression is dependent on environmental conditions, thus making the proteome a very dynamic structure, the original definition was specified: the proteome is the complete protein set of a cell, tissue, or organism considered at a particular moment, in a specific environment. Succeeding to the genomic era, this direct evaluation of protein expression is essential for the analysis of biological system. Indeed, there is no correlation between protein abundance and mRNA levels (Gygi et al., 1999a). This was illustrated by Lorenz et al. (2003a) who have shown a poor correlation between the transcriptome and the proteome modifications in synovial tissue from OA and rheumatoid arthritis (RA) patients. Furthermore, post-translational modifications bring supplementary variations in the proteome that cannot be analyzed at the gene level.
The principle of the method relies on the separation of proteins and their further analysis using either gel-based or gel-free methods. Protein separation methods are coupled to spectrometer for identification by mass spectrometry (MS; Figure 1). Several modes of analysis are available in MS. They differ markedly by the ionization source of the sample. The main sources used in proteomic analysis are matrix-assisted laser desorption/ionization (MALDI) and surface-enhancer laser desorption/ionization (SELDI). These techniques allow a soft ionization of molecules without excessive fragmentation, making the analysis of proteins possible. In the MALDI, the sample is co-crystallized with the matrix and then deposited on a metal support. The source of ionization is a nitrogen laser that bombards the sample. The energy transmitted by the laser is absorbed by the matrix and the input of energy causing it expands in the gas phase with the molecules contained in the sample. MALDI ion source is mainly coupled to an analyzer or time-of-flight (TOF). Its speed, sensitivity, simplicity, and reproducibility make it a very powerful technique for the detection and identification of proteins. However, in many diseases, molecules of interest are often present in very small quantities making them difficult to detect. The SELDI-TOF technology is based on the retention of proteins to small chromatographic surfaces treated to selectively adsorb proteins based on their physicochemical properties. This technique simplifies the protein mixture to be analyzed. Many types of samples such as biological fluids and cell extracts, or cell lysates and histological sections can be analyzed. The proteins of interest are coated to allow their crystallization and facilitate the desorption and ionization generated by the laser in the drive. In single MS mode, the spectrometer quantifies a protein/peptide and determines its structure and its molecular mass. In tandem mode (MS/MS), ions from the first fragmentation are selected and fragmented, allowing identification of the protein/peptide present in the sample.
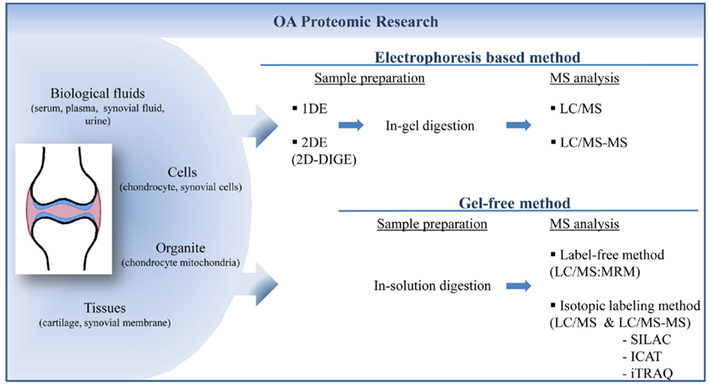
Figure 1. Proteomic strategies to quantify and identify biomarkers of osteoarthritis from different kind of samples. Proteins extracted from samples could be separated by electrophoresis in one (1DE) or two (2DE) dimensions. Proteins are labeled before or after gel migration to perform quantification. After in-gel digestion proteins are identified by mass spectrometry. Gel-free methods involve the separation of digested peptides which could be quantified directly by mass spectrometry analysis. 1DE, one-dimensional electrophoresis; 2DE, two-dimensional electrophoresis; 2D-DIGE, two-dimensional-differential in-gel electrophoresis; LC, liquid chromatography; MS, mass spectrometry; MRM, multiple-reaction monitoring; SILAC, stable isotope labeling by amino acid; ICAT, Isotope-coded affinity tag; iTRAQ, isobaric tag for relative and absolute quantitation.
Applications of proteomic technology may concern three principal objectives in several biomedical or clinical domains of research as in OA: (i) to understand the physiopathology and underlying mechanisms leading to a disease or associated to a particular model, (ii), to find disease-specific biomarker, and (iii) to identify the targets of new therapeutic and their mechanisms of action. Different technologies are available and appropriate to be used in these aspects of investigation. Here, we present different technologies used in proteomic analysis and their main applications and scientific contributions in the field of arthritis research (Table 1).
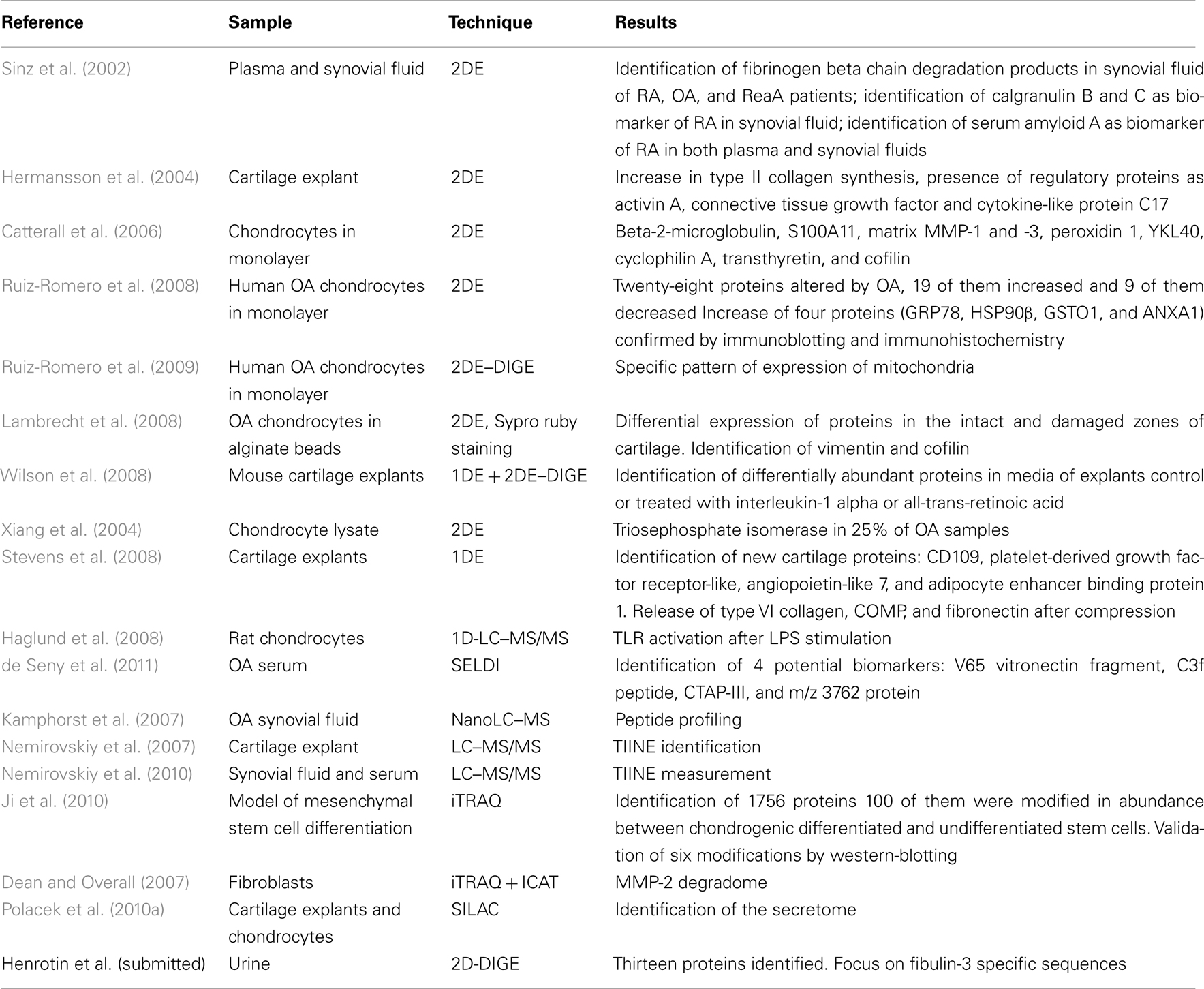
Table 1. Identification of protein in OA samples with proteomic techniques.
Gel-Based Methods
Two-Dimensional Electrophoresis-Based Methods
Two-dimensional electrophoresis (2DE) is the standard proteomic method. It is widely used to compare the proteome of two samples or more. In this technique, proteins extracted from various samples are separated according to their isoelectric point in first dimension and depending on their molecular mass in the second dimension on a polyacrylamide gel. Traditionally, proteins were stained by silver nitrate, Coomassie blue, or fluorescent dye. After an in-gel trypsin digestion, the identity of proteins is determined by MS/MS.
Several studies described below used 2DE in the context of OA research. However, this traditional method showed some limitations in term of reproducibility. Indeed, gel to gel variation leading to significant variability is classically observed. This issue makes difficult to distinguish between the system variations and the induced biological changes. Moreover, available staining methods such as silver nitrate or Coomassie blue, lack of sensitivity and have a poor dynamic range, then limiting the quantitative performance of the technique.
To overcome these technical limitations, 2DE has recently been improved by the introduction of labeling before the migration of proteins, allowing a simultaneous migration of different samples on a single polyacrylamide gel. Two-dimensional difference gel electrophoresis (2D-DIGE) methodology is a powerful tool for the investigation of protein expression profiles in multiple sets of samples (Unlu et al., 1997; Marouga et al., 2005). Samples can be individually labeled with Cy3 or Cy5 CyDye DIGE Fluors, whereas Cy2 CyDye DIGE Fluor is used to label a pooled sample comprising equal amounts of each sample, then acting as an internal standard (Figure 2). Interesting spots with differential fluorescent intensity between Cy3 and Cy5 are removed from the preparative gel after post-staining with Coomassie Blue in order to allow proteins identification by MS analysis.
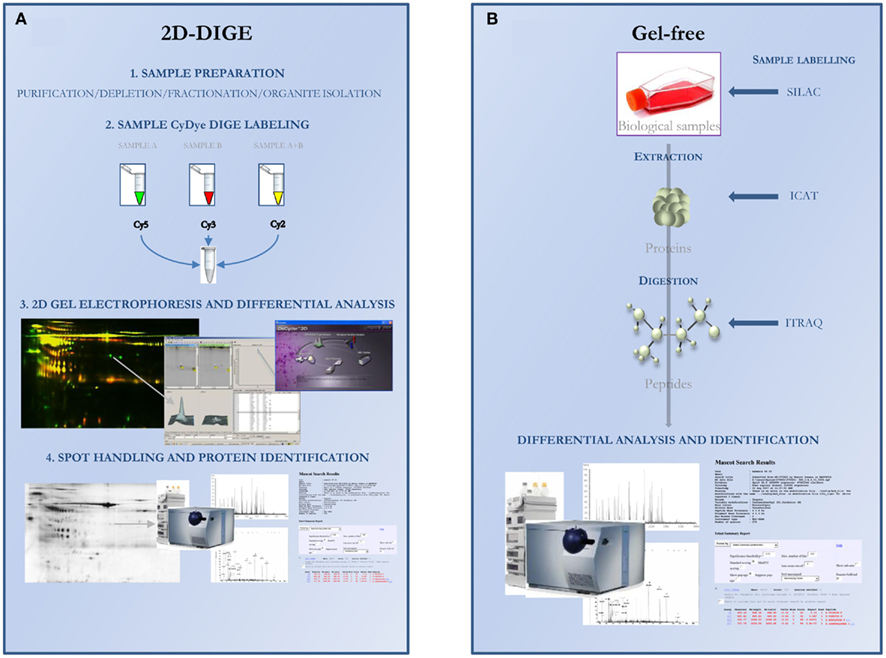
Figure 2. Overview of the proteomic workflow using 2D-DIGE (A) or gel-free (B) approaches for differential analysis. (A) 2D-DIGE approach (1) Proteins are extracted from sample which could be optimized for complexity reduction or particular protein identification; (2) Three fluorescent dyes were used. Samples A and B were fluorescently labeled with either Cy3 or Cy5, and a pooled internal standard is labeled with Cy2; (3) Samples are mixed and resolved in the same 2DE gel. Protein spot pattern could be visualized for each dye by selection of specific wavelength. 2D images are analyzed by specific software and internal standard is used for normalization; (4) Differentially expressed protein spots are excised from a preparative gel and identified by mass spectrometry. (B) Gel-free approach. Proteins or peptides could be labeled at different stages of sample preparation depending on SILAC, ICAT, or iTRAQ technology. Light and heavy forms of isotopic analogs are resolved by 2D-LC and then quantified and identified by mass spectrometry. SILAC: stable isotope labeling by amino acid in cell culture; ICAT: isotope-coded affinity tag; iTRAQ: Isobaric tag for relative and absolute quantitation.
In the context of OA research, 2DE was used to find disease-specific proteins by comparing the protein content of biological fluids, cells (chondrocytes), or other biological material such as tissues (cartilage, synovial membrane) collected from patients and healthy controls. Descriptive applications were also performed to gain insights into arthritic diseases.
2DE applied to biological fluids
By their easy access and their abundant availability, serum and plasma are samples of choice for the identification of new biomarkers. Synovial fluid is in direct contact with the cartilage and may reflect the metabolism of chondrocytes but synovial fluid sampling is difficult. Exchanges between the serum and synovial fluid allow the nutrition of cells but also the release of substances from the joint into the peripheral blood. So, synovial fluid and plasma are often investigated to discover new biomarkers.
Plasma and synovial fluid from patients with OA, RA, or reactive RA (ReA) were studied (Sinz et al., 2002). The authors compared the composition of plasma and synovial fluid from each patient. They showed that the products of degradation of fibrinogen were abundant in synovial fluid with a variable ratio depending on the disease. This study revealed the presence of specific spots containing calgranulin B (S100A9) in the synovial fluid of RA patients. They found the presence of serum amyloid A in synovial fluid and plasma of RA patients, but not in samples from OA patients (Sinz et al., 2002). The modification of the protein S100A9 was also observed in synovial fluid by other team showing a correlation between the level of S100A8/S100A9 complex in plasma and synovial fluid and its capability to discriminate RA from other inflammatory diseases (Drynda et al., 2004).
In urine, we used 2D-DIGE technology to compare the proteome of young healthy volunteers and late-stage OA patients (Henrotin et al., submitted). We identified 13 proteins from spots that exhibited an abundance ratio greater than 1.5 between groups. We also focused on specific sequences of fibulin-3, the only extracellular matrix protein found to be significantly modified in the proteome of urine of OA patients.
The sample preparation represents an important issue for 2DE. High-abundant proteins are present in milligram (mg/ml) quantities particularly in biological fluids and represent more than 95% of the total proteins in plasma. Proteins of interest as potential biomarkers are usually present in samples at levels as low as nanogram (ng/ml) to picogram (pg/ml), making them difficult to detect among abundant proteins. Strategies to remove them consist in the use of immunoaffinity columns retaining the two more abundant proteins, i.e., albumin and immunoglobulins or up to 20 of the most abundant proteins. Some problems linked to these commercial disposable columns reside in their high cost or in their employment. A new method was recently proposed for the depletion of immunoglobulins in serum by thiophilic chromatography (Salgado et al., 2010). The authors applied this technique to compare RA with healthy control sera.
2DE applied to cells and tissues
Articular cartilage is a poorly cellular structure with chondrocyte as the only one cell type. The poor cellularity of human cartilage requires an expansion of cells in culture to obtain enough material to proceed to the proteomic analysis. Another technical challenge is that over 90% of articular volume is majorly composed of large proteins such as collagens or proteoglycans. These proteins have to be removed from sample before 2DE because, as in biological fluids, they hide minor proteins and interfere with the migration of proteins during isoelectric focusing.
A method to analyze the protein secreted by cartilage explants has been developed (Hermansson et al., 2004). They are easily resolved on gels after aggrecan depletion by precipitation with cetylpyridinium chloride. By incorporation of 35Sulfur in culture, the authors identified newly synthesized proteins. They compared the protein pattern of OA and healthy adult cartilage. The results of this study concerned an increase in type II collagen synthesis in OA and the presence of regulatory proteins as activin A, connective tissue growth factor, and cytokine-like protein C17.
In the same way, the proteins secreted by chondrocytes in monolayer culture stimulated by cytokines as interleukin (IL)-1 and oncostatin were analyzed (Catterall et al., 2006). 2DE allowed the identification of low molecular proteins or fragments: beta-2-microglobulin, S100A11, matrix metalloproteinases (MMP)-1 and -3, peroxidin 1, a member of the “mammalian chitinase-like proteins” (YKL40), cyclophilin A, transthyretin, and cofilin. As with the previous investigation, the depletion of abundant proteins was required to improve the resolution in the upper part of the gels. This was achieved using cetylpyridinium chloride precipitation and anion exchange.
The 2DE technique was used to compare the chondrocytic proteome from control and OA patients (Ruiz-Romero et al., 2008). Twenty-eight proteins were showed to be altered by OA, 19 of them were increased and 9 of them were decreased. The increase of four proteins (GRP78, HSP90β, GSTO1, and ANXA1) was confirmed by immunoblotting and immunochemistry. In addition, the authors showed that IL-1 increased not only the gene expression but also the content of the cited proteins by chondrocytes. The same authors further showed by 2D-DIGE technology that a larger number of proteins were concerned by a statistically significant modification in quantity in the analyzed samples. Moreover, the authors improved their investigations by focusing on a particular organite of the cell, i.e., the mitochondria (Ruiz-Romero et al., 2009). The expression of numerous proteins was found to vary in mitochondria of OA cells. Among them, three forms of superoxide dismutase (SOD)-2 were decreased. SOD is an important actor of the oxidative protection pathway. This decrease means that during OA, the cell defense against oxidative stress is impaired. The authors confirmed the observed modifications by western blot (WB), reverse transcription and polymerase chain reaction (RT–PCR), and immunohistochemistry on cartilage tissues. On the contrary, TRAP1 was found to be increased by 2D-DIGE analysis of proteome modification in OA. This protein is related to the protective mechanism against reactive oxygen species (ROS) injury by protecting the cell against oxidative stress-induced apoptosis. The different expression patterns between OA and control and between the different damaged zones of cartilage were further studied. Lambrecht et al. (2008) compared the intracellular content in proteins extracted from healthy and OA chondrocytes cultured in alginate beads. Moreover, they compared intact and damaged zones of cartilage from OA patients. Proteins were extracted from cultured chondrocytes in two different fractions: membrane (hydrophobic) or cytosol. They performed 2DE and stained proteins with Sypro ruby. They identified 16 proteins differentially expressed between control and intact zone of OA patients and 28 proteins between control and damaged zones of OA patients. Finally, 17 proteins were differentially expressed between intact and damaged zones of OA. Among the identified proteins, authors focused on vimentine and confirmed the observed modification by WB. In addition, the authors analyzed the phosphorylation of differentially expressed proteins and focused on cofilin which is involved in severing and depolymerization of actin filaments (DesMarais et al., 2005).
Some investigations were also made in a mouse model of cartilage explants to respond to the increase use of genetically modified mouse model in arthritis research (Wilson et al., 2008). The authors compared effect of retinoic acid (retA) and IL-1 on femoral head cartilage extracts or conditioned media from explant culture. Using one-dimensional electrophoresis (1DE) and 2DE, they showed no modification of protein pattern in cartilage extract while the effect of tested substances was marked on proteins from conditioned media. To confirm and quantify the observed modifications, they used 2D-DIGE technology. The results of this study reported an increase in MMP3, CH3L1, neutrophil gelatinase-associated lipocalin, and haptoglobin content induced by the addition of IL-1; an increase in aggrecan G1 domain, serotransferrin, cartilage oligomeric matrix protein (COMP), matrilin 3, and link protein induced by the addition of retA and a decrease in gelsolin content in both IL-1 and retA conditioned media compared to control. Gelsolin is an actin-capping protein with important roles in extracellular actin scavenging (Vasilopoulos et al., 2007). It is also involved in the regulation of cytoskeletal architecture and cell–matrix interactions in many cell types, including osteoblasts, fibroblasts, and developing chondrocytes (Chellaiah et al., 2000; Djouad et al., 2007; El Sayegh et al., 2007).
Another group has identified 76 proteins in cartilage from collagen-induced arthritis (CIA) mice using 2DE (Lorenz et al., 2003b). In this study, mice were immunized with bovine collagen II which corresponded to a RA model. Five proteins were found significantly changed in expression and three of them have been identified: lymphoid enhancer binding factor 1 was decreased while the ferritin light chain and antioxidant protein 2 were increased.
A novel approach was applied to chondrocytes proteins for the identification of OA-specific antibodies (Xiang et al., 2004). Proteins from chondrocyte lysates were transferred onto a nitrocellulose membrane after 2DE separation and blotted with serum from OA or RA patients in order to investigate autoimmunity profiles. Auto-antigens were found in both OA and RA sera. Several proteins were found to be recognized only by OA serum. Among them, triosephosphate isomerase appeared in almost 25% of the tested samples but only in few controls and in about 6% of RA samples.
The use of chondrocytes as source of proteins for cartilage proteomic analysis requires a cellular expansion in vitro. It is noteworthy to keep in mind that the chondrocyte culture leads to a gradual shift of the cells from chondrocytes to fibroblastic phenotype known as the dedifferentiation process, and to the modification of matrix component synthesis. In fact, chondrocyte dedifferentiation occurs upon the proliferation which can be circumvented by setting up high density non-proliferating cultures. Some authors have compared the protein content in cartilage from healthy and OA knee joint cartilage directly – after removal of collagens and proteoglycans – without the intermediate step of culture (Guo et al., 2008). They used traditional 2DE technology with silver nitrate staining and found 16 proteins of interest. Eight of them were increased while eight others were decreased as annexin A1 whose modification was confirmed by WB analysis on cartilage samples.
Apart from chondrocytes and synovial cells, other teams were interested in other cell types that could probably be involved in the disease pathophysiology. Others cells of interest were bone marrow mesenchymal stem cells. Using 2D-DIGE technology, Rollin et al. (2008) found 38 differentially expressed spots of proteins between OA and control isolated mesenchymal cells. Again, DIGE technology revealed a large number of modifications. This observation was probably due to its sensitivity.
Another approach using 2DE consists in the complete description of most of the proteins found in cells, tissues, or biological fluids in order to obtain a proteomic profile of reference to compare with unknown samples. 2DE of total proteins provides a visual representation of proteome and allows the detection of post-translational modifications of proteins. The proteome of human OA cartilage have been revealed (Vincourt et al., 2006). The authors resolved more than 500 spots on 2D-gel and identified 191 proteins. The proteome of normal chondrocytes isolated from healthy cartilage was also investigated (Ruiz-Romero et al., 2005). The reference map obtained by 2DE was compared with the one obtained of Jurkat cells. They identified some specific proteins more abundant in chondrocytes like cathepsin D, heat shock protein (HSP) 47, mitochondrial superoxide dismutase (SOD), cytoskeleton-related proteins, or members of annexin family.
One-Dimensional Electrophoresis
When proteins are too hard to resolve by isoelectrofocusing, as insoluble proteins, some authors explored protein content using 1DE. Proteins are only resolved on polyacrylamide gel according to their molecular weight. This technique could also be useful to pre-fractionate the sample before liquid chromatography (LC) and MS analysis. Thereby, Gobezie et al. (2007) determined 342 gel slices from 1D-gels to identify proteins modified in OA synovial fluid in comparison to control. They found 18 distinct proteins between control and OA and interestingly two specific subsets of proteins profile in OA group not linked to age, sex, ethnicity, medication, or stage of the disease.
The 1DE investigation was further pursued with cartilage from OA patients or control (Wu et al., 2007). Proteins were directly extracted from cartilage without any culture step. After depletion of abundant matrix proteins like collagens and aggrecans, the authors identified 59 proteins differently expressed between the normal and OA tissues then providing important information on protein expression.
Two approaches of proteomic analysis using MS were compared (Garcia et al., 2006). One the one hand, the proteins from cartilage were digested by trypsin in solution and directly analyzed by MS. On the other hand, proteins were fractionated on 1D-gel and further in-gel-digested by trypsin. Interestingly, the authors showed the significant amelioration of analysis by the introduction of gel fractionation. This technique allowed the detection of more proteins by the simplification of protein mixture. This method was further used on bovine cartilage in order to compare the response of chondrocytes and cartilage matrix to injurious mechanical compression and treatment with IL-1beta and tumor necrosis factor (TNF)-alpha (Stevens et al., 2008). Analyses were made on secreted proteins in culture media of cartilage explants. This analysis retrieved 250 proteins. Among them, new cartilage proteins were identified, i.e., CD109, platelet-derived growth factor receptor–like, angiopoietin-like 7, and adipocyte enhancer binding protein 1. The authors also identified the protein expression induced by the stimulation with IL-1beta and TNF-alpha. They demonstrated that the compression was responsible for the release and proteolysis of type VI collagen, COMP, and fibronectin.
Finally, the analysis of the proteins secreted by chondrocytes after stimulation with lipopolysaccharide soluble (LPS) and the comparison of the obtained pattern with that one of unstimulated cells were performed in rat cells by 1D-LC–MS/MS (Haglund et al., 2008). This study showed the capability of articular chondrocytes to respond to the activation of the toll-like receptor (TLR).
Technical Improvements
As seen with these different studies, the sensitivity of the techniques was often limited by the composition of sample itself. Indeed, very few proteins were found on gels from thousands of different proteins contained in a cell or in biological fluid. In order to improve the sensitivity of proteomic analyses, modifications in the methodology were applied to reduce sample complexity. For example, one approach when working on cells was to focus on one particular organite. Purification of this organite prior to protein extraction strongly improved gel resolution and enhanced proteomic analysis by targeting specific proteins of interest. Ruiz-Romero et al. (2006) established with this technique the mitochondrial proteome of chondrocytes.
Other technical modifications as passive rehydration loading without voltage application have been developed before the first dimension of electrophoresis. It reduces the entry of large proteins in-gel strips and was tested to improve the direct separation of protein from cartilage extract without sample pretreatment (Sanchez et al., 1997; Pecora et al., 2007). Molecular weight cut-off was also evaluated to improve protein load, optimize resolution, and enhance detection of low-abundance proteins (Wilson et al., 2005). In addition, the authors described a method of joint micro-dissection.
Conclusion
Although 2DE-based methods are still widely used in proteomic analysis, this technology has drawbacks that are difficult to circumvent. Limitations concern problems in the detection of low-abundant proteins and in the dynamic range of the protein detection dependent on gel staining. Proteins with extreme characteristics such as very low or high-molecular weight or very acidic or basic isoelectric point like membrane proteins are difficult to resolve on polyacrylamide gel. Moreover, 2DE requires extensive sample handling without easy automation, increasing the risk of bias induced by the manipulation.
Gel-Free Methods or Shotgun Proteomics
To overcome problems inherent to electrophoresis-based methods, others separation methods associated to MS were developed. These techniques were grouped under the general term of shotgun proteomics.
In shotgun proteomics, the proteins are separated by high performance LC (HPLC) before being analyzed by MS. To reduce sample complexity, after digestion of proteins in solution, usually with trypsin, multi-dimensional chromatography is often performed coupling cation exchange with reversed-phase chromatography. Unlike 2DE, this technology is fully automated – separation interfaced directly with the mass spectrometer – and covers a large scale of proteins. An overview of the gel-free methods is presented Figure 2 and the main advantages and disadvantages of gel-free methods in comparison to 2D-DIGE are described in Table 2.
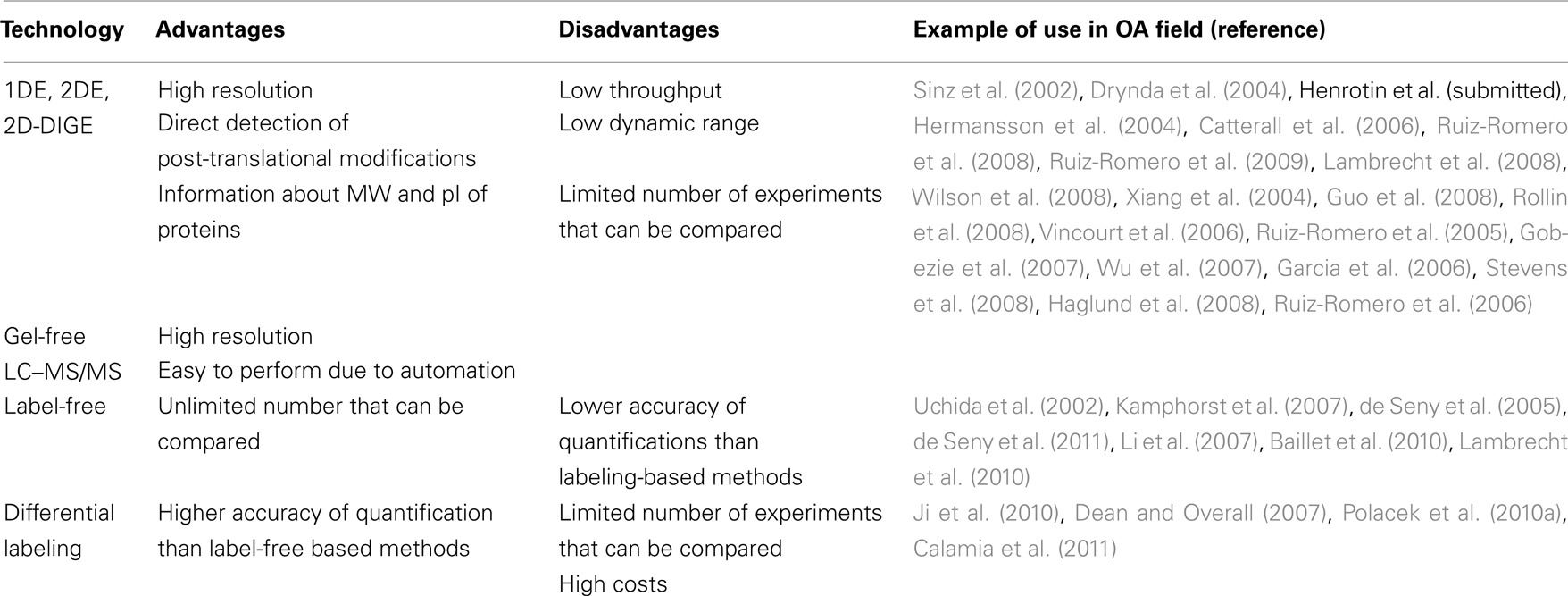
Table 2. Advantages and disadvantages of in-gel and gel-free methods.
Label-Free Methods
In differential analysis, the peptides may be marked with stable isotope at various stages of the analysis process, depending on the used technique (Figure 2). This methodology is applied to identify new potential biomarkers in samples. Label-free methods could be performed to qualify, verify, and quantify a previously discovered biomarker by isotopic-label method with known fragmentation properties in a complex sample. In MS/MS mode, one pair of precursor characteristic of a single peptide is monitored using multiple-reaction monitoring (MRM). This technique provides highly selective, sensitive, and reproductive detection of peptides. MRM was used to quantify level of C-reactive protein (CRP) in serum of RA patients (Kuhn et al., 2004). They used labeled tryptic peptide of CRP as internal standard. They showed that MRM was a useful tool to directly qualify and quantify potential biomarker in clinical samples without a step of antiserum production.
Label-free methods in MS or MS/MS mode were used in OA samples to compare proteomic profile of different populations. The protein profiles of synovial fluid from OA and RA patients were compared using SELDI-MS technique (Uchida et al., 2002). This work reported the presence of specific proteins as myeloid related protein 8 in synovial fluid from RA patient. The concentration of proteins was estimated from the area under the peak of the chromatogram corresponding to the precursor peptide (Bondarenko et al., 2002; Chelius and Bondarenko, 2002). Proteins of interest were identified and their potential as marker was evaluated using MRM technology which allows a quantification using 13C labeled peptide as internal standard (Liao et al., 2004). A methodology has been validated for peptide profiling in synovial fluid (Kamphorst et al., 2007). About 500 peptides from 40 distinct proteins were identified. Hyaluronic acid was removed by ultracentrifugation and solid-phase extraction in order to enrich the sample in low molecular weight peptides. Proteins and peptides were analyzed by shotgun proteomics without trypsin digestion. This approach allowed the evaluation of proteolytic activity by the analysis of cleavage sites. Several chip arrays in SELDI-MS technology were used in order to identify biomarkers of RA (de Seny et al., 2005). They compared the protein content of serum from RA patients with other inflammatory disease such as Crohn’s disease, asthma, psoriatic arthritis, or non-inflammatory control group as OA patients or healthy subjects. Peak corresponding to myeloid related protein 8 appeared discriminant in RA group compared to controls (OA and healthy patients). This technique was recently applied to the serum of OA patients (de Seny et al., 2011). It provided interesting results. It allowed the identification of four potential biomarkers, i.e., V65 vitronectin fragment, C3f peptide, CTAP-III, and m/z 3762 protein. All of them could be involved in OA pathophysiology and could be relevant to reflect inflammation and cartilage and bone turn-over. MS-based technology is also used in targeted approaches. Indeed, shotgun methodology could be applied to the characterization of peptides. More specifically, LC–MS/MS is used to profile directly a peptide or protein suspected to be a useful biomarker. Thus, new epitope of type II collagen (TIINE) degradation by MMP was identified by working on explants and collagen (Nemirovskiy et al., 2007). The authors identified by immunoaffinity a peptide released by MMP13 in human urine and synovial fluid (Nemirovskiy et al., 2010) and developed a method of quantification by immunoaffinity-LC–MS/MS using a deuterated internal standard. The method was clinically validated by the quantification by MRM of TIINE in urine of patients with RA, OA, or polychondritis (Li et al., 2007). TIINE measure in urine was then shown as a potential biomarker of OA. The comparison of synovial fluid and serum content from OA and RA patients and from miscellaneous inflammatory arthritides (MIAs) and RA patients was performed in order to identify new biomarkers of RA. The authors found an overexpression of proteins S100A8, S100A9, and S100A12 in RA synovial fluid compared to OA samples. Versus MIAs, S100A8, S100A9, and alpha defensins -1, -2, -3 discriminates RA populations but with a weakly altered sensitivity and specificity. In serum, none of these markers were found to be modified with disease (Baillet et al., 2010). To improve the discovery of biomarkers in serum, researchers have developed methods for sample preparation before their analysis by shotgun proteomics. As in 2DE, strategies consist in removing abundant proteins to enrich the sample in less abundant proteins and peptides. To this aim, different methods such as membrane filtration cut-off and immunoaffinity are available. A third method using hollow-fiber membrane to remove high-molecular weight proteins and affinity columns to deplete high-abundant proteins has been developed (Tanaka et al., 2006). Moreover, the authors used 3D-LC with the introduction of reverse-phase-chromatographic preparation of sample before trypsin digestion circumventing the overload of MS profiles.
Another application for MS-based technology is the structural analyses of proteins and the study of the post-translational modifications. Zaia et al. (2000) defined the structure of proteins from extracellular matrix and analyzed fragments from matrix turn-over by MALDI. Proteins as COMP, aggrecan, decorin, and biglycan were studied with specific experimental approaches. More recently, 2D-LC–MS/MS was applied to the characterization of the proteome of chondrocyte cultured in alginate beads. They identified 779 unique proteins (Lambrecht et al., 2010). Finally, the culture medium of equine cartilage was studied using 1D-LC–MS/MS after digestion with trypsin (Clutterbuck et al., 2011). Tryptic peptides were analyzed with the aim of identifying biomarker of the early cartilage disease.
Isotopic Labeling-Based Methods
The main developed strategies consist in using isotopic tag for sample labeling. All these methods use the difference of mass as the basis of the quantitation with the measurement of relative peak areas of mass spectra. Quantitative analyses of proteins and peptides are achieved by comparing isotopic light and heavy forms contained in two samples. Peak ratios for isotopic analogs are highly accurate in a same experiment. Methods using stable isotope circumvent the problem of the variation in sample recovery. Different methods were described depending on samples and incorporation method. Stable isotope labeling by amino acid (SILAC) in cell culture was developed by Ong et al. (2002). It consists in the biological incorporation of labeled essential amino acids (e.g., L-leucine or deuterated L-leucine) in amino acid deficient cell culture medium resulting in labeling of all newly synthesized proteins and virtually all proteins after cell doubling population. Precocity of labeling provides a definite advantage to this method. Moreover, there is no requirement of additional purification and it is applicable to living samples on the contrary to Isotope-coded affinity tag (ICAT) method that will be further describe. Labeled isotopic tag can be added to cell culture or directly coupled to protein or peptide according to the moment of enzymatic digestion. ICAT technology was developed in that sense (Gygi et al., 1999b). ICAT reagent is composed of three functional elements: a reactive group, an isotopic coded light or heavy linker group and a biotin affinity tag. In addition to the quantitation of proteins, this method reduces the complexity of samples by selecting labeled proteins using affinity purification. The main limitation of this technique is that ICAT only labels cysteine-containing peptides. Hence, the proteome coverage and the number of peptides labeled per protein are reduced. To circumvent this drawback, isobaric tag for relative and absolute quantitation (iTRAQ) methodology was developed. This method is using multiplexed set of reagents that integrate isobaric mass labels at the N-terminus and lysine side chain of peptides in a digest mixture (Ross et al., 2004). This amine-specific reagent can label all peptides in up to four samples simultaneously.
The different methods using isotopic labeling were applied to OA and RA research. iTRAQ methodology was used in order to find proteins specific of chondrogenesis in a model of mesenchymal stem cell differentiation (Ji et al., 2010). Among the 1756 identified proteins, 17 appeared to be increased and 83 to be decreased. The 17 proteins and enzymes that were increased were involved in the synthesis of cartilage matrix in mature chondrocytes. The ones that were decreased were involved in energy metabolism, chromatin organization, transcription, mRNA processing, signaling transduction, and cytoskeleton. Most of them were newly found to be involved in the chondrogenic process and BTF3l4 and fibulin-5, two novel chondrogenesis-related proteins were also identified in the present study. iTRAQ and ICAT technologies were used in an original work to analyze the degradome of MMP-2 substrates (Dean and Overall, 2007). MMP-2 is known to be involved in OA as its expression is increased in cartilage during the disease process (Kevorkian et al., 2004). The peptides released in culture medium were labeled and identified by shotgun proteomics. New substrates for MMP-2, such as CX(3)CL1 chemokine fractalkine, osteopontin, galectin-1, and HSP90 alpha were identified. In addition, the authors showed a clear difference between iTRAQ and ICAT-labeling and quantitation systems. iTRAQ allowed the identification of eightfold to ninefold more proteins and the precise localization of the cleavage site.
Stable isotope technique also provided information on OA. This technique was recently applied to investigate the secretome of chondrocytes cultured on monolayer or in explants (Polacek et al., 2010a). The authors showed differences between newly synthesized or cell-released proteins depending on the culture conditions. The same group further compared the secretome of articular chondrocytes with the one of mesenchymal stem cells (Polacek et al., 2010b) in order to consider the potential of autologous cell transplant. The same technique was also recently applied to chondrocytes in order to confirm the key role of mitochondria in OA (Calamia et al., 2011). Finally, stable isotope standards and capture by anti-peptide antibodies (SISCAPA) represents another recently developed method for quantitation of peptides in complex mixtures (Anderson et al., 2004). This combines an enrichment of sample in specific peptide by immunoaffinity with the use of internal labeled standard. To date, no application was published in the field of OA research.
Conclusion
Shotgun proteomics offer a set of technologies available for detailed analysis of various samples. They have significant application prospects. They permit the finding of new molecules of interest, the characterization of peptides that can be used in clinical practice or the deepening into the fundamental knowledge. Either one can prefer one technique to another to a specific purpose or one can use the complementary information from several techniques. Isotope labeling methods were used in global profiling strategies to identify biomarker. However, the need of simplification and enrichment of samples in low-abundant proteins and/or peptides have guided new experimental targeted strategies.
General Conclusion
In the recent years, proteomics have faced many challenges that have allowed its technical development and improvement. The identified problems often concerned the reproducibility and repeatability of experiments, particularly with gel electrophoresis. New technologies using more precise standards have then emerged. Another recurrent difficulty encountered in proteomic analysis of complex samples is the step of sample preparation. This is a fortiori the case in the field of osteoarthritis research which involves the study of samples such as cartilage and synovial fluid loaded with bulk proteins. Proteins of interest could be difficult to extract directly in that case. Several methods to overcome this issue or, at least, to reduce this concern have been proposed in the recent years and many proteins of interest have thus been highlighted by different research teams in the field of osteoarthritis.
Recent technological advances allow the exploration in depth of the proteome but also bring a wealth of information. MS-based technologies are able to identify and quantify thousands of proteins and/or peptides and its variants comprising post-translational modifications. In these conditions, the strategies for referencing a complete proteome may appear unrealistic. The examples cited above show that only one proteomic analysis can highlight a multitude of proteins and/or peptides of interest, then opening many possibilities for further investigation. Furthermore, proteomics and advanced technologies are valuable techniques that can be crucial tools to be included in a broader technological scheme in the context of targeted analysis to respond to a research hypothesis. Otherwise, recent technological advances propose new experimental strategies for proteome investigation. They are just beginning to be applied in the study of OA and still offer many opportunities for research application in this area. One still has to learn a lot from these techniques.
Conflict of Interest Statement
M. Gharbi and M. Deberg are employes of Artialis SA. Y. Henrotin is the funder and a shareholder of Artialis SA Company.
Acknowledgments
The authors equally contributed to this review. They would like to thank Christelle Boileau for her kind assistance in the manuscript preparation and edition.
Abbreviations
1DE, one-dimensional electrophoresis; 2DE, two-dimensional electrophoresis; 2D-DIGE, two-dimensional difference gel electrophoresis; CIA, collagen-induced arthritis; COMP, cartilage oligomeric matrix protein; CRP, C-reactive protein; DMOAD, Disease-modifying OA drug; HPLC, high performance liquid chromatography; ICAT, isotope-coded affinity tag; IL, interleukin; iTRAQ, isobaric tag for relative and absolute quantitation; LC, liquid chromatography; LRG, leucine-rich alpha 2 glycoprotein; MALDI, matrix-assisted laser desorption/ionization; MIAs, miscellaneous inflammatory arthritides; MMP, matrix metalloproteinase; MS, mass spectrometry; MRM, multiple-reaction monitoring; OA, osteoarthritis; RA, rheumatoid arthritis; ReaA, reactive arthritis; ROS, reactive oxygen species; RT–PCR, reverse transcriptase and polymerase chain reaction; SELDI, surface-enhancer laser desorption/ionization; SOD, superoxide dismutase; TIINE, new epitope of type II collagen; TLR, toll-like receptor; TNF, tumor necrosis factor; WB, western blot.
References
Anderson, N. L., Anderson, N. G., Haines, L. R., Hardie, D. B., Olafson, R. W., and Pearson, T. W. (2004). Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J. Proteome Res. 3, 235–244.
Baillet, A., Trocme, C., Berthier, S., Arlotto, M., Grange, L., Chenau, J., Quetant, S., Seve, M., Berger, F., Juvin, R., Morel, F., and Gaudin, P. (2010). Synovial fluid proteomic fingerprint: S100A8, S100A9 and S100A12 proteins discriminate rheumatoid arthritis from other inflammatory joint diseases. Rheumatology (Oxford) 49, 671–682.
Berenbaum, F. (2011). Diabetes-induced osteoarthritis: from a new paradigm to a new phenotype. Ann. Rheum. Dis. 70, 1354–1356.
Bondarenko, P. V., Chelius, D., and Shaler, T. A. (2002). Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Chem. 74, 4741–4749.
Burner, T. W., and Rosenthal, A. K. (2009). Diabetes and rheumatic diseases. Curr. Opin. Rheumatol. 21, 50–54.
Calamia, V., Rocha, B., Mateos, J. S., Fernández-Puente, P., Ruiz-Romero, C., and Blanco, F. J. (2011). Metabolic labeling of chondrocytes for the quantitative analysis of the interleukin-1-beta-mediated modulation of their intracellular and extracellular proteomes. J. Proteome Res. 10, 3701–3711.
Catterall, J. B., Rowan, A. D., Sarsfield, S., Saklatvala, J., Wait, R., and Cawston, T. E. (2006). Development of a novel 2D proteomics approach for the identification of proteins secreted by primary chondrocytes after stimulation by IL-1 and oncostatin M. Rheumatology (Oxford) 45, 1101–1109.
Chelius, D., and Bondarenko, P. V. (2002). Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J. Proteome Res. 1, 317–323.
Chellaiah, M., Kizer, N., Silva, M., Alvarez, U., Kwiatkowski, D., and Hruska, K. A. (2000). Gelsolin deficiency blocks podosome assembly and produces increased bone mass and strength. J. Cell Biol. 148, 665–678.
Clutterbuck, A. L., Smith, J. R., Allaway, D., Harris, P., Liddell, S., and Mobasheri, A. (2011). High throughput proteomic analysis of the secretome in an explant model of articular cartilage inflammation. J. Proteomics 74, 704–715.
de Seny, D., Fillet, M., Meuwis, M. A., Geurts, P., Lutteri, L., Ribbens, C., Bours, V., Wehenkel, L., Piette, J., Malaise, M., and Merville, M. P. (2005). Discovery of new rheumatoid arthritis biomarkers using the surface-enhanced laser desorption/ionization time-of-flight mass spectrometry ProteinChip approach. Arthritis Rheum. 52, 3801–3812.
de Seny, D., Sharif, M., Fillet, M., Cobraiville, G., Meuwis, M. A., Maree, R., Hauzeur, J. P., Wehenkel, L., Louis, E., Merville, M. P., Kirwan, J., Ribbens, C., and Malaise, M. (2011). Discovery and biochemical characterisation of four novel biomarkers for osteoarthritis. Ann. Rheum. Dis. 70, 1144–1152.
Dean, R. A., and Overall, C. M. (2007). Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell Proteomics 6, 611–623.
DesMarais, V., Ghosh, M., Eddy, R., and Condeelis, J. (2005). Cofilin takes the lead. J. Cell. Sci. 118(Pt 1), 19–26.
Djouad, F., Delorme, B., Maurice, M., Bony, C., Apparailly, F., Louis-Plence, P., Canovas, F., Charbord, P., Noel, D., and Jorgensen, C. (2007). Microenvironmental changes during differentiation of mesenchymal stem cells towards chondrocytes. Arthritis Res. Ther. 9, R33.
Drynda, S., Ringel, B., Kekow, M., Kuhne, C., Drynda, A., Glocker, M. O., Thiesen, H. J., and Kekow, J. (2004). Proteome analysis reveals disease-associated marker proteins to differentiate RA patients from other inflammatory joint diseases with the potential to monitor anti-TNFalpha therapy. Pathol. Res. Pract. 200, 165–171.
El Sayegh, T. Y., Arora, P. D., Ling, K., Laschinger, C., Janmey, P. A., Anderson, R. A., and McCulloch, C. A. (2007). Phosphatidylinositol-4,5 bisphosphate produced by PIP5KIgamma regulates gelsolin, actin assembly, and adhesion strength of N-cadherin junctions. Mol. Biol. Cell 18, 3026–3038.
Evangelou, E., Chapman, K., Meulenbelt, I., Karassa, F. B., Loughlin, J., Carr, A., Doherty, M., Doherty, S., Gomez-Reino, J. J., Gonzalez, A., Halldorsson, B. V., Hauksson, V. B., Hofman, A., Hart, D. J., Ikegawa, S., Ingvarsson, T., Jiang, Q., Jonsdottir, I., Jonsson, H., Kerkhof, H. J., Kloppenburg, M., Lane, N. E., Li, J., Lories, R. J., van Meurs, J. B., Nakki, A., Nevitt, M. C., Rodriguez-Lopez, J., Shi, D., Slagboom, P. E., Stefansson, K., Tsezou, A., Wallis, G. A., Watson, C. M., Spector, T. D., Uitterlinden, A. G., Valdes, A. M., and Ioannidis, J. P. (2009). Large-scale analysis of association between GDF5 and FRZB variants and osteoarthritis of the hip, knee, and hand. Arthritis Rheum. 60, 1710–1721.
Felson, D. T. (2010). Identifying different osteoarthritis phenotypes through epidemiology. Osteoarthritis Cartilage 18, 601–604.
Garcia, B. A., Platt, M. D., Born, T. L., Shabanowitz, J., Marcus, N. A., and Hunt, D. F. (2006). Protein profile of osteoarthritic human articular cartilage using tandem mass spectrometry. Rapid Commun. Mass Spectrom. 20, 2999–3006.
Gobezie, R., Kho, A., Krastins, B., Sarracino, D. A., Thornhill, T. S., Chase, M., Millett, P. J., and Lee, D. M. (2007). High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 9, R36.
Guo, D., Tan, W., Wang, F., Lv, Z., Hu, J., Lv, T., Chen, Q., Gu, X., Wan, B., and Zhang, Z. (2008). Proteomic analysis of human articular cartilage: identification of differentially expressed proteins in knee osteoarthritis. Joint Bone Spine 75, 439–444.
Gygi, S. P., Rochon, Y., Franza, B. R., and Aebersold, R. (1999a). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730.
Gygi, S. P., Rist, B., Gerber, S. A., Turecek, F., Gelb, M. H., and Aebersold, R. (1999b). Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999.
Haglund, L., Bernier, S. M., Onnerfjord, P., and Recklies, A. D. (2008). Proteomic analysis of the LPS-induced stress response in rat chondrocytes reveals induction of innate immune response components in articular cartilage. Matrix Biol. 27, 107–118.
Hermansson, M., Sawaji, Y., Bolton, M., Alexander, S., Wallace, A., Begum, S., Wait, R., and Saklatvala, J. (2004). Proteomic analysis of articular cartilage shows increased type II collagen synthesis in osteoarthritis and expression of inhibin betaA (activin A), a regulatory molecule for chondrocytes. J. Biol. Chem. 279, 43514–43521.
Hunter, D. J., Demissie, S., Cupples, L. A., Aliabadi, P., and Felson, D. T. (2004). A genome scan for joint-specific hand osteoarthritis susceptibility: The Framingham Study. Arthritis Rheum. 50, 2489–2496.
Ji, Y. H., Ji, J. L., Sun, F. Y., Zeng, Y. Y., He, X. H., Zhao, J. X., Yu, Y., Yu, S. H., and Wu, W. (2010). Quantitative proteomics analysis of chondrogenic differentiation of C3H10T1/2 mesenchymal stem cells by iTRAQ labeling coupled with on-line two-dimensional LC/MS/MS. Mol. Cell Proteomics 9, 550–564.
Kamphorst, J. J., van der Heijden, R., DeGroot, J., Lafeber, F. P., Reijmers, T. H., van El, B., Tjaden, U. R., van der Greef, J., and Hankemeier, T. (2007). Profiling of endogenous peptides in human synovial fluid by NanoLC-MS: method validation and peptide identification. J. Proteome Res. 6, 4388–4396.
Kevorkian, L., Young, D. A., Darrah, C., Donell, S. T., Shepstone, L., Porter, S., Brockbank, S. M., Edwards, D. R., Parker, A. E., and Clark, I. M. (2004). Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 50, 131–141.
Kuhn, E., Wu, J., Karl, J., Liao, H., Zolg, W., and Guild, B. (2004). Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics 4, 1175–1786.
Lambrecht, S., Dhaenens, M., Almqvist, F., Verdonk, P., Verbruggen, G., Deforce, D., and Elewaut, D. (2010). Proteome characterization of human articular chondrocytes leads to novel insights in the function of small heat-shock proteins in chondrocyte homeostasis. Osteoarthr. Cartil. 18, 440–446.
Lambrecht, S., Verbruggen, G., Verdonk, P. C., Elewaut, D., and Deforce, D. (2008). Differential proteome analysis of normal and osteoarthritic chondrocytes reveals distortion of vimentin network in osteoarthritis. Osteoarthr. Cartil. 16, 163–173.
Li, W. W., Nemirovskiy, O., Fountain, S., Rodney Mathews, W., and Szekely-Klepser, G. (2007). Clinical validation of an immunoaffinity LC-MS/MS assay for the quantification of a collagen type II neoepitope peptide: a biomarker of matrix metalloproteinase activity and osteoarthritis in human urine. Anal. Biochem. 369, 41–53.
Liao, H., Wu, J., Kuhn, E., Chin, W., Chang, B., Jones, M. D., O’Neil, S., Clauser, K. R., Karl, J., Hasler, F., Roubenoff, R., Zolg, W., and Guild, B. C. (2004). Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum. 50, 3792–3803.
Lorenz, P., Ruschpler, P., Koczan, D., Stiehl, P., and Thiesen, H. J. (2003a). From transcriptome to proteome: differentially expressed proteins identified in synovial tissue of patients suffering from rheumatoid arthritis and osteoarthritis by an initial screen with a panel of 791 antibodies. Proteomics 3, 991–1002.
Lorenz, P., Bantscheff, M., Ibrahim, S. M., Thiesen, H. J., and Glocker, M. O. (2003b). Proteome analysis of diseased joints from mice suffering from collagen-induced arthritis. Clin. Chem. Lab. Med. 41, 1622–1632.
MacGregor, J., Li, Q., Spector, T. D., and Williams, F. M. (2009). The genetic influence on radiographic osteoarthritis is site specific at the hand, hip and knee. Rheumatology (Oxford) 48, 277–280.
Marouga, R., David, S., and Hawkins, E. (2005). The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 382, 669–678.
Miyamoto, Y., Mabuchi, A., Shi, D., Kubo, T., Takatori, Y., Saito, S., Fujioka, M., Sudo, A., Uchida, A., Yamamoto, S., Ozaki, K., Takigawa, M., Tanaka, T., Nakamura, Y., Jiang, Q., and Ikegawa, S. (2007). A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 39, 529–533.
Nemirovskiy, O., Li, W. W., and Szekely-Klepser, G. (2010). Design and validation of an immunoaffinity LC-MS/MS assay for the quantification of a collagen type II neoepitope peptide in human urine: application as a biomarker of osteoarthritis. Methods Mol. Biol. 641, 253–270.
Nemirovskiy, O. V., Dufield, D. R., Sunyer, T., Aggarwal, P., Welsch, D. J., and Mathews, W. R. (2007). Discovery and development of a type II collagen neoepitope (TIINE) biomarker for matrix metalloproteinase activity: from in vitro to in vivo. Anal. Biochem. 361, 93–101.
Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., and Mann, M. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 1, 376–386.
Pecora, F., Forlino, A., Gualeni, B., Lupi, A., Giorgetti, S., Marchese, L., Stoppini, M., Tenni, R., Cetta, G., and Rossi, A. (2007). A quantitative and qualitative method for direct 2-DE analysis of murine cartilage. Proteomics 7, 4003–4007.
Polacek, M., Bruun, J. A., Johansen, O., and Martinez, I. (2010a). Differences in the secretome of cartilage explants and cultured chondrocytes unveiled by SILAC technology. J. Orthop. Res. 28, 1040–1049.
Polacek, M., Bruun, J. A., Elvenes, J., Figenschau, Y., and Martinez, I. (2010b). The secretory profiles of cultured human articular chondrocytes and mesenchymal stem cells: implications for autologous cell transplantation strategies. Cell Transplant 20, 1381–1393.
Rollin, R., Marco, F., Camafeita, E., Calvo, E., Lopez-Duran, L., Jover, J. A., Lopez, J. A., and Fernandez-Gutierrez, B. (2008). Differential proteome of bone marrow mesenchymal stem cells from osteoarthritis patients. Osteoarthr. Cartil. 16, 929–935.
Rosa, S. C., Goncalves, J., Judas, F., Mobasheri, A., Lopes, C., and Mendes, A. F. (2009). Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res. Ther. 11, R80.
Rosa, S. C., Rufino, A. T., Judas, F. M., Tenreiro, C. M., Lopes, M. C., and Mendes, A. F. (2011). Role of glucose as a modulator of anabolic and catabolic gene expression in normal and osteoarthritic human chondrocytes. J. Cell. Biochem. 112, 2813–2824.
Ross, P. L., Huang, Y. N., Marchese, J. N., Williamson, B., Parker, K., Hattan, S., Khainovski, N., Pillai, S., Dey, S., Daniels, S., Purkayastha, S., Juhasz, P., Martin, S., Bartlet-Jones, M., He, F., Jacobson, A., and Pappin, D. J. (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 3, 1154–1569.
Ruiz-Romero, C., Calamia, V., Mateos, J., Carreira, V., Martinez-Gomariz, M., Fernandez, M., and Blanco, F. J. (2009). Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell Proteomics 8, 172–189.
Ruiz-Romero, C., Carreira, V., Rego, I., Remeseiro, S., Lopez-Armada, M. J., and Blanco, F. J. (2008). Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 8, 495–507.
Ruiz-Romero, C., Lopez-Armada, M. J., and Blanco, F. J. (2005). Proteomic characterization of human normal articular chondrocytes: a novel tool for the study of osteoarthritis and other rheumatic diseases. Proteomics 5, 3048–3059.
Ruiz-Romero, C., Lopez-Armada, M. J., and Blanco, F. J. (2006). Mitochondrial proteomic characterization of human normal articular chondrocytes. Osteoarthr. Cartil. 14, 507–518.
Salgado, F. J., Vazquez, S., Iglesias, A., Perez-Diaz, A., Mera-Varela, A., Arias, P., and Nogueira, M. (2010). Application of thiophilic chromatography to deplete serum immunoglobulins in sample preparation for bidimensional electrophoresis. Anal. Chim. Acta 658, 18–31.
Sanchez, J. C., Rouge, V., Pisteur, M., Ravier, F., Tonella, L., Moosmayer, M., Wilkins, M. R., and Hochstrasser, D. F. (1997). Improved and simplified in-gel sample application using reswelling of dry immobilized pH gradients. Electrophoresis 18, 324–327.
Sinz, A., Bantscheff, M., Mikkat, S., Ringel, B., Drynda, S., Kekow, J., Thiesen, H. J., and Glocker, M. O. (2002). Mass spectrometric proteome analyses of synovial fluids and plasmas from patients suffering from rheumatoid arthritis and comparison to reactive arthritis or osteoarthritis. Electrophoresis 23, 3445–3456.
Stevens, A. L., Wishnok, J. S., Chai, D. H., Grodzinsky, A. J., and Tannenbaum, S. R. (2008). A sodium dodecyl sulfate-polyacrylamide gel electrophoresis-liquid chromatography tandem mass spectrometry analysis of bovine cartilage tissue response to mechanical compression injury and the inflammatory cytokines tumor necrosis factor alpha and interleukin-1beta. Arthritis Rheum. 58, 489–500.
Tanaka, Y., Akiyama, H., Kuroda, T., Jung, G., Tanahashi, K., Sugaya, H., Utsumi, J., Kawasaki, H., and Hirano, H. (2006). A novel approach and protocol for discovering extremely low-abundance proteins in serum. Proteomics 6, 4845–4855.
Uchida, T., Fukawa, A., Uchida, M., Fujita, K., and Saito, K. (2002). Application of a novel protein biochip technology for detection and identification of rheumatoid arthritis biomarkers in synovial fluid. J. Proteome Res. 1, 495–499.
Unlu, M., Morgan, M. E., and Minden, J. S. (1997). Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 18, 2071–2077.
Vasilopoulos, Y., Gkretsi, V., Armaka, M., Aidinis, V., and Kollias, G. (2007). Actin cytoskeleton dynamics linked to synovial fibroblast activation as a novel pathogenic principle in TNF-driven arthritis. Ann. Rheum. Dis. 66(Suppl. 3), iii23–iii28.
Vincourt, J. B., Lionneton, F., Kratassiouk, G., Guillemin, F., Netter, P., Mainard, D., and Magdalou, J. (2006). Establishment of a reliable method for direct proteome characterization of human articular cartilage. Mol. Cell Proteomics 5, 1984–1995.
Wilkins, M. R., Sanchez, J. C., Gooley, A. A., Appel, R. D., Humphery-Smith, I., Hochstrasser, D. F., and Williams, K. L. (1996). Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 13, 19–50.
Wilson, K. E., Marouga, R., Prime, J. E., Pashby, D. P., Orange, P. R., Crosier, S., Keith, A. B., Lathe, R., Mullins, J., Estibeiro, P., Bergling, H., Hawkins, E., and Morris, C. M. (2005). Comparative proteomic analysis using samples obtained with laser microdissection and saturation dye labelling. Proteomics 5, 3851–3858.
Wilson, R., Belluoccio, D., Little, C. B., Fosang, A. J., and Bateman, J. F. (2008). Proteomic characterization of mouse cartilage degradation in vitro. Arthritis Rheum. 58, 3120–3131.
Wu, J., Liu, W., Bemis, A., Wang, E., Qiu, Y., Morris, E. A., Flannery, C. R., and Yang, Z. (2007). Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 56, 3675–3684.
Xiang, Y., Sekine, T., Nakamura, H., Imajoh-Ohmi, S., Fukuda, H., Nishioka, K., and Kato, T. (2004). Proteomic surveillance of autoimmunity in osteoarthritis: identification of triosephosphate isomerase as an autoantigen in patients with osteoarthritis. Arthritis Rheum. 50, 1511–1521.
Keywords: proteomic, osteoarthritis
Citation: Gharbi M, Deberg M and Henrotin Y (2011) Application for proteomic techniques in studying osteoarthritis: a review. Front. Physio. 2:90. doi: 10.3389/fphys.2011.00090
Received: 31 August 2011; Paper pending published: 06 October 2011;
Accepted: 11 November 2011; Published online: 02 December 2011.
Edited by:
Ali Mobasheri, The University of Nottingham, UKReviewed by:
Ali Mobasheri, The University of Nottingham, UKAlexandrina Ferreira Mendes, University of Coimbra, Portugal
Francisco J. Blanco, INIBIC-Hospital Universitario A Coruña, Spain
Copyright: © 2011 Gharbi, Deberg and Henrotin. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Yves Henrotin, Bone and Cartilage Research Unit, University of Liège, Institute of Pathology, Level 5, Centre Hospitalier Universitaire du Sart-Tilman, 4000 Liège, Belgium. e-mail: yhenrotin@ulg.ac.be
†Myriam Gharbi and Yves Henrotin have contributed equally to this work.