
- 1 Department of Physical Performance, Norwegian School of Sport Sciences, Oslo, Norway
- 2 Third Faculty of Medicine, Department of Medicine, Charles University, Prague, Czech Republic
- 3 Protein Phosphorylation Research Group, de Duve Institute, Université Catholique de Louvain, Brussels, Belgium
Glycogen is the storage form of carbohydrates in mammals. In humans the majority of glycogen is stored in skeletal muscles (∼500 g) and the liver (∼100 g). Food is supplied in larger meals, but the blood glucose concentration has to be kept within narrow limits to survive and stay healthy. Therefore, the body has to cope with periods of excess carbohydrates and periods without supplementation. Healthy persons remove blood glucose rapidly when glucose is in excess, but insulin-stimulated glucose disposal is reduced in insulin resistant and type 2 diabetic subjects. During a hyperinsulinemic euglycemic clamp, 70–90% of glucose disposal will be stored as muscle glycogen in healthy subjects. The glycogen stores in skeletal muscles are limited because an efficient feedback-mediated inhibition of glycogen synthase prevents accumulation. De novo lipid synthesis can contribute to glucose disposal when glycogen stores are filled. Exercise physiologists normally consider glycogen’s main function as energy substrate. Glycogen is the main energy substrate during exercise intensity above 70% of maximal oxygen uptake (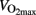 ) and fatigue develops when the glycogen stores are depleted in the active muscles. After exercise, the rate of glycogen synthesis is increased to replete glycogen stores, and blood glucose is the substrate. Indeed insulin-stimulated glucose uptake and glycogen synthesis is elevated after exercise, which, from an evolutional point of view, will favor glycogen repletion and preparation for new “fight or flight” events. In the modern society, the reduced glycogen stores in skeletal muscles after exercise allows carbohydrates to be stored as muscle glycogen and prevents that glucose is channeled to de novo lipid synthesis, which over time will causes ectopic fat accumulation and insulin resistance. The reduction of skeletal muscle glycogen after exercise allows a healthy storage of carbohydrates after meals and prevents development of type 2 diabetes.
) and fatigue develops when the glycogen stores are depleted in the active muscles. After exercise, the rate of glycogen synthesis is increased to replete glycogen stores, and blood glucose is the substrate. Indeed insulin-stimulated glucose uptake and glycogen synthesis is elevated after exercise, which, from an evolutional point of view, will favor glycogen repletion and preparation for new “fight or flight” events. In the modern society, the reduced glycogen stores in skeletal muscles after exercise allows carbohydrates to be stored as muscle glycogen and prevents that glucose is channeled to de novo lipid synthesis, which over time will causes ectopic fat accumulation and insulin resistance. The reduction of skeletal muscle glycogen after exercise allows a healthy storage of carbohydrates after meals and prevents development of type 2 diabetes.
Introduction
Exercise is considered a cornerstone in prevention and treatment of type 2 diabetes and several mechanisms may contribute to the benefits of exercise. Acutely, exercise improves insulin sensitivity in both healthy subjects and insulin resistant people (Heath et al., 1983; Mikines et al., 1988). The improved insulin sensitivity after a single bout of exercise is short-lived but repeated bouts of endurance training improve insulin sensitivity beyond the acute effect of the last training session, and insulin sensitivity correlates with oxidative capacity in skeletal muscles (Koivisto et al., 1986; Bruce et al., 2003). Importantly, the risk for development of type 2 diabetes is reduced by yearlong training (Knowler et al., 2002).
Skeletal muscles are the tissue that transforms chemical energy to mechanical work and therefore uses the majority of energy during exercise; glycogen is the main substrate during high intensity exercise (Hermansen et al., 1967; Romijn et al., 1993). Skeletal muscles are, however, also the major tissue where insulin stimulates glucose uptake to remove glucose from the blood, and the glucose taken up is incorporated into glycogen (DeFronzo et al., 1981b; Shulman et al., 1990). The logic link between glycogen content and insulin sensitivity is also supported experimentally (Jensen et al., 1997).
The flux by which glucose is removed from the blood into skeletal muscle glycogen is the major determinant of insulin sensitivity (Højlund and Beck-Nielsen, 2006). Insulin stimulates glucose uptake via translocation of GLUT4 (Etgen et al., 1996; Larance et al., 2008). Endurance training increases expression of GLUT4 and other proteins involved in insulin signaling and glucose metabolism (Houmard et al., 1993), but the mechanism determining insulin sensitivity remains poorly understood. Nevertheless, the major defect in insulin resistant people is that the non-oxidative glucose disposal (glycogen synthesis) is reduced (Højlund and Beck-Nielsen, 2006). Several reviews have discussed the effect of endurance training on insulin sensitivity from a molecular point of view (Wojtaszewski et al., 2002; Maarbjerg et al., 2011).
Exercise physiologists have performed numerous studies on glycogen utilization during exercise and studied the effects of nutritional supply for optimal glycogen repletion after exercise (Ivy, 2001; Betts and Williams, 2010). Rapid glycogen repletion requires that high rates of blood glucose must be taken up by skeletal muscles, and insulin sensitivity is high after exercise. Diabetes is defined by elevated blood glucose and a major defect is that insulin-stimulated glucose uptake and glycogen synthesis is impaired in skeletal muscle (Shulman et al., 1990). A common point at issue for both diabetologists and exercise physiologists is: How can blood glucose rapidly be converted into skeletal muscle glycogen? In the present review we have taken the view of exercise physiologists to discuss the role of skeletal muscle glycogen in regulation of insulin sensitivity.
Glycogen
Glycogen is the molecular form of carbohydrates stored in humans and other mammals. A glycogen particles in skeletal muscles can contain as much as 50,000 glucose moieties linked with α(1 → 4) bonds and branched by α(1 → 6) bonds (Meléndez et al., 1999). In humans, ∼80% of the glycogen is stored in skeletal muscles, simply because skeletal muscles account for ∼40–50% of body weight in healthy young men and the glycogen concentration is 80–150 mmol kg ww−1 (Ivy et al., 1988; Hawley et al., 1997; Jensen et al., 2011). The liver has a higher glycogen concentration, but as the liver is much smaller (∼1.5 kg) and the total amount of liver glycogen is ∼100 g (Taylor et al., 1996). Other tissue, like the heart and brain contains minor glycogen stores with important physiological function.
A main function of glycogen is to maintain a physiological blood glucose concentration, but only liver glycogen directly contributes to release of glucose into the blood. Skeletal muscles are unable to release glucose (because muscles lack glucose 6-phosphatase) and muscles glycogen is mainly a local energy substrate for exercise, rather than an energy source to maintain blood glucose concentration during fasting. Indeed, muscle glycogen can be broken down to lactate, which can be transported to the liver and via gluconeogenesis in the liver contribute to maintaining euglycemia (Cori cycle). However, humans do not show major decrease in muscle glycogen content during fasting (Nieman et al., 1987; Vendelbo et al., 2011). In contrast, the liver glycogen content decreases rapidly during fasting and the liver glycogen content has decreased by ∼65% after 24 h fasting (Magnusson et al., 1992). So, why is the majority of glycogen stored in muscles?
We believe that the main function of skeletal muscle glycogen, from an evolutional point of view, is to serve as an energy store in “fight or flight” situations. In the heart and the brain, glycogen is also the energy substrate that can generate anaerobic energy during short-term oxygen deficiency contributing to survival (Prebil et al., 2011). Indeed, reduced glycogen content in skeletal muscles increases insulin sensitivity (Jensen et al., 1997), but the increased insulin sensitivity can again be related to the importance to restore glycogen content rapidly for new challenges. Glycogen stored intracellularly is immediately available for energy production, and the rate of energy production far exceeds the flux of glucose into skeletal muscles. Therefore, muscle glycogen may have been important for survival during acute emergencies as substrate for “fight or flight” reactions, whereas accumulated fat has its importance for survival during starvation.
The glycogen content increases slightly by acute intake of large amount of carbohydrates (Hawley et al., 1997). However, an acute bout of glycogen depleting exercise can double glycogen content in skeletal muscles if high amount of carbohydrates are ingested for 3 days (Bergström and Hultman, 1966); this phenomenon is called super compensation. The glycogen content is higher in endurance trained subjects compared to untrained subjects (Hickner et al., 1997), and glycogen content increases in muscles after endurance training (Burgomaster et al., 2005). In contrast, prolonged intake of high amount of carbohydrates does not increase glycogen content in skeletal muscles, and the excess carbohydrate ingested is converted to lipid (Acheson et al., 1988; Jensen, 2009). Therefore, the glycogen content in skeletal muscles from obese and type 2 diabetes subjects is comparable to lean subjects or may even be reduced (Shulman et al., 1990; He and Kelley, 2004). Since exercise increases the glycogen storage capacity in skeletal muscles, it is likely that inactivity will reduce storage capacity. Interestingly, the ratio between glycogen content and oxidative capacity was increased in muscles from obese subjects (He and Kelley, 2004). Is this indicating increased glycogen content relative to the storage capacity in muscles from obese subjects? A reduced glycogen storage capacity in muscles from insulin resistant subjects will cause a stronger feedback inhibition of glycogen synthase at similar glycogen content and deteriorate glucose regulation, and the glycogen content relative to glycogen storage capacity may regulate insulin sensitivity. Indeed, it has been reported that hyperglycemia compensate for impaired insulin-mediated activation of glycogen synthase and glycogen storage in type 2 diabetic subjects (Kelley and Mandarino, 1990; Vaag et al., 1992; Mevorach et al., 1998), but these data also show a defect in regulation of glycogen storage as a higher glucose concentration is required to uphold glycogen synthesis. Such forced glycogen synthesis may increase metabolic stress.
In rats, glycogen content is increased the day after exercise when fed normal chow (Hespel and Richter, 1990; Kawanaka et al., 2000) and increased even more when rats have free access to chow and given drink containing glucose (Hespel and Richter, 1990; Derave et al., 2000). Glycogen content is also increased in epitrochlearis muscles when 24 h fasted rats are fed chow for another 24 h; the glycogen content is twice as high in epitrochlearis muscles from fasted–refed rats compared to rats with free access to chow continuously (Jensen et al., 1997, 2006; Lai et al., 2007). Both exercise and fasting decrease glycogen in the muscle where supercompensation occurs (Hespel and Richter, 1990; Jensen et al., 1997, 2006), but is not understood why glycogen content is increased after glycogen depletion.
Insulin-Stimulated Glucose Uptake
Insulin regulates many biological functions in skeletal muscle and stimulation of skeletal muscle glucose uptake is one of the most important processes regulated by insulin (Taniguchi et al., 2006). Skeletal muscle has been reported to account for 70–75% of insulin-stimulated glucose disposal during hyperinsulinemic clamps and, therefore, represents a principle tissue mediating whole body glucose homeostasis (DeFronzo et al., 1981a; Shulman et al., 1990). After an oral glucose tolerance test, skeletal muscles also dispose a substantial part of the glucose. It has been reported that 30–40% of the glucose is immediately oxidized after an oral glucose tolerance test, and ∼15% of the ingested glucose is stored as muscle glycogen (Kelley et al., 1988). However, after glycogen depleting exercise, more 40% of the ingested glucose can be stored as skeletal muscles glycogen of trained subjects (Hickner et al., 1997; Greiwe et al., 1999). Untrained subjects have lower capacity to store ingested carbohydrates after exercise than endurance trained subjects (Hickner et al., 1997; Greiwe et al., 1999), but exercise will still channel more of the ingested glucose into skeletal muscles glycogen and reduces metabolic stress in untrained subjects.
Insulin stimulates skeletal muscle glucose uptake through an increase of GLUT4 translocation from intracellular storage vesicles to the plasma membrane and transverse tubules (Etgen et al., 1996; Lauritzen et al., 2008). Insulin initiates its effect in skeletal muscle by binding to the insulin receptor, followed by receptor auto-phosphorylation. This induces a series of phosphorylation and protein–protein interactions mediating insulin signaling (Shepherd, 2005). In brief, insulin activates insulin receptor tyrosine kinase activity that increases the tyrosine phosphorylation of insulin receptor substrate (IRS) proteins, which recruit and activates class 1A phosphatidylinositol 3-kinase (PI3K; Figure 1). Activation of PI3K catalyzes the formation of phosphatidylinositol 3,4,5-trisphosphate (PIP3), which recruits both PDK1 and PKB to the phospholipid, and subsequently allows PKB to be activated through phosphorylation by PDK1 at threonine 308 (Alessi and Cohen, 1998). The mammalian target of rapamycin complexed with Rictor (mTORC2) phosphorylates PKB at serine 473, and phosphorylation of both sites is required for full PKB activity (Alessi and Cohen, 1998; Sarbassov et al., 2005). Several lines of evidence have indicated the critical role of PKB phosphorylation and activation in the regulation of insulin-stimulated glucose uptake (Larance et al., 2008). It is the PKBβ isoform that controls whole body glucose homeostasis (Cleasby et al., 2007; Schultze et al., 2011).
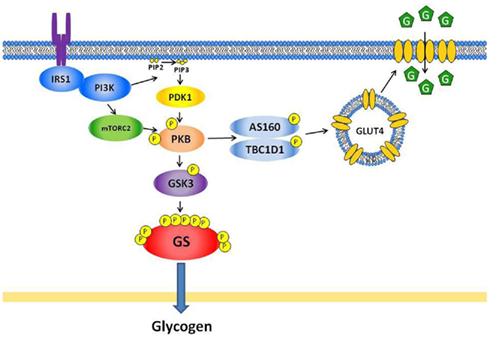
Figure 1. Insulin signaling pathways regulating glucose transport and glycogen synthase in skeletal muscle. Insulin activates protein kinase B (PKB) through phosphatidylinositol 3-kinase (PI3K) and two upstream kinases; namely phosphoinositide-dependent protein kinase-1 (PDK1; phosphorylates PKB at threonine 308) and the mammalian target of rapamycin complexed with Rictor (mTORC2; phosphorylates PKB at serine 473). The activated PKB phosphorylates Akt substrate of 160 kDa (AS160, also called TBC1D4) and TBC1D1, which inhibits Rab GTPase activity and promotes GTP binding to Rabs, thereby allowing GLUT4 translocation. For glycogen synthesis, the activated PKB phosphorylates glycogen synthase kinase-3 (GSK3), which leads to inhibition of GSK3 activity and subsequently dephosphorylation and activation of glycogen synthase (GS). IRS, insulin receptor substrate; PIP2, phosphatidylinositol 4,5-biphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; G, glucose.
PKB-mediated phosphorylation of AS160 and TBC1D1 has recently emerged to regulate insulin-stimulated GLUT4 translocation beyond PKB (Arias et al., 2007; Sakamoto and Holman, 2008). Insulin-stimulated phosphorylation of AS160 and TBC1D1 seems, however, not to be regulated by glycogen content as we did not find correlation between insulin-stimulated glucose uptake and AS160 phosphorylation using the phospho-Akt substrate (PAS) antibody (Lai et al., 2010b).
Insulin also activates glycogen synthase (Cohen, 1993; Jensen and Lai, 2009). Glycogen synthase (GS) is phosphorylated at nine sites and insulin stimulates dephosphorylation of glycogen synthase (Cohen, 1993; Jensen and Lai, 2009). Insulin stimulates dephosphorylation of glycogen synthase via PKB-mediated phosphorylation of GSK3 (McManus et al., 2005; Bouskila et al., 2008; Jensen and Lai, 2009). Phosphorylation of GSK3 decreases kinase activity which will decrease phosphorylation of GS and increase glycogens synthase fractional activity (Lai et al., 2007, 2010b; Jensen and Lai, 2009).
Glycogen synthase is also activated by glucose 6-phosphate and allosteric activation is necessary for normal rate of glycogen synthesis (Jensen and Lai, 2009; Bouskila et al., 2010). Glycogen synthase activity with high concentrations of glucose 6-phosphate (>8 mM) is independent of phosphorylation; activity with high glucose 6-phosphate concentration is called total activity. However, dephosphorylation of glycogen synthase increases affinity for glucose 6-phosphate and glycogen synthase activity with a physiological concentration of glucose 6-phosphate (e.g., 0.17 mM) describes activation of glycogen synthase (Jensen and Lai, 2009).
Recently, a mutated glycogen synthase was developed where phosphorylation-mediated regulation was normal, but allosteric activation by glucose 6-phosphate was abolished (Bouskila et al., 2010). Data achieved with the knockin mice expressing a GS without glucose 6-phophate activation provided seminal information about regulation of glycogen synthase (Brady, 2010). Bouskila et al. (2010) showed that allosteric activation of GS is necessary for regulation of glycogen synthesis in skeletal muscles. Therefore, dephosphorylation of glycogen synthase increases glycogen synthesis mainly by increasing GS affinity for glucose 6-phosphate and allosteric activation. The GS knockin mice without allosteric activation by glucose 6-phosphate also answered the challenging question why AICAR (AMPK activator), which reduces GS fractional activity, increases glycogen content: AICAR stimulates glucose uptake and glucose 6-phosphate mediated GS activation stimulates glycogen synthesis (Hunter et al., 2011).
Impaired insulin-stimulated disposal is a common feature in people with type 2 diabetes, and causes inability to maintain blood glucose in a normal range. Insulin-stimulated glycogen synthesis is reduced in skeletal muscle in insulin resistant people and prevent proper regulation of blood glucose (Shulman et al., 1990) and particularly non-oxidative glucose metabolism is reduced in insulin resistant subjects (Højlund and Beck-Nielsen, 2006). It is also a consistent finding that insulin signaling is reduced at several sites, like PI3K, PKB, GSK3, and GS, in muscle from insulin resistance (Kim et al., 2000; Morino et al., 2005; Højlund and Beck-Nielsen, 2006). Obesity is a strong risk factor for insulin resistance but accumulation of fat per se does not cause insulin resistance, as mice depleted for adipose triglyceride lipase (ATGL) accumulates fat in muscles and heart, but do not develop insulin resistance (Haemmerle et al., 2006). This finding suggest that lipid intermediates like long chain acyl-CoA, diacylglycerol, or ceramides causes insulin resistance (Franch et al., 2002; Samuel et al., 2010).
Effect of Exercise on Insulin Sensitivity and Insulin Signaling
When insulin is administrated immediately after contraction or exercise, there is an additive increase in glucose uptake. This increased glucose uptake immediately after exercise occurs because the effect of muscle contraction on glucose uptake is still present; e.g., AMPK and glycogen synthase remains activated (Franch et al., 1999; Musi et al., 2001). Insulin-mediated activation of the proximal insulin signaling at the level of IRS1 and PI3K is unchanged after exercise (Wojtaszewski et al., 1999; Jessen et al., 2003). Most studies also report that insulin-stimulated PKB activity is unchanged after exercise (Wojtaszewski et al., 1999; Jessen et al., 2003), but some recent studies revealed that prior contractile activity induces higher insulin-stimulated PKB threonine 308 phosphorylation compared to rested muscles, whereas insulin-stimulated PKB phosphorylation at serine 473 was unchanged by exercise (Arias et al., 2007; Lai et al., 2009). Whether this increased site specific PKB phosphorylation contributes to training-enhanced insulin sensitivity is currently unknown. However, insulin-stimulated phosphorylation of GSK3, the critical regulator of GS activity, was not increased after muscle contraction (Lai et al., 2009, 2010b).
Exercise training enhances insulin sensitivity. It is well established that the enhanced insulin sensitivity after training is associated with adaptations in skeletal muscles such as increased expression of key proteins like GLUT4, hexokinase II, and GS, involved in insulin-stimulated glucose metabolism (Dela et al., 1993; Frosig et al., 2007). However, the signaling event that leads to enhanced insulin sensitivity after exercise training is not conclusive. It has been reported that short-term exercise training increased insulin-stimulated PI3K activity (Houmard et al., 1999), but other studies have reported that insulin-stimulated IRS1-associated PI3K activity is unchanged or reduced after training (Christ-Roberts et al., 2004; Frosig et al., 2007). While the training effect on PI3K activity is inconsistent, several studies have reported that enhanced insulin sensitivity was associated with increased PKB phosphorylation and expression (Christ-Roberts et al., 2004; Frosig et al., 2007; Wadley et al., 2007). Consistent with the increased PKB activation after training, it has also been demonstrated that insulin-mediated AS160 phosphorylation is enhanced after training (Frosig et al., 2007; Vind et al., 2011). However, exercise normalized insulin-mediated AS160 phosphorylation in skeletal muscle from type 2 diabetic subjects but without normalizing insulin-stimulated glucose disposal (Vind et al., 2011).
Exercise training also increases insulin-stimulated glucose uptake and GLUT4 translocation in muscles from obese Zucker rats (Etgen et al., 1997). Skeletal muscles from the obese Zucker rats develop severe insulin resistance and impaired insulin signaling (Christ et al., 2002). However, although training increases insulin-stimulated glucose uptake in skeletal from obese Zucker rats, insulin-mediated activation of PI3K and PKB remained low after training (Christ et al., 2002). The signaling mechanisms which increase insulin-stimulated glucose uptake after training remain to be determined.
Glycogen Utilization During Exercise
Energy consumption at rest is low; oxygen uptake at rest is typically ∼0.25 L O2 and carbohydrate oxidation is ∼0.1 g min−1 (Hermansen et al., 1967; van Loon et al., 2001), and the rate of carbohydrate oxidation gradually decreases during fasting. At rest, the rate of carbohydrate oxidation depends mainly on the diet and exercise prior to measurements, and the glycogen utilization in skeletal muscles at rest is low or absent (van Loon et al., 2001).
The utilization of carbohydrate during exercise can easily be calculated from oxygen uptake(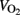 ) and respiratory exchange ratio (RER). Normally carbohydrate oxidation is calculated without taking protein oxidation in consideration; tables and formulae have been published for such calculations (Frayn, 1983; Peronnet and Massicotte, 1991). The relative (as well as absolute) rate of carbohydrate oxidation depends on exercise intensity and well-trained persons have a much higher capacity to metabolize glucose and fat compared to untrained persons. During exercise above 70% the major carbohydrate source is muscle glycogen (Romijn et al., 1993; van Loon et al., 2001).
) and respiratory exchange ratio (RER). Normally carbohydrate oxidation is calculated without taking protein oxidation in consideration; tables and formulae have been published for such calculations (Frayn, 1983; Peronnet and Massicotte, 1991). The relative (as well as absolute) rate of carbohydrate oxidation depends on exercise intensity and well-trained persons have a much higher capacity to metabolize glucose and fat compared to untrained persons. During exercise above 70% the major carbohydrate source is muscle glycogen (Romijn et al., 1993; van Loon et al., 2001).
The physical form of humans are determined by their capacity to oxidize energy substrates (carbohydrates and fat), which is reflected in ability to utilize oxygen. Maximal oxygen uptake is used to describe oxidative capacity, and values of 40–50 ml kg−1 min−1 are common in healthy young men. However,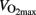 can vary from below 15 ml kg−1 min−1 in elderly people to more than 90 ml kg−1 min−1 in some endurance athletes. Capacity for carbohydrate oxidation varies correspondingly. Although, well-trained people utilize more fat during exercise, there is huge variation in carbohydrate oxidation. Well-trained subjects can more than oxidize 3 g min−1 (Hermansen et al., 1967) which results in oxidation of 180 g carbohydrate during 1 h of intense exercise.
can vary from below 15 ml kg−1 min−1 in elderly people to more than 90 ml kg−1 min−1 in some endurance athletes. Capacity for carbohydrate oxidation varies correspondingly. Although, well-trained people utilize more fat during exercise, there is huge variation in carbohydrate oxidation. Well-trained subjects can more than oxidize 3 g min−1 (Hermansen et al., 1967) which results in oxidation of 180 g carbohydrate during 1 h of intense exercise.
During cycling, ∼20 kg of muscle is active (Boushel et al., 2011) and cycling is the preferred type of activity in exercise physiology. Several studies have investigated glycogen breakdown during cycling and exercise intensity cannot be maintained when the active muscles are depleted for glycogen (Hermansen et al., 1967). Hermansen et al. (1967) reported a glycogen content of only 7 mmol kg ww−1at exhaustion after cycling at 75% of 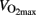 Most studies find low glycogen content at exhaustion, but the degree of depletion depends of the exercise intensity, and the glycogen depletion is most pronounced when cycling to exhaustion at ∼75% of
Most studies find low glycogen content at exhaustion, but the degree of depletion depends of the exercise intensity, and the glycogen depletion is most pronounced when cycling to exhaustion at ∼75% of 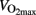 (Saltin and Karlsson, 1971). Most studies report glycogen concentration of 7–20 mmol kg ww−1 in m. vastus lateralis after cycling to exhaustion (Hermansen et al., 1967; Nieman et al., 1987; Hickner et al., 1997). Glycogen concentration in m. vastus lateralis is typically 80–150 mmol kg ww−1 in rested muscles (Coyle et al., 1986; Nieman et al., 1987; Hawley et al., 1997; van Loon et al., 2001).
(Saltin and Karlsson, 1971). Most studies report glycogen concentration of 7–20 mmol kg ww−1 in m. vastus lateralis after cycling to exhaustion (Hermansen et al., 1967; Nieman et al., 1987; Hickner et al., 1997). Glycogen concentration in m. vastus lateralis is typically 80–150 mmol kg ww−1 in rested muscles (Coyle et al., 1986; Nieman et al., 1987; Hawley et al., 1997; van Loon et al., 2001).
During running, the energy consumption is ∼1 kcal kg−1 km−1 (Åstrand and Rodahl, 1992). This means that an 85-kg person will use about 850 kcal during a 10-km run; 850 kcal corresponds to ∼200 g carbohydrate or ∼90 g fat. During exercise, carbohydrates and fat are used simultaneously. During running, a larger muscle mass is used and less glycogen is broken down in the leg muscles and m. gastrocnemius is not depleted for glycogen at exhaustion (Madsen et al., 1990). Cross-country skiing mainly depletes glycogen stores in arms (Ortenblad et al., 2011).
The intensity of exercise, together with duration, determines the amount of energy used in the training session. High intensity intermittent training (HIT) is often performed as 30 s “all-out” cycling in experiments. The power that can be produced during 30 s “all-out” corresponds to ∼250% of 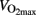 (Gibala et al., 2006) and 3–5 min rest is typically allowed between bouts. The metabolism in skeletal muscles during the moderate intensity training and HIT differs dramatically. During HIT anaerobic provides the major part of energy, which is repaid with aerobic processes in the rest periods. During prolonged continuous exercise energy consumption will be rather stable, and skeletal muscle glycogen content will be reduced by 50–70% after 60 min cycling at 75% of
(Gibala et al., 2006) and 3–5 min rest is typically allowed between bouts. The metabolism in skeletal muscles during the moderate intensity training and HIT differs dramatically. During HIT anaerobic provides the major part of energy, which is repaid with aerobic processes in the rest periods. During prolonged continuous exercise energy consumption will be rather stable, and skeletal muscle glycogen content will be reduced by 50–70% after 60 min cycling at 75% of 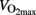 (Hermansen et al., 1967; Saltin and Karlsson, 1971).
(Hermansen et al., 1967; Saltin and Karlsson, 1971).
During high intensity training the power output is high with substantial anaerobic energy turn over and high adrenaline concentration. Jacobs et al. (1982) reported that a single 30 s all-out cycling decreased glycogen content by 22% corresponding to ∼20 mmol kg ww−1. Esbjornsson-Liljedahl et al. (1999) also found that a single 30 s all-out cycling in males and females decreased glycogen content by ∼25% in both type I and type II fibers. Furthermore, three bouts of 30 s all-out cycling with 20 min rest between sprints decreased glycogen content by more than 50% in type II fibers and nearly 50% in type I fibers in both females and males (Esbjornsson-Liljedahl et al., 2002). These data show that high intensity training effectively decreases glycogen content in skeletal muscles.
Adrenaline-Stimulated Glycogen Breakdown
In 1928, Carl and Gerty Cori showed that adrenaline injection into young fasted rats increased glycogen content in the liver whereas carcass glycogen content decreased (Cori and Cori, 1928). It was concluded that “muscle glycogen is an indirect source of blood sugar” (Cori and Cori, 1928); in biochemistry books this metabolism of glucose is called the Cori cycle. The Cori cycle states that skeletal muscles glycogen is broken down during adrenaline stimulation and released as lactate, and converted to glucose in the liver.
It is well-understood that adrenaline stimulates glycogen breakdown via β-adrenergic receptors and phosphorylation (activation) of glycogen phosphorylase (Cohen, 2002). In details, β-adrenergic receptors activate adenylyl cyclase via G-proteins which results in cAMP accumulation and activation of PKA. PKA-mediated phosphorylation of glycogen phosphorylase kinase increases phosphorylation of glycogen phosphorylase (Cohen, 2002). Phosphorylated glycogen phosphorylase is active and catalyzes breakdown of glycogen to glucose 1-phosphate. Skeletal muscles mainly express β2-adrenergic receptors and adrenaline, rather than noradrenaline, stimulates glycogen breakdown (Jensen et al., 1995). Adrenaline-mediated glycogen synthase inactivation also occurs via cAMP and PKA (Cohen, 2002; Jensen et al., 2007, 2008).
The amount of glycogen breakdown in resting muscles during adrenaline stimulation is significant but relatively low compared to glycogen breakdown during intense muscle contraction (Jensen et al., 1989; Jensen and Dahl, 1995; Aslesen and Jensen, 1998; Lai et al., 2007, 2009). In humans, it has also been shown that adrenaline infusion activates glycogen phosphorylase and stimulates glycogen breakdown (Chasiotis et al., 1983). Indeed, not all studies find that adrenaline infusion reduces glycogen content in humans (Laurent et al., 1998), but it has consistently been reported release of lactate from muscles during adrenaline infusion (Simonsen et al., 1992; Qvisth et al., 2007; Gjedsted et al., 2011). In humans, we infused adrenaline for 4 h and found increased plasma lactate concentration and lower glycogen content the following day compared to the day after saline infusion (Jensen et al., 2011). Despite that glycogen content was reduced the day after adrenaline infusion, we did not find elevated insulin-stimulated glucose disposal although there was a tendency (p = 0.14) for an increased insulin sensitivity the day after adrenaline infusion (Jensen et al., 2011).
The energy consumption during adrenaline stimulation is not increased similarly to the activation of glycogen phosphorylase because glycolytic intermediates accumulate and via feedback mechanisms inhibit glycogenolytic flux (Connett and Sahlin, 1996; Jensen, 2009). Adrenaline-stimulated glycogen breakdown in resting muscles is fiber type dependent and occurs only in muscles rich in fast-twitch fibers (Jensen et al., 1989; Jensen and Dahl, 1995). With histochemical analysis, it has been shown that adrenaline stimulates glycogen breakdown significantly in type II fibers (fast-twitch) but not in type I (slow-twitch) muscle fibers (Jensen and Dahl, 1995). In vivo, adrenaline acutely decreases glycogen content, and Nolte et al. (1994) reported a 60% reduction in glycogen content in epitrochlearis 2 h after subcutaneous injection of adrenaline in conjunction with increased insulin sensitivity. We have also found that glycogen content is reduced by ∼50% in fast-twitch epitrochlearis muscles 3 h after subcutaneously adrenaline injection but glycogen content did not decrease significantly in the slow-twitch soleus muscle. Interestingly, adrenaline injection increased insulin-stimulated glucose uptake in epitrochlearis, but not in soleus muscles (Kolnes and Jensen, unpublished observation). Adrenaline infusion with osmotic mini pumps for 24 h also lowered glycogen content and increased insulin sensitivity in epitrochlearis muscles (Jensen et al., 2005). However, glycogen content was normal after 11 days of adrenaline infusion, but insulin sensitivity in epitrochlearis muscles remained elevated (Jensen et al., 2005).
The physiological role of adrenaline-stimulated glycogen breakdown in non-active muscles is debated. However, there is some evidence that adrenaline-mediated glycogen breakdown has physiological role. Taylor et al. (1993) reported that skeletal muscle glycogen content increased after a carbohydrate rich meal reaching a maximum after 4 h for thereafter to decrease. These data suggest that skeletal muscle glycogen is used in rested muscles and adrenaline-mediated glycogen breakdown may be the mechanism.
Glycogen Content and Insulin Sensitiivty
The glycogen content contributes to regulation of glucose uptake during muscle contraction. In epitrochlearis muscles with normal glycogen content, contraction-stimulated glucose uptake correlated with glycogen breakdown when muscles were stimulated at different intensities (Aslesen et al., 2001). Varying the glycogen content prior to muscle contraction also showed that contraction-stimulated glucose uptake inversely correlates with glycogen content prior to muscle contraction (Lai et al., 2010b). The mechanistic link between low glycogen content and high rate of contraction-stimulated glucose uptake has not been determined, but contraction-mediated AMPK activation is higher in muscles with low glycogen content and may cause the higher glucose uptake (Lai et al., 2010b).
The glycogen content also influences insulin action. We have in several studies investigated the role of glycogen content on insulin- and contraction-stimulated glucose uptake, glycogen synthase activation, and activation of signaling proteins in skeletal muscles (Jensen et al., 1997, 2006; Lai et al., 2007, 2009, 2010a,b). In 1997, we demonstrated an inverse relationship between glycogen content and insulin-stimulated glucose uptake in the isolated rat skeletal muscle (Jensen et al., 1997). In that study, we observed that the ability of insulin to stimulate glucose uptake was markedly increased in muscle with low glycogen content, compared to muscle with normal and high glycogen content (Jensen et al., 1997). When the glycogen content was increased acutely by fasting–refeeding, insulin signaling, and insulin-stimulated glucose uptake was unchanged (Jensen et al., 1997, 2006). However, high glycogen content decreased insulin-stimulated glycogen synthesis and increased glycolytic flux (Jensen et al., 2006). Such changed glucose metabolism may over time cause insulin resistance (Jensen, 2009).
Several studies have documented similar relationship between glycogen content and metabolic regulation. It has been shown that GLUT4 protein content on cell surface was inversely correlated with glycogen content during insulin stimulation (Derave et al., 1999), suggesting that insulin-stimulated GLUT4 translocation is regulated by the level of muscle glycogen content. Furthermore, the enhanced insulin-stimulated glucose uptake observed after an acute bout of exercise can be preserved for more than 48 h by carbohydrate deprivation (Cartee et al., 1989), whereas the insulin-stimulated glucose uptake returned to normal when rat were fed chow, which is rich in carbohydrate (Young et al., 1983; Cartee et al., 1989).
Varying glycogen content acutely does not change the early steps of proximal insulin signaling, including insulin receptor tyrosine kinase activity, insulin receptor tyrosine phosphorylation, and PI3K activity (Derave et al., 2000; Kawanaka et al., 2000; Jensen et al., 2006). Interestingly, insulin-stimulated PKB phosphorylation and activity was enhanced in muscle with low glycogen content (Derave et al., 2000; Kawanaka et al., 2000; Jensen et al., 2006; Lai et al., 2010b), which suggests that increased PKB activity may contribute the enhanced insulin-stimulated glucose uptake in muscles with low glycogen content. However, we were unable to find elevated AS160 phosphorylation in muscles with reduced glycogen content despite that PKB phosphorylation was increased (Lai et al., 2010b).
Exercise increases insulin sensitivity but insulin signaling is not consistently improved after exercise (see above). However, a consistent finding is that exercise decreases glycogen content (Bergström et al., 1967; Hermansen et al., 1967; Coyle et al., 1986). Glycogen breakdown has mostly been investigated after prolonged exercise, but high intensity also decreases glycogen content (Esbjornsson-Liljedahl et al., 2002). Interestingly, 2 weeks of HIT training has been reported to increase insulin sensitivity (Richards et al., 2010) and exercise-mediated glycogen breakdown in skeletal muscles may contribute to the increased insulin sensitivity.
Exercise regulates insulin sensitivity via other mechanisms than reducing glycogen content. Training increases GLUT4 content in skeletal muscles, which contributes to improved insulin sensitivity (Houmard et al., 1993). A rather consistent finding is that glycogen content is higher in skeletal muscles from trained subjects and training increases glycogen content (Burgomaster et al., 2008). The glycogen stores are also refilled 24 h after exercise (Costill et al., 1981) whereas insulin sensitivity remains increased 24 h after a bout of exercise. Indeed, the fact that glycogen content is increased in skeletal muscles after training may result from increased insulin sensitivity. From an evolutional point of view such increase in glycogen content may reflect an important adaptation: high skeletal muscles glycogen content improves the chance for survival in emergencies.
Decreasing glycogen content by exercise or fasting stimulates glycogen accumulation to levels above the glycogen content in well-fed conditions (Hespel and Richter, 1990; Jensen et al., 1997; Derave et al., 2000; Lai et al., 2007). It is possible to increase the glycogen content in skeletal muscles if they are exposed to high concentrations of insulin and glucose (Richter et al., 1988; Hoy et al., 2007). Why does glycogen content not increase when high amount of carbohydrates are ingested under normal physiological conditions? Why is the excess carbohydrate ingested converted to lipid without elevation of glycogen content in skeletal muscles?
The glycogen content in skeletal muscles will reflects a balance between available glucose and insulin sensitivity in skeletal muscles. Studies in rats have under controlled conditions shown that training increases expression of GLUT4, but insulin sensitivity is not elevated in skeletal muscles because glycogen content also increases (Kawanaka et al., 1999, 2000). The acute adaptation to training is, therefore, higher glycogen content but stable insulin sensitivity. From an evolutional point of view, this indicates that high glycogen content is more important than high insulin sensitivity.
Prolonged training increases insulin sensitivity beyond the last training session, and insulin sensitivity correlates with oxidative capacity in skeletal muscles (Bruce et al., 2003). GLUT4 expression in skeletal muscles also regulates insulin sensitivity and correlates with rate of glycogen resynthesis (Hickner et al., 1997; Greiwe et al., 1999), which supports that glycogen synthesis is important from an evolutionary point of view. Interestingly, 24 h fasting GLUT4 content was elevated in fast-twitch epitrochlearis muscles where glycogen content was reduced (Jensen et al., 2006; Lai et al., 2007) but refeeding rats for 24 h increased glycogen content rather than increasing insulin sensitivity (Jensen et al., 1997, 2006). In soleus (slow-twitch muscle), glycogen content was minimally affected by 24 h fasting and GLUT4 was unchanged (Lai et al., 2009). These findings support that replenishment of glycogen store is superior to elevated insulin sensitivity.
Blood glucose concentration can be regulated in vivo even when skeletal muscle glycogen synthesis is impaired by short-term overeating (Acheson et al., 1988). Genetic findings support that skeletal muscle glycogen synthesis is not an absolute requirement for regulation of blood glucose concentration. Knockout mice lacking the skeletal muscle isoform of glycogen synthase have normal insulin sensitivity (Pederson et al., 2005). However, it is of note that 90% of the mice homozygotic knockout mice with deleted glycogen synthase die shortly after birth (Pederson et al., 2005). In human, a child without glycogen synthase has been described, and also this person had a normal glucose response to an oral glucose tolerance test (Kollberg et al., 2007).
Glycogen resynthesis is an important part of restitution after training and athletes optimize glycogen synthesis by intake of high amount of carbohydrates immediately after exercise (Ivy, 2001). The energy source for rapid glycogen synthesis is blood glucose and rapid extraction of glucose from the blood is required for high rate of glycogen synthesis. Diabetes subjects have impaired removal of blood glucose, because insulin-stimulated glycogen synthesis is impaired (Shulman et al., 1990; Højlund and Beck-Nielsen, 2006). Exercise-stimulated glycogen breakdown will stimulate skeletal muscle glycogen synthesis and extraction of blood glucose and increase insulin sensitivity. Such increased insulin sensitivity may be secondary to replenishing glycogen stores in the context of survival. However, in the modern society, the increased insulin sensitivity after exercise may have its superior role to prevent development of insulin resistance and type 2 diabetes.
Model for Development of Insulin Resistance
Glycogen content has a strong feedback inhibition of glycogen synthase activity (Danforth, 1965) and the glycogen stores are limited. It is not possible to dispose glucose into glycogen when stores are filled and under such condition, glucose remains in the blood until it is utilized as energy or transformed into lipid. Skeletal muscles have a crucial role for regulation of whole body glucose metabolism, but acute elevation of glycogen does not impair insulin signaling and insulin-stimulated glucose transport may be normal (Jensen et al., 1997, 2006). However, insulin-stimulated glycogen synthesis is decreased, and more glucose is metabolized via glycolysis and we suggest that such increased glucose metabolism in skeletal muscles is unhealthy.
Insulin signaling and insulin-stimulated glucose transport are impaired in muscles from rats and humans showing manifest insulin resistance or type 2 diabetes (Etgen et al., 1996; Ruzzin et al., 2005; Højlund and Beck-Nielsen, 2006; Petersen et al., 2007). However, such insulin resistance develops gradually. The mechanisms for development of insulin resistance in skeletal are not well-understood, but accumulation of lipid and lipid intermediates are likely contributors (Aas et al., 2005). Furthermore, energy surplus increases production of reactive oxidative spices (Hoehn et al., 2009; Hue and Taegtmeyer, 2009). The production of ROS is increased when high amount of glucose and fat is supplied the mitochondria simultaneously and forces electrons into the electron transport chain (Hue and Taegtmeyer, 2009). Preventing ROS production in skeletal muscles protects skeletal muscles form developing insulin resistance (Hoehn et al., 2009) and high glycogen content will favor metabolic stress in skeletal muscles. Insulin resistant muscles are characterized with numerous changes (e.g., expression of signaling proteins and activation of signaling pathways), and the mechanisms for initiation of insulin resistance may vary.
In skeletal muscles with low glycogen, glucose will be stored as muscles glycogen (Ivy, 1991; Hickner et al., 1997; Greiwe et al., 1999; Jensen et al., 2006). A major concern for athletes after strenuous training is to replete the glycogen stores is skeletal muscles preparing for new training sessions or competitions. Skeletal muscles are able to extract blood glucose effectively when high amount of carbohydrate are supplied (Ivy, 2001), and we suggest that glucose disposal into skeletal muscle glycogen is healthy storage of carbohydrates.
Indeed, healthy humans have large capacity to store glucose as lipid (Figure 2). Acheson et al. (1988) overfed people for 7 days in a calorimeter and found that healthy humans were able to convert 475 g carbohydrate to 150 g lipid per day. Importantly, de novo lipid synthesis occurred without development of hyperglycemia, but blood triglyceride content increased 10-fold (Acheson et al., 1988). Accumulation of fat per se does not cause insulin resistance (Haemmerle et al., 2006), but lipid intermediates like long chain acyl-CoA, ceramides, or diacylglycerol will impair insulin signaling and cause insulin resistance (Aas et al., 2005; Samuel et al., 2010).
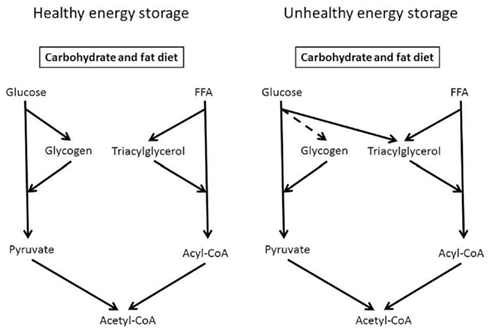
Figure 2. Excess energy intake is stored after meals as glycogen and triacylglycerols. Carbohydrate can be stored as glycogen mainly in skeletal muscles or the liver; fat is manly stores as triacylglycerol in adipose tissue. With filled glycogen stores, glucose can be the substrate for de novo lipid synthesis and stored in adipocytes, muscles, or the liver and cause insulin resistance. Glycogen and fat are important energy substrates during exercise.
Accumulation of lipid intermediates seems to occur secondary to increased glycogen content and acute exercise reduces lipid synthesis during glucose loads (Figure 2). Glucose conversion to lipid was reduced in untrained young healthy males 15 h after cycling 1 h at 65% of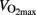 (Mikines et al., 1989). Moreover, it has been reported that insulin resistant subjects stores a larger part of ingested glucose as lipid in skeletal muscles and liver compared to insulin sensitive subjects, whereas skeletal muscles glycogen synthesis is lower in insulin resistant subjects (Petersen et al., 2007). A reduced capacity to store glucose as glycogen promotes de novo lipogenesis, which will deteriorate of insulin sensitivity due to lipid accumulation.
(Mikines et al., 1989). Moreover, it has been reported that insulin resistant subjects stores a larger part of ingested glucose as lipid in skeletal muscles and liver compared to insulin sensitive subjects, whereas skeletal muscles glycogen synthesis is lower in insulin resistant subjects (Petersen et al., 2007). A reduced capacity to store glucose as glycogen promotes de novo lipogenesis, which will deteriorate of insulin sensitivity due to lipid accumulation.
The increased insulin-mediated glycogen synthesis after exercise benefits survival in “fight or flight” situations from an evolutionary perspective. In the modern society, abundant food and inactivity are large challenges for humans, and metabolic diseases related to obesity deteriorate public health. Although the improved insulin sensitivity after glycogen depleting exercise may not have evolved to improve regulation of blood glucose, such effect of exercise may be the mechanism that protect humans from developing type 2 diabetes in the modern society. We suggest that dynamic glycogen metabolism is important for healthy regulation of blood glucose and prevention of insulin resistance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jørgen Jensen’s research is supported by grants from Novo Nordisk Foundation and via participation in COST Action BM0602 (Network supported by EU).
References
Aas, V., Rokling-Andersen, M., Wensaas, A. J., Thoresen, G. H., Kase, E. T., and Rustan, A. C. (2005). Lipid metabolism in human skeletal muscle cells: effects of palmitate and chronic hyperglycaemia. Acta Physiol. Scand. 183, 31–41.
Acheson, K. J., Schutz, Y., Bessard, T., Anantharaman, K., Flatt, J. P., and Jequier, E. (1988). Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am. J. Clin. Nutr. 48, 240–247.
Alessi, D. R., and Cohen, P. (1998). Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev. 8, 55–62.
Arias, E. B., Kim, J., Funai, K., and Cartee, G. D. (2007). Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 292, E1191–E1200.
Aslesen, R., Engebretsen, E. M. L., Franch, J., and Jensen, J. (2001). Glucose uptake and metabolic stress in rat muscles stimulated electrically with different protocols. J. Appl. Physiol. 91, 1237–1244.
Aslesen, R., and Jensen, J. (1998). Effects of epinephrine on glucose metabolism in contracting rat skeletal muscle. Am. J. Physiol. 275, E448–E456.
Åstrand, P. O., and Rodahl, K. (1992). Textbook of Work Physiology. New York: McGraw-Hill Book Company, 1–681.
Bergström, J., Hermansen, L., Hultman, E., and Saltin, B. (1967). Diet, muscle glycogen and physical performance. Acta Physiol. Scand. 71, 140–150.
Bergström, J., and Hultman, E. (1966). Muscle glycogen synthesis after exercise: an enhancing factor localized to the muscle cells in man. Nature 210, 319–310.
Betts, J. A., and Williams, C. (2010). Short-term recovery from prolonged exercise: exploring the potential for protein ingestion to accentuate the benefits of carbohydrate supplements. Sports Med. 40, 941–959.
Boushel, R., Gnaiger, E., Calbet, J. A., Gonzalez-Alonso, J., Wright-Paradis, C., Sondergaard, H., Ara, I., Helge, J. W., and Saltin, B. (2011). Muscle mitochondrial capacity exceeds maximal oxygen delivery in humans. Mitochondrion 11, 303–307.
Bouskila, M., Hirshman, M. F., Jensen, J., Goodyear, L. J., and Sakamoto, K. (2008). Insulin promotes glycogen synthesis in the absence of GSK3 phosphorylation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 294, E28–E35.
Bouskila, M., Hunter, R. W., Ibrahim, A. F., Delattre, L., Peggie, M., van Diepen, J. A., Voshol, P. J., Jensen, J., and Sakamoto, K. (2010). Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab. 12, 456–466.
Brady, M. J. (2010). Allosteric trumps covalent in the control of glycogen synthesis. Cell Metab. 12, 428–430.
Bruce, C. R., Anderson, M. J., Carey, A. L., Newman, D. G., Bonen, A., Kriketos, A. D., Cooney, G. J., and Hawley, J. A. (2003). Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J. Clin. Endocrinol. Metab. 88, 5444–5451.
Burgomaster, K. A., Howarth, K. R., Phillips, S. M., Rakobowchuk, M., MacDonald, M. J., McGee, S. L., and Gibala, M. J. (2008). Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J. Physiol. (Lond.) 586, 151–160.
Burgomaster, K. A., Hughes, S. C., Heigenhauser, G. J., Bradwell, S. N., and Gibala, M. J. (2005). Six sessions of sprint interval training increases muscle oxidative potential and cycle endurance capacity in humans. J. Appl. Physiol. 98, 1985–1990.
Cartee, G. D., Young, D. A., Sleeper, M. D., Zierath, J., Wallberg-Henriksson, H., and Holloszy, J. O. (1989). Prolonged increase in insulin-stimilated glucose transport in muscle after exercise. Am. J. Physiol. 256, E494–E499.
Chasiotis, D., Sahlin, K., and Hultman, E. (1983). Regulation of glycogenolysis in human muscle in response to epinephrine infusion. J. Appl. Physiol. 54, 45–50.
Christ, C. Y., Hunt, D., Hancock, J., Garcia-Macedo, R., Mandarino, L. J., and Ivy, J. L. (2002). Exercise training improves muscle insulin resistance but not insulin receptor signaling in obese Zucker rats. J. Appl. Physiol. 92, 736–744.
Christ-Roberts, C. Y., Pratipanawatr, T., Pratipanawatr, W., Berria, R., Belfort, R., Kashyap, S., and Mandarino, L. J. (2004). Exercise training increases glycogen synthase activity and GLUT4 expression but not insulin signaling in overweight nondiabetic and type 2 diabetic subjects. Metabolism 53, 1233–1242.
Cleasby, M. E., Reinten, T. A., Cooney, G. J., James, D. E., and Kraegen, E. W. (2007). Functional studies of Akt isoform specificity in skeletal muscle in vivo; maintained insulin sensitivity despite reduced insulin receptor substrate-1 expression. Mol. Endocrinol. 21, 215–228.
Cohen, P. (1993). Dissection of the protein phosphorylation cascades involved in insulin and growth factor action. Biochem. Soc. Trans. 214, 555–567.
Connett, R. J., and Sahlin, K. (1996). “Control of glycolysis and glycogen metabolism,” in Handbook of Physiology. Exercise: Regulation and Integration of Multiple System, eds L. B. Rowell and J. T. Shepherd (Bethesda, MD: American Physiological Society), 870–911.
Cori, C. F., and Cori, G. T. (1928). The mechanism of epinephrine action. I. The influence of epinephrine on the carbohydrate metabolism of fasting rats, with a note on new formation of carbohydrates. J. Biol. Chem. 79, 309–319.
Costill, D. L., Sherman, W. M., Fink, W. J., Maresh, C., Witten, M., and Miller, J. M. (1981). The role of dietary carbohydrates in muscle glycogen resynthesis after strenuous running. Am. J. Clin. Nutr. 34, 1831–1836.
Coyle, E. F., Coggan, A. R., Hemmert, M. K., and Ivy, J. L. (1986). Muscle glycogen utilization during prolonged strenuous exercise when fed carbohydrate. J. Appl. Physiol. 61, 165–172.
DeFronzo, R. A., Ferrannini, E., Sato, Y., Felig, P., and Wahren, J. (1981a). Synergistic interaction between exercise and insulin on peripheral glucose uptake. J. Clin. Invest. 68, 1468–1474.
DeFronzo, R. A., Jacot, E., Jequier, E., Maeder, E., Wahren, J., and Felber, J. P. (1981b). The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30, 1000–1007.
Dela, F., Handberg, A., Mikines, K. J., Vinten, J., and Galbo, H. (1993). GLUT 4 and insulin receptor binding and kinase activity in trained human muscle. J. Physiol. (Lond.) 469, 615–624.
Derave, W., Hansen, B. F., Lund, S., Kristiansen, S., and Richter, E. A. (2000). Muscle glycogen content affects insulin-stimulated glucose transport and protein kinase B activity. Am. J. Physiol. 279, E947–E955.
Derave, W., Lund, S., Holman, G. D., Wojtaszewski, J., Pedersen, O., and Richter, E. A. (1999). Contraction-stimulated muscle glucose transport and GLUT-4 surface content are dependent on glycogen concentration. Am. J. Physiol. 277, E1103–E1110.
Esbjornsson-Liljedahl, M., Bodin, K., and Jansson, E. (2002). Smaller muscle ATP reduction in women than in men by repeated bouts of sprint exercise. J. Appl. Physiol. 93, 1075–1083.
Esbjornsson-Liljedahl, M., Sundberg, C. J., Norman, B., and Jansson, E. (1999). Metabolic response in type I and type II muscle fibers during a 30-s cycle sprint in men and women. J. Appl. Physiol. 87, 1326–1332.
Etgen, G. J., Jensen, J., Wilson, C. M., Hunt, D. G., Cushman, S. W., and Ivy, J. L. (1997). Exercise training reverses insulin resistance in muscle by enhanced recruitment of GLUT-4 to the cell surface. Am. J. Physiol. 272, E864–E869.
Etgen, G. J., Wilson, C. M., Jensen, J., Cushman, S. W., and Ivy, J. L. (1996). Glucose transport and cell surface GLUT-4 protein in skeletal muscle of the obese Zucker rat. Am. J. Physiol. 271, E294–E301.
Franch, J., Aslesen, R., and Jensen, J. (1999). Regulation of glycogen synthesis in rat skeletal muscle after glycogen depleting contractile activity: effects of adrenaline on glycogen synthesis and activation of glycogen synthase and glycogen phosphorylase. Biochem. J. 344, 231–235.
Franch, J., Knudsen, J., Ellis, B. A., Pedersen, P. K., Cooney, G. J., and Jensen, J. (2002). Acyl-CoA binding protein expression is fibre type specific and elevated in muscles from obese insulin-resistant Zucker rat. Diabetes 51, 449–454.
Frayn, K. N. (1983). Calculation of substrate oxidation rates in vivo from gaseous exchange. J. Appl. Physiol. 55, 628–634.
Frosig, C., Rose, A. J., Treebak, J. T., Kiens, B., Richter, E. A., and Wojtaszewski, J. F. (2007). Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes 56, 2093–2102.
Gibala, M. J., Little, J. P., van, E. M., Wilkin, G. P., Burgomaster, K. A., Safdar, A., Raha, S., and Tarnopolsky, M. A. (2006). Short-term sprint interval versus traditional endurance training: similar initial adaptations in human skeletal muscle and exercise performance. J. Physiol. (Lond.) 575, 901–911.
Gjedsted, J., Buhl, M., Nielsen, S., Schmitz, O., Vestergaard, E. T., Tonnesen, E., and Moller, N. (2011). Effects of adrenaline on lactate, glucose, lipid and protein metabolism in the placebo controlled bilaterally perfused human leg. Acta Physiol. (Oxf.) 202, 641–648.
Greiwe, J. S., Hickner, R. C., Hansen, P. A., Racette, S. B., Chen, M. M., and Holloszy, J. O. (1999). Effects of endurance exercise training on muscle glycogen accumulation in humans. J. Appl. Physiol. 87, 222–226.
Haemmerle, G., Lass, A., Zimmermann, R., Gorkiewicz, G., Meyer, C., Rozman, J., Heldmaier, G., Maier, R., Theussl, C., Eder, S., Kratky, D., Wagner, E. F., Klingenspor, M., Hoefler, G., and Zechner, R. (2006). Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 734–737.
Hawley, J. A., Schabort, E. J., Noakes, T. D., and Dennis, S. C. (1997). Carbohydrate-loading and exercise performance. An update. Sports Med. 24, 73–81.
He, J., and Kelley, D. E. (2004). Muscle glycogen content in type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 287, E1002–E1007.
Heath, G. W., Gavin, J. R. III, Hinderliter, J. M., Hagberg, J. M., Bloomfield, S. A., and Holloszy, J. O. (1983). Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J. Appl. Physiol. 55, 512–517.
Hermansen, L., Hultman, E., and Saltin, B. (1967). Muscle glycogen during prolonged severe exercise. Acta Physiol. Scand. 71, 129–139.
Hespel, P., and Richter, E. A. (1990). Glucose uptake and transport in contracting, perfused rat muscle with different pre-contraction glycogen concentrations. J. Physiol. (Lond.) 427, 347–359.
Hickner, R. C., Fisher, J. S., Hansen, P. A., Racette, S. B., Mier, C. M., Turner, M. J., and Holloszy, J. O. (1997). Muscle glycogen accumulation after endurance exercise in trained and untrained individuals. J. Appl. Physiol. 83, 897–903.
Hoehn, K. L., Salmon, A. B., Hohnen-Behrens, C., Turner, N., Hoy, A. J., Maghzal, G. J., Stocker, R., Van, R. H., Kraegen, E. W., Cooney, G. J., Richardson, A. R., and James, D. E. (2009). Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 17787–17792.
Højlund, K., and Beck-Nielsen, H. (2006). Impaired glycogen synthase activity and mitochondrial dysfunction in skeletal muscle: markers or mediators of insulin resistance in type 2 diabetes? Curr. Diabetes Rev. 2, 375–395.
Houmard, J. A., Shaw, C. D., Hickey, M. S., and Tanner, C. J. (1999). Effect of short-term exercise training on insulin-stimulated PI 3-kinase activity in human skeletal muscle. Am. J. Physiol. 277, E1055–E1060.
Houmard, J. A., Shinebarger, M. H., Dolan, P. L., Leggettfrazier, N., Bruner, R. K., Mccammon, M. R., Israel, R. G., and Dohm, G. L. (1993). Exercise training increases GLUT-4 protein concentration in previously sedentary middle-aged men. Am. J. Physiol. 264, E896–E901.
Hoy, A. J., Bruce, C. R., Cederberg, A., Turner, N., James, D. E., Cooney, G. J., and Kraegen, E. W. (2007). Glucose infusion causes insulin resistance in skeletal muscle of rats without changes in Akt and AS160 phosphorylation. Am. J. Physiol. Endocrinol. Metab. 33, 1358–1364.
Hue, L., and Taegtmeyer, H. (2009). The Randle cycle revisited: a new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 297, E578–E591.
Hunter, R. W., Treebak, J. T., Wojtaszewski, J. F., and Sakamoto, K. (2011). Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60, 766–774.
Ivy, J. L. (2001). Dietary strategies to promote glycogen synthesis after exercise. Can. J. Appl. Physiol. 26(Suppl.), S236–S245.
Ivy, J. L., Katz, A. L., Cutler, C. L., Sherman, W. M., and Coyle, E. F. (1988). Muscle glycogen synthesis after exercise: effect of time of carbohydrate ingestion. J. Appl. Physiol. 64, 1480–1485.
Jacobs, I., Bar-Or, O., Karlsson, J., Dotan, R., Tesch, P., Kaiser, P., and Inbar, O. (1982). Changes in muscle metabolites in females with 30-s exhaustive exercise. Med. Sci. Sports Exerc. 14, 457–460.
Jensen, J. (2009). “The role of skeletal muscle glycolysis in whole body metabolic regulation and type 2 diabetes,” in Glycolysis: Regulation, Processes and Diseases, ed. H. Lithaw (New York: Nova Science Publishers, Inc.), 65–83.
Jensen, J., Aslesen, R., Ivy, J. L., and Brørs, O. (1997). Role of glycogen concentration and epinephrine on glucose uptake in rat epitrochlearis muscle. Am. J. Physiol. 272, E649–E655.
Jensen, J., Brennesvik, E. O., Lai, Y. C., and Shepherd, P. R. (2007). GSK-3 regulation in skeletal muscles by adrenaline and insulin: evidence that PKA and PKB regulate different pools of GSK-3. Cell. Signal. 19, 204–210.
Jensen, J., Brørs, O., and Dahl, H. A. (1995). Different β-adrenergic receptor density in different rat skeletal muscle fibre types. Pharmacol. Toxicol. 76, 380–385.
Jensen, J., and Dahl, H. A. (1995). Adrenaline stimulated glycogen breakdown in rat epitrochlearis muscles: fibre type specificity and relation to phosphorylase transformation. Biochem. Mol. Biol. Int. 35, 145–154.
Jensen, J., Dahl, H. A., and Opstad, P. K. (1989). Adrenaline-mediated glycogenolysis in different skeletal muscle fibre types in the anaesthetized rat. Acta Physiol. Scand. 136, 229–233.
Jensen, J., Grønning-Wang, L. M., Jebens, E., Whitehead, J. P., Zorec, R., and Shepherd, P. R. (2008). Adrenaline potentiates insulin-stimulated PKB activation in the rat fast-twitch epitrochlearis muscle without affecting IRS-1 associated PI 3-kinase activity. Pflugers Arch. 456, 969–978.
Jensen, J., Jebens, E., Brennesvik, E. O., Ruzzin, J., Soos, M. A., Engebretsen, E. M., O’Rahilly, S., and Whitehead, J. P. (2006). Muscle glycogen inharmoniously regulates glycogen synthase activity, glucose uptake, and proximal insulin signaling. Am. J. Physiol. Endocrinol. Metab. 290, E154–E162.
Jensen, J., and Lai, Y. C. (2009). Regulation of muscle glycogen synthase phosphorylation and kinetic properties by insulin, exercise, adrenaline and role in insulin resistance. Arch. Physiol. Biochem. 115, 13–21.
Jensen, J., Ruge, T., Lai, Y. C., Svensson, M. K., and Eriksson, J. W. (2011). Effects of adrenaline on whole-body glucose metabolism and insulin-mediated regulation of glycogen synthase and PKB phosphorylation in human skeletal muscle. Metabolism 60, 215–226.
Jensen, J., Ruzzin, J., Jebens, E., Brennesvik, E. O., and Knardahl, S. (2005). Improved insulin-stimulated glucose uptake and glycogen synthase activation in rat skeletal muscles after adrenaline infusion: role of glycogen content and PKB phosphorylation. Acta Physiol. Scand. 184, 121–130.
Jessen, N., Pold, R., Buhl, E. S., Jensen, L. S., Schmitz, O., and Lund, S. (2003). Effects of AICAR and exercise on insulin-stimulated glucose uptake, signaling, and GLUT-4 content in rat muscles. J. Appl. Physiol. 94, 1373–1379.
Kawanaka, K., Han, D. H., Nolte, L. A., Hansen, P. A., Nakatani, A., and Holloszy, J. O. (1999). Decreased insulin-stimulated GLUT-4 translocation in glycogen-supercompensated muscles of exercised rats. Am. J. Physiol. 276, E907–E912.
Kawanaka, K., Nolte, L. A., Han, D. H., Hansen, P. A., and Holloszy, J. O. (2000). Mechanisms underlying impaired GLUT-4 translocation in glycogen- supercompensated muscles of exercised rats. Am. J. Physiol. Endocrinol. Metab. 279, E1311–E1318.
Kelley, D., Mitrakou, A., Marsh, H., Schwenk, F., Benn, J., Sonnenberg, G., Arcangeli, M., Aoki, T., Sorensen, J., and Berger, M. (1988). Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J. Clin. Invest. 81, 1563–1571.
Kelley, D. E., and Mandarino, L. J. (1990). Hyperglycemia normalizes insulin-stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J. Clin. Invest. 86, 1999–2007.
Kim, Y. B., Nikoulina, S. E., Ciaraldi, T. P., Henry, R. R., and Kahn, B. B. (2000). Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J. Clin. Invest. 104, 733–741.
Knowler, W., Barret-Connor, E., Fowler, S., Hamman, R., Lachin, J. M., Walker, E., and Nathan, D. (2002). Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 346, 393–403.
Koivisto, V. A., Yki-Järvinen, H., and DeFronzo, R. A. (1986). Physical training and insulin sensitivity. Diabetes Metab. Rev. 1, 445–481.
Kollberg, G., Tulinius, M., Gilljam, T., Ostman-Smith, I., Forsander, G., Jotorp, P., Oldfors, A., and Holme, E. (2007). Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N. Engl. J. Med. 357, 1507–1514.
Lai, Y. C., Lin, F. C., and Jensen, J. (2009). Glycogen content regulates insulin- but not contraction-mediated glycogen synthase activation in the rat slow-twitch soleus muscles. Acta Physiol. (Oxf.) 197, 139–150.
Lai, Y. C., Stuenaes, J. T., Kuo, C. H., and Jensen, J. (2010a). Insulin-stimulated glycogen synthesis and glycogen synthase activation after electrical stimulation of epitrochlearis muscles with different initial glycogen contents. Arch. Physiol. Biochem. 116, 116–127.
Lai, Y. C., Zarrinpashneh, E., and Jensen, J. (2010b). Additive effect of contraction and insulin on glucose uptake and glycogen synthase in muscle with different glycogen contents. J. Appl. Physiol. 108, 1106–1115.
Lai, Y. C., Stuenæs, J. T., Kuo, C. H., and Jensen, J. (2007). Glycogen content and contraction regulate glycogen synthase phosphorylation and affinity for UDP-glucose in rat skeletal muscles. Am. J. Physiol. Endocrinol. Metab. 293, E1622–E1629.
Laurent, D., Petersen, K. F., Russell, R. R., Cline, G. W., and Shulman, G. I. (1998). Effect of epinephrine on muscle glycogenolysis and insulin-stimulated muscle glycogen synthesis in humans. Am. J. Physiol. 274, E130–E138.
Lauritzen, H. P., Ploug, T., Ai, H., Donsmark, M., Prats, C., and Galbo, H. (2008). Denervation and high-fat diet reduce insulin signaling in T-tubules in skeletal muscle of living mice. Diabetes 57, 13–23.
Maarbjerg, S. J., Sylow, L., and Richter, E. A. (2011). Current understanding of increased insulin sensitivity after exercise – emerging candidates. Acta Physiol. (Oxf.) 202, 323–335.
Madsen, K., Pedersen, P. K., Rose, P., and Richter, E. A. (1990). Carbohydrate supercompensation and muscle glycogen utilization during exhaustive running in highly trained athletes. Eur. J. Appl. Physiol. 61, 467–472.
Magnusson, I., Rothman, D. L., Katz, L. D., Shulman, R. G., and Shulman, G. I. (1992). Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J. Clin. Invest. 90, 1323–1327.
McManus, E. J., Sakamoto, K., Armit, L. J., Ronaldson, L., Shpiro, N., Marquez, R., and Alessi, D. R. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583.
Meléndez, R., Meléndez-Hevia, E., and Canela, E. I. (1999). The fractal structure of glycogen: a clever solution to optimize cell metabolism. Biophys. J. 77, 1327–1332.
Mevorach, M., Giacca, A., Aharon, Y., Hawkins, M., Shamoon, H., and Rossetti, L. (1998). Regulation of endogenous glucose production by glucose per se is impaired in type 2 diabetes mellitus. J. Clin. Invest. 102, 744–753.
Mikines, K. J., Sonne, B., Farrell, P. A., Tronier, B., and Galbo, H. (1988). Effect of physical exercise on sensitivity and responsiveness to insulin in humans. Am. J. Physiol. 254, E248–E259.
Mikines, K. J., Sonne, B., Farrell, P. A., Tronier, B., and Galbo, H. (1989). Effect of training on the dose-response relationship for insulin action in men. J. Appl. Physiol. 66, 695–703.
Morino, K., Petersen, K. F., Dufour, S., Befroy, D., Frattini, J., Shatzkes, N., Neschen, S., White, M. F., Bilz, S., Sono, S., Pypaert, M., and Shulman, G. I. (2005). Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Invest. 115, 3587–3593.
Musi, N., Hayashi, T., Fujii, N., Hirshman, M. F., Witters, L. A., and Goodyear, L. J. (2001). AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 280, E677–E684.
Nieman, D. C., Carlson, K. A., Brandstater, M. E., Naegele, R. T., and Blankenship, J. W. (1987). Running endurance in 27-h-fasted humans. J. Appl. Physiol. 63, 2502–2509.
Nolte, L. A., Gulve, E. A., and Holloszy, J. O. (1994). Epinephrine-induced in vivo muscle glycogen depletion enhances insulin sensitivity of glucose transport. J. Appl. Physiol. 76, 2054–2058.
Ortenblad, N., Nielsen, J., Saltin, B., and Holmberg, H. C. (2011). Role of glycogen availability in sarcoplasmic reticulum Ca2+ kinetics in human skeletal muscle. J. Physiol. (Lond.) 589, 711–725.
Pederson, B. A., Schroeder, J. M., Parker, G. E., Smith, M. W., Paoli-Roach, A. A., and Roach, P. J. (2005). Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes 54, 3466–3473.
Peronnet, F., and Massicotte, D. (1991). Table of nonprotein respiratory quotient: an update. Can. J. Sport Sci. 16, 23–29.
Petersen, K. F., Dufour, S., Savage, D. B., Bilz, S., Solomon, G., Yonemitsu, S., Cline, G. W., Befroy, D., Zemany, L., Kahn, B. B., Papademetris, X., Rothman, D. L., and Shulman, G. I. (2007). The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. U.S.A. 104, 12587–12594.
Prebil, M., Jensen, J., Zorec, R., and Kreft, M. (2011). Astrocytes and energy metabolism. Arch. Physiol. Biochem. 117, 64–69.
Qvisth, V., Hagstrom-Toft, E., Moberg, E., Sjoberg, S., and Bolinder, J. (2007). Lactate release from adipose tissue and skeletal muscle in vivo: defective insulin regulation in insulin-resistant obese women. Am. J. Physiol. Endocrinol. Metab. 292, E709–E714.
Richards, J. C., Johnson, T. K., Kuzma, J. N., Lonac, M. C., Schweder, M. M., Voyles, W. F., and Bell, C. (2010). Short-term sprint interval training increases insulin sensitivity in healthy adults but does not affect the thermogenic response to beta-adrenergic stimulation. J. Physiol. (Lond.) 588, 2961–2972.
Richter, E. A., Hansen, S. A., and Hansen, B. F. (1988). Mechanisms limiting glycogen storage in muscle during prolonged insulin stimulation. Am. J. Physiol. 255, E621–E628.
Romijn, J. A., Coyle, E. F., Sidossis, L. S., Gastaldelli, A., Horowitz, J. F., Endert, E., and Wolfe, R. R. (1993). Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am. J. Physiol. 265, E380–E391.
Ruzzin, J., Wagman, A. S., and Jensen, J. (2005). Glucocorticoids-induced insulin resistance: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 48, 2119–2130.
Sakamoto, K., and Holman, G. D. (2008). Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 295, E29–E37.
Saltin, B., and Karlsson, J. (1971). “Muscle glycogen utilization during work of different work intensities,” in Muscle Metabolism During Exercise (Stockholm: Karolinska Institute), 289–299.
Samuel, V. T., Petersen, K. F., and Shulman, G. I. (2010). Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277.
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101.
Schultze, S. M., Jensen, J., Hemmings, B. A., Tschopp, O., and Niessen, M. (2011). Promiscuous affairs of PKB/AKT isoforms in metabolism. Arch. Physiol. Biochem. 117, 70–77.
Shepherd, P. R. (2005). Mechanisms regulating phosphoinositide 3-kinase signalling in insulin-sensitive tissues. Acta Physiol. Scand. 183, 3–12.
Shulman, G. I., Rothman, D. L., Jue, T., Stein, P., DeFronzo, R. A., and Shulman, R. G. (1990). Quantification of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 322, 223–228.
Simonsen, L., Bulow, J., Madsen, J., and Christensen, N. J. (1992). Thermogenic response to epinephrine in the forearm and abdominal subcutaneous adipose tissue. Am. J. Physiol. 263, E850–E855.
Taniguchi, C. M., Emanuelli, B., and Kahn, C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96.
Taylor, R., Magnusson, I., Rothman, D. L., Cline, G. W., Caumo, A., Cobelli, C., and Shulman, G. I. (1996). Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J. Clin. Invest. 97, 126–132.
Taylor, R., Price, T. B., Katz, L. D., Shulman, R. G., and Shulman, G. I. (1993). Direct measurement of change in muscle glycogen concentration after a mixed meal in normal subjects. Am. J. Physiol. 265, E224–E229.
Vaag, A., Damsbo, P., Hother-Nielsen, O., and Beck-Nielsen, H. (1992). Hyperglycaemia compensates for the defects in insulin-mediated glucose metabolism and in the activation of glycogen synthase in the skeletal muscle of patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 35, 80–88.
van Loon, L. J., Greenhaff, P. L., Constantin-Teodosiu, D., Saris, W. H., and Wagenmakers, A. J. (2001). The effects of increasing exercise intensity on muscle fuel utilisation in humans. J. Physiol. (Lond.) 536, 295–304.
Vendelbo, M. H., Clasen, B. F., Treebak, J. T., Moller, L., Krusenstjerna-Hafstrom, T., Madsen, M., Nielsen, T. S., Stodkilde-Jorgensen, H., Pedersen, S. B., Jorgensen, J. O., Goodyear, L. J., Wojtaszewski, J. F., Moller, N., and Jessen, N. (2011). Insulin resistance after a 72 hour fast is associated with impaired AS160 phosphorylation and accumulation of lipid and glycogen in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. PMID: 22028408. [Epub ahead of print].
Vind, B. F., Pehmoller, C., Treebak, J. T., Birk, J. B., Hey-Mogensen, M., Beck-Nielsen, H., Zierath, J. R., Wojtaszewski, J. F., and Hojlund, K. (2011). Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise-training. Diabetologia 54, 157–167.
Wadley, G. D., Konstantopoulos, N., Macaulay, L., Howlett, K. F., Garnham, A., Hargreaves, M., and Cameron-Smith, D. (2007). Increased insulin-stimulated Akt pSer473 and cytosolic SHP2 protein abundance in human skeletal muscle following acute exercise and short-term training. J. Appl. Physiol. 102, 1624–1631.
Wojtaszewski, J. F., Higaki, Y., Hirshman, M. F., Michael, M. D., Dufresne, S. D., Kahn, C. R., and Goodyear, L. J. (1999). Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J. Clin. Invest. 104, 1257–1264.
Wojtaszewski, J. F., Nielsen, J. N., and Richter, E. A. (2002). Invited review: effect of acute exercise on insulin signaling and action in humans. J. Appl. Physiol. 93, 384–392.
Keywords: glycogen phosphorylase, glycogen synthase, exercise, type 2 diabetes, insulin resistance, exercise, de novo lipogenesis
Citation: Jensen J, Rustad PI, Kolnes AJ and Lai Y-C (2011) The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physio. 2:112. doi: 10.3389/fphys.2011.00112
Received: 26 July 2011; Paper pending published: 01 November 2011;
Accepted: 09 December 2011; Published online: 30 December 2011.
Edited by:
Heikki Veli Huikuri, Univeristy of Oulu, FinlandReviewed by:
Francesco Giorgino, Università degli Studi di Bari Aldo Moro, ItalyNiels Jessen, Aarhus University Hospital, Denmark
Copyright: © 2011 Jensen, Rustad, Kolnes and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jørgen Jensen, Department of Physical Performance, Norwegian School of Sport Sciences, P.O. Box 4014, Ullevål Stadion, 0806 Oslo, Norway. e-mail: jorgen.jensen@nih.no