
- Department of Medical Pharmacology and Physiology, University of Missouri, Columbia, MO, USA
Caveolae are cholesterol and glycosphingolipid-rich flask-shaped invaginations of the plasma membrane which are particularly abundant in vascular endothelium and present in all other cell types of the cardiovascular system, including vascular smooth-muscle cells, macrophages, cardiac myocytes, and fibroblasts. Caveolins and the more recently discovered cavins are the major protein components of caveolae. When caveolae were discovered, their functional role was believed to be limited to transport across the endothelial cell barrier. Since then, however, a large body of evidence has accumulated, suggesting that these microdomains are very important in regulating many other important endothelial cell functions, mostly due to their ability to concentrate and compartmentalize various signaling molecules. Over the course of several years, multiple studies involving knockout mouse and small interfering RNA approaches have considerably enhanced our understanding of the role of caveolae and caveolin-1 in regulating many cardiovascular functions. New findings have been reported implicating other caveolar protein components in endothelial cell signaling and function, such as the understudied caveolin-2 and newly discovered cavin proteins. The aim of this review is to focus primarily on molecular and cellular aspects of the role of caveolae, caveolins, and cavins in endothelial cell signaling and function. In addition, where appropriate, the possible implications for the cardiovascular and pulmonary physiology and pathophysiology will be discussed.
Caveolae and Caveolins
Caveolae or “small caves” were originally identified as 50–100 nm flask-shaped, non-clathrin-coated invaginations of the plasma membrane (Palade, 1953, 1961; Yamada, 1953; Palade and Bruns, 1968). These organelles are present in most mammalian cell types and tissues, and are particularly abundant in endothelial cells (ECs), adipocytes, and pneumocytes type I (Anderson, 1993; Fielding and Fielding, 1995; Parton, 1996; Severs, 1988). The originally described functions for caveolae included cholesterol transport (Fielding and Fielding, 1995; Smart et al., 1996), endocytosis (Schnitzer et al., 1996), and potocytosis (Anderson et al., 1992). However, later studies have revealed that this morphologically distinct subset of lipid rafts, highly enriched in cholesterol and sphingolipids, play a pivotal role in regulating cell signaling. Membrane rafts and caveolae concentrate certain membrane proteins and other components involved in transport and signal transduction (Allen et al., 2007; Parton and Simons, 2007; Patel et al., 2008; Insel and Patel, 2009).
A significant advance in understanding the roles of caveolae was made with identification of the coat proteins of caveolae, the caveolins: VIP21/caveolin-1 (Cav-1), caveolin-2 (Cav-2), and caveolin-3 (Cav-3; Glenney and Soppet, 1992; Kurzchalia et al., 1992; Way and Parton, 1995; Scherer et al., 1996; Tang et al., 1996). Cav-1 and Cav-2 are expressed in most cell types including all cell types of the cardiovascular system, while Cav-3 is expressed primarily in cardiac and skeletal muscle. Cav-1 expression is essential for the formation of caveolae, whereas the role of Cav-2 can vary depending on a cell and tissue type (Scheiffele et al., 1998; Fujimoto et al., 2000; Razani et al., 2002; Lahtinen et al., 2003; Sowa et al., 2003).
Caveolae in ECs
All blood vessels are lined by a monolayer of ECs, called the endothelium that helps supply nutrients and oxygen to underlying tissues and organs. Caveolae are most numerous in the microvascular endothelia of the lung and relatively infrequent in the highly restrictive microvascular endothelia of brain, retina, and testes. Interestingly, caveolae are mostly absent in passively leaky blood vessels with sinusoidal endothelia such as the liver (Ogi et al., 2003). It is important to note that caveolae contain all of the components required for vesicle formation, fission, docking, and fusion with target membranes (Schnitzer et al., 1995). Comprehensive proteomic studies revealed many proteins specifically enriched in EC caveolae (Durr et al., 2004). A large number of signaling molecules that regulate vascular ECs localize to lipid rafts/caveolae (Figure 1). These include receptors, e.g., receptor tyrosine kinase (RTK), G-protein-coupled receptors (GPCRs), transforming growth factor-beta (TGF-β) type I and II receptors, certain steroid receptors, low molecular weight and heterotrimeric G-proteins, and “downstream” enzymes and components including endothelial nitric oxide synthase (eNOS; Patel et al., 2008; Insel and Patel, 2009). This review will also discuss how various EC signaling molecules and their functions are regulated by caveolae and their specific coat and adapter protein components, i.e., caveolins and cavins.
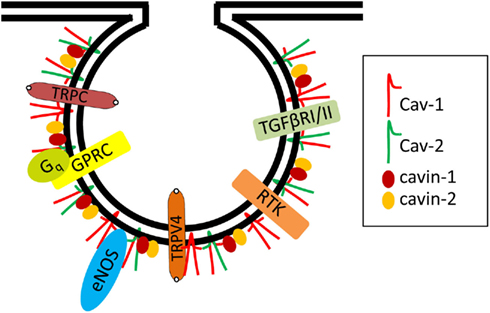
Figure 1. Examples of signaling proteins localized in endothelial cell caveolae. Several signaling molecules were localized in endothelial caveolae and interact with Cav-1 including eNOS, RTK (e.g., VEGF Receptor 2), GPRC (e.g., Bradykinin Receptor 2, Endothelin Receptor, Muscarinic Receptor), heterotrimeric G protein subunits (e.g., Gq), TGF-beta Receptors I and II, calcium channels (e.g., TRPC1 and 4, TRPV4).
Endothelial Cav-1, eNOS, Vascular Reactivity, and Blood Pressure
Endothelial nitric oxide synthase was one of the first non-receptor proteins found to be localized to plasma membrane caveolae (Feron et al., 1996). Since then, many in vitro studies have characterized the interaction between eNOS and Cav-1. Specifically, using co-immunoprecipitation and domain-mapping approaches, several groups have shown that eNOS directly interacts with the scaffolding domain (aa 81–101) of Cav-1 (Garcia-Cardena et al., 1997; Ju et al., 1997). Evidence supporting the functional relevance of this interaction in intact cells has been shown by delivery of a cell-permeable peptide containing the Cav-1 scaffolding domain or by adenoviral overexpression of Cav-1 in living cells or tissues. In all cases, NO release was attenuated, indicating that Cav-1 has a negative role in regulating eNOS activity (Michel et al., 1997; Bucci et al., 2000; Sowa et al., 2001). Moreover, we have shown that Cav-1 present in caveolae but not lipid rafts is able to inhibit eNOS under basal conditions (Sowa et al., 2001), suggesting that caveolae formation and localization is necessary for optimal tonic inhibition of eNOS by Cav-1. Studies involving Cav-1 KO validated the negative regulation of eNOS by caveolae and Cav-1. Specifically, the two independent studies revealed that the basal NO release and cGMP production were both significantly higher in Cav-1 KO than in WT mice (Drab et al., 2001; Razani et al., 2001). These data clearly indicate that loss of Cav-1 and caveolae results in hyperactivation of eNOS and associated NO release. Moreover, in addition to eNOS hyperactivation, the following observations were also reported: a lack of steady contractile tone in aortas isolated from Cav-1 KO mice, as well as an increased relaxation in response to acetylcholine coupled with a lower L-NAME-sensitive steady-state maximal tension in response to phenylephrine (Drab et al., 2001; Razani et al., 2001). These data suggest that Cav-1 and caveolae play important roles in the negative regulation of eNOS activity in vivo. However, these studies examined cerebral arteries and aorta, respectively, none of which can be regarded as resistance arteries. Thus additional studies on systemic small arteries have been performed by at least two independent laboratories (Albinsson et al., 2007; Dubroca et al., 2007). Specifically, in both studies Cav-1 KO mice displayed a reduced myogenic tone, i.e., active constriction induced by pressure. Interestingly, the reduction in myogenic tone was partially independent of increased NOS activity observed in Cav-1 KO mice (Albinsson et al., 2007; Dubroca et al., 2007).
Considering numerous studies showing that Cav-1 KO mice have increased NO production (Drab et al., 2001; Razani et al., 2001; Zhao et al., 2002), and reduced arterial myogenic tone (Drab et al., 2001; Albinsson et al., 2007), a decrease in systemic arterial blood pressure could be predicted. However, out of seven studies examining systemic blood pressure in Cav-1 KO mice five studies did not find a significant difference in systemic blood pressure between Cav-1 KO and WT mice, whereas two studies reported reduced systemic blood pressure (Reviewed in Rahman and Sward, 2009). Based on the fact that majority of these studies, including the most recent one using telemetry (Desjardins et al., 2008), did not report difference and considering that telemetry reliably measures both diastolic and systolic pressures without a dependence on anesthesia, one might conclude that despite increased eNOS activity and reduced myogenic tone, a loss of Cav-1 does not alter mean systemic arterial blood pressure. It is possible however, that prolonged hyperactivation of eNOS due to chronic loss of Cav-1 results in compensatory mechanisms in Cav-1 KO mice (Insel and Patel, 2007). One alternate approach could be using conditional Cav-1 KO or shRNA, allowing for an inducible loss of Cav-1 within a relatively short time frame. Another alternative might use a peptide which could antagonize the effect of Cav-1 on eNOS in vivo. Indeed, in their most recent studies Bernatchez et al. (2011) used a cell-permeable peptide containing Cav-1 scaffolding domain with a single mutation (F92A) named cavnoxin, which blocked a specific interaction between endogenous Cav-1 and eNOS, resulting in eNOS hyperactivation and increased NO release in WT but not Cav-1 KO ECs and mice. Remarkably, cavnoxin reduced vascular tone ex vivo in WT but not Cav-1 KO and eNOS KO aortic blood vessels. In addition, cavnoxin lowered blood pressure in WT but not in eNOS KO mice. Importantly, blood pressure measurements were performed in non-anesthetized mice (Bernatchez et al., 2011). Taken together, these data suggest that cavnoxin-induced acute disruption of Cav-1/eNOS interaction results in hyperactivation of eNOS, increased vasodilation and reduced blood pressure in vivo.
Of note, in addition to NO that stimulates signaling via activation of PKG, prolonged local hyperactivation of eNOS can also lead to S-nitrosylation of proteins (Iwakiri et al., 2006). In contrast to the stimulating properties of free NO, protein nitration has also been shown to play an inhibitory role in cell signaling and function. For example, recent studies have revealed that eNOS hyperactivation observed in Cav-1 KO mice results in excessive peroxynitrite production and inhibitory nitration of PKG, leading to pulmonary hypertension (Zhao et al., 2009).
The Role of Endothelial Caveolae and Cav-1 in Regulating Intracellular Calcium and EDHF Pathway
Numerous molecules involved in calcium-ion translocation have been localized to caveolae and might play a very important role in cell signaling, including activation of eNOS (Pani and Singh, 2009). For example, it has been shown that calcium waves originate in EC caveolae (Isshiki and Anderson, 1999). Moreover, Cav-1 regulates store-operated calcium-ion influx by binding its scaffold domain to transient receptor potential channel-1 (TRPC1) in ECs (Kwiatek et al., 2006). Studies involving Cav-1 KO mice revealed that loss of Cav-1 expression in ECs abrogated calcium-ion entry due to calcium-ion store depletion (Murata et al., 2007b). Mechanistically, the protein–protein interactions between TRPC1 and TRPC4, and their targeting to plasma membrane lipid rafts were impaired in Cav-1 KO ECs. Moreover, re-expression of Cav-1 in Cav-1 KO ECs rescued calcium-ion entry as well as TRPC1 and TRCP4 interaction and targeting to plasma membrane lipid rafts (Murata et al., 2007b), suggesting the critical role of Cav-1 in regulating calcium-ion entry in ECs.
Another member of the TRP channel family, TRPV4, is believed to be a major regulator of vascular tone. TRPV4 channels appear to be a significant calcium-ion entry pathway in ECs because of their high calcium-ion permeability (Nilius et al., 2003; Kohler et al., 2006). These channels can be activated by shear stress and significantly contribute to endothelial mechanotransduction (Hartmannsgruber et al., 2007). Interestingly, recent studies identified TRPV4 as the link between the impairment of calcium entry and the defect in endothelium-derived hyperpolarizing factor (EDHF)-induced relaxation observed in Cav-1 KO mice (Saliez et al., 2008). Specifically, a comparison of relaxation in vessels from Cav-1 KO and WT mice, showed a complete absence of EDHF-mediated vasodilation in isolated mesenteric arteries from Cav-1 KO mice. Interestingly, the loss of Cav-1 and caveolae resulted in impairment of calcium homeostasis in ECs, i.e., decreased activity of calcium-ion permeable TRPV4 cation channels that participate in NO- and EDHF-mediated relaxation. Moreover, morphological characterization of Cav-1 KO and WT arteries revealed fewer gap junctions in vessels from Cav-1 KO mice, associated with a lower expression of connexins 37, 40, and 43 and altered myoendothelial communication. Finally, studies have also shown that TRPV4 channels and connexins colocalize with Cav-1 in plasma membrane caveolar compartments (Saliez et al., 2008). Taken together, these data suggest that loss of Cav-1 leads to impaired intercellular coupling through gap junctions, implying that Cav-1 and caveolae might be critical for a correct membrane location of connexins and gap-junction assembly. Thus Cav-1 and caveolae are also important in regulating EDHF-related relaxation by modulating membrane location and activity of TRPV4 channels and connexins that are involved, at different steps, in the EDHF signaling. The broader significance of impaired EDHF signaling observed in Cav-1 KO mice for systemic vasculature appears to be complex and has been thoroughly discussed in a recent review by Rahman and Sward (2009).
Endothelial Caveolae/Cav-1 and Redox Signaling and Function
It has been recently shown that the NADPH Oxidase (NOX) complex may be preassembled and functional in caveolae, and its enzymatic activity enhanced by recruitment of additional components (Yang and Rizzo, 2007). In ECs, various stimuli may organize NOX components in ceramide-enriched lipid rafts (Li et al., 2007; Jin et al., 2008a). Formation of such complexes can be initiated by pro-apoptotic signals such as Fas ligand, endostatin, and TNF-α (Zhang et al., 2006). In addition to pro-apoptotic signals, lipolysis of triglyceride-rich lipoproteins can promote aggregation of lipid rafts and enhance ROS production in ECs, the latter effect being attenuated by NOX inhibitors (Wang et al., 2008). Cav-1 can serve as a sensor of shear stress in ECs and thereby regulate ROS-mediated signaling via NOX (Milovanova et al., 2008). Thus numerous stimuli affect lipid rafts and caveolae and modulate NOX in ECs. Compartmented generation of NO and superoxide by eNOS and NOX, respectively, may contribute to protein nitration on tyrosine residues. Disruption of lipid raft/caveolae domains with cholesterol-removing drugs dissociates these enzymes from these microdomains and decreases their ability to generate reactive species and to nitrate proteins in bovine aortic ECs (Yang and Rizzo, 2007).
Heme oxygenase (HO), that catalyzes the breakdown of heme to biliverdin, iron, and carbon monoxide (CO), exists as three membrane-bound isoforms: inducible HO-1, constitutive HO-2, and HO-3 which is catalytically inactive (Unno et al., 2007). Among the products generated by HO, CO has signaling potential (Kim et al., 2008). CO has been shown to be regulated by NO and potentially to be involved in regulating vascular function (Durante et al., 1997; Wang et al., 1997). HO-1 in ECs was shown to interact with Cav-1 and Cav-2 and localize in caveolae. HO-1 has also been shown to be negatively regulated by Cav-1 (Kim et al., 2004). Cav-1 KO mice are protected from hyperoxic damage in the lung due to an increased expression and activity of HO-1 (Jin et al., 2008b).
Endothelial Caveolae/Cav-1 and Mechanotransduction
Endothelial cells are normally exposed to mechanical forces which regulate their function (Traub and Berk, 1998). The general notion is that laminar and disturbed flows regulate endothelial function differently. It has been shown that exposure of ECs to shear stress results in an increased number of caveolae (Park et al., 1998) and a rapid NO release due to the dissociation of eNOS from Cav-1 (Rizzo et al., 1998a). Moreover, Rizzo et al. (1998b) have shown that exposure of ECs to shear stress leads to tyrosine phosphorylation of proteins localized to caveolae. Also, the translocation of several signaling molecules into caveolae has been observed, resulting in activation of the Ras-p42/44/MAPK pathway. Furthermore, upon exposure of ECs to laminar shear stress, Cav-1 undergoes translocation to the abluminal side of the cell (Sun et al., 2002), suggesting that relocation of Cav-1 and caveolae in ECs could contribute to the adaptive response in cells exposed to shear stress. More recent studies involving Cav-1 KO mice and ECs from these mice further reinforced the functional significance for Cav-1 in short- and long-term mechanotransduction in the vasculature. In particular, studies by Yu et al. (2006) have examined the role of Cav-1/caveolae in the regulation of flow-induced mechanotransduction in vessels from WT, Cav-1 KO, and Cav-1 KO with Cav-1 re-expressed in ECs. Their results revealed that endothelial Cav-1 and caveolae are necessary for both rapid and long-term mechanotransduction in intact blood vessels, suggesting that Cav-1 and caveolae are important sensors of altered shear stress and associated signaling in ECs (Yu et al., 2006). Most recent mechanistic studies by Yang et al. (2011) involving siRNA approaches in primary bovine aortic ECs and Cav-1 KO ECs determined that p190RhoGAP links integrins and Cav-1/caveolae to RhoA in a mechanotransduction cascade that participates in endothelial adaptation to flow.
Interestingly, although the previously noted studies strongly suggest that caveolae and Cav-1 are essential for mechanotransduction induced by shear stress/flow, there are also studies suggesting that the role of caveolae and Cav-1 in mechanotransduction and its possible implications for systemic vasculature may be more complex. Specifically, studies of Albinsson et al. (2008) revealed that pressure (stretch)-induced vascular smooth-muscle growth and differentiation were unaltered in Cav-1 KO versus WT vessels in vitro, suggesting that unlike sensing of shear stress, the sensing of pressure occurs independently of caveolae and Cav-1. Moreover, another study from the same laboratory has shown no significant difference inflow-mediated dilation in small mesenteric arteries isolated from Cav-1 KO mice, suggesting that impaired mechanosensing by caveolae and Cav-1-deficient endothelium might not be relevant, at least in the specific context of systemic blood pressure control (Albinsson et al., 2007). Alternatively, possible adaptations to continuous loss of Cav-1 and caveolae in mice could mask certain differences. Thus using conditional Cav-1 KO (inducible within a specific time frame), siRNA, or short cell-permeable peptide delivery approaches could shed more light on physiological relevance of the role of caveolae and Cav-1 in mechanotransduction.
Endothelial Caveolae/Cav-1 and Macromolecular Transport/Permeability
The transcytosis of macromolecules was the first function proposed for caveolae (Palade and Bruns, 1968). Caveolae have been suggested to mediate the transport of molecules such as albumin (Ghitescu et al., 1986), iron-transferrin (Soda and Tavassoli, 1984), insulin (King and Johnson, 1985), low-density lipoproteins (LDL; Ghitescu et al., 1986), and chemokines (Ge et al., 2008). The transcytosis pathway could be central for the specific and targeted delivery of molecules to certain organs. For example, this pathway may be crucial for the capillary ECs that form the blood brain barrier. The transport of albumin has been the most extensively studied case of transcytosis (Predescu et al., 2004); it is significant because albumin can carry various small molecules such as fatty acids and steroid hormones. The transport of albumin was suggested to be mediated by the gp60 receptor localized in caveolae (Tiruppathi et al., 1997), and more recently, caveolae have been shown to be involved in the endothelial transcytosis of albumin (Schubert et al., 2001; Mehta et al., 2004). Importantly, in contrast to WT, Cav-1 KO ECs could not transcytose albumin in mice injected with gold-labeled albumin (Schubert et al., 2001).
Although, the physiological role of caveolae in transcellular transport of specific molecules such as LDL (Frank et al., 2008), or at certain anatomical locations including lung microvascular endothelium (Oh et al., 2007) cannot be excluded, the physiological significance of transcytosis and its quantitative importance remain a matter of controversy for most macromolecules (Rippe et al., 2002). In fact, several studies using Cav-1 KO or siRNA approaches in vivo determined that albumin or other macromolecular transport is increased due to loss or reduction of Cav-1 (Schubert et al., 2002; Miyawaki-Shimizu et al., 2006; Rosengren et al., 2006). Although these studies were consistent regarding increased macromolecular transport, the proposed mechanisms are different. Specifically, two of these studies reported opening of the paracellular junctions in endothelia of small veins and capillaries (Schubert et al., 2002; Miyawaki-Shimizu et al., 2006). In contrast, in another study, Rosengren et al. (2006) did not observe an opening of alternative paracellular pathway in Cav-1 KO mice. Instead, they concluded that this increased macromolecular transport could be a result of passive porous transport via an unperturbed two-pore system, presumably at an elevated capillary hydraulic pressure (Rosengren et al., 2006). Regardless of these differences the increased macromolecular transport in Cav-1 KO appears to be mediated through the ability of caveolae and Cav-1 to regulate cell signaling, in particular eNOS. Specifically, Schubert et al. (2002) have shown that treatment of Cav-1 KO mice with eNOS inhibitor reversed the enhancing effect of Cav-1 loss on macromolecular transport, suggesting that Cav-1 might control molecular transport via regulating eNOS activity. The opening of adherens junctions that was observed in Cav-1 KO endothelium (Schubert et al., 2002; Miyawaki-Shimizu et al., 2006) suggests that Cav-1 is necessary for adherens junction assembly or maintenance. Most recently, a plausible mechanistic explanation regarding this phenomenon was provided by Siddiqui et al. (2011), who, using ECs isolated from Cav-1 KO mice, have shown that loss of Cav-1 could activate eNOS and the generation of NO and peroxynitrite. They found that the GTPase-activating protein (GAP) p190RhoGAP-A was selectively nitrated at Tyr 1105, resulting in impaired GAP activity and RhoA activation. Inhibition of eNOS or RhoA restored adherens junction integrity and diminished endothelial hyperpermeability in Cav-1 KO mice. In addition, thrombin also induced nitration of p120-catenin-associated p190RhoGAP-A, suggesting that eNOS-dependent nitration of p190RhoGAP-A is critical for adherens junction disassembly leading to increased endothelial permeability (Siddiqui et al., 2011).
Interestingly, in contrast to the previously discussed increase basal permeability in Cav-1 KO mice, these mice were resistant to hydrogen peroxide-induced pulmonary vascular albumin hyperpermeability and edema formation. Furthermore, the vascular hyperpermeability in response to hydrogen peroxide also observed in Cav-1 KO mouse lung microvessels was rescued by expression of WT but not by the expression of phosphorylation-deficient mutant of Cav-1. The increase in Cav-1 phosphorylation induced by hydrogen peroxide was concentration-dependently coupled to both increased albumin transcytosis and decreased transendothelial electric resistance in pulmonary ECs. Hydrogen peroxide-induced phosphorylation of Cav-1 resulted in the dissociation of vascular endothelial cadherin/beta-catenin complexes and endothelial barrier disruption (Sun et al., 2009). Thus Cav-1 phosphorylation-dependent signaling plays a critical role in oxidative stress-induced pulmonary vascular hyperpermeability via transcellular and paracellular pathways.
In light of the experimental evidence discussed in this section, it is clear that caveolae and Cav-1 play a critical role in regulating microvascular permeability. Moreover, the regulatory role of caveolae and Cav-1 appears to be complex and context-specific, as seen in basal- versus oxidative stress-induced microvascular permeability.
Is Loss of Endothelial Cav-1 Responsible for Cardiac Hypertrophy in Cav-1 KO Mice?
Cardiac hypertrophy is a critical pathology leading to heart failure. Although, ventricular cardiomyocytes express primarily Cav-3, surprisingly numerous studies have reported cardiomyopathy in Cav-1 KO mice (Zhao et al., 2002; Cohen et al., 2003; Wunderlich et al., 2006; Murata et al., 2007a). A detailed discussion of the role of Cav-1 and caveolae in cardiac muscle and cardiac hypertrophy observed in Cav-1 KO mice can be found in a recent review by Rahman and Sward (2009). A very important study, from an endothelial Cav-1 and caveolae perspective conducted by Murata et al. (2007a) revealed that the mechanism leading to cardiac hypertrophy observed in Cav-1 KO mice appears to originate in ECs. Specifically, they showed that selective re-expression of Cav-1 under the control of the preproendothelin-1 promoter completely reversed cardiac hypertrophy and associated coronary arterial remodeling and fibrosis in Cav-1 KO mice (Murata et al., 2007a). These data suggest that loss of endothelial Cav-1 is primarily responsible for the cardiac phenotype observed in Cav-1 KO mice. However, in addition to ECs, endothelin-1 expression has been reported in other cell types such as smooth muscle, adventitial fibroblasts, or epithelial cells (Rahman and Sward, 2009). Therefore, additional studies with Cav-1 re-expression and/or conditional KO approaches, using independent EC-specific promoter(s) such as VE-cadherin or Tie-2, may be needed to further reinforce the importance of endothelial Cav-1 in cardiac pathophysiology.
Caveolae/Cav-1 and Postnatal Angiogenesis
Angiogenesis is the process of new blood vessel formation that takes place in three clearly distinct phases: initiation, proliferation of vascular cells, and morphogenesis. Several important signaling proteins involved in angiogenesis have been localized to caveolae such as the VEGF receptor (VEGFR), the urokinase receptor (uPAR), eNOS, and TGF-β receptors.
Initial studies using Matrigel plugs supplemented with basic fibroblast growth factor implanted into WT and Cav-1 KO mice have shown that angiogenesis in Cav-1 KO mice is markedly reduced (Woodman et al., 2003), suggesting that Cav-1 is necessary for optimal neovascularization. Furthermore, three independent studies using in vivo models of tumor-induced angiogenesis with mouse B16 melanoma cells implanted in Cav-1 KO and WT C57BL/6 mice (Woodman et al., 2003; Chang et al., 2009), and RM-9 prostate cancer cells or human prostate cancer LNCaP cells implanted into nude mice (Tahir et al., 2008), have further reinforced the positive role for Cav-1 in angiogenesis. Specifically, the results of the first study revealed that tumor weight, volume, and vessel density were all reduced in Cav-1 KO mice subcutaneously (sc) injected with B16-F10 melanoma cells (Woodman et al., 2003). A more recent study, using a similar model of tumor-induced angiogenesis, also reported impaired tumor growth and diminished angiogenesis in Cav-1 KO mice (Chang et al., 2009). In another study using an orthotopic RM-9 mouse prostate cancer model both tumor growth and angiogenesis were also reduced in Cav-1 KO mice (Tahir et al., 2008). Furthermore, both tumor volumes and tumor microvessel density were significantly greater in nude mice implanted sc with LNCaP cells in which expression of Cav-1 was induced with doxycycline (Tahir et al., 2008). In addition to in vivo studies, at least two studies involving Cav-1 KO aortic ECs seem to support the pro-angiogenic role for Cav-1. For example, Cav-1 KO aortic ECs displayed impaired VEGF-stimulated signaling and angiogenesis in vitro (Sonveaux et al., 2004). A different study involving Cav-1 KO aortic ECs determined that recombinant Cav-1 could restore specific angiogenic functions in Cav-1 KO aortic ECs (Tahir et al., 2008). Numerous studies using antisense or siRNA approaches, frequently combined with Cav-1 overexpression also support pro-angiogenic role of Cav-1 in vitro. Knockdown of Cav-1 with antisense oligos suppressed capillary tube formation in human umbilical vein EC (HUVEC) shown using a fibrin gel-based angiogenesis assay (Griffoni et al., 2000). Adenoviral overexpression of Cav-1 and treatment with a cell-permeable peptide containing the Cav-1 scaffolding domain enhanced capillary-like tube formation, while downregulation of Cav-1 expression with antisense adenoviral approach reduced this process (Liu et al., 2002). Treatment with the two caveolae-disrupting agents cyclodextrin and filipin, or selective targeting of Cav-1 expression with siRNA, resulted in blocking MT1-MMP function and inhibition of PMA-stimulated HUVEC migration through polycarbonate filters and invasion into type I collagen gel as well as capillary tube formation in matrigel (Galvez et al., 2004). Similarly, siRNA-mediated knockdown of Cav-1 inhibited directional cell migration in HUVEC stimulated with VEGF as shown in a Dunn chamber assay, demonstrating that Cav-1 is crucial for VEGF-induced migration (Beardsley et al., 2005). A considerable amount of evidence has accumulated showing a positive correlation between Cav-1 expression, tumor microvascular density, and often shorter survival in humans with clear cell renal cell carcinoma (Joo et al., 2004), prostate cancer (Yang et al., 2007), meningioma (Barresi et al., 2008), or hepatic cell carcinoma (Zhang et al., 2009). Collectively, basic and clinical research data strongly support a pro-angiogenic function for Cav-1.
Data suggesting an anti-angiogenic role for Cav-1 based on in vivo and in vitro models of angiogenesis have also been reported. However, little clinical evidence has been obtained to date. Negative regulation of tumor-induced angiogenesis in vivo by Cav-1 has been reported using Cav-1 KO mice, Cav-1 overexpression, or delivery of cell-permeable peptide containing Cav-1 scaffolding domain. Transfection of the liposome Cav-1 plasmid complex delayed sc implanted Lewis lung carcinoma (LLC) tumor growth in mice, particularly during later stages (Brouet et al., 2005). In another study, a significantly higher tumor growth rate, angiogenesis, and tumor vascular permeability were observed in Cav-1 KO implanted with LLC tumors (Lin et al., 2007). Furthermore, administration of cell-permeable peptide containing scaffolding domain of Cav-1 through the tail vein of Cav-1 KO mice prevented both the tumor vascular hyperpermeability and increased tumor growth (Lin et al., 2007). Similar to the previous study, a more rapid tumor growth, increased angiogenesis and tumor vessel hypermeability was reported in Cav-1 KO mice implanted sc with B16 melanoma cells (Dewever et al., 2007). An anti-angiogenic role for Cav-1 was also suggested by several studies using in vitro models of angiogenesis, For example, adenoviral overexpression of Cav-1 inhibited the proliferation of HUVEC in response to VEGF as well as the kinase activity of VEGFR2 and the downstream p42/44 MAP kinase (Fang et al., 2007). Another study reported that VEGF-stimulated VEGFR2 tyrosine phosphorylation was more robust and sustained in Cav-1 KO lung ECs. This coincided with a decreased basal and VEGF-stimulated association between VEGFR2 and the VE-cadherin complex as compared with WT ECs (Lin et al., 2007). Overall, these data support anti-angiogenic function of Cav-1.
Thus the experimental evidence has been gathered suggesting that Cav-1 could play a bi-directional role in angiogenesis, i.e., promote or inhibit the process of new blood vessel formation. How can the studies showing positive and negative role of Cav-1 in angiogenesis be reconciled? It is essential to remember that tumor-induced angiogenesis depends on the delicate balance between pro- and anti-angiogenic factors which vary between specific tumor models, stages of tumor growth and angiogenesis, and the genetic backgrounds or ages of animals used. Also, differences between specific in vitro assays of angiogenesis, source of ECs, or pro-angiogenic stimuli could result in different outcomes. Importantly, Cav-1, through its scaffolding domain, can interact with and inhibit activity of numerous signaling proteins such as eNOS, PI3K, Src, PKC, or Erk that could play a role in angiogenesis (see review by Patel et al., 2008). In the absence or presence of low levels of pro-angiogenic stimuli, Cav-1 could play an anti-angiogenic role. The shift from anti- to pro-angiogenic role for Cav-1 could occur once a critical level of pro-angiogenic stimulation is reached. At that point, correct caveolar localization of a pro-angiogenic signaling protein may be essential for optimal signal transduction. Furthermore, Cav-1 protein is crucial for maintaining intact and functional caveolar membranes that sequester many receptors and downstream signaling proteins involved in angiogenesis such as VEGFR2 (Labrecque et al., 2003; Sonveaux et al., 2004), PDGF receptor, Src, eNOS, PI3K, or PKC (de Laurentiis et al., 2007).
Endothelial Caveolae, Cav-1, and Atherosclerosis
Atherosclerosis is the result of inflammatory and fibro-proliferative responses which reflect a complex crosstalk among the vascular wall, circulating cells, and cardiovascular risk factors.
Many studies involving animals and humans have provided the evidence showing that EC dysfunction plays a major role in initiation of the atherosclerotic process (Luscher and Noll, 1994; Brandes et al., 2005). The entrapment of LDL particles in the sub-endothelial space of arteries and their subsequent modification is believed to be one of the key events that ultimately lead to the development of an atheroma (Ross, 1999; Williams and Tabas, 2005).
Recently, direct genetic evidence has been provided supporting the important role of endothelial caveolae and Cav-1 in atherosclerosis. Initially, using Cav-1 KO mice bred to ApoE KO mice, Frank et al. (2004) demonstrated that the loss of Cav-1 markedly inhibits fatty streak lesion formation compared with ApoE KO mice. This decrease was accompanied by lower CD36 expression in the aorta. Also, plasma LDL levels in Cav-1 KO mice were elevated, suggesting a defect in either uptake and/or transfers of LDL to peripheral tissues, consistent with a role of caveolae in the LDL transcytosis process (Frank et al., 2004). Taken together, this data suggest that caveolae and Cav-1 markedly contribute to process of atherosclerosis in mice. However, in addition to ECs, macrophages are also involved in LDL uptake and express Cav-1. Thus it was important to determine a specific contribution of endothelial Cav-1 to the process of atherosclerosis. To address this, an EC-specific re-expression of Cav-1 in Cav-1 KO mice was used (Fernandez-Hernando et al., 2009). The results of these studies revealed that although global loss of Cav-1 in an ApoE KO background inhibited atherosclerotic lesion expansion, endothelial-specific re-expression of Cav-1 restored this process. Mechanistically, loss of Cav-1 decreased LDL infiltration into the arterial wall, promoted NO production, and reduced the expression of leukocyte adhesion molecules such as VCAM-1, ICAM-1, and E-selectin. These effects of global loss of Cav-1 were completely reversed by re-expression of Cav-1 in endothelium (Fernandez-Hernando et al., 2009). Another study from the same laboratory revealed that endothelial-specific overexpression of Cav-1 enhanced the progression of atherosclerosis in mice and was accompanied by reduced EC proliferation, migration, and NO production in vitro and increased expression of VCAM-1 in vivo (Fernandez-Hernando et al., 2010). Taken together, these data support the notion that Cav-1 expressed in ECs plays a pro-atherogenic role in mouse models of atherosclerosis.
The Role of EC Caveolae and Cav-1 in Acute Lung Injury and Inflammation
Primary acute lung injury (ALI) is a direct injury to the lung resulting from pneumonia, ventilation-associated injury, hyperoxic injury, trauma, and contusion (Ingbar, 2000; Mantell and Lee, 2000; Matute-Bello et al., 2008). Secondary ALI is typically caused indirectly by severe sepsis, pancreatitis, or transfusion-related ALI (Ingbar, 2000). Acute inflammation has been associated with the pathological stages of ALI and with enhanced vascular permeability, fibroproliferation, epithelial cell apoptosis, and varying degrees of interstitial fibrosis (Ingbar, 2000; Mantell and Lee, 2000; Matute-Bello et al., 2008).
Experimental evidence suggests that caveolae and Cav-1 expressed in ECs play a critical role during acute inflammation by regulating the transport of macromolecules such as albumin from the blood-space to the tissue-space (Schubert et al., 2002; Hu et al., 2008a,b; Sun et al., 2009). Albumin cannot be endocytosed by Cav-1 KO lung ECs and is retained in the blood vessel lumen (Sun et al., 2009). Previous studies have suggested that caveolae and lipid rafts contribute to non-cardiogenic pulmonary edema during ALI (Hu et al., 2008a,b). Additional studies using cell-permeable peptide containing scaffolding domain of Cav-1 have demonstrated that scaffolding domain of Cav-1 regulates calcium store release-induced calcium influx in ECs, suggesting a potential role in endothelial permeability (Sundivakkam et al., 2009). The results of numerous studies suggest that Cav-1 plays a dual role in regulating microvascular permeability. First, as a caveolae-associated structural protein Cav-1 may control caveolar transcytosis. Second, as a tonic inhibitor of eNOS activity, Cav-1 plays a negative role in regulating paracellular permeability (Sun et al., 2009). As a result of such dual regulation, although the Cav-1 KO mice display vascular and fluid balance abnormalities in the lung, they are resistant to ALI in comparison to WT mice. In addition, Cav-1 KO mice have markedly improved survival during secondary ALI resulting from sepsis induced by lipopolysaccharide (LPS; Garrean et al., 2006; Mirza et al., 2010). Analogous results were reported using hyperoxia (Jin et al., 2008b). Remarkably, Cav-1 KO mice display basal pulmonary edema manifested by elevated extravascular lung fluid. It has been postulated that this opposing tissue pressure may limit further transport and accumulation of pulmonary edema fluid from vascular damage during lung injury (Jin et al., 2011). Furthermore, loss of Cav-1 hyper-activates eNOS and subsequently reduces toll-like receptor 4 signaling, leading to the decreased innate immune response to LPS and thus protecting from LPS-induced inflammation and injury (Mirza et al., 2010). These data are in agreement with the previous studies by Garrean et al. (2006) demonstrating that Cav-1 KO mice have markedly reduced pro-inflammatory response to LPS via NF-κB-mediated pathways.
The Negative Role of Cav-2 in Regulating EC Proliferation
The possibility for the involvement of Cav-2 in regulating EC proliferation and differentiation in vivo was suggested by the observation that Cav-2 KO mice develop a hyperproliferative phenotype in the lungs associated with increased number of VEGFR2 positive cells (Razani et al., 2002). Because VEGFR2 is predominantly expressed in ECs, this observation suggests that Cav-2 may negatively regulate microvascular EC proliferation in the lung. However, due to the complexity of the in vivo system, it is impossible to unequivocally conclude if Cav-2 directly regulates lung microvascular EC proliferation. Thus, we immunoisolated and characterized pure populations of lung ECs from Cav-2 KO and WT mice, and compared their proliferation potential and the expression or phosphorylation levels of cell cycle-associated signaling proteins (Xie et al., 2010). These studies determined that Cav-2 suppresses lung microvascular EC proliferation via inhibition of extracellular signal regulated kinase 1/2 (ERK1/2) phosphorylation, increased expression of cyclin-dependent kinase (cdk) inhibitors p16INK4 and p27Kip1 and activation (hypophosphorylation) of the retinoblastoma (Rb) protein, resulting in a reduced cell cycle progression (Xie et al., 2010). Recently, another group using a combination of miRNA, siRNA, and plasmid overexpression approaches, has confirmed anti-proliferative function of Cav-2 in a rat prostate EC line (YPEN-1; Shatseva et al., 2011), suggesting that in addition to the lung, Cav-2 can also inhibit EC proliferation from other organs such as the prostate.
We are now exploring the mechanistic nature of this negative regulation by Cav-2. Since Cav-2 almost entirely targets to lipid rafts/caveolar microdomains in mouse lung ECs used in our studies (Xie et al., 2011), our data suggest that the inhibitory effect of Cav-2 on EC proliferation is most likely initiated in plasma membrane lipid rafts and caveolae. In addition, the fact that Cav-2 can be serine phosphorylated (Sowa et al., 2003), and that serine 36 phosphorylation of Cav-2 increases in mitotic ECs (Sowa et al., 2008) indirectly suggest involvement of serine phosphorylation in the growth inhibitory function of Cav-2 in ECs. However, more direct studies will be required to determine involvement of serine phosphorylation of Cav-2 in regulating EC proliferation.
Cav-2 Suppresses the TGF-β-Induced Signaling and Anti-Proliferative Function in Lung ECs
Our most recent findings suggest that the role of Cav-2 in regulating lung microvascular EC proliferation is more complex and context-specific than we originally thought (Xie et al., 2011). Specifically, using a combination of WT and Cav-2 KO, along with retroviral re-expression approaches, we have shown that Cav-2 may be a physiological inhibitor of anti-proliferative function and signaling of TGF-β in mouse lung ECs. Remarkably, although treatment with TGF-β resulted only in a marginal inhibitory effect on WT lung ECs, it greatly inhibited proliferation of Cav-2 KO lung ECs. Similar to WT ECs, the anti-proliferative effect of TGF-β was dramatically reduced in the Cav-2 KO ECs re-expressing Cav-2. Mechanistically, Cav-2 inhibits anti-proliferative action of TGF-β by suppressing the Alk5/Smad2/3 pathway manifested by reduced magnitude and length of TGF-β-induced Smad2/3 phosphorylation as well as activation of Alk5/Smad2/3 target genes, plasminogen activator inhibitor-1, and collagen type I in Cav-2-positive ECs. Our preliminary data suggest that expression of Cav-2 neither significantly changes targeting of TGF-β receptors type I, Alk5 and Alk1 nor Smad2/3 to caveolar and lipid raft microdomains. However, additional studies with control versus TGF-β-treated ECs using various subcellular fractionation and immunofluorescence microscopy localization techniques will be needed for a more comprehensive analysis. Cav-1 expression levels are reduced at least by c.a. 50% in Cav-2 KO, relative to WT ECs and thus could possibly contribute to the enhanced anti-proliferative effect of TGF-β in Cav-2 KO ECs. Nevertheless, the levels of re-expressed Cav-2 in Cav-2 KO ECs required to suppress TGF-β-induced signaling and anti-proliferative function are insufficient to upregulate the expression levels of Cav-1 or change its targeting to lipid raft/caveolar microdomains. In addition, just as in the case of endogenous Cav-2 in WT ECs, the re-expressed Cav-2 displays normal targeting to plasma membrane and lipid raft/caveolae. In addition, the re-expressed Cav-2 does not affect subcellular targeting of endogenous Cav-1. Thus the negative regulation of TGF-β signaling and function by Cav-2 is independent of both the expression levels and targeting of Cav-1 to lipid raft/caveolar microdomains. Future studies exploring the detailed mechanisms responsible for inhibitory regulation of TGF-β-induced signaling and function in ECs by Cav-2 will be essential. For example it will be important to examine interactions of Cav-2 and Cav-1 with Alk5 or other components of the TGF-β pathway in the absence and presence of TGF- β. In addition, it will be important to examine possible regulation of subcellular localization of Alk5, TGF-β receptor II, or accessory receptors such as endoglin and β-glycan by Cav-2. Also, it will be important to examine the specific role of previously identified serine and tyrosine phosphorylation of Cav-2 (Lee et al., 2002; Sowa et al., 2003, 2008; Wang et al., 2004). Finally, determining thein vivo significance of our results is clearly warranted.
Could Cav-2 be Considered a Molecular Switch Controlling Lung EC Proliferation?
It is important to reconcile the role for Cav-2 in inhibiting TGF-β-induced signaling and anti-proliferative function (Xie et al., 2011) with the previously described anti-proliferative role of Cav-2 in ECs (Xie et al., 2010). Though, in both cases, Cav-2 acts as an inhibitor, the final outcome depends on the particular context. Specifically, in our previous studies in which Cav-2 had an anti-proliferative effect, EC proliferation was evaluated in the absence of known growth inhibitors (Xie et al., 2010). Under these conditions, Cav-2 diminishes the stimulatory effect of serum and growth factors on EC proliferation. Conversely, in the presence of TGF-β, the role of Cav-2 switches from anti- to pro-proliferative through the negative regulation of the growth inhibitory action of TGF-β/Alk5/Smad2/3 pathway (Xie et al., 2011). Thus, it is conceivable to suggest that Cav-2 could act as a molecular switch neutralizing excessive cell responses to both pro- and anti-proliferative signals. Further studies will be necessary to solidify this newly proposed role for Cav-2 in ECs and possibly other cell types.
Role of Cavins in Caveolae Turnover
Recent studies have revealed that in addition to their coat proteins, caveolae also contain adapter proteins, cavins. Cavin-1 (polymerase transcript release factor, PTRF), cavin-2 (serum deprivation protein response, SDPR), cavin-3 (srd-related gene product that binds to c-kinase, SRBC), and cavin-4 (muscle-restricted coiled-coil protein, MURC) may be important in regulating caveolin expression and caveolar morphology (Hansen and Nichols, 2010; Briand et al., 2011a).
Cavin-1/PTRF can be recruited by caveolins to plasma membrane caveolar domains and is necessary for caveolae formation (Hill et al., 2008; Liu and Pilch, 2008; Figure 2A). Deletion of cavin-1 led to the loss of morphologically distinct caveolae and to decreased protein stability of all three caveolins. Cavin-1 downregulation has been shown to cause increased mobility of plasma membrane Cav-1, leading to its rapid internalization and degradation (Hill et al., 2008). It is believed that cavin-1 contributes to the last steps of caveolae biogenesis as the latter protein only associates with plasma membrane caveolae and not with non-caveolar caveolins (Hill et al., 2008; Hayer et al., 2010). Thus, cavin-1 could be considered as a soluble protein being recruited to caveolae as a scaffold stabilizing the caveolae unit (Figure 2A).
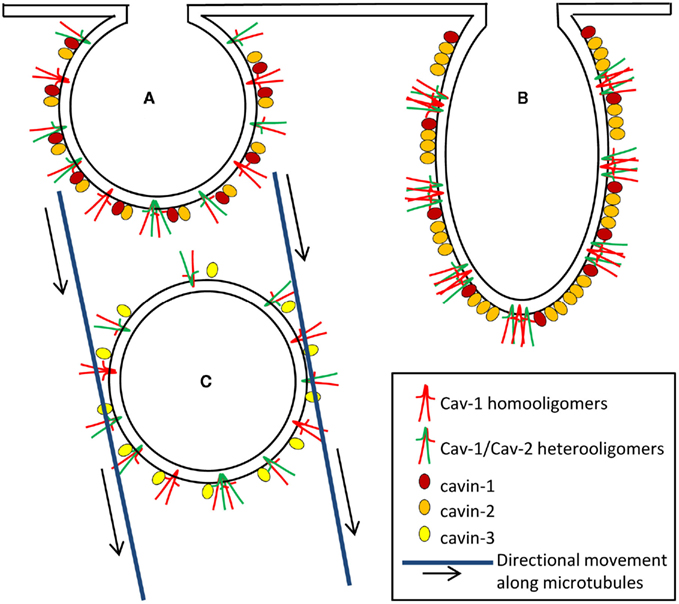
Figure 2. The complex role of caveolar coat an adapter proteins in caveolae formation (A), deformation/elongation (B), and internalization (C). Heterooligomers of the caveolar coat proteins, Cav-1 and Cav-2 and homooligomers of Cav-1 crossing the inner leaflet of plasma membrane are responsible for creating a structure or backbone of caveolae. The adapter proteins of caveolae, cavins provide the scaffold, determine shape, and regulate internalization/budding of caveolae. (A) Cavin-1 is a soluble protein which is recruited to caveolae as a scaffold stabilizing the caveolae unit (Deletion of cavin-1 results in the loss of morphologically distinct caveolae and in decreased protein stability of caveolins). Cavin-2 and cavin-1 interact with each other and cavin-2 promotes recruitment of cavin-1 to caveolae. Cavin-2 downregulation induces loss of cavin-1 and Cav-1 expression and thereby limits caveolae formation. (B) Cavin-2 is thought to be a necessary component for inducing membrane-curvature of caveolae (Overexpression of cavin-2 induces elongated caveolar morphology as well as caveolae-associated tubule formation). (C) Cavin-3 associates with Cav-1 upon caveolae internalization to form vesicles and its absence markedly reduces intracellular Cav-1 traffic along microtubules.
Cavin-2/SDPR downregulation induces loss of cavin-1 and Cav-1 expression and thereby limits caveolae formation (Figure 2A), suggesting that cavin-1, cavin-2, and Cav-1 are functionally inter-dependent (Hansen et al., 2009). Studies using co-immunoprecipitation approaches revealed that cavin-2 and cavin-1 interact with each other and cavin-2 promotes recruitment of cavin-1 to caveolae. However, this interaction does not require Cav-1 (Hansen et al., 2009). In contrast to Cav-1 or cavin-1 respectively, cavin-2 does not increase caveolae number but induces elongated caveolar morphology as well as caveolae-associated tubule formation (Hansen et al., 2009; Figure 2B). Thus cavin-2 may be a necessary component for inducing membrane-curvature of caveolae.
Based on homology with cavin-1, cavin-3 is also considered to play a role in determining caveolae structures. Interestingly, cavin-3 still associates with Cav-1, upon caveolae budding, to form vesicles and intracellular Cav-1 traffic along microtubules is markedly impaired in the absence of cavin-3 (McMahon et al., 2009). Taken together, these data suggest that although it is unclear if cavin-3 is necessary for caveolae formation the latter protein might be involved in coupling caveolae to the intracellular transport machinery (Figure 2C).
Cavin-4/MURC has been previously described as purely cytosolic and able to interact with cavin-2 (Ogata et al., 2008). Cavin-4 was also found to associate with sarcolemmal caveolae of muscle cells and its expression is perturbed in human muscle diseases associated with Cav-3 dysfunction. At this point, the exact role of cavin-4 in muscle caveolae turnover is unknown. However, Cavin-4 was shown to associate with cardiac dysfunction through the modulation of the Rho/ROCK pathway and to be important in muscle biogenesis (Tagawa et al., 2008). This is consistent with the specific expression of cavin-4 in cardiac and muscle tissues, which parallels Cav-3 expression (Bastiani et al., 2009).
Cavins and EC Function
Recently generated Cavin-1 KO mice were shown to lack morphologically distinct caveolae in all tissues/cell types examined, including endothelium. In addition, the expression levels of all three caveolin isoforms were markedly diminished as a result of protein degradation (Liu and Pilch, 2008). These data suggest that cavin-1 is responsible for caveolae formation and caveolin protein stabilization in vivo. At this point it is unknown if these mice develop pulmonary and cardiovascular phenotypes similar to those observed in Cav-1 KO mice, although based on a loss of caveolae and significant reduction of Cav-1, one could predict that this would be the case. This study reported a lipodystrophic phenotype in cavin-1 KO mice, suggesting adipocyte, and muscle dysfunction (Liu and Pilch, 2008). Most recent study by Briand et al. (2011b) suggests that lipodystrophy develops independently of the presence of Cav-1 and caveolae in the endothelium of Cav-1 KO. Clearly, direct studies examining presence of pulmonary and cardiovascular phenotypes which could depend on endothelial cavin-1 and caveolae will be required to determine the potential role of endothelial cavin-1. In addition, these studies may also help to better define the possible role of endothelial Cav-1/Cav-2 independent of cavin-1 and caveolae targeting. Of note, cases of patients with cavin-1 null mutations were recently reported (Rajab et al., 2010; Shastry et al., 2010). These patients presented with multiple pathologies such as generalized lipodystrophy, cardiac arrhythmias, long-QT syndrome, myopathy with muscle rippling, skeletal as well as smooth-muscle hypertrophy. Electron microscopy revealed nearly complete loss of caveolae (to less than 3%; Rajab et al., 2010). Interestingly, unlike general degradation of caveolin proteins reported in cavin-1 KO mice (Liu and Pilch, 2008), loss of cavin-1 did not affect the expression levels of Cav-1 in patient fibroblasts. However, Cav-1 lost its plasma membrane localization in these cells (Rajab et al., 2010). It is unknown if and to what extent loss of endothelial cavin-1, directly or indirectly through loss of endothelial caveolae and Cav-1 mislocalization, could contribute to some of these pathologies, in particular to cardiac defects. Moreover, it would be very interesting to see if these patients present any vascular or pulmonary phenotypes which might depend on endothelial caveolae and caveolins.
Although no studies focused on endothelial cavin in vivo were reported, there is one study which has examined the functional significance of cavin-1 in cultured ECs (Davalos et al., 2010). Specifically, to identify proteins that require Cav-1 for targeting to lipid raft/caveolae domains, a quantitative proteomics analysis using isobaric tagging was performed on lipid raft membranes isolated from WT and Cav-1 KO mice. In three independent experiments, 117 proteins could be consistently identified in lipid raft membranes with the largest differences in the levels of caveolar coat protein Cav-2, and in the caveolar adapter proteins cavin-1 and cavin-2. Because the lung is highly enriched in ECs, the role of the newly described protein cavin-1 was examined in several cardiovascular tissues and in isolated ECs. Cavin-1 was highly expressed in ECs lining blood vessels and in cultured ECs. SiRNA-mediated knockdown of cavin-1 also reduced the levels of Cav-1 and -2 and had only marginal effect on the formation of high molecular weight oligomers containing Cav-1 and -2. Furthermore, silencing of cavin-1 enhanced basal NO release from ECs but blocked pro-angiogenic phenotypes such as EC proliferation, migration, and morphogenesis in vitro. Thus, these data support an important role for cavin-1 as a regulator of caveolar function in ECs (Davalos et al., 2010). Further mechanistic studies will be required to determine how cavin-1 regulates EC function in vitro and possibly in vivo. Also, in addition to cavin-1, studies examining a possible functional role of cavin-2 in ECs in vitro and in vivo are warranted. It will also be interesting to examine if cavin-3 is expressed in ECs and its potential functional significance.
Conclusion
Much of the literature reviewed here suggests that caveolae, caveolins, and cavins play important roles in regulating EC signaling and function and thereby the cardiovascular and pulmonary function at cell and systemic levels. In addition to the physiological role, caveolae and caveolins, in particular Cav-1 expressed in ECs, have been shown to have important regulatory roles in pathological angiogenesis and in vascular disease, for example, in atherosclerosis, cardiac hypertrophy, pulmonary hypertension, and ALI. Caveolae are required for the proper organization of signaling pathways that support numerous signaling events, through sequestration of receptors and downstream signaling regulators. KO and siRNA approaches targeting caveolin proteins revealed that these proteins are directly responsible for many of the regulatory mechanisms attributed to caveolae. In addition, the discovery of cavin proteins should further our understanding of caveolar functions in ECs and in the cardiovascular system. Despite considerable progress, many unresolved issues still remain with respect to caveolae and Cav-1, although their role in regulating EC and vascular systemic function seems unquestioned. In particular, more selective approaches distinguishing between caveolar versus non-caveolar functions of Cav-1 in ECs will be necessary. Moreover the roles of the understudied Cav-2, and the more recently discovered cavin proteins, in EC function are still poorly understood and thus future functional and mechanistic studies a clearly warranted.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author wishes to apologize for any omissions of citing primary research articles due to space limitations. Work in the author’s laboratory on this topic is supported by the grant from the National Institutes of Health (1R01HL081860 to Grzegorz Sowa).
References
Albinsson, S., Nordstrom, I., Sward, K., and Hellstrand, P. (2008). Differential dependence of stretch and shear stress signaling on caveolin-1 in the vascular wall. Am. J. Physiol. Cell Physiol. 294, C271–C279.
Albinsson, S., Shakirova, Y., Rippe, A., Baumgarten, M., Rosengren, B. I., Rippe, C., Hallmann, R., Hellstrand, P., Rippe, B., and Sward, K. (2007). Arterial remodeling and plasma volume expansion in caveolin-1-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1222–R1231.
Allen, J. A., Halverson-Tamboli, R. A., and Rasenick, M. M. (2007). Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140.
Anderson, R. G. (1993). Caveolae: where incoming and outgoing messengers meet. Proc. Natl. Acad. Sci. U.S.A. 90, 10909–10913.
Anderson, R. G., Kamen, B. A., Rothberg, K. G., and Lacey, S. W. (1992). Potocytosis: sequestration and transport of small molecules by caveolae. Science 255, 410–411.
Barresi, V., Cerasoli, S., and Tuccari, G. (2008). Correlative evidence that tumor cell-derived caveolin-1 mediates angiogenesis in meningiomas. Neuropathology 28, 472–478.
Bastiani, M., Liu, L., Hill, M. M., Jedrychowski, M. P., Nixon, S. J., Lo, H. P., Abankwa, D., Luetterforst, R., Fernandez-Rojo, M., Breen, M. R., Gygi, S. P., Vinten, J., Walser, P. J., North, K. N., Hancock, J. F., Pilch, P. F., and Parton, R. G. (2009). MURC/Cavin-4 and cavin family members form tissue-specific caveolar complexes. J. Cell Biol. 185, 1259–1273.
Beardsley, A., Fang, K., Mertz, H., Castranova, V., Friend, S., and Liu, J. (2005). Loss of caveolin-1 polarity impedes endothelial cell polarization and directional movement. J. Biol. Chem. 280, 3541–3547.
Bernatchez, P., Sharma, A., Bauer, P. M., Marin, E., and Sessa, W. C. (2011). A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J. Clin. Invest. 121, 3747–3755.
Briand, N., Dugail, I., and Le Lay, S. (2011a). Cavin proteins: new players in the caveolae field. Biochimie 93, 71–77.
Briand, N., Le Lay, S., Sessa, W. C., Ferre, P., and Dugail, I. (2011b). Distinct roles of endothelial and adipocyte caveolin-1 in macrophage infiltration and adipose tissue metabolic activity. Diabetes 60, 448–453.
Brouet, A., DeWever, J., Martinive, P., Havaux, X., Bouzin, C., Sonveaux, P., and Feron, O. (2005). Antitumor effects of in vivo caveolin gene delivery are associated with the inhibition of the proangiogenic and vasodilatory effects of nitric oxide. FASEB J. 19, 602–604.
Bucci, M., Gratton, J. P., Rudic, R. D., Acevedo, L., Roviezzo, F., Cirino, G., and Sessa, W. C. (2000). In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 6, 1362–1367.
Chang, S. H., Feng, D., Nagy, J. A., Sciuto, T. E., Dvorak, A. M., and Dvorak, H. F. (2009). Vascular permeability and pathological angiogenesis in caveolin-1-null mice. Am. J. Pathol. 175, 1768–1776.
Cohen, A. W., Park, D. S., Woodman, S. E., Williams, T. M., Chandra, M., Shirani, J., Pereira de Souza, A., Kitsis, R. N., Russell, R. G., Weiss, L. M., Tang, B., Jelicks, L. A., Factor, S. M., Shtutin, V., Tanowitz, H. B., and Lisanti, M. P. (2003). Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 284, C457–C474.
Davalos, A., Fernandez-Hernando, C., Sowa, G., Derakhshan, B., Lin, M. I., Lee, J. Y., Zhao, H., Luo, R., Colangelo, C., and Sessa, W. C. (2010). Quantitative proteomics of caveolin-1-regulated proteins: characterization of polymerase i and transcript release factor/CAVIN-1 IN endothelial cells. Mol. Cell Proteomics 9, 2109–2124.
de Laurentiis, A., Donovan, L., and Arcaro, A. (2007). Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem. J. 1, 12–32.
Desjardins, F., Lobysheva, I., Pelat, M., Gallez, B., Feron, O., Dessy, C., and Balligand, J. L. (2008). Control of blood pressure variability in caveolin-1-deficient mice: role of nitric oxide identified in vivo through spectral analysis. Cardiovasc. Res. 79, 527–536.
Dewever, J., Frerart, F., Bouzin, C., Baudelet, C., Ansiaux, R., Sonveaux, P., Gallez, B., Dessy, C., and Feron, O. (2007). Caveolin-1 is critical for the maturation of tumor blood vessels through the regulation of both endothelial tube formation and mural cell recruitment. Am. J. Pathol. 171, 1619–1628.
Drab, M., Verkade, P., Elger, M., Kasper, M., Lohn, M., Lauterbach, B., Menne, J., Lindschau, C., Mende, F., Luft, F. C., Schedl, A., Haller, H., and Kurzchalia, T. V. (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449–2452.
Dubroca, C., Loyer, X., Retailleau, K., Loirand, G., Pacaud, P., Feron, O., Balligand, J. L., Levy, B. I., Heymes, C., and Henrion, D. (2007). RhoA activation and interaction with Caveolin-1 are critical for pressure-induced myogenic tone in rat mesenteric resistance arteries. Cardiovasc. Res. 73, 190–197.
Durante, W., Kroll, M. H., Christodoulides, N., Peyton, K. J., and Schafer, A. I. (1997). Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Circ. Res. 80, 557–564.
Durr, E., Yu, J., Krasinska, K. M., Carver, L. A., Yates, J. R., Testa, J. E., Oh, P., and Schnitzer, J. E. (2004). Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat. Biotechnol. 22, 985–992.
Fang, K., Fu, W., Beardsley, A. R., Sun, X., Lisanti, M. P., and Liu, J. (2007). Overexpression of caveolin-1 inhibits endothelial cell proliferation by arresting the cell cycle at G0/G1 phase. Cell Cycle 6, 199–204.
Fernandez-Hernando, C., Yu, J., Davalos, A., Prendergast, J., and Sessa, W. C. (2010). Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 177, 998–1003.
Fernandez-Hernando, C., Yu, J., Suarez, Y., Rahner, C., Davalos, A., Lasuncion, M. A., and Sessa, W. C. (2009). Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 10, 48–54.
Feron, O., Belhassen, L., Kobzik, L., Smith, T. W., Kelly, R. A., and Michel, T. (1996). Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 271, 22810–22814.
Fielding, P. E., and Fielding, C. J. (1995). Plasma membrane caveolae mediate the efflux of cellular free cholesterol. Biochemistry 34, 14288–14292.
Frank, P. G., Lee, H., Park, D. S., Tandon, N. N., Scherer, P. E., and Lisanti, M. P. (2004). Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 98–105.
Frank, P. G., Pavlides, S., Cheung, M. W., Daumer, K., and Lisanti, M. P. (2008). Role of caveolin-1 in the regulation of lipoprotein metabolism. Am. J. Physiol. Cell Physiol. 295, C242–C248.
Fujimoto, T., Kogo, H., Nomura, R., and Une, T. (2000). Isoforms of caveolin-1 and caveolar structure. J. Cell. Sci. 113(Pt 19), 3509–3517.
Galvez, B. G., Matias-Roman, S., Yanez-Mo, M., Vicente-Manzanares, M., Sanchez-Madrid, F., and Arroyo, A. G. (2004). Caveolae are a novel pathway for membrane-type 1 matrix metalloproteinase traffic in human endothelial cells. Mol. Biol. Cell 15, 678–687.
Garcia-Cardena, G., Martasek, P., Masters, B. S., Skidd, P. M., Couet, J., Li, S., Lisanti, M. P., and Sessa, W. C. (1997). Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 272, 25437–25440.
Garrean, S., Gao, X. P., Brovkovych, V., Shimizu, J., Zhao, Y. Y., Vogel, S. M., and Malik, A. B. (2006). Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J. Immunol. 177, 4853–4860.
Ge, S., Song, L., Serwanski, D. R., Kuziel, W. A., and Pachter, J. S. (2008). Transcellular transport of CCL2 across brain microvascular endothelial cells. J. Neurochem. 104, 1219–1232.
Ghitescu, L., Fixman, A., Simionescu, M., and Simionescu, N. (1986). Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: receptor-mediated transcytosis. J. Cell Biol. 102, 1304–1311.
Glenney, J. R. Jr., and Soppet, D. (1992). Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 89, 10517–10521.
Griffoni, C., Spisni, E., Santi, S., Riccio, M., Guarnieri, T., and Tomasi, V. (2000). Knockdown of caveolin-1 by antisense oligonucleotides impairs angiogenesis in vitro and in vivo. Biochem. Biophys. Res. Commun. 276, 756–761.
Hansen, C. G., Bright, N. A., Howard, G., and Nichols, B. J. (2009). SDPR induces membrane curvature and functions in the formation of caveolae. Nat. Cell Biol. 11, 807–814.
Hansen, C. G., and Nichols, B. J. (2010). Exploring the caves: cavins, caveolins and caveolae. Trends Cell Biol. 20, 177–186.
Hartmannsgruber, V., Heyken, W. T., Kacik, M., Kaistha, A., Grgic, I., Harteneck, C., Liedtke, W., Hoyer, J., and Kohler, R. (2007). Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS ONE 2, e827. doi:10.1371/journal.pone.0000827
Hayer, A., Stoeber, M., Bissig, C., and Helenius, A. (2010). Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic 11, 361–382.
Hill, M. M., Bastiani, M., Luetterforst, R., Kirkham, M., Kirkham, A., Nixon, S. J., Walser, P., Abankwa, D., Oorschot, V. M., Martin, S., Hancock, J. F., and Parton, R. G. (2008). PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124.
Hu, G., Vogel, S. M., Schwartz, D. E., Malik, A. B., and Minshall, R. D. (2008a). Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ. Res. 102, e120–e131.
Hu, G., Ye, R. D., Dinauer, M. C., Malik, A. B., and Minshall, R. D. (2008b). Neutrophil caveolin-1 expression contributes to mechanism of lung inflammation and injury. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L178–L186.
Ingbar, D. H. (2000). Mechanisms of repair and remodeling following acute lung injury. Clin. Chest Med. 21, 589–616.
Insel, P. A., and Patel, H. H. (2007). Do studies in caveolin-knockouts teach us about physiology and pharmacology or instead, the ways mice compensate for ‘lost proteins’? Br. J. Pharmacol. 150, 251–254.
Insel, P. A., and Patel, H. H. (2009). Membrane rafts and caveolae in cardiovascular signaling. Curr. Opin. Nephrol. Hypertens. 18, 50–56.
Isshiki, M., and Anderson, R. G. (1999). Calcium signal transduction from caveolae. Cell Calcium 26, 201–208.
Iwakiri, Y., Satoh, A., Chatterjee, S., Toomre, D. K., Chalouni, C. M., Fulton, D., Groszmann, R. J., Shah, V. H., and Sessa, W. C. (2006). Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. U.S.A. 103, 19777–19782.
Jin, S., Zhang, Y., Yi, F., and Li, P. L. (2008a). Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 28, 485–490.
Jin, Y., Kim, H. P., Chi, M., Ifedigbo, E., Ryter, S. W., and Choi, A. M. (2008b). Deletion of caveolin-1 protects against oxidative lung injury via up-regulation of heme oxygenase-1. Am. J. Respir. Cell Mol. Biol. 39, 171–179.
Jin, Y., Lee, S. J., Minshall, R. D., and Choi, A. M. (2011). Caveolin-1: a critical regulator of lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L151–L160.
Joo, H. J., Oh, D. K., Kim, Y. S., Lee, K. B., and Kim, S. J. (2004). Increased expression of caveolin-1 and microvessel density correlates with metastasis and poor prognosis in clear cell renal cell carcinoma. BJU Int. 93, 291–296.
Ju, H., Zou, R., Venema, V. J., and Venema, R. C. (1997). Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272, 18522–18525.
Kim, H. P., Wang, X., Galbiati, F., Ryter, S. W., and Choi, A. M. (2004). Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 18, 1080–1089.
Kim, S., Lee, Y., Seo, J. E., Cho, K. H., and Chung, J. H. (2008). Caveolin-1 increases basal and TGF-beta1-induced expression of type I procollagen through PI-3 kinase/Akt/mTOR pathway in human dermal fibroblasts. Cell. Signal. 20, 1313–1319.
King, G. L., and Johnson, S. M. (1985). Receptor-mediated transport of insulin across endothelial cells. Science 227, 1583–1586.
Kohler, R., Heyken, W. T., Heinau, P., Schubert, R., Si, H., Kacik, M., Busch, C., Grgic, I., Maier, T., and Hoyer, J. (2006). Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler. Thromb. Vasc. Biol. 26, 1495–1502.
Kurzchalia, T. V., Dupree, P., Parton, R. G., Kellner, R., Virta, H., Lehnert, M., and Simons, K. (1992). VIP21, a 21-kD membrane protein is an integral component of trans-Golgi-network-derived transport vesicles. J. Cell Biol. 118, 1003–1014.
Kwiatek, A. M., Minshall, R. D., Cool, D. R., Skidgel, R. A., Malik, A. B., and Tiruppathi, C. (2006). Caveolin-1 regulates store-operated Ca2+ influx by binding of its scaffolding domain to transient receptor potential channel-1 in endothelial cells. Mol. Pharmacol. 70, 1174–1183.
Labrecque, L., Royal, I., Surprenant, D. S., Patterson, C., Gingras, D., and Beliveau, R. (2003). Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol. Biol. Cell 14, 334–347.
Lahtinen, U., Honsho, M., Parton, R. G., Simons, K., and Verkade, P. (2003). Involvement of caveolin-2 in caveolar biogenesis in MDCK cells. FEBS Lett. 538, 85–88.
Lee, H., Park, D. S., Wang, X. B., Scherer, P. E., Schwartz, P. E., and Lisanti, M. P. (2002). Src-induced phosphorylation of caveolin-2 on tyrosine 19. Phospho-caveolin-2 (Tyr(P)19) is localized near focal adhesions, remains associated with lipid rafts/caveolae, but no longer forms a high molecular mass hetero-oligomer with caveolin-1. J. Biol. Chem. 277, 34556–34567.
Li, P. L., Zhang, Y., and Yi, F. (2007). Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid. Redox Signal. 9, 1457–1470.
Lin, M. I., Yu, J., Murata, T., and Sessa, W. C. (2007). Caveolin-1-deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res. 67, 2849–2856.
Liu, J., Wang, X. B., Park, D. S., and Lisanti, M. P. (2002). Caveolin-1 expression enhances endothelial capillary tubule formation. J. Biol. Chem. 277, 10661–10668.
Liu, L., and Pilch, P. F. (2008). A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J. Biol. Chem. 283, 4314–4322.
Luscher, T. F., and Noll, G. (1994). Endothelium dysfunction in the coronary circulation. J. Cardiovasc. Pharmacol. 24(Suppl. 3), S16–S26.
Mantell, L. L., and Lee, P. J. (2000). Signal transduction pathways in hyperoxia-induced lung cell death. Mol. Genet. Metab. 71, 359–370.
Matute-Bello, G., Frevert, C. W., and Martin, T. R. (2008). Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L379–L399.
McMahon, K. A., Zajicek, H., Li, W. P., Peyton, M. J., Minna, J. D., Hernandez, V. J., Luby-Phelps, K., and Anderson, R. G. (2009). SRBC/cavin-3 is a caveolin adapter protein that regulates caveolae function. EMBO J. 28, 1001–1015.
Mehta, D., Bhattacharya, J., Matthay, M. A., and Malik, A. B. (2004). Integrated control of lung fluid balance. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L1081–L1090.
Michel, J. B., Feron, O., Sacks, D., and Michel, T. (1997). Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 272, 15583–15586.
Milovanova, T., Chatterjee, S., Hawkins, B. J., Hong, N., Sorokina, E. M., Debolt, K., Moore, J. S., Madesh, M., and Fisher, A. B. (2008). Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim. Biophys. Acta 1783, 1866–1875.
Mirza, M. K., Yuan, J., Gao, X. P., Garrean, S., Brovkovych, V., Malik, A. B., Tiruppathi, C., and Zhao, Y. Y. (2010). Caveolin-1 deficiency dampens Toll-like receptor 4 signaling through eNOS activation. Am. J. Pathol. 176, 2344–2351.
Miyawaki-Shimizu, K., Predescu, D., Shimizu, J., Broman, M., Predescu, S., and Malik, A. B. (2006). siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L405–L413.
Murata, T., Lin, M. I., Huang, Y., Yu, J., Bauer, P. M., Giordano, F. J., and Sessa, W. C. (2007a). Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J. Exp. Med. 204, 2373–2382.
Murata, T., Lin, M. I., Stan, R. V., Bauer, P. M., Yu, J., and Sessa, W. C. (2007b). Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J. Biol. Chem. 282, 16631–16643.
Nilius, B., Droogmans, G., and Wondergem, R. (2003). Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium 10, 5–15.
Ogata, T., Ueyama, T., Isodono, K., Tagawa, M., Takehara, N., Kawashima, T., Harada, K., Takahashi, T., Shioi, T., Matsubara, H., and Oh, H. (2008). MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol. Cell. Biol. 28, 3424–3436.
Ogi, M., Yokomori, H., Oda, M., Yoshimura, K., Nomura, M., Ohshima, S., Akita, M., Toda, K., and Ishii, H. (2003). Distribution and localization of caveolin-1 in sinusoidal cells in rat liver. Med. Electron. Microsc. 36, 33–40.
Oh, P., Borgstrom, P., Witkiewicz, H., Li, Y., Borgstrom, B. J., Chrastina, A., Iwata, K., Zinn, K. R., Baldwin, R., Testa, J. E., and Schnitzer, J. E. (2007). Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotechnol. 25, 327–337.
Palade, G. E., and Bruns, R. R. (1968). Structural modulations of plasmalemmal vesicles. J. Cell Biol. 37, 633–649.
Pani, B., and Singh, B. B. (2009). Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 45, 625–633.
Park, H., Go, Y. M., St John, P. L., Maland, M. C., Lisanti, M. P., Abrahamson, D. R., and Jo, H. (1998). Plasma membrane cholesterol is a key molecule in shear stress-dependent activation of extracellular signal-regulated kinase. J. Biol. Chem. 273, 32304–32311.
Parton, R. G., and Simons, K. (2007). The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194.
Patel, H. H., Murray, F., and Insel, P. A. (2008). Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol. Toxicol. 48, 359–391.
Predescu, D., Vogel, S. M., and Malik, A. B. (2004). Functional and morphological studies of protein transcytosis in continuous endothelia. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L895–L901.
Rahman, A., and Sward, K. (2009). The role of caveolin-1 in cardiovascular regulation. Acta Physiol. (Oxf.) 195, 231–245.
Rajab, A., Straub, V., McCann, L. J., Seelow, D., Varon, R., Barresi, R., Schulze, A., Lucke, B., Lutzkendorf, S., Karbasiyan, M., Bachmann, S., Spuler, S., and Schuelke, M. (2010). Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet. 6, e1000874. doi:10.1371/journal.pgen.1000874
Razani, B., Engelman, J. A., Wang, X. B., Schubert, W., Zhang, X. L., Marks, C. B., Macaluso, F., Russell, R. G., Li, M., Pestell, R. G., Di Vizio, D., Hou, H. Jr., Kneitz, B., Lagaud, G., Christ, G. J., Edelmann, W., and Lisanti, M. P. (2001). Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276, 38121–38138.
Razani, B., Wang, X. B., Engelman, J. A., Battista, M., Lagaud, G., Zhang, X. L., Kneitz, B., Hou, H. Jr., Christ, G. J., Edelmann, W., and Lisanti, M. P. (2002). Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol. Cell. Biol. 22, 2329–2344.
Rippe, B., Rosengren, B. I., Carlsson, O., and Venturoli, D. (2002). Transendothelial transport: the vesicle controversy. J. Vasc. Res. 39, 375–390.
Rizzo, V., McIntosh, D. P., Oh, P., and Schnitzer, J. E. (1998a). In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association. J. Biol. Chem. 273, 34724–34729.
Rizzo, V., Sung, A., Oh, P., and Schnitzer, J. E. (1998b). Rapid mechanotransduction in situ at the luminal cell surface of vascular endothelium and its caveolae. J. Biol. Chem. 273, 26323–26329.
Rosengren, B. I., Rippe, A., Rippe, C., Venturoli, D., Sward, K., and Rippe, B. (2006). Transvascular protein transport in mice lacking endothelial caveolae. Am. J. Physiol. Heart Circ. Physiol. 291, H1371–H1377.
Saliez, J., Bouzin, C., Rath, G., Ghisdal, P., Desjardins, F., Rezzani, R., Rodella, L. F., Vriens, J., Nilius, B., Feron, O., Balligand, J. L., and Dessy, C. (2008). Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 117, 1065–1074.
Scheiffele, P., Verkade, P., Fra, A. M., Virta, H., Simons, K., and Ikonen, E. (1998). Caveolin-1 and -2 in the exocytic pathway of MDCK cells. J. Cell Biol. 140, 795–806.
Scherer, P. E., Okamoto, T., Chun, M., Nishimoto, I., Lodish, H. F., and Lisanti, M. P. (1996). Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc. Natl. Acad. Sci. U.S.A. 93, 131–135.
Schnitzer, J. E., Liu, J., and Oh, P. (1995). Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. J. Biol. Chem. 270, 14399–14404.
Schnitzer, J. E., Oh, P., and McIntosh, D. P. (1996). Role of GTP hydrolysis in fission of caveolae directly from plasma membranes. Science 274, 239–242. [Published erratum appears in Science 1996 Nov 15; 274 (5290), 109].
Schubert, W., Frank, P. G., Razani, B., Park, D. S., Chow, C. W., and Lisanti, M. P. (2001). Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J. Biol. Chem. 276, 48619–48622.
Schubert, W., Frank, P. G., Woodman, S. E., Hyogo, H., Cohen, D. E., Chow, C. W., and Lisanti, M. P. (2002). Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J. Biol. Chem. 277, 40091–40098.
Severs, N. J. (1988). Caveolae: static inpocketings of the plasma membrane, dynamic vesicles or plain artifact? J. Cell Sci. 90, 341–348.
Shastry, S., Delgado, M. R., Dirik, E., Turkmen, M., Agarwal, A. K., and Garg, A. (2010). Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am. J. Med. Genet. A 152A, 2245–2253.
Shatseva, T., Lee, D. Y., Deng, Z., and Yang, B. B. (2011). MicroRNA miR-199a-3p regulates cell proliferation and survival by targeting caveolin-2. J. Cell. Sci. 124, 2826–2836.
Siddiqui, M. R., Komarova, Y. A., Vogel, S. M., Gao, X., Bonini, M. G., Rajasingh, J., Zhao, Y. Y., Brovkovych, V., and Malik, A. B. (2011). Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. J. Cell Biol. 193, 841–850.
Smart, E. J., Ying, Y., Donzell, W. C., and Anderson, R. G. (1996). A role for caveolin in transport of cholesterol from endoplasmic reticulum to plasma membrane. J. Biol. Chem. 271, 29427–29435.
Soda, R., and Tavassoli, M. (1984). Transendothelial transport (transcytosis) of iron-transferrin complex in the bone marrow. J. Ultrastruct. Res. 88, 18–29.
Sonveaux, P., Martinive, P., DeWever, J., Batova, Z., Daneau, G., Pelat, M., Ghisdal, P., Gregoire, V., Dessy, C., Balligand, J. L., and Feron, O. (2004). Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ. Res. 95, 154–161.
Sowa, G., Pypaert, M., Fulton, D., and Sessa, W. C. (2003). The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc. Natl. Acad. Sci. U.S.A. 100, 6511–6516.
Sowa, G., Pypaert, M., and Sessa, W. C. (2001). Distinction between signaling mechanisms in lipid rafts vs. caveolae. Proc. Natl. Acad. Sci. U.S.A. 98, 14072–14077.
Sowa, G., Xie, L., Xu, L., and Sessa, W. C. (2008). Serine 23 and 36 phosphorylation of caveolin-2 is differentially regulated by targeting to lipid raft/caveolae and in mitotic endothelial cells. Biochemistry 47, 101–111.
Sun, R. J., Muller, S., Stoltz, J. F., and Wang, X. (2002). Shear stress induces caveolin-1 translocation in cultured endothelial cells. Eur. Biophys. J. 30, 605–611.
Sun, Y., Hu, G., Zhang, X., and Minshall, R. D. (2009). Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 105, 676–685, 15 p following 685.
Sundivakkam, P. C., Kwiatek, A. M., Sharma, T. T., Minshall, R. D., Malik, A. B., and Tiruppathi, C. (2009). Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am. J. Physiol. Cell Physiol. 296, C403–C413.
Tagawa, M., Ueyama, T., Ogata, T., Takehara, N., Nakajima, N., Isodono, K., Asada, S., Takahashi, T., Matsubara, H., and Oh, H. (2008). MURC, a muscle-restricted coiled-coil protein, is involved in the regulation of skeletal myogenesis. Am. J. Physiol. Cell Physiol. 295, C490–C498.
Tahir, S. A., Yang, G., Goltsov, A. A., Watanabe, M., Tabata, K., Addai, J., Fattah el, M. A., Kadmon, D., and Thompson, T. C. (2008). Tumor cell-secreted caveolin-1 has proangiogenic activities in prostate cancer. Cancer Res. 68, 731–739.
Tang, Z., Scherer, P. E., Okamoto, T., Song, K., Chu, C., Kohtz, D. S., Nishimoto, I., Lodish, H. F., and Lisanti, M. P. (1996). Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J. Biol. Chem. 271, 2255–2261.
Tiruppathi, C., Song, W., Bergenfeldt, M., Sass, P., and Malik, A. B. (1997). Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J. Biol. Chem. 272, 25968–25975.
Traub, O., and Berk, B. C. (1998). Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler. Thromb. Vasc. Biol. 18, 677–685.
Unno, M., Matsui, T., and Ikeda-Saito, M. (2007). Structure and catalytic mechanism of heme oxygenase. Nat. Prod. Rep. 24, 553–570.
Wang, L., Sapuri-Butti, A. R., Aung, H. H., Parikh, A. N., and Rutledge, J. C. (2008). Triglyceride-rich lipoprotein lipolysis increases aggregation of endothelial cell membrane microdomains and produces reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 295, H237–H244.
Wang, R., Wang, Z., and Wu, L. (1997). Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br. J. Pharmacol. 121, 927–934.
Wang, X. B., Lee, H., Capozza, F., Marmon, S., Sotgia, F., Brooks, J. W., Campos-Gonzalez, R., and Lisanti, M. P. (2004). Tyrosine phosphorylation of caveolin-2 at residue 27: differences in the spatial and temporal behavior of phospho-Cav-2 (pY19 and pY27). Biochemistry 43, 13694–13706.
Way, M., and Parton, R. G. (1995). M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 376, 108–112. [Corrected and republished in FEBS Lett 1996 Jan 2; 378(1):108–112].
Williams, K. J., and Tabas, I. (2005). Lipoprotein retention – and clues for atheroma regression. Arterioscler. Thromb. Vasc. Biol. 25, 1536–1540.
Woodman, S. E., Ashton, A. W., Schubert, W., Lee, H., Williams, T. M., Medina, F. A., Wyckoff, J. B., Combs, T. P., and Lisanti, M. P. (2003). Caveolin-1 knockout mice show an impaired angiogenic response to exogenous stimuli. Am. J. Pathol. 162, 2059–2068.
Wunderlich, C., Schober, K., Lange, S. A., Drab, M., Braun-Dullaeus, R. C., Kasper, M., Schwencke, C., Schmeisser, A., and Strasser, R. H. (2006). Disruption of caveolin-1 leads to enhanced nitrosative stress and severe systolic and diastolic heart failure. Biochem. Biophys. Res. Commun. 340, 702–708.
Xie, L., Frank, P. G., Lisanti, M. P., and Sowa, G. (2010). Endothelial cells isolated from caveolin-2 knockout mice display higher proliferation rate and cell cycle progression relative to their wild-type counterparts. Am. J. Physiol. Cell Physiol. 298, C693–C701.
Xie, L., Vo-Ransdell, C., Abel, B., Willoughby, C., Jang, S., and Sowa, G. (2011). Caveolin-2 is a negative regulator of anti-proliferative function and signaling of transforming growth factor-beta in endothelial cells. Am. J. Physiol. Cell Physiol. 301, C1161–C1174.
Yamada, E. (1953). The fine structure of the gall bladder epithelium of the mouse. J. Biophys. Biochem. Cytol. 1, 445–458.
Yang, B., Radel, C., Hughes, D., Kelemen, S., and Rizzo, V. (2011). p190 RhoGTPase-activating protein links the beta1 integrin/caveolin-1 mechanosignaling complex to RhoA and actin remodeling. Arterioscler. Thromb. Vasc. Biol. 31, 376–383.
Yang, B., and Rizzo, V. (2007). TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 292, H954–H962.
Yang, G., Addai, J., Wheeler, T. M., Frolov, A., Miles, B. J., Kadmon, D., and Thompson, T. C. (2007). Correlative evidence that prostate cancer cell-derived caveolin-1 mediates angiogenesis. Hum. Pathol. 38, 1688–1695.
Yu, J., Bergaya, S., Murata, T., Alp, I. F., Bauer, M. P., Lin, M. I., Drab, M., Kurzchalia, T. V., Stan, R. V., and Sessa, W. C. (2006). Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J. Clin. Invest. 116, 1284–1291..
Zhang, A. Y., Yi, F., Zhang, G., Gulbins, E., and Li, P. L. (2006). Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 47, 74–80.
Zhang, Z. B., Cai, L., Zheng, S. G., Xiong, Y., and Dong, J. H. (2009). Overexpression of caveolin-1 in hepatocellular carcinoma with metastasis and worse prognosis: correlation with vascular endothelial growth factor, microvessel density and unpaired artery. Pathol. Oncol. Res. 15, 495–502.
Zhao, Y. Y., Liu, Y., Stan, R. V., Fan, L., Gu, Y., Dalton, N., Chu, P. H., Peterson, K., Ross, J. Jr., and Chien, K. R. (2002). Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. U.S.A. 99, 11375–11380.
Zhao, Y. Y., Zhao, Y. D., Mirza, M. K., Huang, J. H., Potula, H. H., Vogel, S. M., Brovkovych, V., Yuan, J. X., Wharton, J., and Malik, A. B. (2009). Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J. Clin. Invest. 119, 2009–2018.
Keywords: endothelial cell, caveolae, caveolin-1, caveolin-2, cavins
Citation: Sowa G (2012) Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front. Physio. 2:120. doi: 10.3389/fphys.2011.00120
Received: 18 October 2011;
Accepted: 19 December 2011;
Published online: 06 January 2012.
Edited by:
Mariappan Muthuchamy, Texas A&M Health Science Center, USAReviewed by:
Xu Peng, Texas A&M Health Science Center, USAUthayashanker Ezekiel, Saint Louis University, USA
Andrea Foskett, Texas A&M Health Science Center, USA
Karl Henrik Swärd, Lund University, Sweden
Copyright: © 2012 Sowa. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Grzegorz Sowa, Department of Medical Pharmacology and Physiology, University of Missouri, 1 Hospital Drive, Room MA 415, Columbia, MO 65212, USA. e-mail: sowag@health.missouri.edu