 Jin Qian1 David J. R. Fulton1,2*
Jin Qian1 David J. R. Fulton1,2*- 1 Vascular Biology Center, Georgia Health Sciences University, Augusta, GA, USA
- 2 Department of Pharmacology, Georgia Health Sciences University, Augusta, GA, USA
Nitric oxide (NO) has many beneficial actions on the vascular wall including suppression of inflammation. The mechanism(s) by which NO antagonizes cytokine signaling are poorly understood, but are thought to involve inhibition of the pro-inflammatory transcription factor, NF-κB. NO represses nuclear translocation of NF-κB via the S-nitrosylation of its subunits which decreases the expression of target genes including adhesion molecules. In previous studies, we have shown that the intracellular location of endothelial nitric oxide synthase (eNOS) can influence the amount of NO produced and that NO levels are paramount in regulating the S-nitrosylation of target proteins. The purpose of the current study was to investigate the significance of subcellular eNOS to NF-κB signaling induced by pro-inflammatory cytokines in human aortic endothelial cells (HAECs). We found that in HAECs stimulated with TNFα, L-NAME did not influence the expression of intercellular adhesion molecule 1 (ICAM-1) or vascular cell adhesion molecular 1 (VCAM-1). In eNOS “knock down” HAECs reconstituted with either plasma membrane or Golgi restricted forms of eNOS, there was no significant effect on the activation of the NF-κB pathway over different times and concentrations of TNFα. Similarly, the endogenous production of NO did not influence the phosphorylation of IκBα. In contrast, higher concentrations of NO derived from the use of the exogenous NO donor, DETA NONOate, effectively suppressed the expression of ICAM-1/VCAM-1 in response to TNFα and induced greater S-nitrosylation of IKKβ and p65. Collectively these results suggest that neither endogenous eNOS nor eNOS location is an important influence on inflammatory signaling via the NF-κB pathway and that higher NO concentrations are required to suppress NF-κB in HAECs.
Introduction
Nitric oxide (NO) is a short-lived, free radical, gaseous signaling molecule which participates in a diverse range of biological processes including the regulation of blood vessel and airway tone, inflammation, neurotransmission, and apoptosis (Nathan and Xie, 1994). The sGC–cGMP pathway is arguably the best recognized and best understood endpoint of NO signaling. The activation of sGC and increased cGMP production occurs at low levels of NO and has been shown to mediate numerous processes including, but not limited to, vascular relaxation, modulation of ion channel patency, gastro intestinal peristalsis, and inhibition of platelet aggregation (Moncada et al., 1991; Radomski et al., 1991; Archer et al., 1994; Nathan and Xie, 1994). However, it is increasingly recognized that there exists alternative pathways of NO signaling. The most prominent of these involves the direct nitrosylation of cysteine sulfhydryls of target proteins by NO (Stamler et al., 2001). Independent of sGC–cGMP signaling, this pathway has been shown to control the activity of a number of important biological processes including intracellular signaling (Gopalakrishna et al., 1993), exocytosis (Matsushita et al., 2003), and inflammation (Marshall et al., 2004). As S-nitrosylation requires higher concentrations of NO, important variables involved in the regulation of target protein modification include the intracellular location of NO synthesis and the amount of NO produced (Qian et al., 2010).
The acute loss of NO production in the blood vessel wall stimulates inflammatory processes including the increased recruitment of leukocytes to the vascular endothelium (Kubes et al., 1991; Ma et al., 1993). Endothelial nitric oxide synthase (eNOS) is the primary isoform responsible for the production of NO within blood vessels (Huang et al., 1995). The loss of eNOS in knockout mice significantly increases the development of chronic inflammatory disorders such as atherosclerosis and further suggests that NO is a buffer against pro-inflammatory mechanisms (Chen et al., 2001). How NO regulates vascular inflammation is poorly understood. NO suppresses the expression of intercellular adhesion molecule 1 (ICAM-1) in the endothelium via a process independent of sGC/cGMP signaling (Niu et al., 1994). One mechanism proposed is through the inhibition of NF-κB (De Caterina et al., 1995; Peng et al., 1995; Matthews et al., 1996). NF-κB is a transcription factor that participates in the cellular response to cellular stress including inflammation, cell survival, and proliferation (Li and Verma, 2002). NF-κB activation is observed in response to cytokines and promotes increased transcription of genes with κB sites in their promoters which include ICAM-1 and vascular cell adhesion molecular 1 (VCAM-1; Janssen-Heininger et al., 2000).
NF-κB is a protein complex that is maintained in an inactive form by sequestration in the cytosol via interaction with inhibitory subunits (IκB). Activation of cytokine receptors and elevated ROS promotes the phosphorylation of IκB, which leads to its ubiquitination and degradation and translocation of the active complex to the nucleus (Baud and Karin, 2001; Mohan et al., 2003). Multiple mechanisms have been proposed to account for the ability of NO to inhibit NF-κB. These include the stabilization of IκB, induction of IκBα mRNA, and reduced nuclear translocation of NF-κB. Given the multiple mechanisms of inhibition, it is not surprising that NO has been shown to S-nitrosylate multiple targets within the NF-κB pathway including Cys62 of p50 which compromises DNA binding (Matthews et al., 1996), Cys38 of p65 which prevents heterodimer formation (Kelleher et al., 2007) and Cys-179 of IKKβ which prevents IκB phosphorylation (Reynaert et al., 2004).
Previously we have shown that the location of eNOS with the endothelial cell is important for enzyme activity as well as the ability to influence the S-nitrosylation of target proteins such as NSF (Fulton et al., 2002; Zhang et al., 2006; Qian et al., 2010). When targeted to the plasma membrane, eNOS produces more NO than eNOS residing on endomembranes of the Golgi and also greater S-nitrosylation of target proteins such as NSF. However, this difference in S-nitrosylation was negated when the amount of NO produced was normalized through the use of calcium-insensitive forms of eNOS and suggests that the amount of NO has a greater influence on the S-nitrosylation of target proteins versus the proximity of NO production. The majority of studies investigating the role of NO in NF-κB activation have used NO donors or induced the high capacity NOS2 isoform. Comparatively little is known about whether endogenously produced NO from eNOS can influence endothelial NF-κB activation and whether the location of eNOS is important for NF-κB activation. In the current study we addressed both of these questions by investigating whether endogenously produced NO, the location of NO synthesis and the amount of NO are important for the regulation of NF-κB signaling and adhesion molecule expression in human aortic endothelial cells (HAECs).
Materials and Methods
Cells and Reagents
TNFα was purchased from Sigma. L-NAME and DETA NONOate were purchased from Cayman. ICAM-1 antibody was purchased from Santa Cruz. eNOS and GAPDH antibody were purchased from BD. IκBα and phosphorylated IκBα antibody were purchased from Cell Signaling. HAECs were purchased from Cascade Biologics and grown in endothelial cell basal medium-2 (EBM-2, Clonetics). eNOS knock down HAECs were generated as described previously (Zhang et al., 2006). Adenoviruses encoding the control viruses red fluorescent protein (RFP) and Golgi and PM targeted eNOS were generated using the pAdDEST adenoviral expression system (Invitrogen) as described (Zhang et al., 2006). Cells were seeded at a density of 2.5 × 105 cells/12 well plate and transduced the next day at a multiplicity of infection (MOI) of 20.
Immunoblotting
Cells were washed twice with phosphate-buffered saline, lysed on ice in 50 mM Tris–HCl, pH 6.8, 2% SDS, 30% glycerol, 6% β-mercaptoethanol, and 0.02% bromphenol blue. After gently sonication, lysates were clarified at 14,000 rpm for 10 min at 4°C and size-fractionated by 10% SDS-polyacrylamide gel electrophoresis and immunoblotted with corresponding antibodies.
No Release
Thirty-six hours post viral transduction, medium (200 μl) containing nitrite and nitrate (primarily 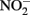 ) was precipitated with ethanol (1:2) to remove proteins and refluxed in sodium iodide/glacial acetic acid to convert to NO for measurement of the basal NO. NO was measured via NO-specific chemiluminescence after reaction with ozone (Sievers NO analyzer; GE Analytical Instruments, Boulder, CO, USA). Net from cells transfected with eNOS or iNOS was calculated after subtracting levels from cells lacking NOS activity (RFP transfected group).
) was precipitated with ethanol (1:2) to remove proteins and refluxed in sodium iodide/glacial acetic acid to convert to NO for measurement of the basal NO. NO was measured via NO-specific chemiluminescence after reaction with ozone (Sievers NO analyzer; GE Analytical Instruments, Boulder, CO, USA). Net from cells transfected with eNOS or iNOS was calculated after subtracting levels from cells lacking NOS activity (RFP transfected group).
Detection of S-Nitrosylation
Human aortic endothelial cells were treated with L-NAME (1 mM) or DETA NONOate (1 mM) for 6 h and cells were lysed and biotin labeled using biotin switch assay with some modification (Jaffrey and Snyder, 2001; Forrester et al., 2009). In brief, 48 h after transfection, cells were washed two times with cold PBS, and lysates prepared by incubation with HEN buffer containing 250 mM HEPES, 1 mM EDTA, 0.1 mM neocuproine (pH 7.7), SDS (2.5% final concentration), and methyl methanethiosulfonate (Sigma-Aldrich) at 50°C for 20 min and vortex every 4 min. Proteins were precipitated with acetone, washed three times with 70% acetone, and mixed with 0.2 mM biotin-HPDP (Pierce) with or without 50 mM ascorbate at ambient temperature for 1 h. Biotinylated proteins were pulled down by using streptavidin–agarose beads (Sigma-Aldrich), separated by SDS-PAGE, and detected by chemiluminescence with anti-p65 (Abcam) or IKKβ (Cell Signaling) antibody.
Cell Viability
Human aortic endothelial cells were plated into 96-well plate and treated with DETA NONOate (1 mM), TNFα (1 ng/ml), and thapsigargin (10 μM) was used as a positive control. Cell viability was determined after 24 h treatment by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT reagent was prepared by dissolving 5 mg/ml in PBS and then diluted with complete media (1:10) in each well. MTT reagent was added to each well (100 μl) and allowed to incubate for 3 h, after that the MTT was removed and DMSO (100 μl) was added to each well and shaken for 15 min. Viability was determined by recording absorbance at λ = 570 nm. The effect was assessed as percent cell viability where vehicle-treated cells (i.e., control) were taken as 100% viable.
Statistical Analysis
Data are expressed as mean ± SEM. Comparisons were made using analysis of variance (ANOVA) with a post hoc test. Differences were considered as significant at p < 0.05.
Results
Our first experimental goal was to determine whether endogenous NO is capable of influencing the ability of TNFα to upregulate the expression of ICAM-1 in HAECs. HAECs were treated with TNFα (1 ng/ml) for 4, 8, 24 h, with or without pre-treatment with the NOS inhibitor L-NAME (1 mM). ICAM-1 expression was robustly upregulated at all time points in a time-dependent manner. As shown in Figure 1A, L-NAME did not influence the level of induction of ICAM-1. We next investigated where NO influences the induction of ICAM-1 to lower concentrations of TNFα and similar effects of L-NAME were observed (Figure 1B). We next increased the concentration of L-NAME to 2 mM, but again found no significant change in the expression of ICAM-1 in response to TNFα (Figure 1C). To exclude the possibility that more stable S-nitrosylation is not reversed with short term inhibition of eNOS, we increased the L-NAME pre-treatment time from 30 min to 24 h. Despite the longer inhibition of NO synthesis, we did not observe any effect on ICAM-1 upregulation or VCAM-1 induction (Figure 1D).
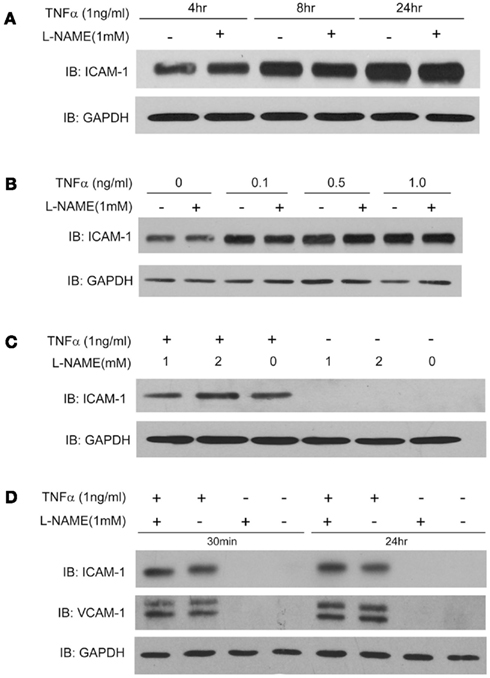
Figure 1. (A) Human aortic endothelial cells were incubated with TNFα (1 ng/ml) for 4, 8, and 24 h, in the presence and absence of L-NAME (1 mM, 30 min pre-treatment). Cell lysates were immunoblotted using an anti-ICAM-1 antibody and GAPDH was used as a loading control. (B) HAECs were pre-treated with L-NAME for 30 min, and stimulated with different concentrations of TNFα (0.5, 0.1, 1 ng/ml) for 24 h. (C) HAECs were treated with different concentrations of L-NAME (1 or 2 mM) for 30 min, and then exposed to TNFα (1 ng/ml) for another 24 h. (D) HAECs were pre-treated with L-NAME (1 mM) for different times (30 min or 24 h) prior to exposure to TNFα (1 ng/ml) for an additional 24 h. Cell lysates were immunoblotted using an anti-ICAM-1 or anti-VCAM-1 antibody and GAPDH was used as a loading control.
We next determined whether the location of eNOS was important in regulating the TNFα-dependent induction of adhesion molecules. To do so, we utilized a strategy of knocking down endogenous eNOS using a stably delivered shRNA and then re-expressing novel forms of eNOS that contain silent mutations to bypass the siRNA knockdown and are specifically targeted to the Golgi (S17) or plasma membrane (CAAX; Zhang et al., 2006). As shown in Figure 2 and consistent with our previous findings, we did not detect any significant difference in ICAM-1 expression between HAECs expressing the Golgi (S17) or PM (CAAX) eNOS versus a control adenovirus (RFP).
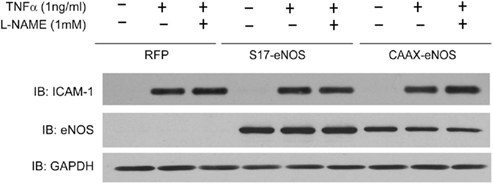
Figure 2. “Endothelial nitric oxide synthase knockdown” HAECs were transduced with adenovirus (MOI of 20) encoding either RFP (control), S17-eNOS (Golgi targeted), or CAAX–eNOS (PM targeted). Twenty-four hours later, cells were treated with L-NAME (1 mM) for 30 min, and TNFα (1 ng/ml) was added for an additional 24 h. Cell lysates were immunoblotted with anti-ICAM-1, anti-eNOS, and anti-GAPDH antibodies.
As endogenous NO failed to inhibit TNFα-stimulated expression of adhesion molecule expression, we next looked at whether exogenous NO could prove more effective. HAECs were treated with vehicle or 1 mM DETA NONOate for 24 h and then challenged with TNFα in the presence and absence of L-NAME. As shown in Figure 3A, DETA NONOate robustly inhibited TNFα stimulated expression of VCAM-1 and also ICAM-1. Consistent with previous results, L-NAME did not have any effect. To exclude the possibility that reduced viability of cells may also lead to the decrease in ICAM-1 or VCAM-1 expression, we perfomed the MTT assay after 24 hr exposure to TNFα and DETA NONOate (Figure 3B). There was no significant difference in viability in cells exposed to DETA NONOate or the combination of TNFα and DETA NONOate. Thapsigargin (10 μM) was used as positive control and showed a robust decrease in cell viability. Therefore the decreased expression of adhesion molecules we observed with DETA NONOate was not due to reduced cell viability. To compare the amount of NO produced from endogenous NO synthases versus that obtained from an exogenous source of NO, we measured NO levels in the media using NO-specific chemiluminescence. As shown in Figure 3C, we found that 100 μM DETA NONOate produced about threefold more NO than that produced by iNOS, fivefold higher than a PM-restricted eNOS and 100-fold higher than Golgi restricted eNOS. The 10-fold higher concentrations of NO donor which are necessary to inhibit NF-κB signaling in HAECs are thus much greater than that obtained from enzymatic sources.
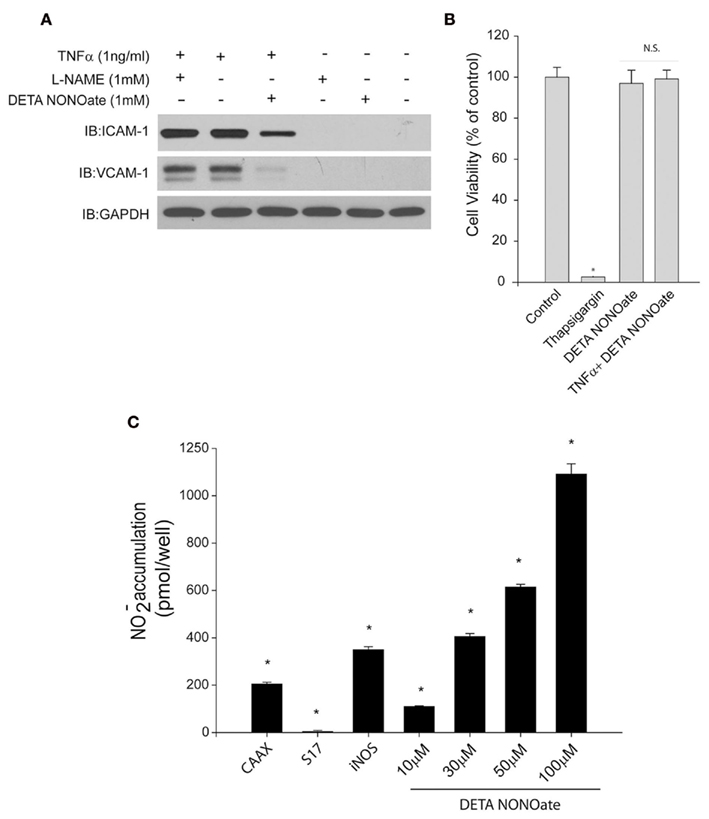
Figure 3. (A) Human aortic endothelial cells were treated with L-NAME (1 mM) or DETA NONOate (1 mM) for 30 min, and then TNFα (1 ng/ml) was added for 24 h. Activation of NF-κB was detected by anti-ICAM-1 or anti-VCAM-1 antibodies. GAPDH was used as a loading control. (B) HAECs were plated into 96-well plate and cells were treated with the same condition as above, cell viability was detected by MTT assay 24 h after treatment. (C) Nitric oxide release from cells expressing RFP, S17-eNOS, CAAX–eNOS, or iNOS or from different concentrations (10, 30, 50, 100 μM) of DETA NONOate over 24 h. Results are presented as mean ± SEM (n = 5). *p < 0.05 versus control (RFP).
To further determine the underlying mechanism of NO inhibition of adhesion molecules, we next monitored the activation of NF-κB by multiple approaches. NO has been reported to inhibit NF-κB activation via the S-nitrosylation and inhibition of IKKβ activity and a corresponding reduction in the phosphorylation of its substrate IκBα. In HAECs, we found that inhibition of endogenous NO with L-NAME did not prevent the increase in IκBα phosphorylation in response to TNFα (Figure 4A). Activation of NF-κB by TNFα was confirmed by an increase in phosphorylated IκBα and the degradation of total IκBα (Figure 4A). To further determine if higher amounts of NO, as generated by DETA NONOate, can inhibit ICAM-1/VCAM-1 expression through the S-nitrosylation of IKKβ (Reynaert et al., 2004) or p65 (Kelleher et al., 2007) as previously reported, we treated HAEC cells with L-NAME (1 mM) or DETA NONOate (1 mM) for 6 h and the S-nitrosylation of both proteins was detected using the biotin switch assay. Endogenous NO, inhibited with L-NAME, did not alter the S-nitrosylation level of IKKβ or p65, while DETA NONOate significantly increased the nitrosylation of both proteins (Figures 4B,C).
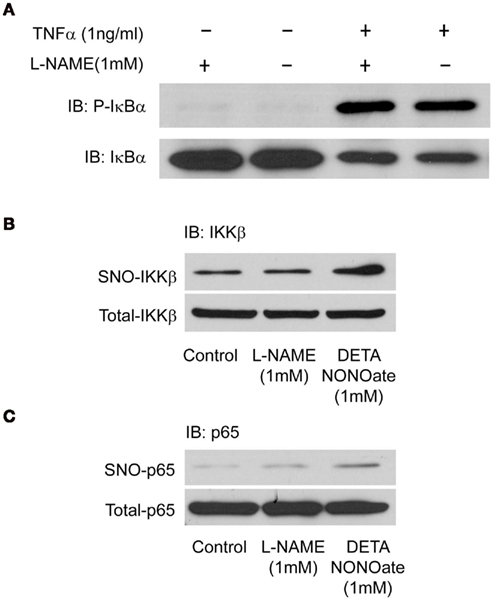
Figure 4. Human aortic endothelial cells were pre-treated with L-NAME (1 mM) for 30 min and stimulated with (A) 1 ng TNFα for 30 min. Cells were lysed and the activation of NF-κB was detected by immunoblotting using anti-phosphorylated IκBα and total IκBα antibodies. (B,C) HAECs were treated with L-NAME (1 mM) or DETA NONOate (1 mM) for 6 h, and cells were lysed for biotin switch assay, biotin labeled samples were immunoblotted with anti-p65 and anti-IKKβ antibodies.
Discussion
In the current study, we investigated the ability of NO to regulate NF-κB signaling and adhesion molecule expression in HAECs. We found that endogenous NO was unable to influence NF-κB activation or the induction of ICAM-1 or VCAM-1 expression either under basal conditions or in response to TNFα in both a time and dose-dependent manner. Selective expression of eNOS at the plasma membrane which results in greater NO release also failed to suppress the induction of ICAM-1. Significantly higher amounts of NO, greater than that produced endogenously, were necessary to observe suppression of adhesion molecule expression in human endothelium.
Previous studies have found that NF-κB is sensitive to both oxidative and nitrosative stress (Marshall et al., 2000; Marshall and Stamler, 2002). NO can suppresses NF-κB signaling via allosteric modification of its subunits including p50, p65, and also IKKβ (Matthews et al., 1996; Reynaert et al., 2004; Kelleher et al., 2007). We found no change in the expression of NF-κB-dependent genes and no change in the phosphorylation of IκBα suggesting that the levels of endogenous NO produced from eNOS are insufficient to influence these pathways. Indeed, the majority of studies that have shown inhibition of NF-κB have relied on higher concentrations of NO, either from an NO donor or from immune cells that expresses high levels of iNOS (Reynaert et al., 2004; Kelleher et al., 2007).
S-nitrosylation, the formation of an S-nitrosothiol (S-NO) by the covalent addition of a NO moiety to the SH-group of accessible cysteine residues requires higher concentrations of NO than does activation of sGC (Gow and Ischiropoulos, 2001). There are several reasons for this including the requirement that NO must first be converted to a nitrosating species such as N2O3 which may not occur uniformly throughout the cell (Espey et al., 2001). Also as NO is a rapidly diffusing gas, the proteins in closest proximity to the source of NO have the best chance of S-nitrosylation versus competition with other molecules such as superoxide or free thiols. Recent studies have suggested that the location of inhibition of NF-κB by NO can vary depending on the cell type studied. In A540 cells, NO operates at the nuclear level and inhibits the binding of NF-κB to DNA through a reversible, redox-based modification of the p50–p65 heterodimer (Kelleher et al., 2007). In Jurkat T cells, NO functions in the cytoplasm by inhibiting IκBα degradation, thus preventing the nuclear translocation of NF-κB. In intact blood vessels and cultured endothelium, eNOS can be found localized to either the perinuclear Golgi or the plasma membrane (Zhang et al., 2006) and NO production and the S-nitrosylation of target molecules can be found concentrated at sites of eNOS localization (Iwakiri et al., 2006). We found that the restricted expression of eNOS at the Golgi or plasma membrane of HAEC had no significant effect on TNFα induced activation of NF-κB. Even at the higher concentrations of NO afforded by the PM-location of eNOS, which could compensate for the reduced basal NO production secondary to the absence of hemodynamic stress in culture conditions, there was no meaningful change in adhesion molecule expression.
When targeted to the plasma membrane, eNOS releases more NO, generates more cGMP in adjacent reporter cells and produces more pronounced endothelium-dependent relaxations when expressed in intact blood vessels from eNOS−/− mice. Expression of eNOS at the plasma membrane also results in greater S-nitrosylation of the redox sensitive fusogen N-ethylmaleimide-sensitive factor (NSF) which functionally translates into a more significant suppression of vWF release from endothelial cells. In contrast to WT–eNOS, expression of calcium-insensitive forms of eNOS at different intracellular locations results in equal production of NO. The equalization of the amount of NO produced by eNOS targeted to the Golgi and plasma membrane resulted in similar increases in cGMP, S-nitrosylation of NSF, and equal suppression of vWF exocytosis (Qian et al., 2010). These results suggest that the amount of NO released is the most influential factor in controlling cellular function via S-nitrosylation. This concept is supported by the data in the current study showing that higher concentrations of NO are necessary to effectively constrain the NF-κB-dependent expression of cellular adhesion molecules. While it is clear that there is significant variety in the sensitivity of substrate proteins to S-nitrosylation and also the sites of S-nitrosylation within those proteins (Hess et al., 2005; Greco et al., 2006), the variables involved are not well defined (Doulias et al., 2010). Our results suggest that, at least in the context of HAECs, the S-nitrosylation of NF-κB requires higher NO concentrations than other more susceptible proteins such as GAPDH and NSF. This is supported by a number of studies mapping the S-nitrosylation proteome which have failed to identify constituents of the NF-κB pathway (Greco et al., 2006; Hao et al., 2006). The reversal of protein S-nitrosylation is a poorly understood process and the rate of reversal is known to vary significantly with the substrate (Paige et al., 2008). Two enzymes have been discovered that can accelerate the loss of S-NO from substrate proteins, S-nitrosoglutathione reductase (GSNOR) or alcohol dehydrogenase 5 (ADH5) and Thioredoxin-1 (Trx1; Jensen et al., 1998; Benhar et al., 2010). To exclude the possibility that in HAECs, NF-κB is a low turnover or stable S-NO protein we preincubated cells with the NOS inhibitor for 0.5–24 h prior to stimulation with TNFα and saw no difference in ICAM-1 induction. While the possibility exists that this was not sufficient time to observe an effect, other reports have shown effects of NO on NF-κB over a more acute time scale. For example, the activity of IKKβ was markedly enhanced 4 h after treatment with the NOS inhibitor, L-NMMA, and occurred contemporaneously with a decrease in IKKβ S-nitrosylation (Reynaert et al., 2004).
It is difficult to extrapolate the quantity of NO made by eNOS in cultured endothelial cells to that made in vivo. The anti-inflammatory effect of eNOS-derived NO has been shown in vivo following the genetic deletion of eNOS (Mashimo and Goyal, 1999). Thus the possibility exists that eNOS in cultured endothelial cells may be less active than in the in vivo setting, although the expression of an active form of eNOS did not have additional effects. Alternatively it might be that endogenous sources of NO (i.e., eNOS) are important in modulating inflammatory signaling but that this occurs under select pathophysiological conditions that have a multitude of mediators such as tissue/cell injury (e.g., I/R injury) versus the single inflammatory cytokine used in the current study. The regulation of inflammatory signaling cascades are complex and exhibit multiple redundant pathways so that TNFα alone may not be sufficiently sensitive to endogenous NO versus the multitude of signaling pathways and cell types involved in vivo.
NF-κB signaling, leading to increased ICAM-1 expression, is also sensitive to oxidative stress. Indeed, compounds such as resveratrol, which have anti-oxidant properties and can also blunt the effects of peroxynitrite, have been shown to inhibit TNFα-mediated activation of NF-κB and ICAM-1 expression (Holthoff et al., 2010). It is therefore possible that peroxynitrite may be a more efficient regulator of NF-κB activation than NO. However, the pursuit of this hypothesis is beyond the scope of the present study and would likely lend further support to our overall finding, that NO is a relatively weak modulator of NF-κB signaling.
The promoter region for ICAM-1 contains binding sites for a number of redox sensitive transcription factors including NF-κB, EGR-1, Sp1, and AP1. Pharmacological or genetic inhibition of NF-κB prevents upregulation of ICAM-1 and VCAM-1 in response to TNFα and suggests an absolute dependence on NF-κB activity (Ledebur and Parks, 1995; Pierce et al., 1997). However, other cytokines such as IL-1β, can upregulate ICAM-1 expression in endothelial cells via NO-mediated inhibition of Sp1 and AP1 (Berendji-Grun et al., 2001). A reasonable assumption of the current study is that the upregulation of ICAM-1 in response to TNFα is, at least in part, NF-κB-dependent; however, we would expect that if Sp1 and AP1 were involved in this response, then they too would be sensitive to NO inhibition. Recently, it was discovered that TNFα stimulated ICAM-1 expression can also occur downstream of p38 MAPK signaling (Zerr et al., 2011). However, NO has been shown to activate p38 MAPK signaling (Wang et al., 2006; Kaddai et al., 2008) which is the opposite of its actions on NF-κB. Although increased p38 MAPK activity could account for some of the reduced NO-sensitivity of adhesion molecular upregulation, it does not explain the lack of sensitivity of IκBα, IKKβ, and p65 to endogenous NO. The mechanism by which NO regulates p38 MAPK signaling and whether S-nitrosylation is involved remains unclear.
Collectively, we have shown that the endogenous production of NO in HAEC is not sufficient to inhibit the upregulation of adhesion molecules in response to cytokine stimulation. Higher concentrations of NO, in the pharmacological range, are required to suppress the induction of ICAM-1 and VCAM-1. Our results reinforce the concepts that NO concentration, susceptibility of target proteins and the cell type are critical factors regulating the control of cellular function by S-nitrosylation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH grants RO1 HL085827, R01HL092446, and P01 HL101902-01A1 and an established investigator award from the AHA.
References
Archer, S. L., Huang, J. M., Hampl, V., Nelson, D. P., Shultz, P. J., and Weir, E. K. (1994). Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 91, 7583–7587.
Baud, V., and Karin, M. (2001). Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 11, 372–377.
Benhar, M., Thompson, J. W., Moseley, M. A., and Stamler, J. S. (2010). Identification of S-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry 49, 6963–6969.
Berendji-Grun, D., Kolb-Bachofen, V., and Kroncke, K. D. (2001). Nitric oxide inhibits endothelial IL-1[beta]-induced ICAM-1 gene expression at the transcriptional level decreasing Sp1 and AP-1 activity. Mol. Med. 7, 748–754.
Chen, J., Kuhlencordt, P. J., Astern, J., Gyurko, R., and Huang, P. L. (2001). Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation 104, 2391–2394.
De Caterina, R., Libby, P., Peng, H. B., Thannickal, V. J., Rajavashisth, T. B., Gimbrone, M. A. Jr., Shin, W. S., and Liao, J. K. (1995). Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. 96, 60–68.
Doulias, P. T., Greene, J. L., Greco, T. M., Tenopoulou, M., Seeholzer, S. H., Dunbrack, R. L., and Ischiropoulos, H. (2010). Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 107, 16958–16963.
Espey, M. G., Miranda, K. M., Thomas, D. D., and Wink, D. A. (2001). Distinction between nitrosating mechanisms within human cells and aqueous solution. J. Biol. Chem. 276, 30085–30091.
Forrester, M. T., Foster, M. W., Benhar, M., and Stamler, J. S. (2009). Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 46, 119–126.
Fulton, D., Fontana, J., Sowa, G., Gratton, J. P., Lin, M., Li, K. X., Michell, B., Kemp, B. E., Rodman, D., and Sessa, W. C. (2002). Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J. Biol. Chem. 277, 4277–4284.
Gopalakrishna, R., Chen, Z. H., and Gundimeda, U. (1993). Nitric oxide and nitric oxide-generating agents induce a reversible inactivation of protein kinase C activity and phorbol ester binding. J. Biol. Chem. 268, 27180–27185.
Gow, A. J., and Ischiropoulos, H. (2001). Nitric oxide chemistry and cellular signaling. J. Cell. Physiol. 187, 277–282.
Greco, T. M., Hodara, R., Parastatidis, I., Heijnen, H. F., Dennehy, M. K., Liebler, D. C., and Ischiropoulos, H. (2006). Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 103, 7420–7425.
Hao, G., Derakhshan, B., Shi, L., Campagne, F., and Gross, S. S. (2006). SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc. Natl. Acad. Sci. U.S.A. 103, 1012–1017.
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166.
Holthoff, J. H., Woodling, K. A., Doerge, D. R., Burns, S. T., Hinson, J. A., and Mayeux, P. R. (2010). Resveratrol, a dietary polyphenolic phytoalexin, is a functional scavenger of peroxynitrite. Biochem. Pharmacol. 80, 1260–1265.
Huang, P. L., Huang, Z., Mashimo, H., Bloch, K. D., Moskowitz, M. A., Bevan, J. A., and Fishman, M. C. (1995). Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377, 239–242.
Iwakiri, Y., Satoh, A., Chatterjee, S., Toomre, D. K., Chalouni, C. M., Fulton, D., Groszmann, R. J., Shah, V. H., and Sessa, W. C. (2006). Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. U.S.A. 103, 19777–19782.
Jaffrey, S. R., and Snyder, S. H. (2001). The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, pl1.
Janssen-Heininger, Y. M., Poynter, M. E., and Baeuerle, P. A. (2000). Recent advances towards understanding redox mechanisms in the activation of nuclear factor κB. Free Radic. Biol. Med. 28, 1317–1327.
Jensen, D. E., Belka, G. K., and Du Bois, G. C. (1998). S-nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem. J. 331(Pt 2), 659–668.
Kaddai, V., Gonzalez, T., Bolla, M., Le Marchand-Brustel, Y., and Cormont, M. (2008). The nitric oxide-donating derivative of acetylsalicylic acid, NCX 4016, stimulates glucose transport and glucose transporters translocation in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 295, E162–E169.
Kelleher, Z. T., Matsumoto, A., Stamler, J. S., and Marshall, H. E. (2007). NOS2 regulation of NF-κB by S-nitrosylation of p65. J. Biol. Chem. 282, 30667–30672.
Kubes, P., Suzuki, M., and Granger, D. N. (1991). Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 88, 4651–4655.
Ledebur, H. C., and Parks, T. P. (1995). Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-κB site and p65 homodimers. J. Biol. Chem. 270, 933–943.
Li, Q., and Verma, I. M. (2002). NF-κB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734.
Ma, X. L., Weyrich, A. S., Lefer, D. J., and Lefer, A. M. (1993). Diminished basal nitric oxide release after myocardial ischemia and reperfusion promotes neutrophil adherence to coronary endothelium. Circ. Res. 72, 403–412.
Marshall, H. E., Hess, D. T., and Stamler, J. S. (2004). S-nitrosylation: physiological regulation of NF-κB. Proc. Natl. Acad. Sci. U.S.A. 101, 8841–8842.
Marshall, H. E., Merchant, K., and Stamler, J. S. (2000). Nitrosation and oxidation in the regulation of gene expression. FASEB J. 14, 1889–1900.
Marshall, H. E., and Stamler, J. S. (2002). Nitrosative stress-induced apoptosis through inhibition of NF-κB. J. Biol. Chem. 277, 34223–34228.
Mashimo, H., and Goyal, R. K. (1999). Lessons from genetically engineered animal models. IV. Nitric oxide synthase gene knockout mice. Am. J. Physiol. 277, G745–G750.
Matsushita, K., Morrell, C. N., Cambien, B., Yang, S. X., Yamakuchi, M., Bao, C., Hara, M. R., Quick, R. A., Cao, W., O’Rourke, B., Lowenstein, J. M., Pevsner, J., Wagner, D. D., and Lowenstein, C. J. (2003). Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115, 139–150.
Matthews, J. R., Botting, C. H., Panico, M., Morris, H. R., and Hay, R. T. (1996). Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res. 24, 2236–2242.
Mohan, S., Hamuro, M., Sorescu, G. P., Koyoma, K., Sprague, E. A., Jo, H., Valente, A. J., Prihoda, T. J., and Natarajan, M. (2003). IκBα-dependent regulation of low-shear flow-induced NF-κB activity: role of nitric oxide. Am. J. Physiol. Cell Physiol. 284, C1039–C1047.
Moncada, S., Palmer, R. M., and Higgs, E. A. (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 43, 109–142.
Nathan, C., and Xie, Q. W. (1994). Nitric oxide synthases: roles, tolls, and controls. Cell 78, 915–918.
Niu, X. F., Smith, C. W., and Kubes, P. (1994). Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 74, 1133–1140.
Paige, J. S., Xu, G., Stancevic, B., and Jaffrey, S. R. (2008). Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem. Biol. 15, 1307–1316.
Peng, H. B., Libby, P., and Liao, J. K. (1995). Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. J. Biol. Chem. 270, 14214–14219.
Pierce, J. W., Schoenleber, R., Jesmok, G., Best, J., Moore, S. A., Collins, T., and Gerritsen, M. E. (1997). Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272, 21096–21103.
Qian, J., Zhang, Q., Church, J. E., Stepp, D. W., Rudic, R. D., and Fulton, D. J. (2010). Role of local production of endothelium-derived nitric oxide on cGMP signaling and S-nitrosylation. Am. J. Physiol. Heart Circ. Physiol. 298, H112–H118.
Radomski, M. W., Palmer, R. M., and Moncada, S. (1991). Modulation of platelet aggregation by an L-arginine-nitric oxide pathway. Trends Pharmacol. Sci. 12, 87–88.
Reynaert, N. L., Ckless, K., Korn, S. H., Vos, N., Guala, A. S., Wouters, E. F., van der Vliet, A., and Janssen-Heininger, Y. M. (2004). Nitric oxide represses inhibitory κB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 101, 8945–8950.
Stamler, J. S., Lamas, S., and Fang, F. C. (2001). Nitrosylation. the prototypic redox-based signaling mechanism. Cell 106, 675–683.
Wang, S., Zhang, J., Theel, S., Barb, J. J., Munson, P. J., and Danner, R. L. (2006). Nitric oxide activation of Erk1/2 regulates the stability and translation of mRNA transcripts containing CU-rich elements. Nucleic Acids Res. 34, 3044–3056.
Zerr, M., Hechler, B., Freund, M., Magnenat, S., Lanois, I., Cazenave, J. P., Léon, C., and Gachet, C. (2011). Major contribution of the P2Yreceptor in purinergic regulation of TNFα-induced vascular inflammation. Circulation 123, 2404–2413.
Keywords: nitric oxide, NF-κB, inflammation, endothelial, S-nitrosylation
Citation: Qian J and Fulton DJR (2012) Exogenous, but not endogenous nitric oxide inhibits adhesion molecule expression in human endothelial cells. Front. Physio. 3:3. doi: 10.3389/fphys.2012.00003
Received: 31 August 2011; Paper pending published: 30 November 2011;
Accepted: 04 January 2012; Published online: 18 January 2012.
Edited by:
Guillermo Garcia-Cardena, Harvard Medical School, USAReviewed by:
Philip M. Bauer, University of Pittsburgh, USAChristopher G. Kevil, LSU Health Sciences Center, USA
Rudolf Lucas, Medical College of Georgia, USA
Wenbo Zhang, The University of Texas Medical Branch, USA
Copyright: © 2012 Qian and Fulton. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David J. R. Fulton, Vascular Biology Center, Georgia Health Sciences University, 1459 Laney Walker Boulevard, Augusta, GA 30912, USA. e-mail: dfulton@georgiahealth.edu