
- Laboratory of Vascular Biology, Division of Physiology, Department of Medicine, Faculty of Science, University of Fribourg, Fribourg, Switzerland
The global population aging is accelerating and age-associated diseases including cardiovascular diseases become more challenging. The underlying mechanisms of aging and age-associated cardiovascular dysfunction remain elusive. There are substantial evidences demonstrating a pivotal role of the mammalian target of rapamycin complex 1 (mTORC1) and its down-stream effector S6K1 signaling in mammalian lifespan regulation and age-related diseases such as type II diabetes mellitus and cancer. The role of mTORC1–S6K1 in age-related cardiovascular diseases is, however, largely unknown and the available experimental results are controversial. This review article primarily summarizes the most recent advances toward understanding the role of mTORC1–S6K1 in cardiovascular aging and discusses the future perspectives of targeting mTORC1–S6K1 signaling as a healthy lifespan extension modality in anti-aging and anti-cardiovascular aging.
Introduction
Global population aging is accelerating, which has been predicted to be a great challenge for our society in the twenty-first century (Christensen et al., 2009). This is due to the steady achievements of medicine and public healthcare over the past century, having resulted in an increased life expectancy, which is mainly attributed to improvement of infant and childhood survival and reduction of mortality in the elderly population (Christensen et al., 2008, 2009; Rau et al., 2008). It has been predicted that the global proportion above age 60 will increase from 10% of the total population in 2000 to 21.8% in 2050 and 32.2% in 2100 (Lutz et al., 2008). With the increased aging population, we are confronted with an increase in age-associated diseases including cardiovascular disease, cancer, diabetes, and neurodegenerative disease (Christensen et al., 2009). Although aging has been proven to be an prominent independent risk factor for cardiovascular disease (Najjar et al., 2005), the mechanisms of aging and age-associated cardiovascular dysfunction are still elusive. There are substantial evidences suggesting that oxidative stress plays a crucial role in cardiovascular aging (Ungvari et al., 2010). However, several prospective clinical trials with various combinations of antioxidants fail to show significant effects on the incidence of major adverse cardiovascular events (Vivekananthan et al., 2003; Sesso et al., 2008). The results suggest that more thorough research on the mechanisms of aging and age-associated cardiovascular diseases is required.
Research in the past years provides compelling evidences showing a potential role of the target of rapamycin (TOR) signaling pathway in lifespan regulation, which is remarkably conserved across various species. In model systems such as yeast, nematodes, fruit flies, and also recently in mice, inhibition of TOR signaling increases lifespan (Evans et al., 2011). A role of deregulated mammalian TOR (mTOR) signaling in mammalian aging and age-related diseases such as type II diabetes mellitus and cancer are demonstrated (Evans et al., 2011). The role of mTOR, especially its down-stream effector S6K1 in age-related cardiovascular diseases or cardiovascular aging is, however, largely unknown. The primary emphasis of this review article is to discuss emerging evidence and future perspectives for a role of mTOR and S6K1 signaling in mammalian aging and age-associated cardiovascular diseases.
Brief Biochemistry of mTOR Signaling
mTOR is a serine/threonine protein kinase which serves as intracellular sensor for energy, nutrients, and stress, regulating cellular and organism growth and metabolism. Therefore, dysfunctional mTOR signaling has been considered a central integral mechanism linking aging, metabolic disorders, and cancer (Zoncu et al., 2011). The detailed molecular signaling network and regulation of mTOR signaling have been reviewed comprehensively in several articles (Dann et al., 2007; Sengupta et al., 2010; Evans et al., 2011; Zoncu et al., 2011). A few important biochemical features of mTOR signaling network are summarized in Figure 1. mTOR, with other molecular components, forms two structurally and functionally distinct complexes namely mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). In these complexes, mTOR functions as the catalytic subunit and its enzymatic activity is regulated and distinguished by its unique accessory proteins Raptor and Rictor, respectively. Raptor and Rictor function as scaffold proteins for assembling mTORC1 and mTORC2 and also for binding substrates and regulators in the respective complex. In addition to their different protein compositions, there are a few major important biochemical characteristics which distinguish the two complexes: (1) sensitivity to the immunosuppressant rapamycin: rapamycin inhibits mTORC1 but not mTORC2 activity, although prolonged treatment with rapamycin has been shown to be capable of inhibiting mTORC2 in certain cell types (Sarbassov et al., 2006); (2) the upstream signals they integrate: while mTORC2 seems to be regulated only by growth factors, mTORC1 is regulated by many stimuli including growth factors, energy status, stressors such as DNA damage, hypoxia, and nutrients. The small GTPase protein Rheb has been identified to be the key end point for mTORC1 activation by stimulating kinase activity of mTORC1. The TSC1–TSC2, a GTPase activating protein (GAP), negatively modulates Rheb by converting the active form Rheb-GTP into its inactive form Rheb-GDP through its GAP activity and thus reduces mTORC1 activity. Except nutrient amino acids, all of the above mentioned stimuli inputs regulate mTORC1 activity through modulation of TSC1–TSC2 activity. Growth factors inactivate TSC1–TSC2 via Akt/ERK1/2, leading to formation of Rheb-GTP and ultimately mTORC1 activation, whereas energy deficit, DNA damage, or hypoxia activate TSC1–TSC2 through activation of AMPK or REDD1 (regulated in development and DNA damage response 1), respectively, resulting in inactivation of Rheb and thus inhibition of mTORC1. Amino acids activate mTORC1 independently of TSC1–TSC2, but through action of Rag GTPases. In the presence of amino acids, Rag GTPases interact with mTORC1 and translocate the complex from cytoplasm to lysosomal membranes where it is activated by Rheb. For more detailed discussion about the regulatory mechanisms of mTORC1 activation by various inputs, please refer to the review article by Zoncu et al. (2011). In contrast to mTORC1, the regulatory mechanisms and functions of mTORC2 signaling are less well characterized; (3) the substrate they regulate and the biological processes they control: mTORC1 enhances protein synthesis through S6K1 and eIF4E-binding protein 1 (4E-BP1). Upon phosphorylation by mTORC1, 4E-BP1 dissociates from eIF4E, relieving its suppressing effect on mRNA translation, while S6K1, when phosphorylated by mTORC1, promotes mRNA translation. In addition, mTORC1 also induces lipogenesis in the liver through activating transcription factors SREBP1 and PPARγ, inhibits autophagy through phosphorylation of the ULK1–Atg13–FIP200 complex, and promotes mitochondrial biogenesis by activating PGC1α/YY1 (Zoncu et al., 2011). mTORC2 exerts its effects on metabolism, stress responses, apoptosis, and cytoskeleton organization through phosphorylation of many AGC kinases including Akt, serum-and glucocorticoid-induced protein kinase (SGK), protein kinase C-α (PKCα), and Rho1 GDP–GTP exchange protein-2 (Rom2; Sarbassov et al., 2005; Frost and Lang, 2011). Since mTORC2 activates Akt that in turn enhances mTORC1 activity through inactivation of TSC1–TSC2, mTORC2 is the upstream of mTORC1 upon stimulation by growth factors (see Figure 1).
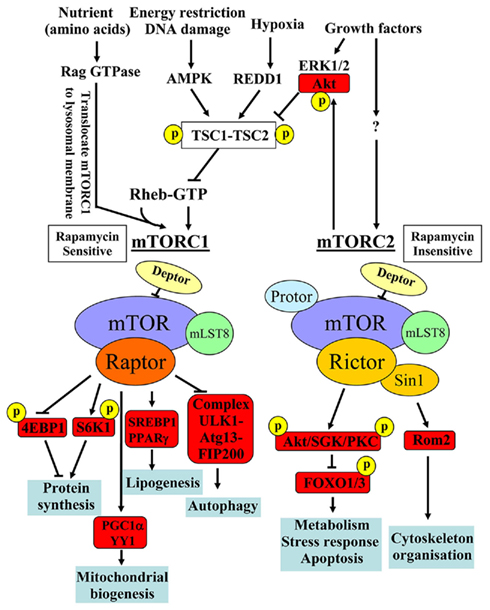
Figure 1. Major distinct characteristics of the two known mTOR signaling complexes, mTORC1 and mTORC2, in composition, rapamycin sensitivity, upstream signals, substrates, and biological functions. Adapted from Foster and Finger (2010) and from Zoncu et al. (2011). 4E-BP1, eIF4E-binding protein 1; AMPK, AMP kinase; Atg, autophagy-related; FIP200, 200 kDa FAK family kinase-interacting protein; FOXO, forkhead box protein O; PPARγ, peroxisome proliferator-activated receptor-γ; REDD1, regulated in development and DNA damage response 1; Rom2, Rho1 GDP–GTP exchange protein-2; SGK, serum- and glucocorticoid-regulated kinase; SREBP, sterol regulatory element-binding protein; TSC, the tuberous sclerosis.
Evidence for a Role of mTORC1–S6K1 in Regulation of Longevity
The role of mTORC1–S6K1 as a master determinant in longevity control stems from experiments in almost all model organisms including yeast, worms, flies, and mice. Genetic inactivation of TOR or core components of TOR signaling including S6K1 or pharmacological inhibition of the signaling pathway with rapamycin showed lifespan extension in yeast and invertebrates and also recently in mice (Chen et al., 2009; Harrison et al., 2009; Selman et al., 2009). These independent studies clearly verified the important role of mTORC1–S6K1 in mammalian aging. No data is available, yet, whether mTORC2 is also involved in regulation of longevity. The lifespan extension experiments with rapamycin may not fully exclude a possible role of mTORC2 in regulation of longevity, since long-term rapamycin treatment is able to inhibit mTORC2 in certain cell types (Sarbassov et al., 2006).
Furthermore, the effect of mTOR signaling inhibition on longevity can be mimicked by caloric restriction, i.e., reduction in calorie intake without malnutrition, across different species (Pan et al., 2007; Evans et al., 2011; Shinmura et al., 2011). A recent report demonstrating that caloric restriction also slows aging in rhesus monkeys (Colman et al., 2009) may suggest that caloric restriction could have anti-aging effects in humans (Kaeberlein and Kennedy, 2009). Indeed, it has been reported that the Okinawan populations who consume 20% less calories compared to the average caloric consumption of the Japanese population live longer and display lower incidences of cancer and cardiovascular diseases in the elderly. The healthier aging status disappears in those who switched to Western diet (Kagawa, 1978; Willcox et al., 2007). A small caloric restriction study in adult humans over 6 years demonstrates less cardiovascular risk factors including lower body mass index, body fat deposition, blood pressure, fasting plasma levels of glucose and insulin, lower inflammation markers, i.e., tumor necrosis factor-α and C-reactive protein, and beneficial lipid profiles (Fontana et al., 2004) and shows better left ventricular diastolic function than healthy age-and sex-matched controls (Meyer et al., 2006). Also, caloric restriction improves cardiac function in patients with type 2 diabetes (Hammer et al., 2008). Whether the beneficial effects of caloric restriction in humans are due to healthier diet and could be translated to lifespan extension in healthy elderly subjects and improve survival in patients requires further investigation. The underlying mechanisms of caloric restriction on longevity may involve reduction of mTORC–S6K1 signaling. In yeast and flies, when TOR is inactivated, caloric restriction does not show further effects on lifespan extension (Kapahi and Zid, 2004; Powers et al., 2006). Whether this holds true also in humans needs to be confirmed.
Another piece of supporting evidence for mTORC1–S6K1 in regulation of longevity derives from experiments showing that down-regulation of insulin and insulin-like growth factor (IGF-1) signaling in mouse models such as haploinsufficiency of IGF-1 receptor and global deletion of insulin receptor substrate-1 (IRS-1) are associated with reduced mTORC1–S6K1 signaling and increased longevity in mice (Holzenberger et al., 2003; Selman et al., 2008) as in yeast, nematodes, and fruit flies (Stanfel et al., 2009). Moreover, caloric restriction prolongs lifespan in animal models, which is associated with reduced mTORC1–S6K1 mediated by IGF-1 signaling pathway (Estep et al., 2009; Fontana et al., 2010). These results further strengthen the role of mTORC1–S6K1 in lifespan regulation in mammals. The exact mechanisms of reduced signaling of mTORC1–S6K1 and IGF-1 under caloric restriction, however, remain elusive. It seems that AMPK and Sirt1, whose activities are augmented by caloric restriction, are involved in negative regulation of mTORC1–S6K1 signaling and in turn regulate aging process (Cohen et al., 2004; Canto et al., 2009; Shackelford and Shaw, 2009; Figure 1), although direct evidence for it in mammals is still lacking (Herranz and Serrano, 2010). It is also noteworthy that loss of S6K1 in mice reciprocally results in activation of AMPK which has been proposed to mediate the lifespan extension (Selman et al., 2009). Intriguingly, persistent activation of AMPK seems to play an important role in vascular endothelial cell senescence (Zu et al., 2010). A working model of reciprocal regulatory effects of Sirt1 and AMPK on cellular senescence and aging is discussed by Wang et al. (2011). The molecular mechanisms of the reciprocal interplay between mTORC1–S6K1 and AMPK and Sirt1 are not clear. A mutual inhibition of mTORC1–S6K1 and AMPK has been reported (Lee et al., 2010).
Although the role of inhibition of mTORC1–S6K1 in lifespan extension is consistently demonstrated in animal models, very little information is available about the role of mTORC1–S6K1 in cardiovascular aging or age-associated cardiovascular diseases.
mTORC1–S6K1 Signaling in Cardiovascular Aging
Cardiac Aging
Age-associated cardiovascular diseases are accompanied by structural and functional changes in heart and blood vessels. These aging-associated changes in cardiovascular system are referred to as cardiovascular aging phenotypes. Cardiac aging is manifested by maladaptation to stress, cardiac dysfunction, and heart failure. Cardiac aging process involves cardiomyocytes and other cell types in the heart, such as interstitial fibroblasts and vascular cells. The morphological and functional changes of these cells with aging lead to cardiac hypertrophy and dilation, cardiac fibrosis due to chronic deposition, and remodeling of extracellular matrix produced mainly by fibroblasts. Moreover, a decreased regenerative capacity of cardiac stem cells, possibly due to impaired cell division and accelerated cell senescence and an increased cardiac myocyte death due to necrosis and apoptosis with aging, may also contribute to cardiac dysfunction in elderly. These pathological and clinical aspects of cardiac remodeling in aging have been recently reviewed in great details (Shih et al., 2011).
There are compelling evidences demonstrating that mTORC1–S6K1 signaling is involved in cardiac hypertrophy under various pathological conditions including diabetes and hypertension (Sadoshima and Izumo, 1995; Boluyt et al., 1997; Tu et al., 2002; McMullen et al., 2004; Soesanto et al., 2009; Kurdi and Booz, 2011; Sung et al., 2011). The underlying mechanisms have been proposed to be attributed to both an increase in protein synthesis upon stress stimulation and a decrease in protein degradation due to inhibition of autophagy by mTORC1 signaling (Hands et al., 2009). However, evidence for this hypothesis needs to be demonstrated. Inhibition of mTORC1–S6K1 pathway by rapamycin or activation of AMPK by 5-aminoimidazole-4-carboxamide riboside (AICAR) to negatively regulate mTORC1 signaling has been shown to attenuate pressure overload-induced cardiac hypertrophy in vivo (Li et al., 2007). Conversely, deficiency in AMPK enhances mTORC1–S6K1 signaling and exacerbates myocardial hypertrophy in response to pressure overload (Zhang et al., 2008).
Besides pressure overload, many hormonal factors, e.g., angiotensin-II, insulin, endothelin-1, catecholamine, etc., that are elevated in plasma and/or tissues under various cardiovascular pathologies and in aging, have been shown to stimulate mTORC1–S6K1 signaling in the cardiovascular system (Moschella et al., 2007; Muniyappa et al., 2007; Kim et al., 2012). Activation of mTORC1–S6K1 by the hormones, e.g., angiotensin-II and also by overnutrition (Glazer et al., 2009) participates in cardiac hypertrophy and vascular remodeling, and causes insulin resistance, an important cardiovascular risk (Reaven, 2011), through phosphorylation of IRS-1 at serine residues (Kim et al., 2012). Importantly, under the condition of cardiovascular insulin resistance, there is a selective inhibition of the metabolic pathway, i.e., Akt–eNOS in response to insulin, while the growth pathway, i.e., p44/p42ERKs remains active (Muniyappa et al., 2007). This selective insulin resistance may play an important role in decreased vascular relaxation due to impaired eNOS activation and enhanced cardiovascular remodeling in metabolic disorders such as type II diabetes (Muniyappa et al., 2007). mTORC1–S6K1 pathway is therefore considered as an important molecular link between metabolic stress and cardiovascular abnormalities.
Despite the evidences for a role of mTORC1–S6K1 signaling in agonist or pressure-induced cardiac hypertrophy, only little information is available about the signaling of mTORC1–S6K1 in physiological process of aging heart. An early study using microarray analyses reported that gene expression pattern associated with mTOR is suppressed in aging heart of Fischer 344 rats (Linford et al., 2007), while a recent study shows no difference in mTORC1–S6K1 activity (measured by phosphorylated mTOR and S6K1 levels) in the heart between 8 and 30 month old rats of the same strain (Shinmura et al., 2011). There are two points that should be considered for interpretation of the inconsistent results. First, microarray has its limitation in elucidating signaling pathways, because signaling molecules are mainly enzymes whose activities could not be investigated with this experimental approach; second, aging-associated kinetics of mTORC1–S6K1 signaling may exist, meaning an enhanced signaling with time followed by a decreased signaling in old age. Indeed, a decreased mTOR signaling has been shown to play a role in sarcopenia in advanced age (Sakuma and Yamaguchi, 2010). Whether this also occurs in the heart requires investigation. Nevertheless, caloric restriction in the old rats showed improved diastolic function associated with reduced cell senescence and mTORC1–S6K1 signaling in the heart compared to the old animals fed ad libitum (Shinmura et al., 2011), suggesting that mTORC1–S6K1 is involved in caloric restriction-induced improvement of heart function in aging. It is also conceivable that the anti-cellular senescence effect and reduced mTORC1–S6K1 signaling contribute to the beneficial effects of caloric restriction on human heart function (Meyer et al., 2006; Hammer et al., 2008).
Vascular Aging
Endothelial dysfunction and inflammatory activation
In the vasculature, increased arterial wall thickening and generalized vascular stiffness occur with aging. This vascular aging phenotype is attributed to vascular calcification, increased collagen content and elastin breakdown, and elevated levels of advanced glycation end products. Vascular inflammation, oxidative stress, endothelial dysfunction, endothelial progenitor cell dysfunction, vascular cell apoptosis all intertwine with each other to affect vascular aging process, which accelerates coronary heart disease, heart failure, stroke, and dementia (Ungvari et al., 2010).
In the past decades, much attention has been devoted to endothelial dysfunction in aging. Evidence demonstrates that besides the enhanced production of endothelium-derived vasoconstrictor prostanoids, which is due to augmented expression and/or activity of cyclo-oxygenases in endothelial cells during aging (Vanhoutte et al., 2009), decreased endothelial nitric oxide (NO) bioavailability is a major characteristic of vascular aging, which is independent of other cardiovascular risk factors (Lakatta, 2001). Endothelial NO causes vascular relaxation, inhibits platelet aggregation, and leukocyte adhesion (Yang and Ming, 2006). Clinical studies provide evidence that endothelial dysfunction is not only highly associated with cardiovascular disease, it also predicts future cardiac events (Schachinger et al., 2000; Halcox et al., 2002; Bugiardini et al., 2004; Huang et al., 2007). eNOS dysfunction seems causally involved in cardiovascular aging, since eNOS−/− male mice have a significantly shorter lifespan than their wild type controls and exhibit accelerated cardiac dysfunction with age (Wei, 2004). Endothelial cells with dysfunctional eNOS in aging are also more vulnerable to apoptotic stimuli (Hoffmann et al., 2001; Csiszar et al., 2004). There is increasing evidence suggesting that the number and regenerative capacity of circulating endothelial progenitor cells which play a role in reendothelialization and repair after vascular injury are decreased in patients with cardiovascular diseases and risk factors including aging (Vasa et al., 2001; Hill et al., 2003; Rauscher et al., 2003; Werner et al., 2005). The underlying mechanisms of eNOS dysfunction in aging are multifactorial and have not been fully understood, yet. Decreased eNOS gene expression (Csiszar et al., 2002; Tanabe et al., 2003) or increased eNOS gene expression with impaired enzymatic activity due to oxidative stress are important mechanisms (Stockklauser-Farber et al., 2000; van der Loo et al., 2000; Ming et al., 2004; Desrois et al., 2010). The up-regulation of eNOS gene in aging and also in other vascular disease may represent a compensatory mechanism counteracting oxidative stress (Drummond et al., 2000; Stockklauser-Farber et al., 2000; Hink et al., 2001).
Oxidative stress has been proposed as the culminant mechanism impairing endothelial function via quenching of NO, i.e., inactivation of NO by increased production of 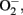 which leads to formation of peroxynitrite, a strong oxidant that can further damage endothelial cells in aging (van der Loo et al., 2000; Brandes et al., 2005). Among other sources, eNOS itself produces significant amount of
which leads to formation of peroxynitrite, a strong oxidant that can further damage endothelial cells in aging (van der Loo et al., 2000; Brandes et al., 2005). Among other sources, eNOS itself produces significant amount of 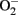 in endothelial cells, when “eNOS uncoupling” occurs – that is, eNOS generates
in endothelial cells, when “eNOS uncoupling” occurs – that is, eNOS generates 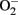 instead of NO (Landmesser et al., 2003; Forstermann and Munzel, 2006; Rajapakse et al., 2011). It seems that normal function of eNOS requires homodimerization of the enzyme which is stabilized by the cofactor BH4. The eNOS reductase domain generates electron flow from NADPH through FAD and FMN flavins, which are then transferred to the oxidase domain of other monomers in which L-arginine is metabolized to NO at the heme group in the active site (Forstermann and Munzel, 2006). In the absence of BH4 due to oxidative inactivation, eNOS dimer/monomer ratio is decreased and the catalytic activity becomes uncoupled – that is – uncoupling of NADPH oxidation and NO synthesis, with oxygen instead of L-arginine as terminal electron acceptor, resulting in
instead of NO (Landmesser et al., 2003; Forstermann and Munzel, 2006; Rajapakse et al., 2011). It seems that normal function of eNOS requires homodimerization of the enzyme which is stabilized by the cofactor BH4. The eNOS reductase domain generates electron flow from NADPH through FAD and FMN flavins, which are then transferred to the oxidase domain of other monomers in which L-arginine is metabolized to NO at the heme group in the active site (Forstermann and Munzel, 2006). In the absence of BH4 due to oxidative inactivation, eNOS dimer/monomer ratio is decreased and the catalytic activity becomes uncoupled – that is – uncoupling of NADPH oxidation and NO synthesis, with oxygen instead of L-arginine as terminal electron acceptor, resulting in 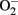 generation (Forstermann and Munzel, 2006). In addition, limited specific pool of intracellular L-arginine bioavailability due to enhanced arginase activity (Csiszar et al., 2002; Berkowitz et al., 2003; Tanabe et al., 2003) or production of endogenous eNOS inhibitor asymmetric dimethylarginine (ADMA; Sydow and Munzel, 2003; Antoniades et al., 2009) have been also reported to contribute to endothelial dysfunction in aging. The finding that eNOS enzymatic dysfunction is the major mechanism for endothelial dysfunction in aging indicates that one should therapeutically focus on improving eNOS enzymatic function instead of increasing eNOS gene expression in blood vessels. Indeed, over-expression of eNOS under disease conditions for example in atherosclerosis prone ApoE−/− mice has been shown to accelerate atherosclerosis (Ozaki et al., 2002). Endothelial specific eNOS transgenic mice have enhanced
generation (Forstermann and Munzel, 2006). In addition, limited specific pool of intracellular L-arginine bioavailability due to enhanced arginase activity (Csiszar et al., 2002; Berkowitz et al., 2003; Tanabe et al., 2003) or production of endogenous eNOS inhibitor asymmetric dimethylarginine (ADMA; Sydow and Munzel, 2003; Antoniades et al., 2009) have been also reported to contribute to endothelial dysfunction in aging. The finding that eNOS enzymatic dysfunction is the major mechanism for endothelial dysfunction in aging indicates that one should therapeutically focus on improving eNOS enzymatic function instead of increasing eNOS gene expression in blood vessels. Indeed, over-expression of eNOS under disease conditions for example in atherosclerosis prone ApoE−/− mice has been shown to accelerate atherosclerosis (Ozaki et al., 2002). Endothelial specific eNOS transgenic mice have enhanced 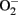 generation which can be inhibited by the eNOS inhibitor L-NAME or by endothelial-targeted GTP cyclohydrolase 1 over-expression to increase BH4 production (Bendall et al., 2005). The results further implicate that too much eNOS under pathological conditions where the enzyme is uncoupled, is detrimental. A recent study reveals that oxidative stress causes cysteine S-glutathionylation of the reductase domain of eNOS, resulting eNOS uncoupling (Chen et al., 2010). The eNOS S-glutathionylation level is increased in spontaneously hypertensive rats as compared to normotensive animals accompanied with impaired endothelium-dependent relaxations that can be reversed after the S-glutathionylation of eNOS is removed by thiol-specific reducing agents (Chen et al., 2010). Whether S-glutathionylation of eNOS also plays a role in endothelial dysfunction in aging requires further investigation.
generation which can be inhibited by the eNOS inhibitor L-NAME or by endothelial-targeted GTP cyclohydrolase 1 over-expression to increase BH4 production (Bendall et al., 2005). The results further implicate that too much eNOS under pathological conditions where the enzyme is uncoupled, is detrimental. A recent study reveals that oxidative stress causes cysteine S-glutathionylation of the reductase domain of eNOS, resulting eNOS uncoupling (Chen et al., 2010). The eNOS S-glutathionylation level is increased in spontaneously hypertensive rats as compared to normotensive animals accompanied with impaired endothelium-dependent relaxations that can be reversed after the S-glutathionylation of eNOS is removed by thiol-specific reducing agents (Chen et al., 2010). Whether S-glutathionylation of eNOS also plays a role in endothelial dysfunction in aging requires further investigation.
Another important feature of endothelial aging is enhanced expression of inflammatory adhesion molecules such as ICAM-1 and VCAM-1 (Gorgoulis et al., 2003; Zhou et al., 2006; Ungvari et al., 2010), which leads to enhanced monocyte–endothelial interaction and accelerated atherogenesis. There are considerable evidences suggesting that oxidative stress is a major mechanism for promoting eNOS dysfunction as well as vascular inflammation which causes further endothelial dysfunction and aging in a positive-feedback manner (Herrera et al., 2010). However, evidence for a causal role of oxidative stress in vascular aging is still lacking, although there is no doubt that oxidative stress contributes to aging-associated vascular dysfunctions. Some authors suggest that mTOR signaling instead of oxidative stress might be the more powerful driving force for organismal aging (Blagosklonny, 2008).
Causal role of mTORC1–S6K1 signaling in endothelial aging
Although evidence for a role of mTOR–S6K1 signaling in regulation of organism lifespan has been well demonstrated (Evans et al., 2011; Zoncu et al., 2011), it is, however, not known, whether mTORC1–S6K1 signaling pathway participates in vascular aging. In contrast to the aging heart, an increased basal activity of mTORC1–S6K1 has been demonstrated in aortas of old Fischer 344xBrown Norway F1 hybrid rats (Rice et al., 2005), which implicates a potential role of mTORC1–S6K1 signaling in vascular aging. However, a study reported that inhibition of mTORC1–S6K1 by rapamycin and everolimus induces endothelial cellular senescence in culture (Ota et al., 2009), which does not support the role of mTORC1–S6K1 in endothelial aging. One has to take into account that the former study shows correlation between increased mTORC1–S6K1 signaling and vascular aging, while the latter used pharmacological inhibitors in young endothelial cells. The drugs may exert some non-specific effects which could interfere with the interpretation. Moreover, the function of mTORC1–S6K1 in young and old cells or organisms might be different. A regulated function of mTORC1–S6K1 signaling is necessary for organism development, while a persistent non-regulated mTORC1–S6K1 signaling is detrimental.
By pharmacological and genetic approaches, we demonstrate a hyperactive S6K1 activity in two aging model systems, i.e., in cultured senescent endothelial cells and in aortas of naturally aging rats as compared to young cells and young animals (Rajapakse et al., 2011). A persistent activation of mTORC1–S6K1 signaling is also shown to be associated with hematopoietic stem cells in old mice, which is responsible for decreased regenerative capacity of the stem cells in aging (Chen et al., 2009). Inhibition of mTORC1–S6K1 pathway either with rapamycin or with S6K1 silencing improves NO production and inhibits 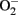 production in senescent cells and old rat aortas. The enhanced
production in senescent cells and old rat aortas. The enhanced 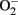 production in senescent cells and old rat aortas can be eliminated by the eNOS inhibitor L-NAME, demonstrating eNOS uncoupling in aging (Rajapakse et al., 2011). Conversely, over-expression of a S6K1 active mutant in young endothelial cells causes eNOS uncoupling and endothelial senescence, which provides the first evidence for a causative role of S6K1 in eNOS uncoupling and endothelial aging. How S6K1 drives endothelial senescence or aging and causes eNOS uncoupling, and whether S6K1 causes endothelial aging through eNOS uncoupling remain to be investigated.
production in senescent cells and old rat aortas can be eliminated by the eNOS inhibitor L-NAME, demonstrating eNOS uncoupling in aging (Rajapakse et al., 2011). Conversely, over-expression of a S6K1 active mutant in young endothelial cells causes eNOS uncoupling and endothelial senescence, which provides the first evidence for a causative role of S6K1 in eNOS uncoupling and endothelial aging. How S6K1 drives endothelial senescence or aging and causes eNOS uncoupling, and whether S6K1 causes endothelial aging through eNOS uncoupling remain to be investigated.
Endothelial aging is also associated with increased expression of the coagulation factor tissue factor (TF) as well as adhesion molecules ICAM-1 and VCAM-1 (Csiszar et al., 2008), which may promote thrombosis and vascular inflammation in elderly subjects. Numerous studies including our own report the inhibitory effect of mTORC1 on TF expression based on the observation that rapamycin or silencing mTOR enhances TF expression in endothelial cells (Camici et al., 2010; Ming et al., 2010). However, silencing S6K1 reduces TF protein level in endothelial cells stimulated with thrombin or TNFα without affecting TF mRNA expression. Conversely, over-expression of a constitutively active S6K1 mutant enhances TF protein level even in the mTOR-silenced cells (Ming et al., 2010). The results reveal an unexpected opposing effect of mTOR and S6K1 on endothelial TF expression and are best explained by the mechanisms that the markedly enhanced TF mRNA expression under the condition of mTORC1 inhibition is translated by S6K1-independent pathways such as RhoA, NF-κB, and p38mapk (Ming et al., 2010), since blockade of RhoA, NF-κB, and p38mapk either pharmacologically or genetically is able to reduce the up-regulation of TF protein level (Figure 2). The uncoupling effect of mTOR and S6K1 has also been reported in skeletal muscle cells (Cunningham et al., 2007a). These findings may have potential clinical implications. TF is highly expressed in cells within atherosclerotic plaques (Hatakeyama et al., 1997). It has been reported that vascular injury after coronary intervention increases circulating TF activity in patients (Tutar et al., 2003), which favors thrombus formation after coronary intervention. With rapamycin (sirolimus)-eluting stents, TF expression in the vasculature and release from vascular cells including endothelial cells, smooth muscle cells, and macrophages might be exaggerated because of the stimulating effect of TF expression by the drug. This effect of rapamycin, may increase thrombotic risk, despite its property of preventing vascular restenosis in patients with coronary artery disease (Luscher et al., 2007), although inhibition of endothelial regeneration and in turn endothelialization of stent surface by drug-eluting stents may play a more prominent role in stent thrombosis (Inoue et al., 2011). It would be interesting to test whether inhibition of S6K1 alone, rather than mTORC1 as achieved by rapamycin, proves sufficient to prevent vascular restenosis and be superior in reducing thrombotic propensity by developing specific S6K1 inhibitors. The same consideration may be applied for anti-aging therapy. Moreover, silencing S6K1 in endothelial cells is able to prevent up-regulation of E-selectin induced by TNFα (Ming et al., 2009), implicating a possible role of S6K1 in regulation of endothelial–leukocyte interaction. Whether S6K1 is involved in TF as well as adhesion molecule expression in aged endothelial cells and blood vessels warrants further investigation. The effects of mTORC1–S6K1 on cardiovascular functions are illustrated in Figure 2.
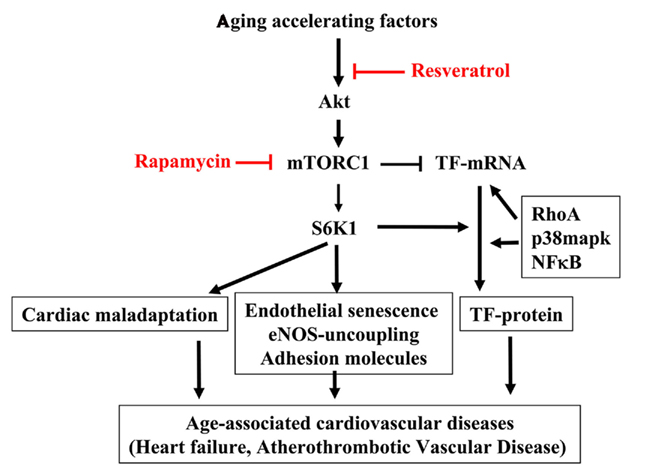
Figure 2. mTORC1–S6K1 signaling in cardiovascular aging. Aging accelerating factors cause persistent activation of the Akt–mTORC1–S6K1 signaling. The hyperactive S6K1 has pleiotropic effects in cardiovascular system. In heart, it causes cardiomyocytes hypertrophy leading to cardiac maladaptation. In endothelium, it induces eNOS uncoupling, endothelial senescence, and adhesion molecule expression as well as TF protein expression. All of these effects contribute to age-associated cardiovascular diseases such as heart failure, atherosclerotic vascular disease. Thus inhibiting S6K1 with resveratrol or mTORC1 inhibitor rapamycin may have beneficial effect in treatment of age-associated cardiovascular diseases in clinic. It is important to recognize that there are some uncoupled biological function(s) of mTORC1 and S6K1 as reported for their opposing effect in regulation of TF expression. While mTOR suppresses endothelial TF mRNA expression, S6K1 is required for TF protein translation, which works in concert with other signaling pathways such as RhoA, p38mapk, and NF-κB. Given this finding, drugs more specifically inhibiting S6K1 rather than mTORC1 should be developed and the effects of targeting mTORC1 or S6K1 on aging-related cardiovascular diseases and cardiovascular aging and beyond should be investigated.
Resveratrol inhibits mTORC1–S6K1 and prevents endothelial aging
It is well demonstrated that resveratrol, a naturally occurring polyphenol, slows aging process and exerts protective effects on aging-associated pathologies including vascular diseases, type II diabetes in animal models (Baur et al., 2006; Lagouge et al., 2006; Barger et al., 2008; Pearson et al., 2008; Miller et al., 2011). Numerous studies including ours demonstrate that resveratrol inhibits ICAM-1 and VCAM-1 expression in endothelial cells in response to high glucose and TNFα (Csiszar et al., 2006; Park et al., 2009; Rajapakse et al., 2009). Interestingly, resveratrol is capable of recoupling eNOS, leading to inhibition of superoxide generation and increase in endothelial NO production in senescent endothelial cells and in aged rat aortas partly through inhibition of mTORC1–S6K1 signaling (Rajapakse et al., 2011), since NO production stimulated by acetylcholine is only partly restored by resveratrol and rapamycin in old rats. Additional defects in eNOS dysfunction as discussed must be present.
It also remains to be investigated whether resveratrol’s inhibitory effect on mTORC1–S6K1–eNOS-uncoupling is mediated through activation of the NAD+-dependent deacetylase Sirt1 (Lagouge et al., 2006; Zang et al., 2006; Baur, 2010), whose activation has been shown to exhibit protection against age-associated diseases including diabetes and atherosclerosis (Lagouge et al., 2006; Zang et al., 2006; Pearson et al., 2008), although current studies do not show life extending effect in mammals (Lagouge et al., 2006; Barger et al., 2008; Pearson et al., 2008; Miller et al., 2011). Recent studies provide evidence showing that resveratrol also exerts pleiotrophic effects independently of Sirt1 (Zhang, 2006; Pirola and Frojdo, 2008; Kitada et al., 2011). It has also been shown that resveratrol may indirectly activate Sirt1 through AMPK, which has been demonstrated to inhibit mTORC1–S6K1 pathway in different cell types (Ruderman et al., 2010). The underlying mechanisms by which resveratrol inhibits mTORC1–S6K1 in aging endothelial cells are not clear, yet. Since the hyperactive Akt, an upstream signaling of mTORC1–S6K1 pathway, is observed in senescent endothelial cells and is inhibited by resveratrol (Rajapakse et al., 2011), it seems that resveratrol negatively regulates mTORC1–S6K1 pathway through inhibition of mTORC2. The persistent activation of Akt–mTORC1–S6K1 signaling in endothelial aging observed in our study is consistent with the finding that a hyperactive Akt plays a role in endothelial cell senescence (Miyauchi et al., 2004). Further studies need to establish a role of mTORC2 in endothelial senescence and in regulation of longevity.
Rapamycin and Resveratrol as Anti-Aging and Anti-Cardiovascular Aging Drugs in Humans?
Although caloric restriction seems the most promising approach to slow aging and onset of age-related diseases in humans with almost no recognizable adverse effects (Fontana et al., 2010), it is difficult to implement as a routine and long-term preventive or treatment modality in humans. The primary target of anti-aging strategy should focus on treatment of age-related diseases and not longevity, i.e., improvement of “healthy” lifespan. By treatment of age-related diseases, healthy lifespan extension, and improvement of life quality in elderly are expected.
In animal models, rapamycin is able to extend lifespan and prevents many age-related diseases such as cancer (Hudes et al., 2007), obesity (Um et al., 2004), cardiovascular diseases (Sadoshima and Izumo, 1995; Elloso et al., 2003; Waksman et al., 2003; McMullen et al., 2004; Pakala et al., 2005; Adelman, 2010). Moreover, a recent study reports that in cultured fibroblasts isolated from patients suffering Hutchinson–Gilford Progeria Syndrome (HGPS), a lethal genetic disease characterized by premature aging and death in adolescence or the teen years, rapamycin postpones cell senescence by activating autophagy, a process by which cells clear junk protein and trashed organelles and is inhibited by hyperactive mTORC1–S6K1 (Cao et al., 2011). These findings suggest an additional mechanism for the beneficial effects of rapamycin on aging.
Interestingly, many of the beneficial effects of rapamycin have been shared with resveratrol (Jang et al., 1997; Baur et al., 2006; Zang et al., 2006; Chan et al., 2008; Smoliga et al., 2011), although the lifespan extending effect could be demonstrated with rapamycin but not with resveratrol in mice (Pearson et al., 2008; Harrison et al., 2009; Miller et al., 2011). Both rapamycin and resveratrol inhibit hyperactive mTORC1–S6K1 signaling, improve endothelial function in senescent cells and aging rat aortas (Rajapakse et al., 2011), and also improve bone marrow-derived progenitor cell function and senescence (Chen et al., 2009; Huang et al., 2010). All the results suggest that rapamycin and resveratrol may be used as anti-aging therapeutics in humans. Rapamycin and analogs are indeed used in patients with organ transplantation and cancer and show clinical benefits (Kauffman et al., 2005; Law, 2005; Stallone et al., 2005; Zmonarski et al., 2005; Campistol et al., 2006). However, there is concern about some undesirable effects of rapamycin which may limit its systemic use as anti-aging or anti-cardiovascular aging drug. In contrast to the initial major concern about immunosuppressive effects, rapamycin has been shown to improve immune function and regenerative capacity of bone marrow stem cells in old mice (Chen et al., 2009). However, early studies in animal models treated with rapamycin reported multiple adverse effects including deregulated glucose homeostasis, hyperlipidemia (Cunningham et al., 2007b; Chang et al., 2009). In humans, rapamycin increases blood triglyceride and cholesterol resulting from lipolysis (Morrisett et al., 2002; Ribes et al., 2005), which may have negative impact on cardiovascular functions and metabolic homeostasis, although rapamycin has been shown to reduce atherosclerosis in mouse models (Basso et al., 2003; Elloso et al., 2003; Waksman et al., 2003; Pakala et al., 2005). The different effects of rapamycin in mice may be related to when the rapamycin therapy is initiated (early or late in life) or how long the therapy persists, or whether chronic persistent or pulsed therapy is instrumented. Finally, it may depend on whether young or old, healthy or diseased animals are treated with the drug. It is important to point out that rapamycin may be beneficial under the condition when mTORC1–S6K1 signaling is inappropriately consistently elevated, for example in aging, but detrimental if its activity under physiological conditions is abolished. Furthermore, taking into account that an opposing and uncoupling effect of mTORC1 and S6K1 has been demonstrated such as in regulation of endothelial TF expression (Ming et al., 2010), and that both rapamycin and resveratrol inhibit mTORC1 at the level and upstream of mTORC1, respectively (Figure 2), it would be worth to test whether targeting S6K1 directly would be a better approach than targeting mTORC1 as achieved by rapamycin or resveratrol. For this purpose, specific inhibitor of S6K1 should be developed. At the molecular level, since off-target effects of the drugs could not be excluded, more specific experimental approaches such as mutants (active or dominant negative), RNA interference, or targeted gene disruption of mTORC1–S6K1 signaling should be applied to evaluate their roles in cardiovascular systems.
So far, almost no undesirable effects of resveratrol have been reported, which might be due to the fact that this drug is newly investigated as compared to rapamycin. The first recently published human study with resveratrol confirmed metabolic beneficial effects which were previously observed in animals (Timmers et al., 2011). The study showed that treatment of healthy, obese men with 150 mg/day resveratrol for 30 days mimicked the effects of calorie restriction, significantly improved metabolic profiles on circulating glucose, triglycerides, liver lipid content, and decreased inflammation markers and systolic blood pressure (Timmers et al., 2011). The effects of resveratrol on cardiovascular diseases in patients remain to be demonstrated. Further, dosage dependent side effects of resveratrol should be investigated, especially for the newly developed derivatives which show much stronger effects than resveratrol on Sirt1 activity (Milne et al., 2007). The potency of a drug is usually associated with toxicity due to non-specific off-target effects.
Conclusion and Perspectives
Emerging evidence demonstrates that targeting mTORC1–S6K1 signaling could be a promising therapeutic modality to slow aging process and treat cardiovascular disease in aging. Future work should further elucidate the mechanisms of persistent hyperactive mTORC–S6K1 and the mechanisms of cardiovascular aging driven by mTORC1–S6K1. Since mTORC1–S6K1 signaling is also essential for normal development and skeletal muscle mass growth, undesirable side effects of targeting mTOCR1–S6K1 may be avoided by short treatment instead long-term treatment. In addition, drugs more specifically inhibiting S6K1 rather than mTORC1 should be developed and the effects of targeting mTORC1 or S6K1 on aging-related cardiovascular diseases and cardiovascular aging and beyond should be investigated.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The original own studies cited by this article were supported by the Swiss National Science Foundation (310000-120435) and the Swiss Heart Foundation.
References
Adelman, S. J. (2010). Sirolimus and its analogs and its effects on vascular diseases. Curr. Pharm. Des. 16, 4002–4011.
Antoniades, C., Shirodaria, C., Leeson, P., Antonopoulos, A., Warrick, N., Van-Assche, T., Cunnington, C., Tousoulis, D., Pillai, R., Ratnatunga, C., Stefanadis, C., and Channon, K. M. (2009). Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur. Heart J. 30, 1142–1150.
Barger, J. L., Kayo, T., Vann, J. M., Arias, E. B., Wang, J., Hacker, T. A., Wang, Y., Raederstorff, D., Morrow, J. D., Leeuwenburgh, C., Allison, D. B., Saupe, K. W., Cartee, G. D., Weindruch, R., and Prolla, T. A. (2008). A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE 3, e2264. doi:10.1371/journal.pone.0002264
Basso, M. D., Nambi, P., and Adelman, S. J. (2003). Effect of sirolimus on the cholesterol content of aortic arch in ApoE knockout mice. Transplant. Proc. 35, 3136–3138.
Baur, J. A. (2010). Resveratrol, sirtuins, and the promise of a DR mimetic. Mech. Ageing Dev. 131, 261–269.
Baur, J. A., Pearson, K. J., Price, N. L., Jamieson, H. A., Lerin, C., Kalra, A., Prabhu, V. V., Allard, J. S., Lopez-Lluch, G., Lewis, K., Pistell, P. J., Poosala, S., Becker, K. G., Boss, O., Gwinn, D., Wang, M., Ramaswamy, S., Fishbein, K. W., Spencer, R. G., Lakatta, E. G., Le, C. D., Shaw, R. J., Navas, P., Puigserver, P., Ingram, D. K., de, C. R., and Sinclair, D. A. (2006). Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342.
Bendall, J. K., Alp, N. J., Warrick, N., Cai, S., Adlam, D., Rockett, K., Yokoyama, M., Kawashima, S., and Channon, K. M. (2005). Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 97, 864–871.
Berkowitz, D. E., White, R., Li, D., Minhas, K. M., Cernetich, A., Kim, S., Burke, S., Shoukas, A. A., Nyhan, D., Champion, H. C., and Hare, J. M. (2003). Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 108, 2000–2006.
Boluyt, M. O., Zheng, J. S., Younes, A., Long, X., O’Neill, L., Silverman, H., Lakatta, E. G., and Crow, M. T. (1997). Rapamycin inhibits alpha 1-adrenergic receptor-stimulated cardiac myocyte hypertrophy but not activation of hypertrophy-associated genes. Evidence for involvement of p70 S6 kinase. Circ. Res. 81, 176–186.
Bugiardini, R., Manfrini, O., Pizzi, C., Fontana, F., and Morgagni, G. (2004). Endothelial function predicts future development of coronary artery disease: a study of women with chest pain and normal coronary angiograms. Circulation 109, 2518–2523.
Camici, G. G., Steffel, J., Amanovic, I., Breitenstein, A., Baldinger, J., Keller, S., Luscher, T. F., and Tanner, F. C. (2010). Rapamycin promotes arterial thrombosis in vivo: implications for everolimus and zotarolimus eluting stents. Eur. Heart J. 31, 236–242.
Campistol, J. M., Eris, J., Oberbauer, R., Friend, P., Hutchison, B., Morales, J. M., Claesson, K., Stallone, G., Russ, G., Rostaing, L., Kreis, H., Burke, J. T., Brault, Y., Scarola, J. A., and Neylan, J. F. (2006). Sirolimus therapy after early cyclosporine withdrawal reduces the risk for cancer in adult renal transplantation. J. Am. Soc. Nephrol. 17, 581–589.
Canto, C., Gerhart-Hines, Z., Feige, J. N., Lagouge, M., Noriega, L., Milne, J. C., Elliott, P. J., Puigserver, P., and Auwerx, J. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060.
Cao, K., Graziotto, J. J., Blair, C. D., Mazzulli, J. R., Erdos, M. R., Krainc, D., and Collins, F. S. (2011). Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in hutchinson-gilford progeria syndrome cells. Sci. Transl. Med. 3, 89ra58.
Chan, A. Y., Dolinsky, V. W., Soltys, C. L., Viollet, B., Baksh, S., Light, P. E., and Dyck, J. R. (2008). Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J. Biol. Chem. 283, 24194–24201.
Chang, G. R., Wu, Y. Y., Chiu, Y. S., Chen, W. Y., Liao, J. W., Hsu, H. M., Chao, T. H., Hung, S. W., and Mao, F. C. (2009). Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin. Pharmacol. Toxicol. 105, 188–198.
Chen, C., Liu, Y., Liu, Y., and Zheng, P. (2009). mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2, ra75.
Chen, C. A., Wang, T. Y., Varadharaj, S., Reyes, L. A., Hemann, C., Talukder, M. A., Chen, Y. R., Druhan, L. J., and Zweier, J. L. (2010). S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468, 1115–1118.
Christensen, K., Doblhammer, G., Rau, R., and Vaupel, J. W. (2009). Ageing populations: the challenges ahead. Lancet 374, 1196–1208.
Christensen, K., McGue, M., Petersen, I., Jeune, B., and Vaupel, J. W. (2008). Exceptional longevity does not result in excessive levels of disability. Proc. Natl. Acad. Sci. U.S.A. 105, 13274–13279.
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., Howitz, K. T., Gorospe, M., de, C. R., and Sinclair, D. A. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392.
Colman, R. J., Anderson, R. M., Johnson, S. C., Kastman, E. K., Kosmatka, K. J., Beasley, T. M., Allison, D. B., Cruzen, C., Simmons, H. A., Kemnitz, J. W., and Weindruch, R. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204.
Csiszar, A., Smith, K., Labinskyy, N., Orosz, Z., Rivera, A., and Ungvari, Z. (2006). Resveratrol attenuates TNF-alpha-induced activation of coronary arterial endothelial cells: role of NF-kappaB inhibition. Am. J. Physiol. Heart Circ. Physiol. 291, H1694–H1699.
Csiszar, A., Ungvari, Z., Edwards, J. G., Kaminski, P., Wolin, M. S., Koller, A., and Kaley, G. (2002). Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 90, 1159–1166.
Csiszar, A., Ungvari, Z., Koller, A., Edwards, J. G., and Kaley, G. (2004). Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genomics 17, 21–30.
Csiszar, A., Wang, M., Lakatta, E. G., and Ungvari, Z. (2008). Inflammation and endothelial dysfunction during aging: role of NF-kappaB. J. Appl. Physiol. 105, 1333–1341.
Cunningham, J. T., Rodgers, J. T., Arlow, D. H., Vazquez, F., Mootha, V. K., and Puigserver, P. (2007a). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740.
Cunningham, J. T., Rodgers, J. T., Arlow, D. H., Vazquez, F., Mootha, V. K., and Puigserver, P. (2007b). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740.
Dann, S. G., Selvaraj, A., and Thomas, G. (2007). mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 13, 252–259.
Desrois, M., Clarke, K., Lan, C., Dalmasso, C., Cole, M., Portha, B., Cozzone, P. J., and Bernard, M. (2010). Upregulation of eNOS and unchanged energy metabolism in increased susceptibility of the aging type 2 diabetic GK rat heart to ischemic injury. Am. J. Physiol. Heart Circ. Physiol. 299, H1679–H1686.
Drummond, G. R., Cai, H., Davis, M. E., Ramasamy, S., and Harrison, D. G. (2000). Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 86, 347–354.
Elloso, M. M., Azrolan, N., Sehgal, S. N., Hsu, P. L., Phiel, K. L., Kopec, C. A., Basso, M. D., and Adelman, S. J. (2003). Protective effect of the immunosuppressant sirolimus against aortic atherosclerosis in apo E-deficient mice. Am. J. Transplant. 3, 562–569.
Estep, P. W. III, Warner, J. B., and Bulyk, M. L. (2009). Short-term calorie restriction in male mice feminizes gene expression and alters key regulators of conserved aging regulatory pathways. PLoS ONE 4, e5242. doi:10.1371/journal.pone.0005242
Evans, D. S., Kapahi, P., Hsueh, W. C., and Kockel, L. (2011). TOR signaling never gets old: aging, longevity and TORC1 activity. Ageing Res. Rev. 10, 225–237.
Fontana, L., Meyer, T. E., Klein, S., and Holloszy, J. O. (2004). Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. U.S.A. 101, 6659–6663.
Fontana, L., Partridge, L., and Longo, V. D. (2010). Extending healthy life span – from yeast to humans. Science 328, 321–326.
Forstermann, U., and Munzel, T. (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113, 1708–1714.
Foster, K. G., and Finger, D. C. (2010). Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077.
Frost, R. A., and Lang, C. H. (2011). mTor signaling in skeletal muscle during sepsis and inflammation: where does it all go wrong? Physiology (Bethesda) 26, 83–96.
Glazer, H. P., Osipov, R. M., Clements, R. T., Sellke, F. W., and Bianchi, C. (2009). Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle 8, 1738–1746.
Gorgoulis, V. G., Zacharatos, P., Kotsinas, A., Kletsas, D., Mariatos, G., Zoumpourlis, V., Ryan, K. M., Kittas, C., and Papavassiliou, A. G. (2003). p53 activates ICAM-1 (CD54) expression in an NF-kappaB-independent manner. EMBO J. 22, 1567–1578.
Halcox, J. P., Schenke, W. H., Zalos, G., Mincemoyer, R., Prasad, A., Waclawiw, M. A., Nour, K. R., and Quyyumi, A. A. (2002). Prognostic value of coronary vascular endothelial dysfunction. Circulation 106, 653–658.
Hammer, S., Snel, M., Lamb, H. J., Jazet, I. M., van der Meer, R. W., Pijl, H., Meinders, E. A., Romijn, J. A., de, R. A., and Smit, J. W. (2008). Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J. Am. Coll. Cardiol. 52, 1006–1012.
Hands, S. L., Proud, C. G., and Wyttenbach, A. (2009). mTOR’s role in ageing: protein synthesis or autophagy? Aging (Albany NY) 1, 586–597.
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., Nadon, N. L., Wilkinson, J. E., Frenkel, K., Carter, C. S., Pahor, M., Javors, M. A., Fernandez, E., and Miller, R. A. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395.
Hatakeyama, K., Asada, Y., Marutsuka, K., Sato, Y., Kamikubo, Y., and Sumiyoshi, A. (1997). Localization and activity of tissue factor in human aortic atherosclerotic lesions. Atherosclerosis 133, 213–219.
Herranz, D., and Serrano, M. (2010). SIRT1: recent lessons from mouse models. Nat. Rev. Cancer 10, 819–823.
Herrera, M. D., Mingorance, C., Rodriguez-Rodriguez, R., and Alvarez de, S. M. (2010). Endothelial dysfunction and aging: an update. Ageing Res. Rev. 9, 142–152.
Hill, J. M., Zalos, G., Halcox, J. P., Schenke, W. H., Waclawiw, M. A., Quyyumi, A. A., and Finkel, T. (2003). Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 348, 593–600.
Hink, U., Li, H., Mollnau, H., Oelze, M., Matheis, E., Hartmann, M., Skatchkov, M., Thaiss, F., Stahl, R. A., Warnholtz, A., Meinertz, T., Griendling, K., Harrison, D. G., Forstermann, U., and Munzel, T. (2001). Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 88, E14–E22.
Hoffmann, J., Haendeler, J., Aicher, A., Rossig, L., Vasa, M., Zeiher, A. M., and Dimmeler, S. (2001). Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: important role of nitric oxide. Circ. Res. 89, 709–715.
Holzenberger, M., Dupont, J., Ducos, B., Leneuve, P., Geloen, A., Even, P. C., Cervera, P., and Le, B. Y. (2003). IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182–187.
Huang, P. H., Chen, J. W., Lu, T. M., Yu-An, D. P., and Lin, S. J. (2007). Combined use of endothelial function assessed by brachial ultrasound and high-sensitive C-reactive protein in predicting cardiovascular events. Clin. Cardiol. 30, 135–140.
Huang, P. H., Chen, Y. H., Tsai, H. Y., Chen, J. S., Wu, T. C., Lin, F. Y., Sata, M., Chen, J. W., and Lin, S. J. (2010). Intake of red wine increases the number and functional capacity of circulating endothelial progenitor cells by enhancing nitric oxide bioavailability. Arterioscler. Thromb. Vasc. Biol. 30, 869–877.
Hudes, G., Carducci, M., Tomczak, P., Dutcher, J., Figlin, R., Kapoor, A., Staroslawska, E., Sosman, J., McDermott, D., Bodrogi, I., Kovacevic, Z., Lesovoy, V., Schmidt-Wolf, I. G., Barbarash, O., Gokmen, E., O’Toole, T., Lustgarten, S., Moore, L., and Motzer, R. J. (2007). Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 356, 2271–2281.
Inoue, T., Croce, K., Morooka, T., Sakuma, M., Node, K., and Simon, D. I. (2011). Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc. Interv. 4, 1057–1066.
Jang, M., Cai, L., Udeani, G. O., Slowing, K. V., Thomas, C. F., Beecher, C. W., Fong, H. H., Farnsworth, N. R., Kinghorn, A. D., Mehta, R. G., Moon, R. C., and Pezzuto, J. M. (1997). Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275, 218–220.
Kagawa, Y. (1978). Impact of Westernization on the nutrition of Japanese: changes in physique, cancer, longevity and centenarians. Prev. Med. 7, 205–217.
Kapahi, P., and Zid, B. (2004). TOR pathway: linking nutrient sensing to life span. Sci. Aging Knowledge Environ. 2004, E34.
Kauffman, H. M., Cherikh, W. S., Cheng, Y., Hanto, D. W., and Kahan, B. D. (2005). Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation 80, 883–889.
Kim, J. A., Jang, H. J., Martinez-Lemus, L. A., and Sowers, J. R. (2012). Activation of mTOR/p70S6 kinase by ANG II inhibits insulin stimulated endothelial nitric oxide synthase and vasodilation. Am. J. Physiol. Endocrinol. Metab. 302, E201–E208.
Kitada, M., Kume, S., Imaizumi, N., and Koya, D. (2011). Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes 60, 634–643.
Kurdi, M., and Booz, G. W. (2011). Three 4-letter words of hypertension-related cardiac hypertrophy: TRPC, mTOR, and HDAC. J. Mol. Cell. Cardiol. 50, 964–971.
Lagouge, M., Argmann, C., Gerhart-Hines, Z., Meziane, H., Lerin, C., Daussin, F., Messadeq, N., Milne, J., Lambert, P., Elliott, P., Geny, B., Laakso, M., Puigserver, P., and Auwerx, J. (2006). Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127, 1109–1122.
Landmesser, U., Dikalov, S., Price, S. R., McCann, L., Fukai, T., Holland, S. M., Mitch, W. E., and Harrison, D. G. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209.
Law, B. K. (2005). Rapamycin: an anti-cancer immunosuppressant? Crit. Rev. Oncol. Hematol. 56, 47–60.
Lee, J. H., Budanov, A. V., Park, E. J., Birse, R., Kim, T. E., Perkins, G. A., Ocorr, K., Ellisman, M. H., Bodmer, R., Bier, E., and Karin, M. (2010). Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327, 1223–1228.
Li, H. L., Yin, R., Chen, D., Liu, D., Wang, D., Yang, Q., and Dong, Y. G. (2007). Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J. Cell. Biochem. 100, 1086–1099.
Linford, N. J., Beyer, R. P., Gollahon, K., Krajcik, R. A., Malloy, V. L., Demas, V., Burmer, G. C., and Rabinovitch, P. S. (2007). Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell 6, 673–688.
Luscher, T. F., Steffel, J., Eberli, F. R., Joner, M., Nakazawa, G., Tanner, F. C., and Virmani, R. (2007). Drug-eluting stent and coronary thrombosis: biological mechanisms and clinical implications. Circulation 115, 1051–1058.
Lutz, W., Sanderson, W., and Scherbov, S. (2008). The coming acceleration of global population ageing. Nature 451, 716–719.
McMullen, J. R., Sherwood, M. C., Tarnavski, O., Zhang, L., Dorfman, A. L., Shioi, T., and Izumo, S. (2004). Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 109, 3050–3055.
Meyer, T. E., Kovacs, S. J., Ehsani, A. A., Klein, S., Holloszy, J. O., and Fontana, L. (2006). Long-term caloric restriction ameliorates the decline in diastolic function in humans. J. Am. Coll. Cardiol. 47, 398–402.
Miller, R. A., Harrison, D. E., Astle, C. M., Baur, J. A., Boyd, A. R., de, C. R., Fernandez, E., Flurkey, K., Javors, M. A., Nelson, J. F., Orihuela, C. J., Pletcher, S., Sharp, Z. D., Sinclair, D., Starnes, J. W., Wilkinson, J. E., Nadon, N. L., and Strong, R. (2011). Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 66, 191–201.
Milne, J. C., Lambert, P. D., Schenk, S., Carney, D. P., Smith, J. J., Gagne, D. J., Jin, L., Boss, O., Perni, R. B., Vu, C. B., Bemis, J. E., Xie, R., Disch, J. S., Ng, P. Y., Nunes, J. J., Lynch, A. V., Yang, H., Galonek, H., Israelian, K., Choy, W., Iffland, A., Lavu, S., Medvedik, O., Sinclair, D. A., Olefsky, J. M., Jirousek, M. R., Elliott, P. J., and Westphal, C. H. (2007). Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716.
Ming, X. F., Barandier, C., Viswambharan, H., Kwak, B. R., Mach, F., Mazzolai, L., Hayoz, D., Ruffieux, J., Rusconi, S., Montani, J. P., and Yang, Z. (2004). Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation 110, 3708–3714.
Ming, X. F., Rajapakse, A. G., Carvas, J. M., Ruffieux, J., and Yang, Z. (2009). Inhibition of S6K1 accounts partially for the anti-inflammatory effects of the arginase inhibitor L-norvaline. BMC Cardiovasc. Disord. 9, 12. doi:10.1186/1471-2261-9-12
Ming, X. F., Rajapakse, A. G., Carvas, J. M., Ruffieux, J., and Yang, Z. (2010). Opposing and uncoupling effects of mTOR and S6K1 in the regulation of endothelial tissue factor expression. FEBS Lett. 584, 135–140.
Miyauchi, H., Minamino, T., Tateno, K., Kunieda, T., Toko, H., and Komuro, I. (2004). Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 23, 212–220.
Morrisett, J. D., Abdel-Fattah, G., Hoogeveen, R., Mitchell, E., Ballantyne, C. M., Pownall, H. J., Opekun, A. R., Jaffe, J. S., Oppermann, S., and Kahan, B. D. (2002). Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J. Lipid Res. 43, 1170–1180.
Moschella, P. C., Rao, V. U., McDermott, P. J., and Kuppuswamy, D. (2007). Regulation of mTOR and S6K1 activation by the nPKC isoforms, PKCepsilon and PKCdelta, in adult cardiac muscle cells. J. Mol. Cell. Cardiol. 43, 754–766.
Muniyappa, R., Montagnani, M., Koh, K. K., and Quon, M. J. (2007). Cardiovascular actions of insulin. Endocr. Rev. 28, 463–491.
Najjar, S. S., Scuteri, A., and Lakatta, E. G. (2005). Arterial aging: is it an immutable cardiovascular risk factor? Hypertension 46, 454–462.
Ota, H., Eto, M., Ako, J., Ogawa, S., Iijima, K., Akishita, M., and Ouchi, Y. (2009). Sirolimus and everolimus induce endothelial cellular senescence via sirtuin 1 down-regulation: therapeutic implication of cilostazol after drug-eluting stent implantation. J. Am. Coll. Cardiol. 53, 2298–2305.
Ozaki, M., Kawashima, S., Yamashita, T., Hirase, T., Namiki, M., Inoue, N., Hirata, K., Yasui, H., Sakurai, H., Yoshida, Y., Masada, M., and Yokoyama, M. (2002). Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J. Clin. Invest. 110, 331–340.
Pakala, R., Stabile, E., Jang, G. J., Clavijo, L., and Waksman, R. (2005). Rapamycin attenuates atherosclerotic plaque progression in apolipoprotein E knockout mice: inhibitory effect on monocyte chemotaxis. J. Cardiovasc. Pharmacol. 46, 481–486.
Pan, K. Z., Palter, J. E., Rogers, A. N., Olsen, A., Chen, D., Lithgow, G. J., and Kapahi, P. (2007). Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 6, 111–119.
Park, J. S., Kim, K. M., Kim, M. H., Chang, H. J., Baek, M. K., Kim, S. M., and Jung, Y. D. (2009). Resveratrol inhibits tumor cell adhesion to endothelial cells by blocking ICAM-1 expression. Anticancer Res. 29, 355–362.
Pearson, K. J., Baur, J. A., Lewis, K. N., Peshkin, L., Price, N. L., Labinskyy, N., Swindell, W. R., Kamara, D., Minor, R. K., Perez, E., Jamieson, H. A., Zhang, Y., Dunn, S. R., Sharma, K., Pleshko, N., Woollett, L. A., Csiszar, A., Ikeno, Y., Le, C. D., Elliott, P. J., Becker, K. G., Navas, P., Ingram, D. K., Wolf, N. S., Ungvari, Z., Sinclair, D. A., and de, C. R. (2008). Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 8, 157–168.
Powers, R. W. III, Kaeberlein, M., Caldwell, S. D., Kennedy, B. K., and Fields, S. (2006). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184.
Rajapakse, A. G., Ming, X. F., Carvas, J. M., and Yang, Z. (2009). The hexosamine biosynthesis inhibitor azaserine prevents endothelial inflammation and dysfunction under hyperglycemic condition through antioxidant effects. Am. J. Physiol. Heart Circ. Physiol. 296, H815–H822.
Rajapakse, A. G., Yepuri, G., Carvas, J. M., Stein, S., Matter, C. M., Scerri, I., Ruffieux, J., Montani, J. P., Ming, X. F., and Yang, Z. (2011). Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS ONE 6, e19237. doi:10.1371/journal.pone.0019237
Rau, R., Soroko, E., Jasilionis, D., and Vaupel, J. W. (2008). Continued reductions in mortality at advanced ages. Popul. Dev. Rev. 34, 747–768.
Rauscher, F. M., Goldschmidt-Clermont, P. J., Davis, B. H., Wang, T., Gregg, D., Ramaswami, P., Pippen, A. M., Annex, B. H., Dong, C., and Taylor, D. A. (2003). Aging, progenitor cell exhaustion, and atherosclerosis. Circulation 108, 457–463.
Reaven, G. M. (2011). Insulin resistance: the link between obesity and cardiovascular disease. Med. Clin. North Am. 95, 875–892.
Ribes, D., Kamar, N., Esposito, L., and Rostaing, L. (2005). Combined use of tacrolimus and sirolimus in de novo renal transplant patients: current data. Transplant. Proc. 37, 2813–2816.
Rice, K. M., Kinnard, R. S., Harris, R., Wright, G. L., and Blough, E. R. (2005). Effects of aging on pressure-induced MAPK activation in the rat aorta. Pflugers Arch. 450, 192–199.
Ruderman, N. B., Xu, X. J., Nelson, L., Cacicedo, J. M., Saha, A. K., Lan, F., and Ido, Y. (2010). AMPK and SIRT1: a long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 298, E751–E760.
Sadoshima, J., and Izumo, S. (1995). Rapamycin selectively inhibits angiotensin II-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6 kinase in angiotensin II-induced cardiac hypertrophy. Circ. Res. 77, 1040–1052.
Sakuma, K., and Yamaguchi, A. (2010). Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr. Aging Sci. 3, 90–101.
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., Markhard, A. L., and Sabatini, D. M. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168.
Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101.
Schachinger, V., Britten, M. B., and Zeiher, A. M. (2000). Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 101, 1899–1906.
Selman, C., Lingard, S., Choudhury, A. I., Batterham, R. L., Claret, M., Clements, M., Ramadani, F., Okkenhaug, K., Schuster, E., Blanc, E., Piper, M. D., Al-Qassab, H., Speakman, J. R., Carmignac, D., Robinson, I. C., Thornton, J. M., Gems, D., Partridge, L., and Withers, D. J. (2008). Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 22, 807–818.
Selman, C., Tullet, J. M., Wieser, D., Irvine, E., Lingard, S. J., Choudhury, A. I., Claret, M., Al-Qassab, H., Carmignac, D., Ramadani, F., Woods, A., Robinson, I. C., Schuster, E., Batterham, R. L., Kozma, S. C., Thomas, G., Carling, D., Okkenhaug, K., Thornton, J. M., Partridge, L., Gems, D., and Withers, D. J. (2009). Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326, 140–144.
Sengupta, S., Peterson, T. R., and Sabatini, D. M. (2010). Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40, 310–322.
Sesso, H. D., Buring, J. E., Christen, W. G., Kurth, T., Belanger, C., MacFadyen, J., Bubes, V., Manson, J. E., Glynn, R. J., and Gaziano, J. M. (2008). Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA 300, 2123–2133.
Shackelford, D. B., and Shaw, R. J. (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575.
Shih, H., Lee, B., Lee, R. J., and Boyle, A. J. (2011). The aging heart and post-infarction left ventricular remodeling. J. Am. Coll. Cardiol. 57, 9–17.
Shinmura, K., Tamaki, K., Sano, M., Murata, M., Yamakawa, H., Ishida, H., and Fukuda, K. (2011). Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J. Mol. Cell. Cardiol. 50, 117–127.
Smoliga, J. M., Baur, J. A., and Hausenblas, H. A. (2011). Resveratrol and health – a comprehensive review of human clinical trials. Mol. Nutr. Food Res. 0.55, 1129–1141.
Soesanto, W., Lin, H. Y., Hu, E., Lefler, S., Litwin, S. E., Sena, S., Abel, E. D., Symons, J. D., and Jalili, T. (2009). Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension 54, 1321–1327.
Stallone, G., Schena, A., Infante, B., Di, P. S., Loverre, A., Maggio, G., Ranieri, E., Gesualdo, L., Schena, F. P., and Grandaliano, G. (2005). Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N. Engl. J. Med. 352, 1317–1323.
Stanfel, M. N., Shamieh, L. S., Kaeberlein, M., and Kennedy, B. K. (2009). The TOR pathway comes of age. Biochim. Biophys. Acta 1790, 1067–1074.
Stockklauser-Farber, K., Ballhausen, T., Laufer, A., and Rosen, P. (2000). Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim. Biophys. Acta 1535, 10–20.
Sung, M. M., Koonen, D. P., Soltys, C. L., Jacobs, R. L., Febbraio, M., and Dyck, J. R. (2011). Increased CD36 expression in middle-aged mice contributes to obesity-related cardiac hypertrophy in the absence of cardiac dysfunction. J. Mol. Med. 89, 459–469.
Tanabe, T., Maeda, S., Miyauchi, T., Iemitsu, M., Takanashi, M., Irukayama-Tomobe, Y., Yokota, T., Ohmori, H., and Matsuda, M. (2003). Exercise training improves ageing-induced decrease in eNOS expression of the aorta. Acta Physiol. Scand. 178, 3–10.
Timmers, S., Konings, E., Bilet, L., Houtkooper, R. H., van de Weijer, T., Goossens, G. H., Hoeks, J., van der Krieken, S., Ryu, D., Kersten, S., Moonen-Kornips, E., Hesselink, M. K. C., Kunz, I., Schrauwen-Hinderling, V. B., Blaak, E. E., Auwer, J., and Schrauwen, P. (2011). Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 14, 612–622.
Tu, V. C., Bahl, J. J., and Chen, Q. M. (2002). Signals of oxidant-induced cardiomyocyte hypertrophy: key activation of p70 S6 kinase-1 and phosphoinositide 3-kinase. J. Pharmacol. Exp. Ther. 300, 1101–1110.
Tutar, E., Ozcan, M., Kilickap, M., Gulec, S., Aras, O., Pamir, G., Oral, D., Dandelet, L., and Key, N. S. (2003). Elevated whole-blood tissue factor procoagulant activity as a marker of restenosis after percutaneous transluminal coronary angioplasty and stent implantation. Circulation 108, 1581–1584.
Um, S. H., Frigerio, F., Watanabe, M., Picard, F., Joaquin, M., Sticker, M., Fumagalli, S., Allegrini, P. R., Kozma, S. C., Auwerx, J., and Thomas, G. (2004). Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205.
Ungvari, Z., Kaley, G., de, C. R., Sonntag, W. E., and Csiszar, A. (2010). Mechanisms of vascular aging: new perspectives. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1028–1041.
van der Loo, B., Labugger, R., Skepper, J. N., Bachschmid, M., Kilo, J., Powell, J. M., Palacios-Callender, M., Erusalimsky, J. D., Quaschning, T., Malinski, T., Gygi, D., Ullrich, V., and Luscher, T. F. (2000). Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 192, 1731–1744.
Vanhoutte, P. M., Shimokawa, H., Tang, E. H., and Feletou, M. (2009). Endothelial dysfunction and vascular disease. Acta Physiol. (Oxf.) 196, 193–222.
Vasa, M., Fichtlscherer, S., Aicher, A., Adler, K., Urbich, C., Martin, H., Zeiher, A. M., and Dimmeler, S. (2001). Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 89, E1–E7.
Vivekananthan, D. P., Penn, M. S., Sapp, S. K., Hsu, A., and Topol, E. J. (2003). Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 361, 2017–2023.
Waksman, R., Pakala, R., Burnett, M. S., Gulick, C. P., Leborgne, L., Fournadjiev, J., Wolfram, R., and Hellinga, D. (2003). Oral rapamycin inhibits growth of atherosclerotic plaque in apoE knock-out mice. Cardiovasc. Radiat. Med. 4, 34–38.
Wang, Y., Liang, Y., and Vanhoutte, P. M. (2011). SIRT1 and AMPK in regulating mammalian senescence: a critical review and a working model. FEBS Lett. 585, 986–994.
Wei, L. (2004). Lysophospholipid signaling in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 36, 465–468.
Werner, N., Kosiol, S., Schiegl, T., Ahlers, P., Walenta, K., Link, A., Bohm, M., and Nickenig, G. (2005). Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 353, 999–1007.
Willcox, B. J., Willcox, D. C., Todoriki, H., Fujiyoshi, A., Yano, K., He, Q., Curb, J. D., and Suzuki, M. (2007). Caloric restriction, the traditional Okinawan diet, and healthy aging: the diet of the world’s longest-lived people and its potential impact on morbidity and life span. Ann. N. Y. Acad. Sci. 1114, 434–455.
Yang, Z., and Ming, X. F. (2006). Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin. Med. Res. 4, 53–65.
Zang, M., Xu, S., Maitland-Toolan, K. A., Zuccollo, A., Hou, X., Jiang, B., Wierzbicki, M., Verbeuren, T. J., and Cohen, R. A. (2006). Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 55, 2180–2191.
Zhang, J. (2006). Resveratrol inhibits insulin responses in a SirT1-independent pathway. Biochem. J. 397, 519–527.
Zhang, P., Hu, X., Xu, X., Fassett, J., Zhu, G., Viollet, B., Xu, W., Wiczer, B., Bernlohr, D. A., Bache, R. J., and Chen, Y. (2008). AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension 52, 918–924.
Zhou, X., Perez, F., Han, K., and Jurivich, D. A. (2006). Clonal senescence alters endothelial ICAM-1 function. Mech. Ageing Dev. 127, 779–785.
Zmonarski, S. C., Boratynska, M., Rabczynski, J., Kazimierczak, K., and Klinger, M. (2005). Regression of Kaposi’s sarcoma in renal graft recipients after conversion to sirolimus treatment. Transplant. Proc. 37, 964–966.
Zoncu, R., Efeyan, A., and Sabatini, D. M. (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35.
Keywords: aging, endothelial senescence, eNOS, mTOR, S6K1, oxidative stress, rapamycin, resveratrol
Citation: Ming X-F, Montani J-P and Yang Z (2012) Perspectives of targeting mTORC1–S6K1 in cardiovascular aging. Front. Physio. 3:5. doi: 10.3389/fphys.2012.00005
Received: 17 November 2011;
Paper pending published: 12 December 2011;
Accepted: 09 January 2012;
Published online: 25 January 2012.
Edited by:
Ana Paula Dantas, Institut d’Investigacions Biomediques August Pi i Sunyer, SpainReviewed by:
Xu Peng, Texas A&M Health Science Center, USAMercè Roqué, University of Barcelona, Spain
Rita Tostes, University of Sao Paulo, Brazil
Copyright: © 2012 Ming, Montani and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Xiu-Fen Ming and Zhihong Yang, Laboratory of Vascular Biology, Department of Medicine, University of Fribourg, Rue du Musée 5, CH-1700 Fribourg, Switzerland. e-mail: xiu-fen.ming@unifr.ch; zhihong.yang@unifr.ch