
- German Center for Neurodegenerative Diseases (DZNE), Munich, Germany
The β-site APP cleaving enzyme 1 (BACE1) is a transmembrane aspartyl protease involved in Alzheimer’s disease (AD) pathogenesis and in myelination. BACE1 initiates the generation of the pathogenic amyloid β-peptide, which makes BACE1 a major drug target for AD. BACE1 also cleaves and activates neuregulin 1, thereby contributing to postnatal myelination, in particular in the peripheral nervous system. Additional proteins are also cleaved by BACE1, but less is known about the physiological consequences of their cleavage. Recently, new phenotypes were described in BACE1-deficient mice. Although it remains unclear through which BACE1 substrates they are mediated, the phenotypes suggest a versatile role of this protease for diverse physiological processes. This review summarizes the enzymatic and cellular properties of BACE1 as well as its regulation by lipids, by transcriptional, and by translational mechanisms. The main focus will be on the recent progress in understanding BACE1 function and its implication for potential mechanism-based side effects upon therapeutic inhibition.
Introduction: APP Processing and Alzheimer’s Disease
Alzheimer’s disease (AD) represents the leading cause of dementia worldwide. It is the most common neurodegenerative disease and is characterized by two defining pathological hallmarks, extracellular amyloid plaques and intracellular neurofibrillary tangles (Ballard et al., 2011). The amyloid hypothesis states that the main constituent of amyloid plaques, the 39–43 amino acid long amyloid-β-peptide (Aβ), is responsible for the initiation of a neurotoxic cascade that ultimately leads to neuronal death and dementia (Hardy and Selkoe, 2002; Haass and Selkoe, 2007). Aβ itself is liberated from the large type I transmembrane amyloid precursor protein (APP) in a stepwise process referred to as regulated intramembrane proteolysis (RIP). RIP in general controls intra- and intercellular communication through the release of extracellular and cytosolic proteolytic fragments that may serve as versatile signaling molecules or are further degraded (Brown et al., 2000; Lichtenthaler et al., 2011). Ectodomain shedding, the first step of RIP, results in the release of the soluble APP ectodomain (APPs) into the extracellular space and is carried out by either one of two alternative protease activities. The molecular identity of these protease activities was unknown for many years, which led to the still established terminology of α- and β-secretase.
In the amyloidogenic, β-secretase dependent pathway, shedding occurs within the ectodomain of APP in close proximity to the membrane and is mediated by the membrane-bound aspartyl protease β-site APP cleaving enzyme 1 (BACE1, β-secretase, memapsin-2, ASP-2; Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000; Figure 1). The remaining membrane-bound APP C-terminal fragment (CTF) bears the N-terminus of Aβ and is processed in a second step within its transmembrane domain by the GxGD-type aspartyl protease γ-secretase. The γ-secretase complex consists of four integral membrane proteins, including the catalytic subunit presenilin, and is able to cleave its substrates within the hydrophobic environment of the lipid bilayer (Steiner et al., 2008). This cleavage generates the Aβ C-terminus and liberates the Aβ peptide into the extracellular space, whereas the APP intracellular domain (ICD) is released into the cytosol. Due to the heterogeneous nature of γ-secretase cleavage, Aβ species of different length are produced, including the pathogenic 42-amino acid long Aβ42, whose toxic properties have been summarized in a recent review (O’Brien and Wong, 2011). In the non-amyloidogenic pathway, a third proteolytic activity, referred to as α-secretase (Lichtenthaler et al., 2011), precludes Aβ formation by cleaving APP within the Aβ domain, and releasing the soluble APPsα fragment into the extracellular space (Esch et al., 1990; Figure 1). ADAM10, a member of the ADAM (a disintegrin and metalloprotease) family of proteases, has α-secretase activity in vitro and in vivo and has been identified as the constitutively cleaving α-secretase in neurons (Lammich et al., 1999; Postina et al., 2004; Jorissen et al., 2010; Kuhn et al., 2010). Other metalloproteases may also act as α-secretases, but appear to cleave APP only after pharmacological activation or overexpression (Buxbaum et al., 1998; Koike et al., 1999; Naus et al., 2006). BACE1 is the rate-limiting and principal enzyme responsible for Aβ generation in neurons. Deletion of BACE1 via homologous recombination in mouse models of AD completely abolishes Aβ generation in the brain (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001). Its therapeutic inhibition should therefore prevent the formation of all Aβ species and be beneficial for the treatment of AD. Besides BACE1, additional proteases with minor, regulated β-secretase activity, such as the cysteine-protease cathepsin B, may contribute to Aβ levels in the brain, suggesting that their inhibition may be of additional therapeutic benefit (Haque et al., 2008; Hook et al., 2008, 2011). However, much less is known about the pathophysiological relevance of cathepsin B compared to BACE1.
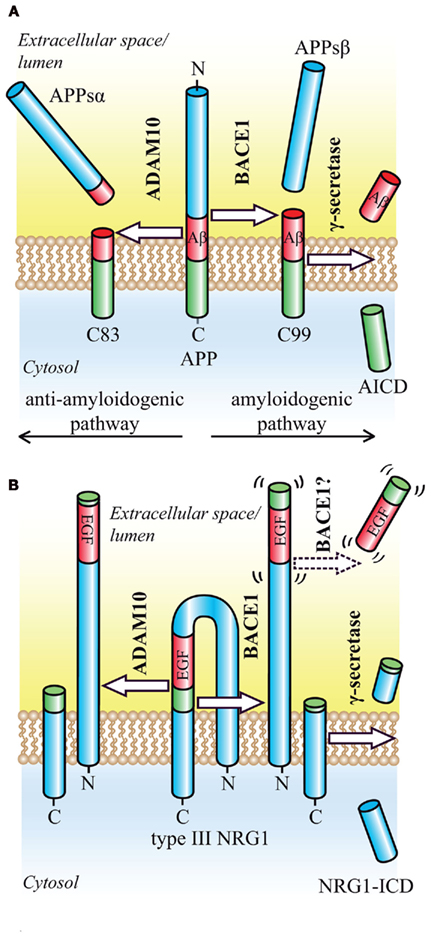
Figure 1. Proteolytic processing of the BACE1 substrates APP and NRG1. (A) Regulated intramembrane proteolysis of APP occurs by two alternative pathways. In the amyloidogenic pathway, ectodomain shedding by BACE1 releases the soluble APP ectodomain and generates the N-terminus of the Aβ peptide. The membrane-bound CTF (C99) is then further processed by γ-secretase, which releases the Aβ peptide into the extracellular space and the APP intracellular domain (AICD) into the cytosol where it is rapidly degraded. In the alternative, anti-amyloidogenic pathway, ectodomain shedding is mediated by ADAM10. Cleavage occurs in the middle of the Aβ domain and yields the soluble APP ectodomain and a CTF (C83), which is further processed by γ-secretase to generate the secreted p3 peptide (not shown). (B) Ectodomain shedding of type III NRG1 by BACE1 or ADAM10 occurs C-terminally of the EGF-like domain and generates a large NTF as well as a shorter CTF. Cleavage by BACE1 leads to a slightly larger NTF, that exerts signaling function. Cleavage within the EGF-like domain by ADAM17 appears to render the resulting NTF inactive (not shown). The remaining CTF is further processed by γ-secretase in order to yield the NRG1-ICD.
Since its discovery in 1999 by five independent research groups (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000), BACE1 is a major drug target for AD. Extensive efforts toward an understanding of BACE1 physiology and pathophysiology as well as the development of potent BACE1 inhibitors have been made. This review focuses on the latest findings and current developments in BACE1 research. We will first describe the role of BACE1 in AD as well as the basic cell biology of this protease. Next we summarize the transcriptional and translational regulation of BACE1 in health and disease. After discussing the role of lipid microdomains on BACE1 activity, we will turn to BACE1 substrates with a focus on the functional consequences of the generated proteolytic fragments. In order to safely reduce Aβ levels in AD, a detailed understanding of BACE1 substrate processing and its implications upon therapeutic inhibition is necessary. Different approaches toward therapeutic inhibition as well as future perspectives of BACE1 research will be addressed in the last part of our review.
BACE1 Cell Biology
The 501 amino acid long type I transmembrane protein BACE1, together with its homolog BACE2, belongs to the family of pepsin and retroviral aspartic proteases. The single transmembrane domain, a unique feature amongst aspartic proteases, exposes the active site toward the luminal side and links the ectodomain to the 21 amino acid long cytosolic tail (Figure 2). The active site features the critical aspartic acid residues within two characteristic D-T/S-G-T/S motifs. Like other aspartyl proteases, BACE1 requires both aspartates for its activity (Hussain et al., 1999; Vassar et al., 1999; Bennett et al., 2000b). However, because BACE1 forms dimers, it appears possible that the two monomers provide one aspartate each. Evidence for this scenario comes from a study showing that mutation of the aspartate within the C-terminal DSGT motif does not affect BACE1 activity (Schmechel et al., 2004). In the endoplasmic reticulum (ER), BACE1 is initially synthesized as a zymogen and subjected to N-glycosylation, palmitoylation, transient acetylation, and disulfide bridge formation (Haniu et al., 2000; Benjannet et al., 2001; Costantini et al., 2007; Ko and Puglielli, 2009; Vetrivel et al., 2009; Figure 2). Full maturation occurs in the Golgi compartment, where complex glycosylation and removal of the prodomain by furin prohormone protein convertases lead to the 70 kDa form (Bennett et al., 2000b; Capell et al., 2000; Benjannet et al., 2001). The zymogen form already possesses significant proteolytic activity, which is enhanced about twofold after removal of the prodomain (Creemers et al., 2001; Benjannet et al., 2004). Rather than suppressing enzymatic activity, the prodomain facilitates correct folding of the protease domain (Shi et al., 2001).
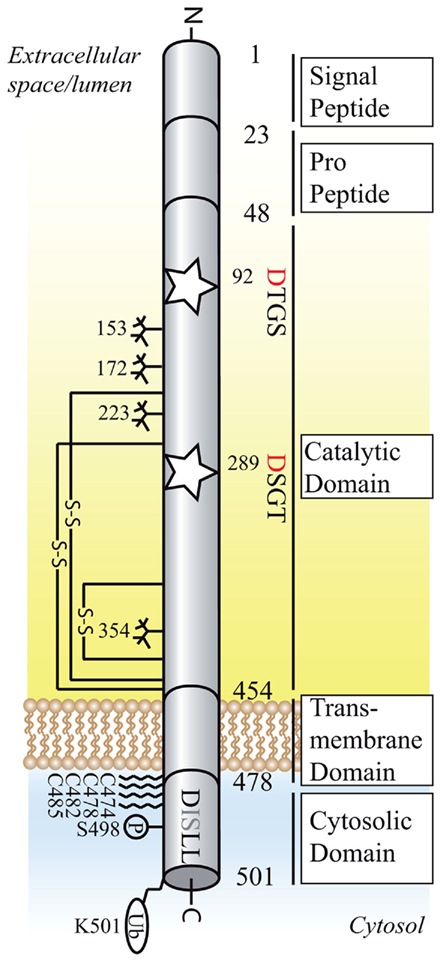
Figure 2. Structural organization of BACE1 (active isoform of 501 amino acids). Schematic diagram of the domain structure, post-translational modifications and interaction motifs of BACE1.The active sites motifs DTGS and DSGT at position 92–95 and 289–292 are marked as stars, the critical aspartic residues are highlighted in red. The DISLL motif at the cytosolic domain is recognized by GGA proteins and serves as an endosomal targeting signal. Glycosylation sites are depicted as sugar “trees,” palmitoylation sites as fatty acid chains. S–S, disulfide bond (connecting amino acids 216–420, 278–443, 330–380); P, phosphorylation site; Ub, ubiquitination site.
After transport to the plasma membrane, the dileucine motif DISLL at position 496–500 in the cytoplasmic domain promotes BACE1 trafficking to early endosomal compartments (Huse et al., 2000). A recent study suggests that this sorting event is controlled by the small GTPase ARF6 in a clathrin-independent fashion, in contrast to the clathrin-dependent endosomal sorting of APP (Sannerud et al., 2011). Phosphorylation of the dileucine motif at serine 498 and interaction with Golgi-localized γ-ear-containing ARF-binding proteins (GGAs) is responsible for the retrieval from early endosomes to late endosomes and the trans-Golgi-network (TGN; He et al., 2002, 2003, 2005; von Arnim et al., 2004, 2006; Wahle et al., 2005). In addition, retrograde BACE1 trafficking depends on the cytosolic domain of the Vps10p homolog Sortilin, most likely via retromer complex or GGA dependent sorting mechanisms (Finan et al., 2011). Recycling of BACE1 from the TGN and endosomal compartments to the plasma membrane contributes to the relatively slow turnover rate of BACE1 (Walter et al., 2001; He et al., 2005; Wahle et al., 2005). Finally, monoubiquitination at lysine 501 promotes lysosomal degradation, a process that depends on the recognition of the ubiquitinated lysine residue by GGA3 (Kang et al., 2010). Depletion of GGA3 enhances BACE1 activity and Aβ generation in vitro and reduced GGA3 protein levels correlate inversely with BACE1 protein levels in AD patients (Tesco et al., 2007; Santosa et al., 2011). GGA3 depletion and subsequent stabilization of BACE1 have been proposed to follow the reduced expression of the neuroprotective protein seladin-1 under stress conditions (Sarajarvi et al., 2009). Shedding by ADAMs, which releases the BACE1 ectodomain into extracellular space, and proteasomal degradation, could be responsible for a minor part of BACE1 turnover (Benjannet et al., 2001; Hussain et al., 2003; Qing et al., 2004). However, the inhibition of BACE1 shedding after treatment with metalloprotease inhibitors does not influence Aβ generation (Hussain et al., 2003).
Enzymatic Properties and Cellular Localization
β-Site APP cleaving enzyme 1 has an acidic pH-optimum (Vassar et al., 1999; Gruninger-Leitch et al., 2002; Shimizu et al., 2008) and cleaves its substrates mostly in the acidic early endosomal and trans-Golgi compartments, where also Aβ generation takes place (Haass et al., 1993; Koo and Squazzo, 1994; Vassar et al., 1999; Capell et al., 2000; Gruninger-Leitch et al., 2002). After internalization from the plasma membrane, wild-type APP is processed in early endosomes. As indicated above, endocytosis and endosomal transport of APP and BACE1 occur via distinct pathways. Interfering with either one, e.g., by knockdown of ARF6 or modulation of APP endocytosis via clathrin-dependent mechanisms, alters the shedding of APP by BACE1 (Chyung and Selkoe, 2003; Neumann et al., 2006; Schobel et al., 2006, 2008; Sannerud et al., 2011). In contrast, the processing of Swedish APP (APPSWE), a rare genetic double mutation leading to familial early-onset AD (Mullan et al., 1992) occurs along the secretory pathway (Haass et al., 1995). The responsible amino acid changes at position-1 and -2 of Aβ enhance the proteolytic activity of BACE1 and therefore Aβ production (Citron et al., 1992). Although the secreted form of BACE1 processes peptide substrates in vitro, efficient processing of APP by BACE1 requires the membrane-bound form, ensuring the proper spatial interaction between protease and substrate at the membrane surface (Yan et al., 2001; Gruninger-Leitch et al., 2002). In mouse and human brain, native BACE1 occurs as a dimer. Dimerization via the ectodomain is dependent on membrane attachment and increases BACE1 affinity and turnover rate toward APPSWE-like peptides when compared to the monomeric, soluble form (Schmechel et al., 2004; Westmeyer et al., 2004). The different enzymatic properties of monomeric and dimeric BACE1 need to be considered in future drug screening and development processes.
BACE1 Structure
The protease domain of BACE1 features the typical bilobal structure of aspartic proteases, showing a high degree of conservation with pepsin and BACE2. The active site cleft is located between the N- and C-terminal lobes and partially shielded by an antiparallel hairpin loop known as “flap,” which controls substrate access and proteolytic specificity (Hong et al., 2000; Ostermann et al., 2006). The main structural feature that sets BACE1 and BACE2 aside from other aspartic proteases is the enlarged molecular boundary of the catalytic domain. This is due to four helical or loop-like insertions and a carboxyterminal extension in the C-terminal lobe, possibly mediating associations with other cell surface molecules. Overall the BACE1 active cleft is broader, less hydrophobic, and more open when compared to other aspartic proteases, allowing the accommodation of up to 11 substrate residues (Hong et al., 2000; Turner et al., 2005). The unusually large active site poses a major challenge in the development of potent small molecule inhibitors in drug development.
Expression Pattern of BACE1
Consistent with its role as β-secretase, the highest expression levels and enzymatic activity of BACE1 are found in brain (Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999). This is mainly due to neuronal expression, as expression levels in glial cells are very low (Laird et al., 2005; Harada et al., 2006; Zhao et al., 2007). In mouse brain, BACE1 expression peaks during early postnatal stages, with highest expression levels found in the cortex, hippocampus, and cerebellum (Irizarry et al., 2001; Willem et al., 2006). Disruption of entorhinal cortex afferents to the hippocampus and immunofluorescence studies suggest that BACE1 localizes to axonal membranes within neurons and is actively transported to synaptic nerve terminals, resulting in the release of Aβ at the synaptic cleft (Lazarov et al., 2002; Sheng et al., 2003). In contrast, one study reports mainly somatodendritic localization of APP and BACE1 within cultured polarized hippocampal neurons and finds that the bulk of APPsβ and Aβ fragments is recovered from somatodendritic and not axonal compartments (Sannerud et al., 2011). Besides brain tissue, high BACE1 mRNA levels are also found in the pancreas. Due to alternative splicing and post-transcriptional inactivation enzymatic activity is comparatively low (Sinha et al., 1999; Bodendorf et al., 2001; Ehehalt et al., 2002; Molinari et al., 2002; Mowrer and Wolfe, 2008). The pancreatic isoforms of BACE1 may not cleave APP, but other substrates instead, such as enteropeptidase, which was recently identified as a possible BACE1 substrate in pancreas (Bodendorf et al., 2001; Hoffmeister et al., 2009). Low expression levels are found in the remainder of the peripheral organs, which is in agreement with the observation that Aβ is produced by most cell lines, regardless of their tissue of origin.
Transcriptional and Translational Regulation of BACE1 Activity
A complex interplay of transcriptional, translational, and post-translational mechanisms regulates BACE1 activity. At the transcriptional level, numerous mechanisms regulating BACE1 activity have been described. Hypoxic and oxidative cell stress, mediated by hypoxia-inducible factor 1α (HIF1α), the lipid peroxidation product hydroxy-non-enal (HNE), the c-JUN N-terminal kinase (JNK)/c-JUN pathway, and others, has been shown to increase BACE1 mRNA levels (Yasojima et al., 2001; Holsinger et al., 2002; Tamagno et al., 2005, 2008, 2009; Zhang et al., 2007). Not all previous work on the transcriptional and translational regulation of BACE1 can be cited at this point and we refer the reader to two excellent reviews for further information (Rossner et al., 2006; Stockley and O’Neill, 2008). However, most of these studies are based on in vitro observations. The physiological relevance for AD pathogenesis remains unclear, as BACE1 transcript levels are not significantly altered in AD brains (Preece et al., 2003; Matsui et al., 2007; Hebert et al., 2008). On the contrary, BACE1 protein levels are increased two- to fivefold in about 30% of patients with sporadic AD, supporting the assumption of disease-relevant post-transcriptional regulatory mechanisms (Fukumoto et al., 2002; Holsinger et al., 2002; Preece et al., 2003; Yang et al., 2003; Li et al., 2004; Hebert et al., 2008).
Indeed, three distinct mechanisms have been described controlling the translation of BACE1. First, the large 5′ untranslated region (5′ UTR) of the BACE1 transcript represses translation in a constitutive fashion, as demonstrated in several cell culture studies (De Pietri Tonelli et al., 2004; Lammich et al., 2004; Rogers et al., 2004; Zhou and Song, 2006; Mihailovich et al., 2007). Mutagenesis studies identified the GC-rich strand of the 5′ UTR as an effective translational barrier, preventing ribosomal binding (Lammich et al., 2004). Further mechanisms such as ribosomal shunting by binding of ribosomes to upstream ORFs and subsequent premature ribosomal dissociation have been proposed (Rogers et al., 2004). It is currently unknown whether regulatory proteins bind to and therefore modulate the extent of 5′ UTR mediated translational repression. However, a recent study demonstrated that the translational block can be partly relieved under conditions of energy deprivation in the brain, which suggests a direct link between reduced brain metabolism and sporadic AD (Velliquette et al., 2005). Post-transcriptionally elevated BACE1 and Aβ levels were found upon acute pharmacologic reduction of energy metabolism in mice overexpressing APPSWE. In vitro and in vivo, phosphorylation of the translation initiation factor eIF2a results in the increased translation of BACE1 mRNA, in a mechanism that bypasses the repressive features of the 5′ UTR and that is typical of genes involved in the cellular stress response (Schroder and Kaufman, 2006; O’Connor et al., 2008). A pathogenic increase in BACE1 activity could therefore stem from a physiologic role in the neuronal stress response.
A second mechanism of BACE1 translational regulation is based on studies of miRNA expression profiles in patients with sporadic AD, where a link between the downregulation of the miRNA cluster miR-29a/b-1 and miRNA-107 and a corresponding increase in BACE1 protein levels was suggested. A direct relationship is supported by the presence of binding sites for the identified miRNAs within the BACE1 3′ UTR and cell culture reporter assays (Hebert et al., 2008; Wang et al., 2008).
A third mechanism regulating BACE1 translation in vitro and in vivo is a non-coding BACE1 antisense transcript (BACE1-AS). BACE1-AS is elevated in AD and surprisingly stabilizes the BACE1 mRNA, resulting in increased BACE1 protein levels and enhanced Aβ42 production (Faghihi et al., 2008). Furthermore, Aβ42 stimulates BACE1-AS expression, resulting in a feed-forward loop that potentially drives disease progression.
The Role of the Lipid Environment on BACE1 Activity
In vivo, active BACE1 localizes to endosomal and trans-Golgi compartments. However, the precise localization within the membrane microenvironment and the related consequences toward BACE1 activity remain controversial. On the one hand, BACE1 and APP in vivo and in vitro partially localize to lipid raft domains enriched in cholesterol and sphingolipids, and enhanced targeting of BACE1 to these microdomains via addition of a GPI-anchor enhances Aβ production (Riddell et al., 2001; Tun et al., 2002; Cordy et al., 2003; Ehehalt et al., 2003; Kawarabayashi et al., 2004; Crameri et al., 2006; Hattori et al., 2006). Feeding mice a high cholesterol diet, which promotes lipid raft formation, increases Aβ levels, whereas the disruption of lipid rafts upon depletion of cellular cholesterol levels has the opposite effect (Simons et al., 1998; Fassbender et al., 2001; Refolo et al., 2001; Cordy et al., 2003). On the other hand, a study focusing on endogenous APP and BACE1 in hippocampal neurons suggests that the main pool of APP resides outside of lipid rafts, unable to be accessed by BACE1 (Abad-Rodriguez et al., 2004). Moderate reductions in cholesterol levels increased Aβ production, probably by disturbing raft architecture and therefore enhancing BACE1 and APP contact. However, strong reductions in cholesterol decrease Aβ production presumably as result of direct BACE1/γ-secretase inhibition (Kaether and Haass, 2004).
Lipid raft association of BACE1 is dependent on the S-palmitoylation of four cysteine (Cys474/478/482/485) residues at the boundary between the cytoplasmic and transmembrane domain (Benjannet et al., 2001; Vetrivel et al., 2009). After mutation of these residues, raft association is lost, without affecting protein stability, subcellular localization, and proteolytic activity toward APP in neuronal and non-neuronal cultured cells, suggesting that BACE1 efficiently cleaves APP in raft and non-raft environments. These results challenge the previous studies that are based on cholesterol depletion (Simons et al., 1998; Fassbender et al., 2001; Refolo et al., 2001; Cordy et al., 2003), which has pleiotropic effects on membrane morphology and vesicular trafficking (Rodal et al., 1999; Wang et al., 2000; Hao et al., 2004). More in vivo studies focusing on endogenous APP and selectively disturbing the lipid raft association of BACE1 are needed to resolve these contradictory issues.
A direct effect on BACE1 activity by the active lipid metabolite sphingosine-1-phosphate has been shown. Sphingosine is phosphorylated by sphingosine kinase 1 and 2 to generate sphingosine-1-phosphate that ultimately binds to the C-terminal and transmembrane region of BACE1, activating its proteolytic activity (Takasugi et al., 2011). Knockdown or inhibition of sphingosine kinase 2 as well as overexpression of S1P degrading enzyme reduced Aβ levels in cultured cells. In line with in vitro results, stereotaxic injection of sphingosine kinase inhibitors reduced brain Aβ loads in wild-type and APP transgenic mouse lines. Furthermore, levels of sphingosine kinase 2 were found to be elevated in AD brain.
BACE1 Substrates
Two years after the discovery of BACE1, the first knockout mouse lines were generated. These mice do not produce C99 or Aβ, further validating BACE1 as the sole β-secretase in vivo. Initially, overt developmental or adult phenotypes in BACE1−/− mice were not detected, encouraging treatment of AD by therapeutic inhibition of BACE1 without significant adverse effects (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001; Dominguez et al., 2005). However, later studies uncovered multiple developmental and behavioral changes, suggesting that BACE1 has diverse physiological roles. As the function of a protease is defined by its substrates, the identification and characterization of novel BACE1 substrates remains one of the key challenges in current AD research, providing insights into BACE1 physiology, substrate specificity, potential side effects upon therapeutic inhibition, and biomarker development. So far, all known BACE1 substrates have been identified by candidate approaches, some of them under overexpressing conditions, where BACE1 may cleave substrates that are not processed in vivo, for example proteins that reside in the ER. Known BACE1 substrates (summarized in Table 1) will be discussed below, in order of evidence regarding their in vivo cleavage and the related functional consequences of the resulting proteolytic fragments. APP, type III neuregulin 1, the β-subunits 2 and 4 of voltage-gated sodium channels (VGSCs), and the β-galactoside α2, 6-sialyltransferase belong to the group of BACE1 substrates where the functional relevance of substrate cleavage has been demonstrated in vivo.
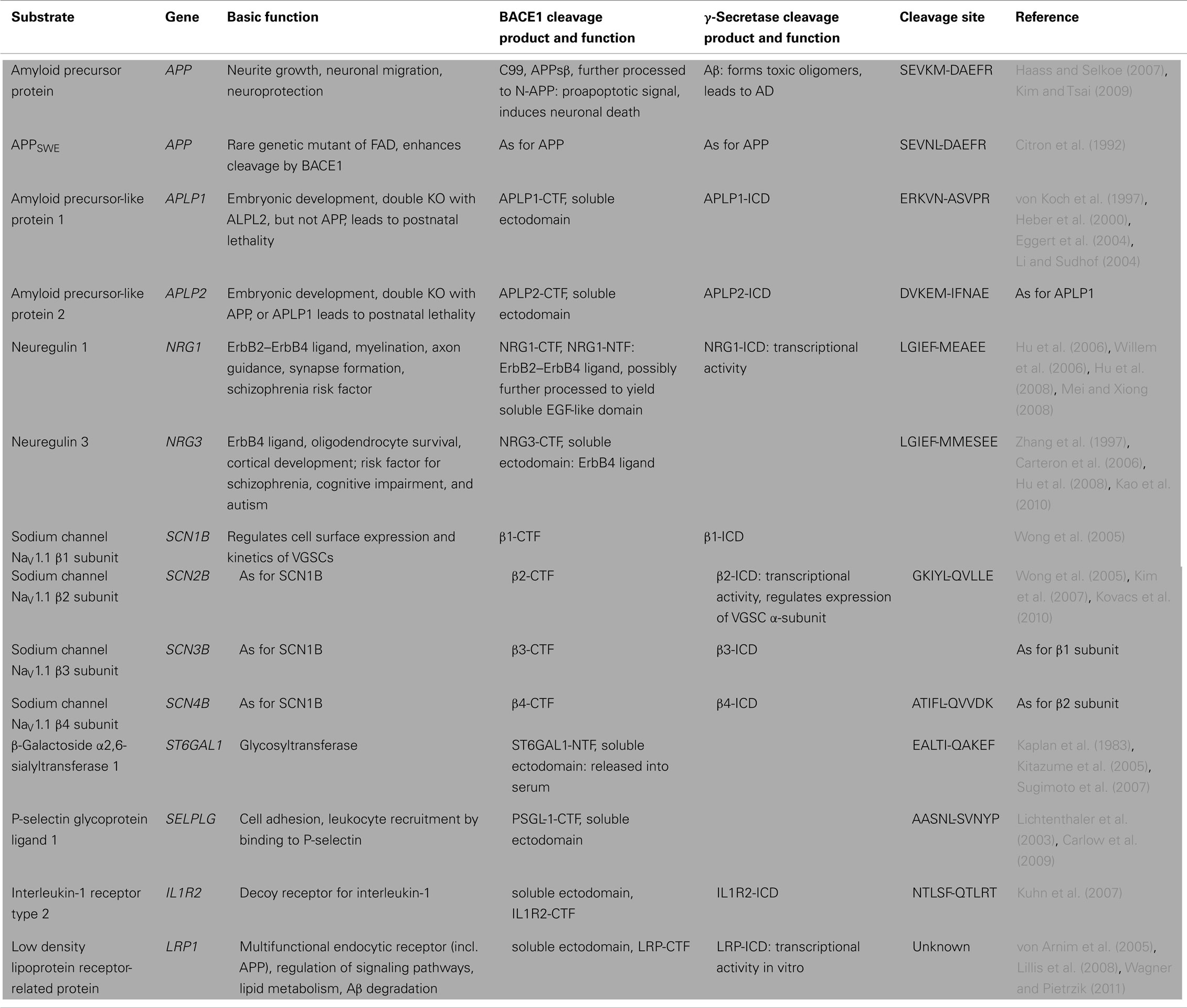
Table 1. Cleavage sites, proteolytic fragments, and molecular function of known BACE1 substrates.
Despite being a prominent substrate, the physiological function of APP and its shedding by BACE1 remains unclear. Genetic deletion of full length APP in mice leads to a reduction in brain and body weight, memory deficits and reduced grip strength, and can be rescued by APPsα (Ring et al., 2007). The additional knockout of the APP homolog amyloid precursor protein 2 (APLP2) results in perinatal lethality and neuromuscular synapse defects (Muller et al., 1994; von Koch et al., 1997). APPsβ actively regulates transthyretin and Klotho expression levels, but fails to rescue the lethality and neuromuscular synapse defects present in APP/APLP2 knockout mice (Li et al., 2010). Upon trophic withdrawal, APPsβ is processed by a yet unidentified mechanism to yield a 35 kDa fragment (N-APP) that binds to death receptor 6 and triggers a widespread proapoptotic signal that may speed up neurodegeneration (Nikolaev et al., 2009). Thus, APPsα and APPsβ apparently have different functions. Future studies on the biology of APP and its homologs could provide links to the complex phenotype of BACE1−/− mice. Besides APP, APLP1 and 2 are also processed by BACE1 in vivo and in vitro (Yanagida et al., 2009; Sala Frigerio et al., 2010; Hogl et al., 2011). The physiological function of APLP1 and APLP2 shedding by BACE1 is currently unknown. Wild-type APP and its homologs are considered relatively poor substrates, as BACE1 binds preferably to APPSWE-like peptides, which feature bulky hydrophobic residues such as leucine and phenylalanine at position P1 (reviewed in Stockley and O’Neill, 2008). However, this assumption is based on in vitro assays using peptide substrates and neglects the proper spatial orientation of the full length substrate and protease within the lipid bilayer.
BACE1 Substrates: Type III Neuregulin 1
The most consistent link between a BACE1 substrate and the related phenotype of BACE1−/− mice has been established for type III neuregulin 1 (NRG1). BACE1−/− mice show prominent hypomyelination in the peripheral nervous system, phenocopying mice haploinsufficient in the EGF-like growth factor NRG1 (Hu et al., 2006; Willem et al., 2006). NRG1 signaling via its cognate ErbB2–ErbB4 receptors is essential for the initiation of myelination and the regulation of myelin sheath thickness during development. Full activation may require the proteolytic liberation of the luminal EGF-like domain (Falls, 2003; Sagane et al., 2005; Birchmeier and Nave, 2008; Brinkmann et al., 2008), which is located in the loop connecting the two transmembrane domains of type III NRG1 and complementing the hairpin shaped structure (Figure 1). An initial shedding event occurs at the juxtamembrane region C-terminal of the EGF-like domain and generates a shorter C-terminal stub and a larger N-terminal fragment (NTF), which now exposes the EGF-like domain for juxtacrine signaling (Mei and Xiong, 2008). A second shedding event might then release the EGF-like domain into the extracellular space, allowing for paracrine signaling, as shown for type I NRG1(reviewed in Willem et al., 2009; Figure 1). The initial shedding of type III NRG1 is mediated by BACE1 and ADAM10, with the BACE1 cleavage sites eight amino acids downstream of the ADAM 10 cleavage site, therefore in inverse order when compared to the proteolytic processing of APP. Interestingly, upon knockdown and inhibitor treatment of ADAM10 and BACE1, only the loss of the NRG1-NTF generated by BACE1 has a significant impact on myelination in a Schwann cell and neuron co-culture system (Luo et al., 2011). Since full length NRG1 accumulates in BACE1−/− mice, the physiologic role of BACE1 in myelination is presumably mediated through NRG1 activation via ectodomain shedding (Hu et al., 2006; Willem et al., 2006).
BACE1-driven myelination is counteracted by ADAM17, which partially cleaves in the middle of the EGF-like domain. Reduction of ADAM17 activity in vivo is sufficient to rescue the hypomyelination phenotype of type III NRG1 haploinsufficient mice (La Marca et al., 2011). Whether NRG1-dependent myelination in the central nervous system is also altered in BACE1−/− mice remains uncertain, as this was reported by one group but not by others (Hu et al., 2006; Willem et al., 2006). One study suggests that BACE1 in conjunction with type III NRG1 also regulates remyelination after sciatic nerve crush injury and therefore may be of importance for myelin maintenance in adults (Hu et al., 2008). Unexpectedly, one group reports accelerated axonal regeneration in BACE1−/− mice after sciatic nerve crush (Farah et al., 2011). No differences were found during the early stages of Wallerian degeneration, but faster neurite outgrowth and an increased number of regenerating sprouts were present in BACE1−/− when compared to wild-type littermates. Clearance of myelin debris from damaged nerve sites sets the stage for efficient axonal regeneration (Vargas et al., 2010) and is increased in BACE1-deficient mice, due to increased phagocytosis of BACE1-deficient macrophages (Farah et al., 2011). Summed up, these studies suggest a myelination-independent role of BACE1 in axonal regeneration by regulating two different pathways, the internal growth state of damaged neurons as well as myelin clearance by macrophages.
In addition to its role in myelination, type III NRG1 is a risk factor for schizophrenia (Stefansson et al., 2002; Williams et al., 2003). This establishes an interesting link to behavioral phenotypes of BACE1−/− mice that are considered rodent analogs of schizophrenia, such as impaired prepulse inhibition, novelty-induced hyperactivity, and cognitive deficits (Savonenko et al., 2008). Supporting the hypothesis of NRG1-dependent behavioral changes in BACE1−/− mice, these animals respond to the antipsychotic drug clozapine in a similar fashion as type III NRG1+/− mice and feature a reduction in cortical ErB4–PSD95 complex formation, an important downstream event for successful type III NRG1 signaling (Hahn et al., 2006; Savonenko et al., 2008). Furthermore, NRG1 regulates expression levels of another schizophrenia susceptibility factor, disrupted-in-schizophrenia-1 (DISC1), in an ErbB2–ErbB3 dependent manner. Decreased DISC1 expression levels are found in NRG1- as well as BACE1-knockout mice and could explain shared behavioral phenotypes with mouse models harboring DISC1 mutations (Kvajo et al., 2008; Seshadri et al., 2010). These observations render BACE1 a putative drug target for schizophrenia, while raising concerns for adverse side effects upon therapeutic inhibition in AD. Notably, effective inhibition of BACE1 in adult mice lowered Aβ levels but had no effect on NRG1 processing (Sankaranarayanan et al., 2008). Neuregulin 3 is also processed by BACE1 in vivo, but no functional consequences have been reported yet (Hu et al., 2008).
BACE1 Substrates: β-Subunits 2 and 4 of Voltage-Gated Sodium Channels
The β-subunits 2 and 4 (Navβ2, Navβ4) of VGSCs are also known to be processed by BACE1 under physiologic conditions. Besides NRG1 they are amongst the best characterized BACE1 substrates (reviewed in Kovacs et al., 2010). Navβs covalently bind to the channel-forming α-subunits (Nav1) of VGSCs and regulate their surface expression levels and inactivation kinetics (Catterall, 2000; Isom, 2001). Cleavage of these β-subunits by BACE1 generates a CTF that is further processed by γ-secretase, yielding an ICD that is released into the cytosol (Wong et al., 2005). The ICD of Navβ2 translocates to the nucleus and increases mRNA and protein levels of α-subunits, which in return alters sodium current densities at the cell surface (Kim et al., 2007). Under overexpression conditions in vitro, Navβ1–4 are subject to RIP by BACE1 and resulting increases in Nav1.1 protein levels lead to intracellular accumulation of α-subunits in a HSP70 positive compartment. This prevents in an unknown fashion the transport of functional channels to the plasma membrane and leads to a decrease of total VGSC surface levels. In vivo, CTFs of Navβ2 and Navβ4 are reduced in the brains of BACE1-knockout mice, whereas increased Navβ2 processing and increased total levels Nav1.1 are found in AD patients with elevated BACE1 activity (Wong et al., 2005; Kim et al., 2007). BACE1-deficient mice show reduced total and surface expression levels of Nav1.1 in hippocampal neurons, with a compensatory increase of Nav1.2 surface levels reported by two groups, but not by another (Hitt et al., 2010; Hu et al., 2010; Kim et al., 2011). Therefore, the regulation of Nav1.1 surface expression by BACE1 in vivo seems to be regulated by the decrease of total Nav1.1 levels. Distinct epilepsy syndromes are caused by mutations in the Nav1.1 channel gene SCN1A and mice lacking Nav1.1 suffer from a severe lethal seizure phenotype (Claes et al., 2001; Mulley et al., 2005; Yu et al., 2006). Whether changes in sodium channel current density in BACE1−/− mice relate to the spontaneous epileptiform abnormalities observed in 20–30% of these animals remains unclear, as reports from different groups remain contradictory, due to different extraction methods and measurement of total, or surface levels of different Nav1 subtypes (Hitt et al., 2010; Hu et al., 2010; Kim et al., 2011). Further complexity is added by the fact that Navβs have been proposed to interact with various other proteins to modulate cell–cell adhesion and migration, therefore altering the neuronal environment beyond sodium current density (Isom, 2001, 2002; Brackenbury et al., 2008).
BACE1 Substrates: α2, 6-Sialyltransferase
The TGN-resident β-galactoside α2, 6-sialyltransferase (ST6Gal1) was the first identified BACE1 substrate that possesses a type II transmembrane topology (Kitazume et al., 2001). It is mainly expressed in the liver and released into various body fluids, such as serum and milk, with enhanced secretion during acute phase reactions (Kaplan et al., 1983; Weinstein et al., 1987; Sarnesto et al., 1992; Kitagawa and Paulson, 1994). Hepatic BACE1 is the major protease responsible for the release of ST6Gal1 into serum, as ST6Gal1 serum levels are reduced to ∼30% in BACE1−/− mice, with a ∼25% decrease in total sialylation of plasma glycoproteins (Kitazume et al., 2005; Sugimoto et al., 2007). A rat model of Wilson’s Disease that features spontaneous hepatitis shows an upregulation of hepatic BACE1 mRNA and increased ST6Gal1 serum levels, without affecting ST6Gal1 mRNA levels, suggesting that hepatic damage may lead to increased BACE1 activity and therefore secretion of ST6Gal1 (Kitazume et al., 2005). Unfortunately, the physiological significance of soluble glycosyltransferases in the serum remains enigmatic.
Processing of Other Known BACE1 Substrates
The second group of BACE1 substrates is known to be processed in vivo, whereas the physiologic function of the shedding event remains unclear. The P-selectin glycoprotein ligand-1 (PSGL-1) is expressed on most leukocytes and mediates adhesion to endothelial cells by binding to P-selectin during inflammation. This ensures efficient leukocyte recruitment and subsequent transmigration into damaged or infected tissue in the CNS and peripheral organs (reviewed in Chen and Geng, 2006; Carlow et al., 2009). The type I transmembrane protein PSGL-1 has been shown to be shed by BACE1 and ADAM10, in a similar fashion to type III NRG1, with the BACE1 cleavage site being closer to the membrane (Lichtenthaler et al., 2003). According to this study, PSGL-1 is cleaved in HEK293 cells and immortalized monocytes under endogenous conditions, whereas CTFs of PSGL-1 are not produced in neurons of BACE1−/− mice overexpressing PSGL-1. However, in an acute model of peritonitis the composition and number of leukocytes transmigrating into inflamed tissue did not differ between BACE1−/− and control mice, arguing against major perturbations in leukocyte recruitment (Dominguez et al., 2005). A third inflammatory protein besides ST6Gal1 and PSGL-1, the interleukin-1 receptor II, is also shed by BACE1 (Kuhn et al., 2007). Currently it is not known whether shedding of this interleukin-1 decoy receptor occurs under endogenous condition as very similar and therefore indistinguishable CTFs might be produced by metalloproteases in a compensatory fashion upon knockout of BACE1.
The low density lipoprotein receptor-related protein (LRP) coimmunoprecipitates with BACE1 in human brain tissue and shedding of overexpressed LRP in HEK cells is decreased after BACE1 inhibitor treatment (von Arnim et al., 2005). No endogenous processing by LRP has been demonstrated so far.
The short list of known BACE1 substrates may explain a minor part of the observed phenotypes in BACE1−/− mice, but the mechanisms for the deficits in cognitive and emotional performance, hyperactive behavior, schizophrenia like phenotypes, and epileptic seizures are far from being understood (Harrison et al., 2003; Hu et al., 2006, 2010; Willem et al., 2006; Savonenko et al., 2008). Changes in myelination and epileptic seizure thresholds may be only partially due to improper type III NRG1 and Navβ processing, with other unknown substrates potentially contributing to this phenotype. The analysis of BACE1 substrates and the related phenotypes is further complicated by the fact that many substrates may be shared with other proteases, primarily metalloproteases, leading to alternative shedding events. This could explain the sometimes only subtle phenotypic changes, as metalloproteases may mediate the major part of substrate processing or even compensate for the lack of BACE1 dependent shedding, as seen for the increased α-shedding of APP upon BACE1 inhibition or knockout (Vassar et al., 1999; Sankaranarayanan et al., 2008; Sala Frigerio et al., 2010).
In addition, BACE1 seems to play a major role outside the central and peripheral nervous system. One group reports a fatality rate of ∼40% in BACE1−/− mice, with a bimodal distribution 1 and 4 weeks after birth, while animals that survive to adulthood weigh ∼30% less and are accordingly smaller than their wild-type littermates (Dominguez et al., 2005). Additional deletion of BACE2 increased the fatality rate to ∼60%. The lean phenotype of BACE1−/− mice is associated with increased insulin sensitivity as measured by increased phosphorylation of protein kinase B in skeletal muscle and liver, improved whole body glucose disposal, and resistance to diet induced obesity (Meakin et al., 2012). High fat diet increased hepatic and muscular BACE1 expression in wild-type and BACE1+/− mice and was accompanied by increased insulin resistance. It is unknown how BACE1 regulates insulin sensitivity, but this study provides further evidence for physiologic BACE1 functions outside the central and peripheral nervous system.
To sum up, a large variety of unexplained phenotypes of the nervous system as well as in peripheral tissues exist, and raise concerns toward mechanism-based side effects upon therapeutic inhibition. Unbiased, objective approaches toward the identification of novel BACE1 substrates are needed, with focus on the CNS and peripheral organs involved in glucose metabolism, such as pancreas, liver, and muscle. A shotgun proteomics approach in HEK293 and HeLa cells uncovered several additional putative BACE1 substrates (Hemming et al., 2009). Although the screen identified several known BACE1 substrates along with numerous putative new substrates, including type II transmembrane and GPI-anchored proteins, it was based on overexpression of BACE1 in non-neuronal cell lines. Further validation of identified putative substrates is therefore necessary, as well as objective screening for additional BACE1 substrates under endogenous conditions in primary cells, e.g., cortical neurons or pancreatic cells. This may ultimately aid in the elucidation of phenotypes observed upon BACE1-knockout or treatment with BACE1 inhibitors.
BACE2 Substrates
The BACE1 homolog BACE2 features the same structural and catalytic properties as BACE1, but differs in regard to expression patterns and substrate specificity (reviewed in Stockley and O’Neill, 2008). The highest expression levels are found in the β-cells of pancreatic islets, whereas BACE2 activity in brain is very low (Bennett et al., 2000a; Dominguez et al., 2005; Esterhazy et al., 2011). Although BACE2 cleaves APP at the β-site to a minor extent, the majority of APP cleavage occurs within the Aβ region in an α-secretase-like fashion (Farzan et al., 2000; Hussain et al., 2000). Whereas no Aβ could be detected in neurons, glial cells derived from BACE1−/− mice still secrete an Aβ-like peptide that is no longer detectable in BACE1/BACE2−/− mice, suggesting a contribution of BACE2 to the total Aβ pool (Dominguez et al., 2005). However, almost no Aβ is found in APP transgenic mice lacking BACE1, arguing against a significant contribution of BACE2 toward Aβ generation (Luo et al., 2001; Ohno et al., 2004). BACE2 in AD is thus mainly regarded as an anti-amyloidogenic protease. BACE2 and BACE1 share a set of type I transmembrane proteins as substrates, albeit with mostly distinct cleavage sites, as described for APP and IL1R2 (Farzan et al., 2000; Fluhrer et al., 2002; Kuhn et al., 2007). The physiologic relevance of substrate shedding by BACE2 is largely unknown, as BACE2−/− mice appear healthy. However, the increased lethality of BACE1/BACE2−/− mice compared to BACE1−/− mice suggest essential common substrates amongst these two proteases or at least activation of the same pathways upon substrate processing. In addition, differences in tissue specific expression levels, subcellular localization, and cleavage sites between BACE1 and BACE2 point to the existence of exclusive substrates for either homolog.
The proproliferative type I transmembrane protein TMEM27 regulates the growth of pancreatic islets and has recently been identified as the first physiologically relevant BACE2 substrate (Esterhazy et al., 2011). Interestingly, TMEM27 is not processed by BACE1, which is in contrast to the majority of known BACE2 substrates. Full length TMEM27 acts in a proproliferative fashion and increases pancreatic β-cell mass upon overexpression in vivo. Ectodomain shedding inactivates the protein, as neither the secreted ectodomain nor the remaining CTF display signaling activity (Akpinar et al., 2005). BACE2−/− mice show a similar metabolic phenotype as mice overexpressing TMEM27 and accumulate full length TMEM27 in their pancreatic islets (Esterhazy et al., 2011). This suggests that increases in β-cell mass, insulin secretion and improved glucose response in BACE2−/− mice are at least partially due to increased TMEM27 protein levels and related signaling events. Full length TMEM27 was also enriched in pancreatic islets of BACE1−/− mice, but with a parallel increase in secreted ectodomains, arguing for a cleavage-independent mechanism. Interestingly, BACE1 and BACE2 both regulate insulin metabolism, however on the opposite ends of the same signaling axis. BACE1 regulates insulin sensitivity, whereas BACE2 regulates insulin secretion. To conclude, our current knowledge about BACE2 is still in its infancy, but the activity of most BACE1 inhibitors toward BACE2 stresses the importance of future research on BACE2 function and the related mechanism-based side effects upon at least partial inactivation.
Therapeutic Inhibition of BACE1
Reducing BACE1 activity by RNAi or complete deletion of the BACE1 gene by homologous recombination leads to a dramatic reduction in Aβ plaque load and improves memory deficits and cholinergic dysfunction in APP transgenic mouse lines, validating the potential therapeutic value of BACE1 inhibition in AD (Ohno et al., 2004; Singer et al., 2005). The BACE1 crystal structure in complex with a transition-state inhibitor based on APPSWE promoted the design of an ever increasing quantity of active site inhibitors, which cannot be listed here in their entirety (Hong et al., 2000; Ghosh et al., 2001; Gruninger-Leitch et al., 2002; Turner et al., 2002). These small molecules feature oral bioavailability and low production costs, but the lack of specificity and the inefficient penetration of the blood–brain barrier (BBB) have prevented major breakthroughs in the past. Successful pre-clinical, oral administration of a BACE1 inhibitor has been demonstrated in two studies, one reporting a reduction of soluble Aβ in mouse brain, the other a reduction in CSF Aβ levels in non-human primates (Sankaranarayanan et al., 2009; Fukumoto et al., 2010). A recent study reports for the first time the efficient inhibition of BACE1 with an orally available, non-peptidic inhibitor in man, as measured by the decrease of Aβ and APPsβ in CSF (May et al., 2011). Unfortunately, this proof of principle study had to be stopped during the clinical phase I trial due to retinal pathology observed upon long-term administration of the inhibitor in a pre-clinical mouse model. The pathology was recapitulated in BACE1−/− mice, speaking against mechanism-based toxicity. However, 11 years after the initiation of BACE1 inhibitor development, designing drugs that are lipophilic and small enough (<700 Da) to cross the BBB while still being able to block the unusually large catalytic site of BACE1 remains one of the main challenges in the field. In order to improve the efficiency of small molecule active site inhibitors, one group reports the design of a transition-state inhibitor linked to a sterol moiety, resulting in increased local membrane concentration and inhibitor potency (Rajendran et al., 2008).
A rather novel approach exploits the selectivity of therapeutic antibodies directed against BACE1. The reported antibodies recognize unique, non-catalytical exosites of BACE1 and inhibit its proteolytic activity in vitro as well as in mice and non-human primates (Atwal et al., 2011; Zhou et al., 2011). To further increase the therapeutic value of these antibodies, a bispecific antibody was generated, with one arm consisting of a high-affinity BACE1 antibody and the other arm consisting of a low-affinity anti-transferrin receptor antibody, thus speeding up receptor-mediated transcytosis across the BBB and increasing the target antibody concentration in the CNS (Yu et al., 2011). In order to bypass the high costs and regular parenteral delivery of passive immunization, one study reports active immunization with BACE1 in APP transgenic mice that led to a significant reduction of brain Aβ without perceivable inflammatory side effects (Chang et al., 2007). An unusual approach toward BACE1 inhibition targets the cellular environment of BACE1 rather than inhibiting the enzyme directly. Bepridil and amiodarone, two approved calcium antagonists that contain weakly basic amino groups, raise the membrane-proximal, endosomal pH above the optimum pH value for BACE1 activity, thus slowing Aβ production (Mitterreiter et al., 2010). Innovative approaches that do not rely on BACE1 active site inhibition may speed up the discovery of drugs that are small enough to penetrate the BBB.
Conclusion and Outlook
BACE1 is the rate-limiting enzyme for Aβ generation in neurons and thus a prime drug target in AD. The complex regulation of BACE1 activity, the increasing number of observed phenotypes in knockout mice as well as the processing of multiple substrates besides APP speak for the versatile role of this protease during development and adulthood but raise concerns toward mechanism-based side effects. Despite extensive efforts, it remains to be shown whether BACE1 inhibition may alleviate or reverse symptoms of AD, as no BACE1 inhibitor has endured a randomized human clinical trial. Combinatorial therapies, where compounds targeting BACE1, γ-secretase and Aβ are combined in a two or three drug regimen may be more efficient and at the same time offer less side effects, as partial inhibition of the individual secretases may be sufficient. It remains unclear, which side effects may be of concern during the long-term administration of BACE1 inhibitors that would be required to treat AD patients. The generation of conditional BACE1−/− mice, where the complete knockout could be initiated during adulthood, could help to address this issue. Additionally, as with other Aβ lowering strategies, it is currently unknown whether treatment must be initiated in the prodromal phase of the disease or whether therapeutic intervention during manifest AD is sufficient to alter cognitive decline. BACE1 is a rationale target in AD drug development, but there is still much to be learned about the physiologic function of BACE1, its precise localization in polarized cells such as neurons, and the role of the lipid microenvironment on enzymatic activity. The identification of novel BACE1 substrates as well as detailed mechanistic studies on existing BACE1 substrates will be of importance to uncover mechanism-based side effects as well as discovering new biomarkers for AD. The latter part is of utmost importance, as therapeutic intervention even before the onset of clinical symptoms may be necessary to alter disease progression.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by grants DFG SFB596 and BMBF KNDD.
References
Abad-Rodriguez, J., Ledesma, M. D., Craessaerts, K., Perga, S., Medina, M., Delacourte, A., Dingwall, C., De Strooper, B., and Dotti, C. G. (2004). Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 167, 953–960.
Akpinar, P., Kuwajima, S., Krutzfeldt, J., and Stoffel, M. (2005). Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metab. 2, 385–397.
Atwal, J. K., Chen, Y., Chiu, C., Mortensen, D. L., Meilandt, W. J., Liu, Y., Heise, C. E., Hoyte, K., Luk, W., Lu, Y., Peng, K., Wu, P., Rouge, L., Zhang, Y., Lazarus, R. A., Scearce-Levie, K., Wang, W., Wu, Y., Tessier-Lavigne, M., and Watts, R. J. (2011). A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci. Transl. Med. 3, 84ra43.
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer’s disease. Lancet 377, 1019–1031.
Benjannet, S., Cromlish, J. A., Diallo, K., Chretien, M., and Seidah, N. G. (2004). The metabolism of beta-amyloid converting enzyme and beta-amyloid precursor protein processing. Biochem. Biophys. Res. Commun. 325, 235–242.
Benjannet, S., Elagoz, A., Wickham, L., Mamarbachi, M., Munzer, J. S., Basak, A., Lazure, C., Cromlish, J. A., Sisodia, S., Checler, F., Chretien, M., and Seidah, N. G. (2001). Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 276, 10879–10887.
Bennett, B. D., Babu-Khan, S., Loeloff, R., Louis, J. C., Curran, E., Citron, M., and Vassar, R. (2000a). Expression analysis of BACE2 in brain and peripheral tissues. J. Biol. Chem. 275, 20647–20651.
Bennett, B. D., Denis, P., Haniu, M., Teplow, D. B., Kahn, S., Louis, J. C., Citron, M., and Vassar, R. (2000b). A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta -secretase. J. Biol. Chem. 275, 37712–37717.
Birchmeier, C., and Nave, K. A. (2008). Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia 56, 1491–1497.
Bodendorf, U., Fischer, F., Bodian, D., Multhaup, G., and Paganetti, P. (2001). A splice variant of beta-secretase deficient in the amyloidogenic processing of the amyloid precursor protein. J. Biol. Chem. 276, 12019–12023.
Brackenbury, W. J., Djamgoz, M. B., and Isom, L. L. (2008). An emerging role for voltage-gated Na+ channels in cellular migration: regulation of central nervous system development and potentiation of invasive cancers. Neuroscientist 14, 571–583.
Brinkmann, B. G., Agarwal, A., Sereda, M. W., Garratt, A. N., Muller, T., Wende, H., Stassart, R. M., Nawaz, S., Humml, C., Velanac, V., Radyushkin, K., Goebbels, S., Fischer, T. M., Franklin, R. J., Lai, C., Ehrenreich, H., Birchmeier, C., Schwab, M. H., and Nave, K. A. (2008). Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 59, 581–595.
Brown, M. S., Ye, J., Rawson, R. B., and Goldstein, J. L. (2000). Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391–398.
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J., Johnson, R. S., Castner, B. J., Cerretti, D. P., and Black, R. A. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767.
Cai, H., Wang, Y., Mccarthy, D., Wen, H., Borchelt, D. R., Price, D. L., and Wong, P. C. (2001). BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 4, 233–234.
Capell, A., Steiner, H., Willem, M., Kaiser, H., Meyer, C., Walter, J., Lammich, S., Multhaup, G., and Haass, C. (2000). Maturation and pro-peptide cleavage of beta-secretase. J. Biol. Chem. 275, 30849–30854.
Carlow, D. A., Gossens, K., Naus, S., Veerman, K. M., Seo, W., and Ziltener, H. J. (2009). PSGL-1 function in immunity and steady state homeostasis. Immunol. Rev. 230, 75–96.
Carteron, C., Ferrer-Montiel, A., and Cabedo, H. (2006). Characterization of a neural-specific splicing form of the human neuregulin 3 gene involved in oligodendrocyte survival. J. Cell Sci. 119, 898–909.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Chang, W. P., Downs, D., Huang, X. P., Da, H., Fung, K. M., and Tang, J. (2007). Amyloid-beta reduction by memapsin 2 (beta-secretase) immunization. FASEB J. 21, 3184–3196.
Chen, M., and Geng, J. G. (2006). P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch. Immunol. Ther. Exp. (Warsz.) 54, 75–84.
Chyung, J. H., and Selkoe, D. J. (2003). Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid beta-protein at the cell surface. J. Biol. Chem. 278, 51035–51043.
Citron, M., Oltersdorf, T., Haass, C., Mcconlogue, L., Hung, A. Y., Seubert, P., Vigo-Pelfrey, C., Lieberburg, I., and Selkoe, D. J. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360, 672–674.
Claes, L., Del-Favero, J., Ceulemans, B., Lagae, L., Van Broeckhoven, C., and De Jonghe, P. (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 68, 1327–1332.
Cordy, J. M., Hussain, I., Dingwall, C., Hooper, N. M., and Turner, A. J. (2003). Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 100, 11735–11740.
Costantini, C., Ko, M. H., Jonas, M. C., and Puglielli, L. (2007). A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 407, 383–395.
Crameri, A., Biondi, E., Kuehnle, K., Lutjohann, D., Thelen, K. M., Perga, S., Dotti, C. G., Nitsch, R. M., Ledesma, M. D., and Mohajeri, M. H. (2006). The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J. 25, 432–443.
Creemers, J. W., Ines Dominguez, D., Plets, E., Serneels, L., Taylor, N. A., Multhaup, G., Craessaerts, K., Annaert, W., and De Strooper, B. (2001). Processing of beta-secretase by furin and other members of the proprotein convertase family. J. Biol. Chem. 276, 4211–4217.
De Pietri Tonelli, D., Mihailovich, M., Di Cesare, A., Codazzi, F., Grohovaz, F., and Zacchetti, D. (2004). Translational regulation of BACE-1 expression in neuronal and non-neuronal cells. Nucleic Acids Res. 32, 1808–1817.
Dominguez, D., Tournoy, J., Hartmann, D., Huth, T., Cryns, K., Deforce, S., Serneels, L., Camacho, I. E., Marjaux, E., Craessaerts, K., Roebroek, A. J., Schwake, M., D’Hooge, R., Bach, P., Kalinke, U., Moechars, D., Alzheimer, C., Reiss, K., Saftig, P., and De Strooper, B. (2005). Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J. Biol. Chem. 280, 30797–30806.
Eggert, S., Paliga, K., Soba, P., Evin, G., Masters, C. L., Weidemann, A., and Beyreuther, K. (2004). The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J. Biol. Chem. 279, 18146–18156.
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123.
Ehehalt, R., Michel, B., De Pietri Tonelli, D., Zacchetti, D., Simons, K., and Keller, P. (2002). Splice variants of the beta-site APP-cleaving enzyme BACE1 in human brain and pancreas. Biochem. Biophys. Res. Commun. 293, 30–37.
Esch, F. S., Keim, P. S., Beattie, E. C., Blacher, R. W., Culwell, A. R., Oltersdorf, T., Mcclure, D., and Ward, P. J. (1990). Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 248, 1122–1124.
Esterhazy, D., Stutzer, I., Wang, H., Rechsteiner, M. P., Beauchamp, J., Dobeli, H., Hilpert, H., Matile, H., Prummer, M., Schmidt, A., Lieske, N., Boehm, B., Marselli, L., Bosco, D., Kerr-Conte, J., Aebersold, R., Spinas, G. A., Moch, H., Migliorini, C., and Stoffel, M. (2011). Bace2 is a beta cell-enriched protease that regulates pancreatic beta cell function and mass. Cell Metab. 14, 365–377.
Faghihi, M. A., Modarresi, F., Khalil, A. M., Wood, D. E., Sahagan, B. G., Morgan, T. E., Finch, C. E., St Laurent, G. III, Kenny, P. J., and Wahlestedt, C. (2008). Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 14, 723–730.
Falls, D. L. (2003). Neuregulins: functions, forms, and signaling strategies. Exp. Cell Res. 284, 14–30.
Farah, M. H., Pan, B. H., Hoffman, P. N., Ferraris, D., Tsukamoto, T., Nguyen, T., Wong, P. C., Price, D. L., Slusher, B. S., and Griffin, J. W. (2011). Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J. Neurosci. 31, 5744–5754.
Farzan, M., Schnitzler, C. E., Vasilieva, N., Leung, D., and Choe, H. (2000). BACE2, a beta -secretase homolog, cleaves at the beta site and within the amyloid-beta region of the amyloid-beta precursor protein. Proc. Natl. Acad. Sci. U.S.A. 97, 9712–9717.
Fassbender, K., Simons, M., Bergmann, C., Stroick, M., Lutjohann, D., Keller, P., Runz, H., Kuhl, S., Bertsch, T., Von Bergmann, K., Hennerici, M., Beyreuther, K., and Hartmann, T. (2001). Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 5856–5861.
Finan, G. M., Okada, H., and Kim, T. W. (2011). BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J. Biol. Chem. 286, 12602–12616.
Fluhrer, R., Capell, A., Westmeyer, G., Willem, M., Hartung, B., Condron, M. M., Teplow, D. B., Haass, C., and Walter, J. (2002). A non-amyloidogenic function of BACE-2 in the secretory pathway. J. Neurochem. 81, 1011–1020.
Fukumoto, H., Cheung, B. S., Hyman, B. T., and Irizarry, M. C. (2002). Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389.
Fukumoto, H., Takahashi, H., Tarui, N., Matsui, J., Tomita, T., Hirode, M., Sagayama, M., Maeda, R., Kawamoto, M., Hirai, K., Terauchi, J., Sakura, Y., Kakihana, M., Kato, K., Iwatsubo, T., and Miyamoto, M. (2010). A noncompetitive BACE1 inhibitor TAK-070 ameliorates Abeta pathology and behavioral deficits in a mouse model of Alzheimer’s disease. J. Neurosci. 30, 11157–11166.
Ghosh, A. K., Bilcer, G., Harwood, C., Kawahama, R., Shin, D., Hussain, K. A., Hong, L., Loy, J. A., Nguyen, C., Koelsch, G., Ermolieff, J., and Tang, J. (2001). Structure-based design: potent inhibitors of human brain memapsin 2 (beta-secretase). J. Med. Chem. 44, 2865–2868.
Gruninger-Leitch, F., Schlatter, D., Kung, E., Nelbock, P., and Dobeli, H. (2002). Substrate and inhibitor profile of BACE (beta-secretase) and comparison with other mammalian aspartic proteases. J. Biol. Chem. 277, 4687–4693.
Haass, C., Hung, A. Y., Schlossmacher, M. G., Oltersdorf, T., Teplow, D. B., and Selkoe, D. J. (1993). Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann. N. Y. Acad. Sci. 695, 109–116.
Haass, C., Lemere, C. A., Capell, A., Citron, M., Seubert, P., Schenk, D., Lannfelt, L., and Selkoe, D. J. (1995). The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1, 1291–1296.
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112.
Hahn, C. G., Wang, H. Y., Cho, D. S., Talbot, K., Gur, R. E., Berrettini, W. H., Bakshi, K., Kamins, J., Borgmann-Winter, K. E., Siegel, S. J., Gallop, R. J., and Arnold, S. E. (2006). Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 12, 824–828.
Haniu, M., Denis, P., Young, Y., Mendiaz, E. A., Fuller, J., Hui, J. O., Bennett, B. D., Kahn, S., Ross, S., Burgess, T., Katta, V., Rogers, G., Vassar, R., and Citron, M. (2000). Characterization of Alzheimer’s beta -secretase protein BACE. A pepsin family member with unusual properties. J. Biol. Chem. 275, 21099–21106.
Hao, M., Mukherjee, S., Sun, Y., and Maxfield, F. R. (2004). Effects of cholesterol depletion and increased lipid unsaturation on the properties of endocytic membranes. J. Biol. Chem. 279, 14171–14178.
Haque, A., Banik, N. L., and Ray, S. K. (2008). New insights into the roles of endolysosomal cathepsins in the pathogenesis of Alzheimer’s disease: cathepsin inhibitors as potential therapeutics. CNS Neurol. Disord. Drug Targets 7, 270–277.
Harada, H., Tamaoka, A., Ishii, K., Shoji, S., Kametaka, S., Kametani, F., Saito, Y., and Murayama, S. (2006). Beta-site APP cleaving enzyme 1 (BACE1) is increased in remaining neurons in Alzheimer’s disease brains. Neurosci. Res. 54, 24–29.
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356.
Harrison, S. M., Harper, A. J., Hawkins, J., Duddy, G., Grau, E., Pugh, P. L., Winter, P. H., Shilliam, C. S., Hughes, Z. A., Dawson, L. A., Gonzalez, M. I., Upton, N., Pangalos, M. N., and Dingwall, C. (2003). BACE1 (beta-secretase) transgenic and knockout mice: identification of neurochemical deficits and behavioral changes. Mol. Cell. Neurosci. 24, 646–655.
Hattori, C., Asai, M., Onishi, H., Sasagawa, N., Hashimoto, Y., Saido, T. C., Maruyama, K., Mizutani, S., and Ishiura, S. (2006). BACE1 interacts with lipid raft proteins. J. Neurosci. Res. 84, 912–917.
He, X., Chang, W. P., Koelsch, G., and Tang, J. (2002). Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 524, 183–187.
He, X., Li, F., Chang, W. P., and Tang, J. (2005). GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J. Biol. Chem. 280, 11696–11703.
He, X., Zhu, G., Koelsch, G., Rodgers, K. K., Zhang, X. C., and Tang, J. (2003). Biochemical and structural characterization of the interaction of memapsin 2 (beta-secretase) cytosolic domain with the VHS domain of GGA proteins. Biochemistry 42, 12174–12180.
Heber, S., Herms, J., Gajic, V., Hainfellner, J., Aguzzi, A., Rülicke, T., von Kretschmar, H., von Koch, C., Sisodia, S., Tremml, P., Lipp, H. P., Wolfer, D. P., and Müller, U. (2000). Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 20, 7951–7963.
Hebert, S. S., Horre, K., Nicolai, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., Kauppinen, S., Delacourte, A., and De Strooper, B. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420.
Hemming, M. L., Elias, J. E., Gygi, S. P., and Selkoe, D. J. (2009). Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS ONE 4, e8477. doi:10.1371/journal.pone.0008477
Hitt, B. D., Jaramillo, T. C., Chetkovich, D. M., and Vassar, R. (2010). BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol. Neurodegener. 5, 31.
Hoffmeister, A., Dietz, G., Zeitschel, U., Mössner, J., Rossner, S., and Stahl, T. (2009). BACE1 is a newly discovered protein secreted by the pancreas which cleaves enteropeptidase in vitro. JOP 10, 481–484.
Hogl, S., Kuhn, P. H., Colombo, A., and Lichtenthaler, S. F. (2011). Determination of the proteolytic cleavage sites of the amyloid precursor-like protein 2 by the proteases ADAM10, BACE1 and gamma-secretase. PLoS ONE 6, e21337. doi:10.1371/journal.pone.0021337
Holsinger, R. M., Mclean, C. A., Beyreuther, K., Masters, C. L., and Evin, G. (2002). Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann. Neurol. 51, 783–786.
Hong, L., Koelsch, G., Lin, X., Wu, S., Terzyan, S., Ghosh, A. K., Zhang, X. C., and Tang, J. (2000). Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 290, 150–153.
Hook, G., Hook, V., and Kindy, M. (2011). The cysteine protease inhibitor, E64d, reduces brain amyloid-beta and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, beta-secretase activity. J. Alzheimers Dis. 26, 387–408.
Hook, V. Y., Kindy, M., and Hook, G. (2008). Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem. 283, 7745–7753.
Hu, X., He, W., Diaconu, C., Tang, X., Kidd, G. J., Macklin, W. B., Trapp, B. D., and Yan, R. (2008). Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 22, 2970–2980.
Hu, X., Hicks, C. W., He, W., Wong, P., Macklin, W. B., Trapp, B. D., and Yan, R. (2006). Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 9, 1520–1525.
Hu, X., Zhou, X., He, W., Yang, J., Xiong, W., Wong, P., Wilson, C. G., and Yan, R. (2010). BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 30, 8819–8829.
Huse, J. T., Pijak, D. S., Leslie, G. J., Lee, V. M., and Doms, R. W. (2000). Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J. Biol. Chem. 275, 33729–33737.
Hussain, I., Hawkins, J., Shikotra, A., Riddell, D. R., Faller, A., and Dingwall, C. (2003). Characterization of the ectodomain shedding of the beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1). J. Biol. Chem. 278, 36264–36268.
Hussain, I., Powell, D., Howlett, D. R., Tew, D. G., Meek, T. D., Chapman, C., Gloger, I. S., Murphy, K. E., Southan, C. D., Ryan, D. M., Smith, T. S., Simmons, D. L., Walsh, F. S., Dingwall, C., and Christie, G. (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 14, 419–427.
Hussain, I., Powell, D. J., Howlett, D. R., Chapman, G. A., Gilmour, L., Murdock, P. R., Tew, D. G., Meek, T. D., Chapman, C., Schneider, K., Ratcliffe, S. J., Tattersall, D., Testa, T. T., Southan, C., Ryan, D. M., Simmons, D. L., Walsh, F. S., Dingwall, C., and Christie, G. (2000). ASP1 (BACE2) cleaves the amyloid precursor protein at the beta-secretase site. Mol. Cell. Neurosci. 16, 609–619.
Irizarry, M. C., Locascio, J. J., and Hyman, B. T. (2001). beta-site APP cleaving enzyme mRNA expression in APP transgenic mice: anatomical overlap with transgene expression and static levels with aging. Am. J. Pathol. 158, 173–177.
Jorissen, E., Prox, J., Bernreuther, C., Weber, S., Schwanbeck, R., Serneels, L., Snellinx, A., Craessaerts, K., Thathiah, A., Tesseur, I., Bartsch, U., Weskamp, G., Blobel, C. P., Glatzel, M., De Strooper, B., and Saftig, P. (2010). The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 30, 4833–4844.
Kaether, C., and Haass, C. (2004). A lipid boundary separates APP and secretases and limits amyloid beta-peptide generation. J. Cell Biol. 167, 809–812.
Kang, E. L., Cameron, A. N., Piazza, F., Walker, K. R., and Tesco, G. (2010). Ubiquitin regulates GGA3-mediated degradation of BACE1. J. Biol. Chem. 285, 24108–24119.
Kao, W. T., Wang, Y., Kleinman, J. E., Lipska, B. K., Hyde, T. M., Weinberger, D. R., and Law, A. J. (2010). Common genetic variation in Neuregulin 3 (NRG3) influences risk for schizophrenia and impacts NRG3 expression in human brain. Proc. Natl. Acad. Sci. 107, 15619–15624.
Kaplan, H. A., Woloski, B. M., Hellman, M., and Jamieson, J. C. (1983). Studies on the effect of inflammation on rat liver and serum sialyltransferase. Evidence that inflammation causes release of Gal beta 1 leads to 4GlcNAc alpha 2 leads to 6 sialyltransferase from liver. J. Biol. Chem. 258, 11505–11509.
Kawarabayashi, T., Shoji, M., Younkin, L. H., Wen-Lang, L., Dickson, D. W., Murakami, T., Matsubara, E., Abe, K., Ashe, K. H., and Younkin, S. G. (2004). Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 24, 3801–3809.
Kim, D. Y., Carey, B. W., Wang, H., Ingano, L. A., Binshtok, A. M., Wertz, M. H., Pettingell, W. H., He, P., Lee, V. M., Woolf, C. J., and Kovacs, D. M. (2007). BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat. Cell Biol. 9, 755–764.
Kim, D. Y., Gersbacher, M. T., Inquimbert, P., and Kovacs, D. M. (2011). Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J. Biol. Chem. 286, 8106–8116.
Kitagawa, H., and Paulson, J. C. (1994). Differential expression of five sialyltransferase genes in human tissues. J. Biol. Chem. 269, 17872–17878.
Kitazume, S., Nakagawa, K., Oka, R., Tachida, Y., Ogawa, K., Luo, Y., Citron, M., Shitara, H., Taya, C., Yonekawa, H., Paulson, J. C., Miyoshi, E., Taniguchi, N., and Hashimoto, Y. (2005). In vivo cleavage of alpha2,6-sialyltransferase by Alzheimer beta-secretase. J. Biol. Chem. 280, 8589–8595.
Kitazume, S., Tachida, Y., Oka, R., Shirotani, K., Saido, T. C., and Hashimoto, Y. (2001). Alzheimer’s beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc. Natl. Acad. Sci. U.S.A. 98, 13554–13559.
Ko, M. H., and Puglielli, L. (2009). Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 284, 2482–2492.
Koike, H., Tomioka, S., Sorimachi, H., Saido, T. C., Maruyama, K., Okuyama, A., Fujisawa-Sehara, A., Ohno, S., Suzuki, K., and Ishiura, S. (1999). Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 343(Pt 2), 371–375.
Koo, E. H., and Squazzo, S. L. (1994). Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389.
Kovacs, D. M., Gersbacher, M. T., and Kim, D. Y. (2010). Alzheimer’s secretases regulate voltage-gated sodium channels. Neurosci. Lett. 486, 68–72.
Kuhn, P. H., Marjaux, E., Imhof, A., De Strooper, B., Haass, C., and Lichtenthaler, S. F. (2007). Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J. Biol. Chem. 282, 11982–11995.
Kuhn, P. H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., Kremmer, E., Rossner, S., and Lichtenthaler, S. F. (2010). ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032.
Kvajo, M., Mckellar, H., Arguello, P. A., Drew, L. J., Moore, H., Macdermott, A. B., Karayiorgou, M., and Gogos, J. A. (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. U.S.A. 105, 7076–7081.
La Marca, R., Cerri, F., Horiuchi, K., Bachi, A., Feltri, M. L., Wrabetz, L., Blobel, C. P., Quattrini, A., Salzer, J. L., and Taveggia, C. (2011). TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci. 14, 857–865.
Laird, F. M., Cai, H., Savonenko, A. V., Farah, M. H., He, K., Melnikova, T., Wen, H., Chiang, H. C., Xu, G., Koliatsos, V. E., Borchelt, D. R., Price, D. L., Lee, H. K., and Wong, P. C. (2005). BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 25, 11693–11709.
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., Haass, C., and Fahrenholz, F. (1999). Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927.
Lammich, S., Schobel, S., Zimmer, A. K., Lichtenthaler, S. F., and Haass, C. (2004). Expression of the Alzheimer protease BACE1 is suppressed via its 5′-untranslated region. EMBO Rep. 5, 620–625.
Lazarov, O., Lee, M., Peterson, D. A., and Sisodia, S. S. (2002). Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J. Neurosci. 22, 9785–9793.
Li, H., Wang, B., Wang, Z., Guo, Q., Tabuchi, K., Hammer, R. E., Sudhof, T. C., and Zheng, H. (2010). Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc. Natl. Acad. Sci. U.S.A. 107, 17362–17367.
Li, Q., and Südhof, T. C. (2004). Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE1. J. Biol. Chem. 279, 10542–10550.
Li, R., Lindholm, K., Yang, L. B., Yue, X., Citron, M., Yan, R., Beach, T., Sue, L., Sabbagh, M., Cai, H., Wong, P., Price, D., and Shen, Y. (2004). Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. U.S.A. 101, 3632–3637.
Lichtenthaler, S. F., Dominguez, D. I., Westmeyer, G. G., Reiss, K., Haass, C., Saftig, P., De Strooper, B., and Seed, B. (2003). The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J. Biol. Chem. 278, 48713–48719.
Lichtenthaler, S. F., Haass, C., and Steiner, H. (2011). Regulated intramembrane proteolysis – lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796.
Lillis, A. P., Van Duyn, L. B., Murphy-Ullrich, J. E., and Strickland, D. K. (2008). LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918.
Lin, X., Koelsch, G., Wu, S., Downs, D., Dashti, A., and Tang, J. (2000). Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 97, 1456–1460.
Luo, X., Prior, M., He, W., Hu, X., Tang, X., Shen, W., Yadav, S., Kiryu-Seo, S., Miller, R., Trapp, B. D., and Yan, R. (2011). Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J. Biol. Chem. 286, 23967–23974.
Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., Fan, W., Kha, H., Zhang, J., Gong, Y., Martin, L., Louis, J. C., Yan, Q., Richards, W. G., Citron, M., and Vassar, R. (2001). Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4, 231–232.
Matsui, T., Ingelsson, M., Fukumoto, H., Ramasamy, K., Kowa, H., Frosch, M. P., Irizarry, M. C., and Hyman, B. T. (2007). Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 1161, 116–123.
May, P. C., Dean, R. A., Lowe, S. L., Martenyi, F., Sheehan, S. M., Boggs, L. N., Monk, S. A., Mathes, B. M., Mergott, D. J., Watson, B. M., Stout, S. L., Timm, D. E., Smith Labell, E., Gonzales, C. R., Nakano, M., Jhee, S. S., Yen, M., Ereshefsky, L., Lindstrom, T. D., Calligaro, D. O., Cocke, P. J., Greg Hall, D., Friedrich, S., Citron, M., and Audia, J. E. (2011). Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J. Neurosci. 31, 16507–16516.
Meakin, P. J., Harper, A. J., Hamilton, D. L., Gallagher, J., Mcneilly, A. D., Burgess, L. A., Vaanholt, L. M., Bannon, K. A., Latcham, J., Hussain, I., Speakman, J. R., Howlett, D. R., and Ashford, M. L. (2012). Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 441, 285–296.
Mei, L., and Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452.
Mihailovich, M., Thermann, R., Grohovaz, F., Hentze, M. W., and Zacchetti, D. (2007). Complex translational regulation of BACE1 involves upstream AUGs and stimulatory elements within the 5′ untranslated region. Nucleic Acids Res. 35, 2975–2985.
Mitterreiter, S., Page, R. M., Kamp, F., Hopson, J., Winkler, E., Ha, H. R., Hamid, R., Herms, J., Mayer, T. U., Nelson, D. J., Steiner, H., Stahl, T., Zeitschel, U., Rossner, S., Haass, C., and Lichtenthaler, S. F. (2010). Bepridil and amiodarone simultaneously target the Alzheimer’s disease beta- and gamma-secretase via distinct mechanisms. J. Neurosci. 30, 8974–8983.
Molinari, M., Galli, C., Piccaluga, V., Pieren, M., and Paganetti, P. (2002). Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J. Cell Biol. 158, 247–257.
Mowrer, K. R., and Wolfe, M. S. (2008). Promotion of BACE1 mRNA alternative splicing reduces amyloid beta-peptide production. J. Biol. Chem. 283, 18694–18701.
Mullan, M., Crawford, F., Axelman, K., Houlden, H., LIlius, L., Winblad, B., and Lannfelt, L. (1992). A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of b-amyloid. Nat. Genet. 1, 345–347.
Muller, U., Cristina, N., Li, Z. W., Wolfer, D. P., Lipp, H. P., Rulicke, T., Brandner, S., Aguzzi, A., and Weissmann, C. (1994). Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 79, 755–765.
Mulley, J. C., Scheffer, I. E., Petrou, S., Dibbens, L. M., Berkovic, S. F., and Harkin, L. A. (2005). SCN1A mutations and epilepsy. Hum. Mutat. 25, 535–542.
Naus, S., Reipschlager, S., Wildeboer, D., Lichtenthaler, S. F., Mitterreiter, S., Guan, Z., Moss, M. L., and Bartsch, J. W. (2006). Identification of candidate substrates for ectodomain shedding by the metalloprotease-disintegrin ADAM8. Biol. Chem. 387, 337–346.
Neumann, S., Schobel, S., Jager, S., Trautwein, A., Haass, C., Pietrzik, C. U., and Lichtenthaler, S. F. (2006). Amyloid precursor-like protein 1 influences endocytosis and proteolytic processing of the amyloid precursor protein. J. Biol. Chem. 281, 7583–7594.
Nikolaev, A., Mclaughlin, T., O’Leary, D. D., and Tessier-Lavigne, M. (2009). APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989.
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204.
O’Connor, T., Sadleir, K. R., Maus, E., Velliquette, R. A., Zhao, J., Cole, S. L., Eimer, W. A., Hitt, B., Bembinster, L. A., Lammich, S., Lichtenthaler, S. F., Hebert, S. S., De Strooper, B., Haass, C., Bennett, D. A., and Vassar, R. (2008). Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009.
Ohno, M., Sametsky, E. A., Younkin, L. H., Oakley, H., Younkin, S. G., Citron, M., Vassar, R., and Disterhoft, J. F. (2004). BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41, 27–33.
Ostermann, N., Eder, J., Eidhoff, U., Zink, F., Hassiepen, U., Worpenberg, S., Maibaum, J., Simic, O., Hommel, U., and Gerhartz, B. (2006). Crystal structure of human BACE2 in complex with a hydroxyethylamine transition-state inhibitor. J. Mol. Biol. 355, 249–261.
Postina, R., Schroeder, A., Dewachter, I., Bohl, J., Schmitt, U., Kojro, E., Prinzen, C., Endres, K., Hiemke, C., Blessing, M., Flamez, P., Dequenne, A., Godaux, E., Van Leuven, F., and Fahrenholz, F. (2004). A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464.
Preece, P., Virley, D. J., Costandi, M., Coombes, R., Moss, S. J., Mudge, A. W., Jazin, E., and Cairns, N. J. (2003). Beta-secretase (BACE) and GSK-3 mRNA levels in Alzheimer’s disease. Brain Res. Mol. Brain Res. 116, 155–158.
Qing, H., Zhou, W., Christensen, M. A., Sun, X., Tong, Y., and Song, W. (2004). Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 18, 1571–1573.
Rajendran, L., Schneider, A., Schlechtingen, G., Weidlich, S., Ries, J., Braxmeier, T., Schwille, P., Schulz, J. B., Schroeder, C., Simons, M., Jennings, G., Knolker, H. J., and Simons, K. (2008). Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 320, 520–523.
Refolo, L. M., Pappolla, M. A., Lafrancois, J., Malester, B., Schmidt, S. D., Thomas-Bryant, T., Tint, G. S., Wang, R., Mercken, M., Petanceska, S. S., and Duff, K. E. (2001). A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 8, 890–899.
Riddell, D. R., Christie, G., Hussain, I., and Dingwall, C. (2001). Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 11, 1288–1293.
Ring, S., Weyer, S. W., Kilian, S. B., Waldron, E., Pietrzik, C. U., Filippov, M. A., Herms, J., Buchholz, C., Eckman, C. B., Korte, M., Wolfer, D. P., and Muller, U. C. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817–7826.
Roberds, S. L., Anderson, J., Basi, G., Bienkowski, M. J., Branstetter, D. G., Chen, K. S., Freedman, S. B., Frigon, N. L., Games, D., Hu, K., Johnson-Wood, K., Kappenman, K. E., Kawabe, T. T., Kola, I., Kuehn, R., Lee, M., Liu, W., Motter, R., Nichols, N. F., Power, M., Robertson, D. W., Schenk, D., Schoor, M., Shopp, G. M., Shuck, M. E., Sinha, S., Svensson, K. A., Tatsuno, G., Tintrup, H., Wijsman, J., Wright, S., and Mcconlogue, L. (2001). BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 10, 1317–1324.
Rodal, S. K., Skretting, G., Garred, O., Vilhardt, F., Van Deurs, B., and Sandvig, K. (1999). Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell 10, 961–974.
Rogers, G. W. Jr., Edelman, G. M., and Mauro, V. P. (2004). Differential utilization of upstream AUGs in the beta-secretase mRNA suggests that a shunting mechanism regulates translation. Proc. Natl. Acad. Sci. U.S.A. 101, 2794–2799.
Rossner, S., Sastre, M., Bourne, K., and Lichtenthaler, S. F. (2006). Transcriptional and translational regulation of BACE1 expression – implications for Alzheimer’s disease. Prog. Neurobiol. 79, 95–111.
Sagane, K., Hayakawa, K., Kai, J., Hirohashi, T., Takahashi, E., Miyamoto, N., Ino, M., Oki, T., Yamazaki, K., and Nagasu, T. (2005). Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci. 6, 33. doi:10.1186/1471-2202-6-33
Sala Frigerio, C., Fadeeva, J. V., Minogue, A. M., Citron, M., Van Leuven, F., Staufenbiel, M., Paganetti, P., Selkoe, D. J., and Walsh, D. M. (2010). beta-Secretase cleavage is not required for generation of the intracellular C-terminal domain of the amyloid precursor family of proteins. FEBS J. 277, 1503–1518.
Sankaranarayanan, S., Holahan, M. A., Colussi, D., Crouthamel, M. C., Devanarayan, V., Ellis, J., Espeseth, A., Gates, A. T., Graham, S. L., Gregro, A. R., Hazuda, D., Hochman, J. H., Holloway, K., Jin, L., Kahana, J., Lai, M. T., Lineberger, J., Mcgaughey, G., Moore, K. P., Nantermet, P., Pietrak, B., Price, E. A., Rajapakse, H., Stauffer, S., Steinbeiser, M. A., Seabrook, G., Selnick, H. G., Shi, X. P., Stanton, M. G., Swestock, J., Tugusheva, K., Tyler, K. X., Vacca, J. P., Wong, J., Wu, G., Xu, M., Cook, J. J., and Simon, A. J. (2009). First demonstration of cerebrospinal fluid and plasma A beta lowering with oral administration of a beta-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates. J. Pharmacol. Exp. Ther. 328, 131–140.
Sankaranarayanan, S., Price, E. A., Wu, G., Crouthamel, M. C., Shi, X. P., Tugusheva, K., Tyler, K. X., Kahana, J., Ellis, J., Jin, L., Steele, T., Stachel, S., Coburn, C., and Simon, A. J. (2008). In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased alpha-secretase processing of amyloid precursor protein without effect on neuregulin-1. J. Pharmacol. Exp. Ther. 324, 957–969.
Sannerud, R., Declerck, I., Peric, A., Raemaekers, T., Menendez, G., Zhou, L., Veerle, B., Coen, K., Munck, S., De Strooper, B., Schiavo, G., and Annaert, W. (2011). ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl. Acad. Sci. U.S.A. 108, E559–E568.
Santosa, C., Rasche, S., Barakat, A., Bellingham, S. A., Ho, M., Tan, J., Hill, A. F., Masters, C. L., Mclean, C., and Evin, G. (2011). Decreased expression of GGA3 Protein in Alzheimer’s disease frontal cortex and increased co-distribution of BACE with the amyloid precursor protein. Neurobiol. Dis. 43, 176–183.
Sarajarvi, T., Haapasalo, A., Viswanathan, J., Makinen, P., Laitinen, M., Soininen, H., and Hiltunen, M. (2009). Down-regulation of seladin-1 increases BACE1 levels and activity through enhanced GGA3 depletion during apoptosis. J. Biol. Chem. 284, 34433–34443.
Sarnesto, A., Kohlin, T., Hindsgaul, O., Thurin, J., and Blaszczyk-Thurin, M. (1992). Purification of the secretor-type beta-galactoside alpha 1 – 2-fucosyltransferase from human serum. J. Biol. Chem. 267, 2737–2744.
Savonenko, A. V., Melnikova, T., Laird, F. M., Stewart, K. A., Price, D. L., and Wong, P. C. (2008). Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. U.S.A. 105, 5585–5590.
Schmechel, A., Strauss, M., Schlicksupp, A., Pipkorn, R., Haass, C., Bayer, T. A., and Multhaup, G. (2004). Human BACE forms dimers and colocalizes with APP. J. Biol. Chem. 279, 39710–39717.
Schobel, S., Neumann, S., Hertweck, M., Dislich, B., Kuhn, P. H., Kremmer, E., Seed, B., Baumeister, R., Haass, C., and Lichtenthaler, S. F. (2008). A novel sorting nexin modulates endocytic trafficking and alpha-secretase cleavage of the amyloid precursor protein. J. Biol. Chem. 283, 14257–14268.
Schobel, S., Neumann, S., Seed, B., and Lichtenthaler, S. F. (2006). Expression cloning screen for modifiers of amyloid precursor protein shedding. Int. J. Dev. Neurosci. 24, 141–148.
Schroder, M., and Kaufman, R. J. (2006). Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr. Mol. Med. 6, 5–36.
Seshadri, S., Kamiya, A., Yokota, Y., Prikulis, I., Kano, S., Hayashi-Takagi, A., Stanco, A., Eom, T. Y., Rao, S., Ishizuka, K., Wong, P., Korth, C., Anton, E. S., and Sawa, A. (2010). Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc. Natl. Acad. Sci. U.S.A. 107, 5622–5627.
Sheng, J. G., Price, D. L., and Koliatsos, V. E. (2003). The beta-amyloid-related proteins presenilin 1 and BACE1 are axonally transported to nerve terminals in the brain. Exp. Neurol. 184, 1053–1057.
Shi, X. P., Chen, E., Yin, K. C., Na, S., Garsky, V. M., Lai, M. T., Li, Y. M., Platchek, M., Register, R. B., Sardana, M. K., Tang, M. J., Thiebeau, J., Wood, T., Shafer, J. A., and Gardell, S. J. (2001). The pro domain of beta-secretase does not confer strict zymogen-like properties but does assist proper folding of the protease domain. J. Biol. Chem. 276, 10366–10373.
Shimizu, H., Tosaki, A., Kaneko, K., Hisano, T., Sakurai, T., and Nukina, N. (2008). Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol. Cell. Biol. 28, 3663–3671.
Simons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464.
Singer, O., Marr, R. A., Rockenstein, E., Crews, L., Coufal, N. G., Gage, F. H., Verma, I. M., and Masliah, E. (2005). Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat. Neurosci. 8, 1343–1349.
Sinha, S., Anderson, J. P., Barbour, R., Basi, G. S., Caccavello, R., Davis, D., Doan, M., Dovey, H. F., Frigon, N., Hong, J., Jacobson-Croak, K., Jewett, N., Keim, P., Knops, J., Lieberburg, I., Power, M., Tan, H., Tatsuno, G., Tung, J., Schenk, D., Seubert, P., Suomensaari, S. M., Wang, S., Walker, D., Zhao, J., Mcconlogue, L., and John, V. (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402, 537–540.
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., Brynjolfsson, J., Gunnarsdottir, S., Ivarsson, O., Chou, T. T., Hjaltason, O., Birgisdottir, B., Jonsson, H., Gudnadottir, V. G., Gudmundsdottir, E., Bjornsson, A., Ingvarsson, B., Ingason, A., Sigfusson, S., Hardardottir, H., Harvey, R. P., Lai, D., Zhou, M., Brunner, D., Mutel, V., Gonzalo, A., Lemke, G., Sainz, J., Johannesson, G., Andresson, T., Gudbjartsson, D., Manolescu, A., Frigge, M. L., Gurney, M. E., Kong, A., Gulcher, J. R., Petursson, H., and Stefansson, K. (2002). Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892.
Steiner, H., Fluhrer, R., and Haass, C. (2008). Intramembrane proteolysis by gamma-secretase. J. Biol. Chem. 283, 29627–29631.
Stockley, J. H., and O’Neill, C. (2008). Understanding BACE1: essential protease for amyloid-beta production in Alzheimer’s disease. Cell. Mol. Life Sci. 65, 3265–3289.
Sugimoto, I., Futakawa, S., Oka, R., Ogawa, K., Marth, J. D., Miyoshi, E., Taniguchi, N., Hashimoto, Y., and Kitazume, S. (2007). Beta-galactoside alpha2,6-sialyltransferase I cleavage by BACE1 enhances the sialylation of soluble glycoproteins. A novel regulatory mechanism for alpha2,6-sialylation. J. Biol. Chem. 282, 34896–34903.
Takasugi, N., Sasaki, T., Suzuki, K., Osawa, S., Isshiki, H., Hori, Y., Shimada, N., Higo, T., Yokoshima, S., Fukuyama, T., Lee, V. M., Trojanowski, J. Q., Tomita, T., and Iwatsubo, T. (2011). BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 31, 6850–6857.
Tamagno, E., Guglielmotto, M., Aragno, M., Borghi, R., Autelli, R., Giliberto, L., Muraca, G., Danni, O., Zhu, X., Smith, M. A., Perry, G., Jo, D. G., Mattson, M. P., and Tabaton, M. (2008). Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 104, 683–695.
Tamagno, E., Guglielmotto, M., Giliberto, L., Vitali, A., Borghi, R., Autelli, R., Danni, O., and Tabaton, M. (2009). JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol. Aging 30, 1563–1573.
Tamagno, E., Parola, M., Bardini, P., Piccini, A., Borghi, R., Guglielmotto, M., Santoro, G., Davit, A., Danni, O., Smith, M. A., Perry, G., and Tabaton, M. (2005). Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 92, 628–636.
Tesco, G., Koh, Y. H., Kang, E. L., Cameron, A. N., Das, S., Sena-Esteves, M., Hiltunen, M., Yang, S. H., Zhong, Z., Shen, Y., Simpkins, J. W., and Tanzi, R. E. (2007). Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 54, 721–737.
Tun, H., Marlow, L., Pinnix, I., Kinsey, R., and Sambamurti, K. (2002). Lipid rafts play an important role in A beta biogenesis by regulating the beta-secretase pathway. J. Mol. Neurosci. 19, 31–35.
Turner, R. T. III, Hong, L., Koelsch, G., Ghosh, A. K., and Tang, J. (2005). Structural locations and functional roles of new subsites S5, S6, and S7 in memapsin 2 (beta-secretase). Biochemistry 44, 105–112.
Turner, R. T. III, Loy, J. A., Nguyen, C., Devasamudram, T., Ghosh, A. K., Koelsch, G., and Tang, J. (2002). Specificity of memapsin 1 and its implications on the design of memapsin 2 (beta-secretase) inhibitor selectivity. Biochemistry 41, 8742–8746.
Vargas, M. E., Watanabe, J., Singh, S. J., Robinson, W. H., and Barres, B. A. (2010). Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc. Natl. Acad. Sci. U.S.A. 107, 11993–11998.
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S., Fuller, J., Edenson, S., Lile, J., Jarosinski, M. A., Biere, A. L., Curran, E., Burgess, T., Louis, J. C., Collins, F., Treanor, J., Rogers, G., and Citron, M. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741.
Velliquette, R. A., O’Connor, T., and Vassar, R. (2005). Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 25, 10874–10883.
Vetrivel, K. S., Meckler, X., Chen, Y., Nguyen, P. D., Seidah, N. G., Vassar, R., Wong, P. C., Fukata, M., Kounnas, M. Z., and Thinakaran, G. (2009). Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 284, 3793–3803.
von Arnim, C. A., Kinoshita, A., Peltan, I. D., Tangredi, M. M., Herl, L., Lee, B. M., Spoelgen, R., Hshieh, T. T., Ranganathan, S., Battey, F. D., Liu, C. X., Bacskai, B. J., Sever, S., Irizarry, M. C., Strickland, D. K., and Hyman, B. T. (2005). The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J. Biol. Chem. 280, 17777–17785.
von Arnim, C. A., Spoelgen, R., Peltan, I. D., Deng, M., Courchesne, S., Koker, M., Matsui, T., Kowa, H., Lichtenthaler, S. F., Irizarry, M. C., and Hyman, B. T. (2006). GGA1 acts as a spatial switch altering amyloid precursor protein trafficking and processing. J. Neurosci. 26, 9913–9922.
von Arnim, C. A., Tangredi, M. M., Peltan, I. D., Lee, B. M., Irizarry, M. C., Kinoshita, A., and Hyman, B. T. (2004). Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J. Cell. Sci. 117, 5437–5445.
von Koch, C. S., Zheng, H., Chen, H., Trumbauer, M., Thinakaran, G., Van Der Ploeg, L. H., Price, D. L., and Sisodia, S. S. (1997). Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 18, 661–669.
Wagner, T., and Pietrzik, C. U. (2011). The role of lipoprotein receptors on the physiological function of APP. Exp. Brain Res. (in press).
Wahle, T., Prager, K., Raffler, N., Haass, C., Famulok, M., and Walter, J. (2005). GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol. Cell. Neurosci. 29, 453–461.
Walter, J., Fluhrer, R., Hartung, B., Willem, M., Kaether, C., Capell, A., Lammich, S., Multhaup, G., and Haass, C. (2001). Phosphorylation regulates intracellular trafficking of beta-secretase. J. Biol. Chem. 276, 14634–14641.
Wang, W. X., Rajeev, B. W., Stromberg, A. J., Ren, N., Tang, G., Huang, Q., Rigoutsos, I., and Nelson, P. T. (2008). The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 28, 1213–1223.
Wang, Y., Thiele, C., and Huttner, W. B. (2000). Cholesterol is required for the formation of regulated and constitutive secretory vesicles from the trans-Golgi network. Traffic 1, 952–962.
Weinstein, J., Lee, E. U., Mcentee, K., Lai, P. H., and Paulson, J. C. (1987). Primary structure of beta-galactoside alpha 2,6-sialyltransferase. Conversion of membrane-bound enzyme to soluble forms by cleavage of the NH2-terminal signal anchor. J. Biol. Chem. 262, 17735–17743.
Westmeyer, G. G., Willem, M., Lichtenthaler, S. F., Lurman, G., Multhaup, G., Assfalg-Machleidt, I., Reiss, K., Saftig, P., and Haass, C. (2004). Dimerization of beta-site beta-amyloid precursor protein-cleaving enzyme. J. Biol. Chem. 279, 53205–53212.
Willem, M., Garratt, A. N., Novak, B., Citron, M., Kaufmann, S., Rittger, A., Destrooper, B., Saftig, P., Birchmeier, C., and Haass, C. (2006). Control of peripheral nerve myelination by the beta-secretase BACE1. Science 314, 664–666.
Willem, M., Lammich, S., and Haass, C. (2009). Function, regulation and therapeutic properties of beta-secretase (BACE1). Semin. Cell Dev. Biol. 20, 175–182.
Williams, N. M., Preece, A., Spurlock, G., Norton, N., Williams, H. J., Zammit, S., O’Donovan, M. C., and Owen, M. J. (2003). Support for genetic variation in neuregulin 1 and susceptibility to schizophrenia. Mol. Psychiatry 8, 485–487.
Wong, H. K., Sakurai, T., Oyama, F., Kaneko, K., Wada, K., Miyazaki, H., Kurosawa, M., De Strooper, B., Saftig, P., and Nukina, N. (2005). beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J. Biol. Chem. 280, 23009–23017.
Yan, R., Bienkowski, M. J., Shuck, M. E., Miao, H., Tory, M. C., Pauley, A. M., Brashier, J. R., Stratman, N. C., Mathews, W. R., Buhl, A. E., Carter, D. B., Tomasselli, A. G., Parodi, L. A., Heinrikson, R. L., and Gurney, M. E. (1999). Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402, 533–537.
Yan, R., Han, P., Miao, H., Greengard, P., and Xu, H. (2001). The transmembrane domain of the Alzheimer’s beta-secretase (BACE1) determines its late Golgi localization and access to beta -amyloid precursor protein (APP) substrate. J. Biol. Chem. 276, 36788–36796.
Yanagida, K., Okochi, M., Tagami, S., Nakayama, T., Kodama, T. S., Nishitomi, K., Jiang, J., Mori, K., Tatsumi, S., Arai, T., Ikeuchi, T., Kasuga, K., Tokuda, T., Kondo, M., Ikeda, M., Deguchi, K., Kazui, H., Tanaka, T., Morihara, T., Hashimoto, R., Kudo, T., Steiner, H., Haass, C., Tsuchiya, K., Akiyama, H., Kuwano, R., and Takeda, M. (2009). The 28-amino acid form of an APLP1-derived Abeta-like peptide is a surrogate marker for Abeta42 production in the central nervous system. EMBO Mol. Med. 1, 223–235.
Yang, L. B., Lindholm, K., Yan, R., Citron, M., Xia, W., Yang, X. L., Beach, T., Sue, L., Wong, P., Price, D., Li, R., and Shen, Y. (2003). Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4.
Yasojima, K., Mcgeer, E. G., and Mcgeer, P. L. (2001). Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 919, 115–121.
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., Spain, W. J., Mcknight, G. S., Scheuer, T., and Catterall, W. A. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149.
Yu, Y. J., Zhang, Y., Kenrick, M., Hoyte, K., Luk, W., Lu, Y., Atwal, J., Elliott, J. M., Prabhu, S., Watts, R. J., and Dennis, M. S. (2011). Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 3, 84ra44.
Zhang, D., Sliwkowski, M. X., Mark, M., Frantz, G., Akita, R., Sun, Y., Hillan, K., Crowley, C., Brush, J., and Godowski, P. J. (1997). Neuregulin-3 (NRG3): a novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. 94, 9562–9567.
Zhang, X., Zhou, K., Wang, R., Cui, J., Lipton, S. A., Liao, F. F., Xu, H., and Zhang, Y. W. (2007). Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 282, 10873–10880.
Zhao, J., Fu, Y., Yasvoina, M., Shao, P., Hitt, B., O’Connor, T., Logan, S., Maus, E., Citron, M., Berry, R., Binder, L., and Vassar, R. (2007). Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J. Neurosci. 27, 3639–3649.
Zhou, L., Chavez-Gutierrez, L., Bockstael, K., Sannerud, R., Annaert, W., May, P. C., Karran, E., and De Strooper, B. (2011). Inhibition of beta-secretase in vivo via antibody binding to unique loops (D and F) of BACE1. J. Biol. Chem. 286, 8677–8687.
Keywords: neurodegeneration, amyloid-beta, regulated intramembrane proteolysis, beta-secretase, aspartic protease
Citation: Dislich B and Lichtenthaler SF (2012) The membrane-bound aspartyl protease BACE1: molecular and functional properties in Alzheimer’s disease and beyond. Front. Physio. 3:8. doi: 10.3389/fphys.2012.00008
Received: 21 December 2011;
Accepted: 11 January 2012;
Published online: 17 February 2012.
Edited by:
Raquel Marin, Universidad de La Laguna, SpainReviewed by:
Giuseppina Tesco, Tufts University, USAMikko Hiltunen, University of Eastern Finland, Finland
Copyright: © 2012 Dislich and Lichtenthaler. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Stefan F. Lichtenthaler, German Center for Neurodegenerative Diseases (DZNE), site Munich, Schillerstrasse 44, 80336 Munich, Germany. e-mail: stefan.lichtenthaler@dzne.lmu.de