
- Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
A broad array of evidence indicates that T lymphocytes make significant contributions to vascular inflammation in the setting of atherosclerotic disease, hypertension, autoimmune vasculitis, and other disorders. Experimental data show that costimulatory and coinhibitory pathways involving molecules of the B7-CD28 and TNF–TNFR families regulate T cell responses that promote vascular disease. Antigen presenting cells (APCs) display both peptide–major histocompatibility complex antigen and costimulators or coinhibitors to T cells. Two major types of APCs, dendritic cells (DCs) and macrophages, are present in significant numbers in the walls of arteries affected by atherosclerosis and arteritis, and some DCs are present in normal arteries. Costimulatory and coinhibitory molecules expressed by these vascular APCs can contribute to the activation or inhibition of effector T cells within the arterial wall. Vascular DCs may also be involved in transport of antigens to secondary lymphoid organs, where they activate or tolerize naïve T cells, depending on the balance of costimulators and coinhibitors they express. Costimulatory blockade is already an approved therapeutic approach to treat autoimmune disease and prevent transplant rejection. Preclinical models suggest that costimulatory blockade may also be effective in treating vascular disease. Experiential data in mice show that DCs pulsed with the appropriate antigens and treated in a way that reduces costimulatory capacity can reduce atherosclerotic disease, presumably by inducing T cell tolerance. Progress in treating vascular disease by immune modulation will require a more complete understanding of the functions of different costimulatory and coinhibitory pathways and the different subsets of vascular APCs involved.
Introduction
This review will discuss the current knowledge on costimulatory and coinhibitory interactions between DC or macrophage antigen presenting cells (APCs) and T cells in the context of vascular disease. There is great interest in costimulatory and coinhibitory pathways because they have profound influences on T cell mediated immunity, and there are drugs targeting these pathways already approved or in trials for the purpose of therapeutically inhibiting or enhancing immune responses. The review will begin with a primer on costimulatory and coinhibitory molecules and how they contribute to DC and macrophage interactions with T cells. Next, evidence for the importance of these pathways in diseases of blood vessels will be reviewed, emphasizing atherosclerosis, but also considering other disorders. Finally, the potential for treating vascular disease by blocking or stimulating these pathways will be addressed.
Antigen Presenting Cell Function of Dendritic Cells and Macrophages and the Role of Costimulation and Coinhibition
The antigen receptors of T lymphocytes (TCR) recognize molecular complexes displayed on the surface of other cells called APCs. The molecular complex that most T cells recognize is comprised of a peptide fragment of a protein non-covalently bound to a major histocompatibility complex (MHC) protein (Abbas et al., 2011). A numerically minor subset of T cells called NKT cells, recognize lipids bound to a class I MHC like protein called CD1. Both dendritic cells (DCs) and macrophages, which are found in arterial walls in various disease conditions, perform APC functions, including the sampling of foreign and self proteins, the proteolytic processing of these proteins into peptide fragments that can bind to MHC proteins, and the display of the peptide–MHC complexes on the cell surface for possible recognition by T lymphocytes. CD4+ T cells recognize peptide–class II MHC complexes, and CD8+ T cells recognize peptide–class I MHC complexes. A related process for display of lipid/CD1 complexes is also performed by DCs. DCs and macrophages are important APCs in protective T cell responses to infectious pathogens (Heath and Carbone, 2009; Murray and Wynn, 2011), and in pathogenic T cell responses against self or altered self proteins in immune/inflammatory diseases. DCs likely also play a role inducing T cell tolerance to self by presenting peptides from normal self proteins to T cells under non-inflammatory conditions (Figure 1A) (Maldonado and von Andrian, 2010).
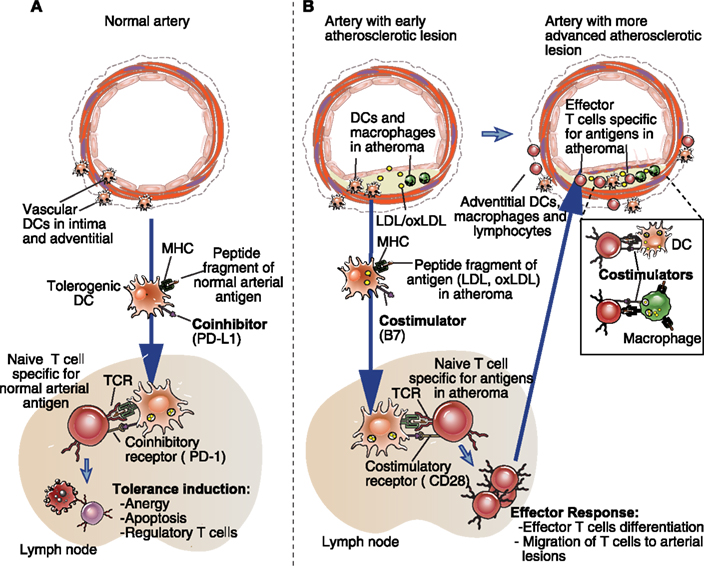
Figure 1. Possible roles of costimulators and coinhibitors on vascular dendritic cells and macrophages in health and disease. (A) Dendritic cells are present in low numbers in normal arterial walls, in the adventitia, and in the intima. Based on what is known about other tissues, these resting or immature DCs may have some migratory activity to secondary lymphoid organs (e.g., lymph node), where they could induce T cell tolerance to normal vascular antigens. Mechanisms of peripheral T cell tolerance induction include anergy or apoptosis of the self-antigen specific naïve T cells, and the induction of regulatory T cells. The lack of costimulators (e.g., CD80, CD86), and the presence coinhibitors (e.g., CD274, CD273) on these DC are likely to be important characteristic of their tolerogenic DC phenotype. (B) In atherosclerosis, both macrophages and DCs accumulate in the arterial adventitia and in intimal lesions, along with T cells. Innate immune stimuli related to the early retention and oxidation of LDL in the arterial intima may induce vascular DC maturation, expression of costimulatory molecules and uptake, and/or intracellular generation of protein antigens. These DCs could carry the antigens from the early atheromas to lymph nodes, and present them, in the form of peptide–MHC complexes, to naïve T cells specific for those antigens. The costimulators expressed on these mature DCs are required for productive naïve T cell activation and differentiation into effector T cells. The effector T cells then migrate out of the lymphoid tissues, and some will home to the atherosclerotic lesion. Both macrophages and DCs in the lesion may process and present the same atheroma-antigens (e.g., ApoB100 derived peptides) to the effector T cells and activate the T cells to perform inflammatory effector functions that promote arterial disease. Costimulators present on the DCs and macrophages may enhance the effector T cell responses. Note that CD4+ T cell responses are depicted in the figure, but the same principals apply for CD8+ T cell responses. A similar pattern of lymphoid tissue-based, costimulator-dependent priming and arterial wall based effector T cell responses is likely to be relevant to other vascular inflammatory diseases, as discussed in the text.
In addition to displaying peptide–MHC antigens to T cells, APCs also express cell surface molecules called costimulators, which engage costimulatory receptors on T cells concurrently with TCR recognition of peptide–MHC complexes (Gotsman et al., 2008; Sharpe, 2009; Bour-Jordan et al., 2011). Both the TCR with its associated signaling molecules (CD3 and ζ proteins) and the costimulatory receptors are recruited into specialized lipid rafts that comprise the T cell side of an immune synapse with the APCs. When costimulators on APCs bind to their receptors on the T cell, signals are generated in the T cell that work synergistically with the signals generated by TCR binding of antigen to activate functional responses of the T cell. There are many molecules that have been characterized as costimulators or T cell costimulatory receptors; some of the best characterized examples are shown in Table 1. The type of APC that activates a T cell, the particular costimulatory molecules and receptors that are engaged, and the nature of the functional responses of the T cells all vary with the history of antigen exposure of the T cell, and the particular subset the T cell belongs to. Naïve T cells (both CD4+ and CD8+), which have no prior antigen experience after emerging from the thymus, are efficiently activated only by DCs within secondary lymphoid organs (e.g., lymph nodes, spleen). DCs express both class I MHC, needed to display antigen to CD8+ T cells, and class II MHC needed to display antigen to CD4+ T cells. In addition, DCs express a variety of costimulatory molecules, especially when they are exposed to innate immune stimuli such as Toll-like receptor (TLR) ligands. Naïve T cells have stringent requirements for costimulation by members of the B7 family of proteins (CD80, CD86), which bind to CD28 on the T cell. Costimulation by CD275 (ICOS ligand) on APCs binding to CD278 (ICOS) on T cells, or TNF superfamily costimulators (CD137 ligand, CD252, CD70) on APCs binding to TNF receptor superfamily proteins (CD137, CD134, CD27) on T cells, likely occur mainly after the initiation of T cell responses (Ishii et al., 2010; Gerdes and Zirlik, 2011). This is because the expression of CD278, CD137, CD134, and CD27 on T cells is usually induced or upregulated in response to TCR and CD28 signaling, and their expression on naïve or resting memory T cells is very low. The response of naïve T cells to antigen recognition plus costimulation includes clonal expansion, i.e., multiple rounds of mitosis, and differentiation into effector T cells and memory T cells. Effector cells include CD4+ helper T cells and CD8+ cytotoxic T lymphocytes (CTL), which survive for a short time (days–weeks), while memory T cells may survive for many years. Compared to naïve T cells, antigen experienced T cells (effector and memory T cells) can be activated by a wider variety of APCs, as demonstrated by the many cell types that can present antigen to and thereby become targets of CTL. Although it is commonly assumed that antigen experienced T cells have less stringent requirements for costimulation, TNF family costimulators appear to be particularly important for CD4+ and CD8+ effector/memory T cell responses (Sharpe, 2009), and there is evidence that CD28-dependent costimulation is required for optimal memory CD8+ T cell responses to some viral infections in vivo (Boesteanu and Katsikis, 2009). CD4+ effector and memory T cells respond to peptide–class II MHC antigen presented by B cells within secondary lymphoid tissues, or presented by macrophages and DCs in non-lymphoid and lymphoid tissues. Some evidence indicates that human microvascular endothelial cells, which express class II MHC, may also present antigen to effector and memory T cells. Almost any class I MHC expressing cell can serve as an activating APC for CD8+ cytotoxic T cells. Thus antigen experienced T cells are relatively promiscuous in responding to various APC types in various locations. Nonetheless, there is ample evidence that the responses of antigen experienced human and mouse T cells in vitro and in vivo are enhanced by costimulatory molecules (Gotsman et al., 2008; Sharpe, 2009). In particular, costimulatory pathways that are most important for antigen experienced T cells involve receptors that are only induced after activation of a naïve T cell, such as CD278. The types of responses that are dependent on induced costimulators are the specialized effector functions of the T cells, such as germinal center formation and B cell help in the case of ICOS, rather than generic clonal expansion seen in all naïve T cell response.
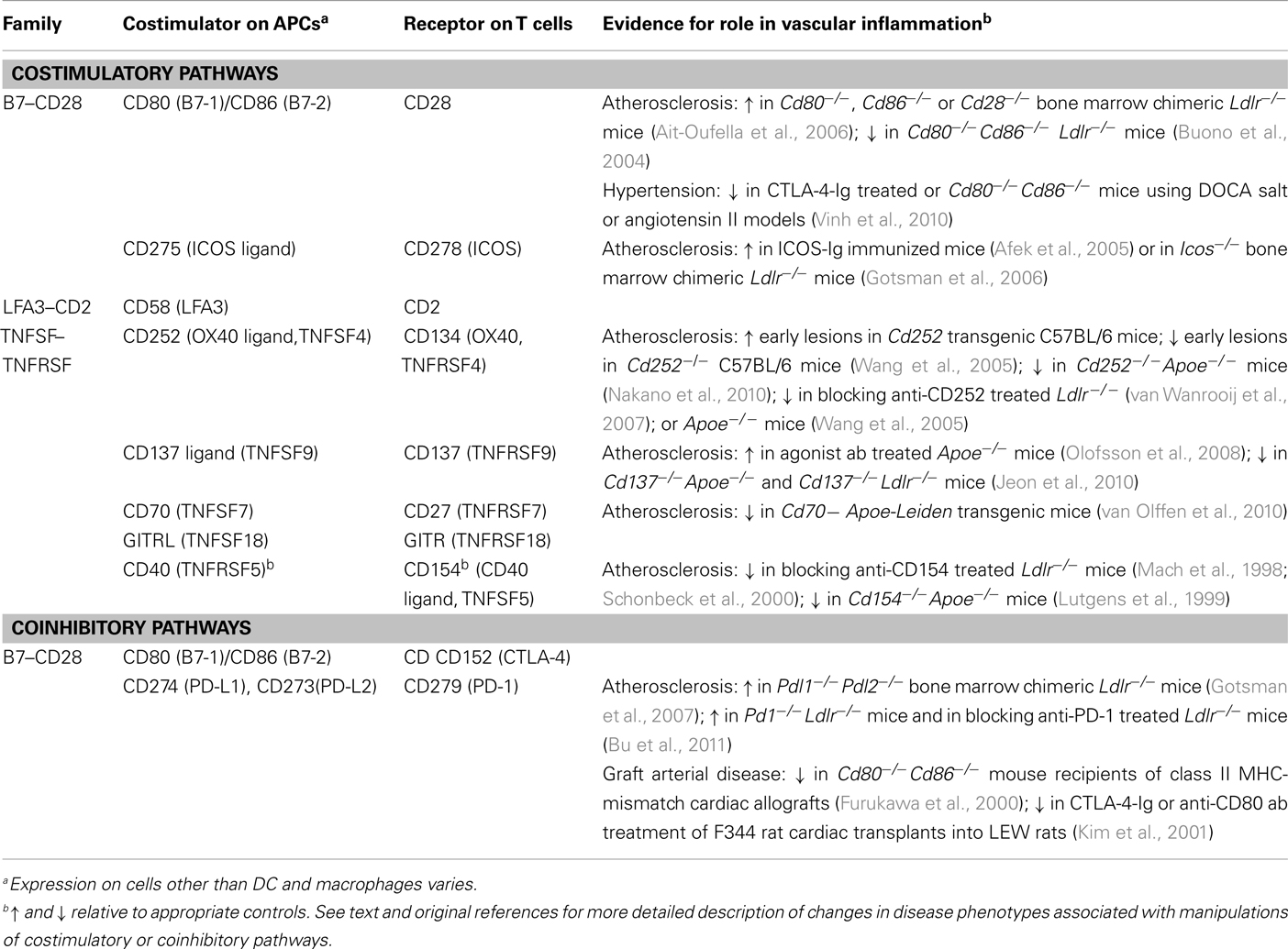
Table 1. Selected major T cell costimulatory and coinhibitory pathways.
The ability of APCs to costimulate T cells is enhanced by the CD154:CD40 pathway. CD154 (CD40 ligand) is a membrane bound TNF superfamily protein that is inducibly expressed on effector T cells after activation by antigen presentation. CD40L binds to the TNF receptor superfamily protein CD40 expressed constitutively by DCs, macrophages, B cells, and other cell types. Ligated CD40 generates signals that activate many functional responses of each of these cell types. One of the responses is upregulation of costimulatory molecules, such as CD80 and CD86, thereby enhancing the ability of these APCs to activate naïve T cells (Grewal and Flavell, 1998; Miga et al., 2000). Therefore, CD40L:CD40 interactions may not directly provide costimulation, but rather enhance the ability of APCs to costimulate T cells. Nonetheless, there is some evidence suggesting that CD40 expressed on CD8+ T cells functions as a costimulatory signaling receptor (Munroe, 2009).
Coinhibitory pathways are mediated by proteins expressed on APC membranes that engage receptors on T cells concurrently with antigen recognition, like costimulatory pathways (see Table 1). However, the functional consequence of coinhibitory receptor engagement is to inhibit T cell activation. These pathways are best designated by the receptors on T cells. The only well defined coinhibitory receptors are CTLA-4 (CD152) and PD-1 (CD279), both members of the CD28 family of proteins, and both expressed on T cell surfaces only after activation by antigen. CTLA-4 binds to either CD80 or CD86 (the same proteins that costimulate T cells by binding to CD28). PD-1 binds both PD-L2 (CD273) and PD-L1 (CD274), which are closely related members of the B7 family. PD-L2 is expressed by DCs, macrophages, and B cells. PD-L1 is more broadly expressed on APCs and non-hematopoietically derived tissue cells, especially after stimulation by TLR ligand or type 1 or type II interferons (Francisco et al., 2010). Of particular relevance to this review, PD-L1 can be expressed by vascular endothelial cells in vitro and in vivo (Rodig et al., 2003; Grabie et al., 2007), and PD-L1 is reported to be expressed on vascular smooth muscle cells in vitro (Koga et al., 2004). The importance of these pathways in regulating T cell responses was first demonstrated in gene knockout mouse models, which showed an absolute requirement for CTLA-4 to prevent lethal systemic lymphoproliferative/autoimmune disease (Tivol et al., 1995; Waterhouse et al., 1995), and a more subtle requirement of PD-1 or its ligands to prevent autoimmunity or dysregulated immune response to infections (Francisco et al., 2010). The presumed physiological function of these coinhibitory pathways is to down-regulate or limit appropriate immune responses to pathogens, and to prevent autoimmune responses. Blockade of CTLA-4 or PD-1 with mAbs in order to boost T cell responses holds promise as a way to promote anti-tumor immunity in patients with metastatic cancers (Topalian et al., 2011).
So what is the relevance of costimulatory pathways to vascular inflammation? Ample evidence exists to show that different T cell subsets contribute to the regulation or progression of atherosclerosis, and compelling emerging evidence indicates that T lymphocytes contribute to vascular pathology in essential hypertension, as discussed in other reviews in this volume. Vascular damage in the context of a subset of vasculitidies (such a giant cell arteritis) is primarily caused by T lymphocytes, and other antibody mediated vasculitidies are dependent on T cell-dependent helper functions for the production of pathogenic antibodies. In all these examples, it is clear that effector T cells with specificity for antigens expressed in blood vessel walls are being activated and causing damage to the vessels. Given our understanding of the progression of T cell mediated immune responses, an early step in the pathogenesis of these vascular diseases is likely to be DC antigen presentation to, and costimulation of, naïve T cells specific for vascular antigens (Figure 1B). Furthermore, macrophages and DCs are present within the inflammatory infiltrates in the affected vessel walls in these diseases, often adjacent to infiltrating T cells. Therefore, DC and macrophage costimulatory signals could enhance the pathologic functions of the effector T cells in the blood vessel wall. Based on these assumptions, is it likely that costimulatory and coinhibitory molecules expressed on DCs will influence activation of naïve T cells that have the potential to become pathogenic effectors in vascular disease, while the same molecules expressed on macrophages are likely to influence only antigen experienced T cells in the vessel walls. Otherwise, there is neither useful data nor adequate in vivo methods available that resolve different functional consequences to a T cell after engagement of a particular costimulatory molecule on a DC vs. a macrophage.
Costimulation by DCs and Macrophages in Atherosclerosis
Indirect evidence consistent with a role for T cell costimulators in atherosclerosis includes the demonstration of their expression in human and mouse lesions. For example, CD80 and CD86 were detected on lesional macrophages of human arterial plaques detected by double color immunohistochemistry (de Boer et al., 1997), and in lesions of Ldlr−/− mice by single color immunohistochemistry (Buono et al., 2004). The B7 family costimulator CD275 (ICOS ligand) and its CD28 family receptor CD278 (ICOS) were readily detected by immunohistochemistry in the aortic lesions of Apoe−/− mice (Afek et al., 2005). The TNFSF T cell costimulator CD252 (OX40 ligand) was detected on human carotid plaque macrophages (CD68+ cells) by double-label immunofluorescence staining (Olofsson et al., 2009) and in Apoe−/− mouse aortic lesions by single color immunohistochemistry (Wang et al., 2005). In each of these cases, the receptors for these costimulators were detected on lesional T cells. Additionally, GITR ligand (another TNF family molecule with putative costimulatory activity) colocalized with macrophages (CD68+ cells) in human atheroma using two color immunohistochemistry (Kim et al., 2006). Definitive data that demonstrates the expression of costimulatory molecules on DCs within atherosclerotic lesion is still lacking.
More direct evidence that supports the hypothesis that T cell costimulation promotes atherosclerotic vascular disease comes largely from experiments testing if costimulatory blockade by gene deletion or pharmacologic agents alters lesion development in mouse models of atherosclerosis. Global genetic deletion of both CD80 and CD86 resulted in decreased development of lesions in the Ldlr−/− mouse model of atherosclerosis, as well as a reduction in the hypercholesterolemia-dependent priming of CD4+ T cells specific for the atherosclerosis-associated antigen HSP60 (Buono et al., 2004). Targeted deletion or transgenic over-expression of CD252 in B6 mice (without Apoe or Ldlr mutations) resulted in reduced or enhanced aortic root early lesion development, respectively (Wang et al., 2005), and targeted deletion of CD252 in Apoe−/− mice resulted in reduced lesion development and decreased adventitial neovascularization (Nakano et al., 2010). Likewise, antibody blockade of CD252 in Ldlr−/− mice (van Wanrooij et al., 2007) or Apoe−/− mice (Wang et al., 2005) significantly reduced aortic lesion development. Mouse studies also indicate that CD137 (TNFRSF9), a costimulatory receptor expressed on T lymphocytes, contributes to pro-atherosclerotic T cell responses. Agonist anti-CD137 antibody treatment of Apoe−/− mice increased atherosclerosis lesion development and lesion inflammation (Olofsson et al., 2008), and targeted deletion of CD137 in both Apoe−/− and Ldlr−/− mice reduced lesion development and inflammation (Jeon et al., 2010). One study addressed the impact of constitutive CD27 signaling on atherosclerosis, by transgenic over-expression of CD70 on B cells in hypercholesterolemic ApoE*-Leiden mice (van Olffen et al., 2010). Paradoxically, these mice were resistant to atherosclerosis lesion development, although the mechanisms remain unclear.
Although most of the mouse studies discussed above convincingly show that costimulators in the B7 and TNF protein families can have pro-atherogenic effects, they neither implicate nor rule out the importance of vascular DCs or macrophages. The presence DCs in the normal and atherosclerotic arterial walls has been reported by several laboratories, as discussed in other reviews in this volume, but proof that these DCs present antigen and costimulate T cells within lesions is still quite limited. Live cell two-photon microscopic imaging studies show that endogenous DCs in atherosclerotic aortas of Apoe−/− mice are able to present antigen to and activate previously stimulated CD4+ T cells (Klaus Ley, La Jolla Institute of Immunology, personal communication). The interrogation of costimulatory pathways using this approach will be of great interest.
Emerging evidence for heterogeneity of DC subtypes within arterial walls suggests that the effects of antigen presentation and costimulation on vascular pathology are complex. One study making use of CCL17 and CD11c fluorescent reporter mice, showed that CCL17 expressing CD11c+ DCs accumulate in intimal lesions of ApoE−/− mice, and these DCs form close contacts with lesional CD4+ T cells (Weber et al., 2011). Although costimulator expression on these DCs was not shown in situ, reporter gene detectable CCL17 producing CD11c cells from lymph nodes did express CD80 and CD86. The DC-derived CCL17 repressed Treg responses and enhanced pro-atherogenic CD4+ T effector cell responses in Apoe−/− mice, but whether or not the effect of the DCs was localized to lesions or lymphoid tissues was not resolved. In another study, intimal DCs were detected in normal mouse aortas, and they belonged to two developmental subsets, classical Flt3–Flt3L signaling-dependent, CD103+CD11b- DCs, and macrophage-colony stimulating factor-dependent CD14+CD11b+DCSIGN+ monocyte-derived DCs (Choi et al., 2011). No analyses of costimulatory molecule expression on the aortic DCs of either subset were reported. Both subsets increased in numbers in atherosclerotic lesions of Ldlr−/− mice. The loss of the classical subset in Flt3−/− × Ldlr−/− mice resulted in enhanced atherosclerosis with reduced aortic Treg (in contrast to the effect of the loss of CCL17+ DC described above). However, lymph node and splenic Treg were also reduced in the FLt3-null mice, and therefore the relative importance of Flt3–Flt3L signaling-dependent DC regulation of pro-atherogenic T cell responses in the blood vessel wall vs. in lymphoid tissues cannot be distinguished by this study either.
Vascular DCs could promote pro-atherogenic T cells responses by picking up atheroma-associated antigens, migrating to lymphoid tissues, and presenting the antigens to recirculating naïve T cells (Figure 1B). However, it is likely that some of the antigens that are recognized by pathogenic effector T cells within vessel walls are also present systemically. Therefore, the primary activation of naïve T cells specific for these antigens within lymphoid tissues may not require that the antigen be carried to lymph by DCs originating in the arterial walls. For example, modified or native lipoproteins that are recognized by pro-atherogenic effector T cells are present systemically in dyslipidemic individuals. The costimulatory molecules interrogated in the atherosclerosis studies discussed above are expressed on DCs and macrophages in lymphoid organs where naïve T cells are activated, and the targeted costimulatory receptors are expressed by naïve T cells (e.g., CD28) or induced early after activation (e.g., CD278, CD134).
The highly abnormal extracellular and intracellular lipid environments in atherosclerotic lesions have direct impacts on DCs and macrophages, the most obvious of which is the formation of foam cells loaded with cholesterol and cholesteryl esters after uptake of oxidatively modified LDL particles that have been trapped by intimal matrix molecules (Tabas et al., 2007; Moore and Tabas, 2011). Although monocyte derive macrophages have long been known to comprise a significant fraction of lesional foam cells, more recent work has shown that DCs within arterial intima can also take up lipid and appear as foam cells (Packard et al., 2008; Cybulsky and Jongstra-Bilen, 2010; Paulson et al., 2010). The overall functional impact of lipid accumulation on macrophage and DC antigen presentation function remain unresolved. The expression of costimulators on APCs is induced by innate inflammatory stimuli, via pattern recognition receptors including TLRs. Oxidatively modified LDL (ox-LDL), which is the major source of cholesterol taken up by macrophages and DCs during foam cell formation in arterial lesions, can act as an innate immune stimulus (Bae et al., 2009; Choi et al., 2009; Chavez-Sanchez et al., 2010; Miller et al., 2011). The abnormal accumulation of lipids in the skin of hypercholesterolemic Apoe−/− mice was shown to inhibit DC migration to draining lymph nodes (Angeli et al., 2004), leading to the speculation that a similar impairment of DC and macrophage migration out of the vascular wall would enhance local lesion formation and inflammation (Randolph, 2008). The current knowledge about the retention and migration of DCs and macrophages out of atherosclerotic lesions is discussed in more detail in other reviews in this volume. Suffice it to say here that one cannot exclude the possibility of vascular DCs transporting T cell antigens to secondary lymphoid organs based on the limited data available, and impairment of migration may not be significant early in lesion development, when priming of pro-atherogenic T cells is perhaps most relevant. There is also data indicating that ox-LDL blocks DC activation and antigen presenting functions. Myeloid DCs from the spleens of high fat diet fed Apoe−/− mice had impaired activation responses to TLR-ligands, including impaired upregulation of CD80 and CD86 (Shamshiev et al., 2007). Systemic T cell defense against an intracellular pathogen was also shown to be impaired in the dyslipidemic mice. In that study, ox-LDL was identified as the component responsible for uncoupling TLR-mediated signaling in the DCs. In another study, impaired antigen processing functions, but not impaired costimulator expression, was seen in triglyceride loaded DCs in tumor bearing mice (Herber et al., 2010). In contrast, splenic DC antigen presenting functions and CD80 and CD86 expression were not affected by hypercholesterolemia in either Ldlr−/− or Apoe−/− mice (Packard et al., 2008). In vitro experiments have also shown that ox-LDL can induce human monocyte and DC upregulation of B7 family costimulators (Fortun et al., 2001; Nickel et al., 2009). Overall, given the robust evidence of T cell activation in atherosclerotic plaques, and evidence of systemic T cell responses to atherosclerosis-associated antigens in hypercholesterolemic mice and humans (Ketelhuth and Hansson, 2011), it is seems unlikely that vascular APCs nor APCs in secondary lymphoid organs have significant defects in costimulatory function in individuals with atherosclerotic disease. Furthermore, there is little evidence for immune surveillance defects in dyslipidemic humans, which is consistent with normal costimulatory functions of APCs.
Ample experimental evidence indicates that the CD40–CD40 ligand pathway of cellular activation promotes atherosclerotic lesion development and the inflammatory phenotype of lesions. This was originally shown by antibody blockade or targeted mutation of CD154 in atherosclerosis prone mice (Mach et al., 1998; Lutgens et al., 1999; Schonbeck et al., 2000). However the expression of CD154 on platelets, mast cells, B cells, and non-hematopoietic cells including vascular smooth muscle cells endothelial cells, and the expression of CD40 on endothelial cells, macrophages, and DCs makes it difficult to determine the mechanisms of the in vivo effects of CD40 ligand blockade on atherosclerosis (Lievens et al., 2009). In fact, despite the large number of studies examining the relationship of CD154 and CD40 in atherosclerotic disease in humans and mice, there is virtually no data addressing if enhancement of T cell costimulation is a key mechanism. Experiments that examine CD154 blockade in the absence of T cell costimulation or T lineage selective ablation of CD154 expression in atherosclerosis prone mice, could provide some insights into this question.
Coinhibition by DCs and Macrophages in Atherosclerosis
Mouse models have also established a role for T cell coinhibition in regulating pro-atherogenic T cell responses, and implicate but do not prove that vascular DCs and/or macrophages are involved in this mode of regulation. CD11c+ and Mac3+ cells within aortic lesions of Ldlr−/− mice express PD-L1 and hypercholesterolemia enhances PD-L1 expression on splenic macrophages and DCs from these mice (Gotsman et al., 2007). Targeted deletion of both PD-L1 and PD-L2 or of PD-1 in Ldlr−/− mice lead to increased atherosclerotic lesion development, and marked increases in CD4+ and CD8+ T cells within lesions (Gotsman et al., 2007; Bu et al., 2011). Furthermore, T cells isolated from hypercholesterolemic PD-L1/2 or PD-1 deficient Ldlr−/− mice are more activated than T cells from Ldlr−/− control mice. Treatment of Ldlr−/− mice with a blocking anti-PD-1 antibody also causes increased lesion development and T cell infiltration (Bu et al., 2011). The robust lesional CD8+ T cell responses seen in Ldlr−/− mice lacking the PD-1:PD-L1/2 pathway and the enhanced ability of PD-1 deficient CD8+ T cells to kill vascular cells are both consistent with an important role for coinhibition in suppressing effector CD8+ T cells in the vascular wall. There is little published data addressing the contribution of the PD-1/PD-L1/2 pathway in human atherosclerotic disease. PD-1 and PD-L1 expression is reported to be significantly reduced on blood T cells and myeloid DCs in coronary artery disease patients compared to in healthy controls (Lee et al., 2009).
The impact of CTLA-4 on atherosclerosis has not been studied in mice, mainly because of the lethal lymphoproliferative and autoimmune phenotype of CTLA-4 knockout mice (Tivol et al., 1995; Waterhouse et al., 1995). Blocking anti-CTLA-4 antibody treatment is approved for treatment of some cancers, and blocking anti-PD-1 antibodies are being used in clinical trials also for cancers treatment (Topalian et al., 2011). If this type of therapy becomes more established, there will be an opportunity to study the impact of coinhibitory blockade on atherosclerotic disease in humans, albeit those with metastatic cancer. To date, there has been limited success in developing agonist reagents that can be used to test the possible therapeutic impact of pharmacologically induced PD-1 or CTLA-4 signaling on atherosclerosis in animal models.
In addition to coinhibitory pathways, regulatory T cells (Treg) are essential to prevent autoimmunity and to regulate effector T cell responses to microbes (Josefowicz et al., 2012). The relationship between Treg and costimulatory or coinhibitory pathways is complex and not fully understood. Most Treg are CD4+ CD3+TCRαβ+ CD25+ CD127loGITR+ CTLA-4+ FoxP3+ cells. FoxP3 is a transcription factor required for Treg development and function. The critical importance of Treg to immunoregulation is evident from the lethal autoimmune phenotype of patients with FoxP3 mutations (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome), and of mice with similar mutations (SCURFY mice) or targeted FoxP3 gene deletion. Natural Treg develop in the thymus and populate secondary lymphoid tissues. Induced or adaptive Treg differentiate from naïve CD4+ T cells in secondary lymphoid organs in response to antigen stimulation in the presence of certain soluble stimuli including TGFβ and retinoic acid. Tregs suppress naïve and effector T cell activation, likely by several different mechanisms including the secretion of immunosuppressive cytokines (IL-10, TGFβ, IL-35) and by direct contact with T cells or APCs via CTLA-4 and PD-L1 (Shevach, 2009). One currently favored mechanisms of Treg suppression is by the binding of CTLA-4 on the surface of Treg to CD80 and CD86 on the surface of DCs and the downregulation of removal of CD80 and CD86 from the DC membrane (Onishi et al., 2008; Wing et al., 2011). Thus, Treg may block the ability of the CD80/CD806 proteins on DCs to bind to CD28 on naïve T cells, thereby impairing initiation of T cell immune responses. A similar mode of action of Treg in peripheral tissues including vessels walls may regulate effector T cell activation by APCs. Treg development in the thymus is at least partially dependent on CD80, CD86, CD28, and CD278. Under certain conditions, such as after bone marrow transplantation in mice, or in autoimmune prone mice (e.g., NOD mice) the lack CD80 and CD86 or CD28 results in profound Treg deficiency and autoimmunity (Bour-Jordan et al., 2004; Ait-Oufella et al., 2006). In mouse models of atherosclerosis, hematopoietic cell deficiency of the CD80/86:CD28 or CD275:CD278 pathways, created by bone marrow transplantation of gene knockout marrow into recipients wild type for those genes, results in reduced Treg numbers and function and enhanced atherosclerotic lesion development and inflammation (Ait-Oufella et al., 2006; Gotsman et al., 2006). Immunization of ApoE−/− with an ICOS-Ig fusion protein with the intent of inducing a blocking anti-ICOS antibody responses also increased atherosclerosis and increased T cell IFNγ expression (Afek et al., 2005). Whether or not Treg responses were impaired in the ICOS-immunized mice was not determined. Coinhibition appears to influence induced Treg responses as well. For example, PD-L1deficeiny markedly impaired differentiation and maintenance of induced Treg (Francisco et al., 2009). Thus both costimulatory and coinhibitory pathways are required for both effector and regulatory T cell responses, and the net effects of blocking or enhancing these coregulatory pathways for therapeutic benefit is likely to vary with timing, tissue, and disease process.
Costimulation and Coinhibition by DCs and Macrophages in Other Vascular Diseases
The possibility that inflammation and immune responses contribute to hypertensive disease has been addressed in various studies over many years, and recent work using modern immunological models has established an important role for T cell responses in this disease (Harrison et al., 2010), as discussed in detail in another review in this volume. One of the compelling recent studies linking T cell responses with hypertension focused on costimulation, and showed that pharmacologic blockade or genetic deficiency of CD80 and CD86 in mice rendered them resistant to development of hypertension using two different models (Vinh et al., 2010). Furthermore, perivascular T cell infiltrates that accumulate in the hypertensive mice were significantly reduced when costimulation was impaired. As in the atherosclerosis studies discussed above, it remains to be proven that blockade of T cell costimulation by APC within the affected blood vessels was relevant to the anti-hypertensive effect.
Graft arterial disease (transplant arteriopathy, graft arteriosclerosis) is characterized by chronic remodeling of allograft arteries by intimal smooth muscle cell accumulation, progressing to luminal obstruction, and ischemic damage to the graft. This process is the major cause of long term failure of transplanted hearts and kidneys. Graft arterial disease is believed to be initiated, and may or may not be sustained, by graft recipient alloreactive T cells (Mitchell and Libby, 2007). In rodent models of cardiac allograft rejection, blocking, or genetic deficiency of B7–CD28 costimulatory pathways reduces development of graft arterial disease (Furukawa et al., 2000; Kim et al., 2001). However, there is no published evidence for a role of vascular macrophages or DCs in this disease, and in fact DC or macrophages have not been found in significant numbers in affected vessels. Thus, the effectiveness of costimulatory blockade in preventing graft arterial disease is more likely to reflect interference with the priming of alloreactive T cells in secondary lymphoid organs.
The pathogenesis of certain forms of arteritis, including giant cell arteritis, Takayasu’s disease, and Kawasaki disease, involve T cell mediated damage to the vessel wall. DCs are present within the media of the affected vessels in these diseases, and their interaction with infiltrating T cells has been documented. The influence of costimulatory pathways in either the lymphoid tissue-based priming of the vasculopathogenic T cells, or in the local activation of these T cells in the arteries, is largely unexplored. In giant cell arteritis, a population of CD83+ CD86+ DCs infiltrates into the media of the artery from the adventitia and colocalize with T cells (Krupa et al., 2002). In coronary arteries of patients with Kawasaki disease, mature myeloid DCs identified by expression of BDC1, HLA-DR, and the costimulator CD83 accumulate in the intima and colocalize with T cells (Yilmaz et al., 2007). In Takayasu’s disease, immunohistochemical S-100+ cells adjacent to CD3+ cells in the adventitia of arterial specimens was interpreted as showing DC interactions with T cells (Inder et al., 2000a,b).
Mouse models of T cell mediated arteritis have been developed and may be useful to explore the contribution of costimulatory pathways to these disorders and the therapeutic potential of blocking these pathways. In a humanized mouse model of giant cell arteritis, human artery segments are implanted into SCID mice, and human allogeneic T cells from healthy donors or from giant cell arteritis patients are adoptively transferred into the mice. After LPS injection, a T cell rich inflammatory infiltrate and medial infiltration of vascular DCs develops in the arteries (Ma-Krupa et al., 2004). A mouse model of coronary arteritis resembling Kawasaki disease has also been described, in which mice are injected with a cell wall extract isolated from Lactobacillus casei (Lehman et al., 1985). The coronary arteritis that develops in this model resembles that seen in the human disease, and is dependent on T cells (Schulte et al., 2009). Interestingly, F4/80+ macrophages, activated myeloid DCs, and plasmacytoid DCs are present in the arterial lesions, and CD3+ T cells colocalize with the myeloid DCs. It will be of interest to explore the impact of costimulator blocking agents or costimulatory gene knockout approaches on disease development in these models.
Therapeutic Targeting of the Costimulatory and Coinhibitory Functions of Vascular APCs
Given the evidence for a significant contribution of effector T cells to the pathogenesis of various inflammatory diseases of blood vessels, and the knowledge that costimulation of T cells by DC and/or macrophages is required for generation and full activation of effector T cells, it is logical investigate costimulatory blockade as a therapeutic approach to vascular inflammation. So far, one form of costimulatory blockade, namely CTLA-4-Ig, has been shown to be effective in clinical trials leading to FDA approval for particular disorders. CTLA-4-Ig is a soluble fusion protein consisting of the extracellular CD80/CD86 binding portion of CTLA-4 coupled to a modified Fc region of IgG1 to achieve optimal pharmacokinetics properties. CTLA-4 binds to CD80 and CD86 with much higher affinity than CD28, and thus CTLA-4-Ig functions as a competitive inhibitor of costimulation by CD80 and CD86 expressing APCs. Two forms of CTLA-4-Ig, abatacept, and belatacept, which differ in only two amino acids and thereby differ in their affinities for CD80 and CD86, are approved for the treatment of rheumatoid arthritis and prevention of renal allograft rejection, respectively (Ford and Larsen, 2009). (CTLA-4-Ig should not be confused with anti-CTLA-4 antibody, e.g., Ipilimumab, which has the opposite effect of enhancing T cell mediated responses and is approved for treatment of metastatic melanoma.)
There are significant challenges to the development of safe and effective therapies for vascular diseases based on costimulatory blockade. Because these approaches are immunosuppressive and unlikely to be specific for pathologic immune responses targeting blood vessels, there will be some risks of opportunistic infections. So far, the experience with CTLA-4-Ig treatment of rheumatoid arthritis patients has indicated such risks are low compared to other immunosuppressive biologic drugs (Curtis and Singh, 2011). Atherosclerosis and hypertension, the two most common vascular diseases for which there is preclinical evidence supporting a costimulatory blockade strategy, are chronic diseases in which activation of T cells may be contributing to disease progression over many years, and chronic immunosuppression may not be an acceptable approach. In each of these diseases, however, there are acute phases that pose significantly elevated risk of morbidity and mortality, and these acute exacerbations may be related to increased immune mediated inflammation. Thus, as we refine the use of biomarkers to predict the onset of acute coronary syndrome and hypertensive crises, we may be able to effectively intervene with temporary but robust costimulatory blockade. Another obstacle to highly effective costimulatory blockade for the treatment of vascular disease, as well as other autoimmune diseases, is the contribution of memory T cells. Memory T cell responses may be resistant to costimulatory blockade for two reasons. First, as discussed earlier, memory T cells have less stringent requirements for costimulation. Second, after a pathologic T cell mediated immune response has been established for some time, there will be a high frequency of memory T cells specific for the target tissue antigens compared to the few naive T cells present before the disease developed. If costimulation enhances the response of these memory T cells, it may be difficult to block enough APC-memory T cell costimulatory interactions to achieve therapeutic benefit.
Another possible complication of targeting the B7:CD28 pathway to treat established diseases is the role played by CD4+ effector T cells that lack CD28. Increased numbers of circulating CD4+CD28− T cells are found in the blood of patients with various disorders, including rheumatoid arthritis (Schmidt et al., 1996), diabetes (Giubilato et al., 2011), and significantly for vascular pathology, acute coronary syndrome (Zal et al., 2004; Liuzzo et al., 2007). These cells produce inflammatory cytokines (interferon γ and TNF) and they have cytolytic functions. Whether or not they contribute to atherosclerotic lesion inflammation and acute plaque changes is not know. Other molecules may serve as costimulators for these CD28− T cell, such as activating receptors found typically on NK cells (Snyder et al., 2004).
T cell tolerance to self proteins is maintained in part by the presentation of peptides of these proteins to T cells in the absence of innate inflammatory signals that usually accompany infection or tissue injury (see Figure 1A). Tolerizing DCs carrying self antigens from healthy tissues to secondary lymphoid organs are considered key to this process. These DCs are capable of presenting peptide–MHC antigen to naïve T cells without providing sufficient costimulatory signals to lead to clonal expansion and effector T cell differentiation. Tolerizing DCs may also express more coinhibitors that activating DCs. The consequence of antigen presentation by these DCs is deletion, anergy, or Treg differentiation. The emerging understanding that DCs are present in normal blood vessels may explain how T cell tolerance to blood vessel specific protein antigens is maintained. There has been a long held view that peripheral APC-mediated induction of T cell tolerance could be achieved therapeutically by costimulatory blockade, especially in the context of transplantation (Wekerle et al., 2002). The basic concept of tolerance induction has been to block costimulation at a time when the relevant target antigens are being presented to naïve T cells, for example in allograft tolerance around the time the transplant is performed. Thus T cells would receive antigen receptor signals in the absence of costimulation. Immunologic tolerance induction has the advantage of a limited time of pharmacologic intervention and limited non-specific immunosuppression. In studies performed more than 15 years ago in rodent models, short term costimulatory blockade of the B7–CD28 pathway in combination with anti-CD154 treatment induced long term acceptance of allografts, and donor antigen specific tolerance was apparently achieved (Larsen et al., 1996). This strategy has not proven successful in inducing tolerance in preclinical primate models. Potential thrombosis/hemostasis complications of targeting CD154 related to expression of the protein on platelets has hampered progress in the strategy of targeting both B7:CD28 and CD154:CD40 pathways in humans. With the development of new reagents, and better, albeit still incomplete, understanding of the signaling pathways that mediate costimulation and coinhibition, effective strategies for tolerance induction based on costimulatory blockade appears to be an achievable goal (Ford and Larsen, 2009). With regard to vascular diseases, a major challenge in adopting tolerance induction strategies will be the identification of the relevant antigens. In the case of atherosclerosis, most attention had been paid to the heterogeneous collection of antigens generated by LDL oxidation, based on the isolation of ox-LDL reactive T cells from human plaques (Stemme et al., 1995) as well as the presence of T-dependent anti-ox-LDL antibodies in atherosclerotic patients and hypercholesterolemic animals. However, more recently, evidence from studies of mice immunized with ox-LDL indicated that the T cells that were activated and exerted pro-atherogenic responses were specific for native LDL and unmodified ApoB100 peptides (Hermansson et al., 2010). Furthermore, adoptive transfer of myeloid DCs pulsed with native human ApoB100 and rendered tolerogenic by in vitro treatment with IL-10 significantly reduced atherosclerosis in Ldlr−/− mice expressing transgenic human ApoB100 (Hermansson et al., 2011). The atheroprotection was correlated with induction of Treg and reduction in plaque T cell infiltration in the treated mice. That study confirms that DCs can modulate atherosclerosis in an antigen specific manner, and suggests that DC-induced peripheral T cell tolerance can ameliorate inflammatory vascular diseases.
More advanced animal models, improved immunophenotyping of human arterial disease, and studies of vascular disease in patients being treated with drugs that target costimulatory and coinhibitory molecules will all be required in order to more fully understand the role of different costimulatory and coinhibitory pathways. This will permit the development specialized treatments to target the relevant molecules on the relevant cell types, in the relevant locations.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by National Institutes of Health grant R01HL087282.
References
Abbas, A. K., Lichtman, A. H., and Pillai, S. (2011). Cellular and Molecular Immunology, 7th Edn. Philadephia: Elsevier, Inc.
Afek, A., Harats, D., Roth, A., Keren, G., and George, J. (2005). A functional role for inducible costimulator (ICOS) in atherosclerosis. Atherosclerosis 183, 57–63.
Ait-Oufella, H., Salomon, B. L., Potteaux, S., Robertson, A. K., Gourdy, P., Zoll, J., Merval, R., Esposito, B., Cohen, J. L., Fisson, S., Flavell, R. A., Hansson, G. K., Klatzmann, D., Tedgui, A., and Mallat, Z. (2006). Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180.
Angeli, V., Llodra, J., Rong, J. X., Satoh, K., Ishii, S., Shimizu, T., Fisher, E. A., and Randolph, G. J. (2004). Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity 21, 561–574.
Bae, Y. S., Lee, J. H., Choi, S. H., Kim, S., Almazan, F., Witztum, J. L., and Miller, Y. I. (2009). Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ. Res. 104, 210–218.
Boesteanu, A. C., and Katsikis, P. D. (2009). Memory T cells need CD28 costimulation to remember. Semin. Immunol. 21, 69–77.
Bour-Jordan, H., Esensten, J. H., Martinez-Llordella, M., Penaranda, C., Stumpf, M., and Bluestone, J. A. (2011). Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol. Rev. 241, 180–205.
Bour-Jordan, H., Salomon, B. L., Thompson, H. L., Szot, G. L., Bernhard, M. R., and Bluestone, J. A. (2004). Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J. Clin. Invest. 114, 979–987.
Bu, D. X., Tarrio, M., Maganto-Garcia, E., Stavrakis, G., Tajima, G., Lederer, J., Jarolim, P., Freeman, G. J., Sharpe, A. H., and Lichtman, A. H. (2011). Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 1100–1107.
Buono, C., Pang, H., Uchida, Y., Libby, P., Sharpe, A. H., and Lichtman, A. H. (2004). B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation 109, 2009–2015.
Chavez-Sanchez, L., Madrid-Miller, A., Chavez-Rueda, K., Legorreta-Haquet, M. V., Tesoro-Cruz, E., and Blanco-Favela, F. (2010). Activation of TLR2 and TLR4 by minimally modified low-density lipoprotein in human macrophages and monocytes triggers the inflammatory response. Hum. Immunol. 71, 737–744.
Choi, J. H., Cheong, C., Dandamudi, D. B., Park, C. G., Rodriguez, A., Mehandru, S., Velinzon, K., Jung, I. H., Yoo, J. Y., Oh, G. T., and Steinman, R. M. (2011). Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 35, 819–831.
Choi, S. H., Harkewicz, R., Lee, J. H., Boullier, A., Almazan, F., Li, A. C., Witztum, J. L., Bae, Y. S., and Miller, Y. I. (2009). Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ. Res. 104, 1355–1363.
Curtis, J. R., and Singh, J. A. (2011). Use of biologics in rheumatoid arthritis: current and emerging paradigms of care. Clin. Ther. 33, 679–707.
Cybulsky, M. I., and Jongstra-Bilen, J. (2010). Resident intimal dendritic cells and the initiation of atherosclerosis. Curr. Opin. Lipidol. 21, 397–403.
de Boer, O. J., Hirsch, F., van der Wal, A. C., van der Loos, C. M., Das, P. K., and Becker, A. E. (1997). Costimulatory molecules in human atherosclerotic plaques: an indication of antigen specific T lymphocyte activation. Atherosclerosis 133, 227–234.
Ford, M. L., and Larsen, C. P. (2009). Translating costimulation blockade to the clinic: lessons learned from three pathways. Immunol. Rev. 229, 294–306.
Fortun, A., Khalil, A., Gagne, D., Douziech, N., Kuntz, C., Jay-Gerin, J. P., Dupuis, G., and Fulop, T. Jr. (2001). Monocytes influence the fate of T cells challenged with oxidised low density lipoproteins towards apoptosis or MHC-restricted proliferation. Atherosclerosis 156, 11–21.
Francisco, L. M., Sage, P. T., and Sharpe, A. H. (2010). The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242.
Francisco, L. M., Salinas, V. H., Brown, K. E., Vanguri, V. K., Freeman, G. J., Kuchroo, V. K., and Sharpe, A. H. (2009). PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029.
Furukawa, Y., Mandelbrot, D. A., Libby, P., Sharpe, A. H., and Mitchell, R. N. (2000). Association of B7-1 co-stimulation with the development of graft arterial disease. Studies using mice lacking B7-1, B7-2, or B7-1/B7-2. Am. J. Pathol. 157, 473–484.
Gerdes, N., and Zirlik, A. (2011). Co-stimulatory molecules in and beyond co-stimulation – tipping the balance in atherosclerosis? Thromb. Haemost. 106, 804–813.
Giubilato, S., Liuzzo, G., Brugaletta, S., Pitocco, D., Graziani, F., Smaldone, C., Montone, R. A., Pazzano, V., Pedicino, D., Biasucci, L. M., Ghirlanda, G., and Crea, F. (2011). Expansion of CD4+CD28null T-lymphocytes in diabetic patients: exploring new pathogenetic mechanisms of increased cardiovascular risk in diabetes mellitus. Eur. Heart J. 32, 1214–1226.
Gotsman, I., Grabie, N., Dacosta, R., Sukhova, G., Sharpe, A., and Lichtman, A. H. (2007). Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Invest. 117, 2974–2982.
Gotsman, I., Grabie, N., Gupta, R., Dacosta, R., MacConmara, M., Lederer, J., Sukhova, G., Witztum, J. L., Sharpe, A. H., and Lichtman, A. H. (2006). Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation 114, 2047–2055.
Gotsman, I., Sharpe, A. H., and Lichtman, A. H. (2008). T-cell costimulation and coinhibition in atherosclerosis. Circ. Res. 103, 1220–1231.
Grabie, N., Gotsman, I., DaCosta, R., Pang, H., Stavrakis, G., Butte, M. J., Keir, M. E., Freeman, G. J., Sharpe, A. H., and Lichtman, A. H. (2007). Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation 116, 2062–2071.
Grewal, I. S., and Flavell, R. A. (1998). CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 16, 111–135.
Harrison, D. G., Vinh, A., Lob, H., and Madhur, M. S. (2010). Role of the adaptive immune system in hypertension. Curr. Opin. Pharmacol. 10, 203–207.
Heath, W. R., and Carbone, F. R. (2009). Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244.
Herber, D. L., Cao, W., Nefedova, Y., Novitskiy, S. V., Nagaraj, S., Tyurin, V. A., Corzo, A., Cho, H. I., Celis, E., Lennox, B., Knight, S. C., Padhya, T., McCaffrey, T. V., McCaffrey, J. C., Antonia, S., Fishman, M., Ferris, R. L., Kagan, V. E., and Gabrilovich, D. I. (2010). Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–886.
Hermansson, A., Johansson, D. K., Ketelhuth, D. F., Andersson, J., Zhou, X., and Hansson, G. K. (2011). Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation 123, 1083–1091.
Hermansson, A., Ketelhuth, D. F., Strodthoff, D., Wurm, M., Hansson, E. M., Nicoletti, A., Paulsson-Berne, G., and Hansson, G. K. (2010). Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J. Exp. Med. 207, 1081–1093.
Inder, S. J., Bobryshev, Y. V., Cherian, S. M., Lord, R. S., Masuda, K., and Yutani, C. (2000a). Accumulation of lymphocytes, dendritic cells, and granulocytes in the aortic wall affected by Takayasu’s disease. Angiology 51, 565–579.
Inder, S. J., Bobryshev, Y. V., Cherian, S. M., Wang, A. Y., Lord, R. S., Masuda, K., and Yutani, C. (2000b). Immunophenotypic analysis of the aortic wall in Takayasu’s arteritis: involvement of lymphocytes, dendritic cells and granulocytes in immuno-inflammatory reactions. Cardiovasc. Surg. 8, 141–148.
Ishii, N., Takahashi, T., Soroosh, P., and Sugamura, K. (2010). OX40-OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv. Immunol. 105, 63–98.
Jeon, H. J., Choi, J. H., Jung, I. H., Park, J. G., Lee, M. R., Lee, M. N., Kim, B., Yoo, J. Y., Jeong, S. J., Kim, D. Y., Park, J. E., Park, H. Y., Kwack, K., Choi, B. K., Kwon, B. S., and Oh, G. T. (2010). CD137 (4-1BB) deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation 121, 1124–1133.
Josefowicz, S. Z., Lu, L. F., and Rudensky, A. Y. (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. PMID: 22224781. [Epub ahead of print].
Ketelhuth, D. F., and Hansson, G. K. (2011). Cellular immunity, low-density lipoprotein and atherosclerosis: break of tolerance in the artery wall. Thromb. Haemost. 106, 779–786.
Kim, K. S., Denton, M. D., Chandraker, A., Knoflach, A., Milord, R., Waaga, A. M., Turka, L. A., Russell, M. E., Peach, R., and Sayegh, M. H. (2001). CD28-B7-mediated T cell costimulation in chronic cardiac allograft rejection: differential role of B7-1 in initiation versus progression of graft arteriosclerosis. Am. J. Pathol. 158, 977–986.
Kim, W. J., Bae, E. M., Kang, Y. J., Bae, H. U., Hong, S. H., Lee, J. Y., Park, J. E., Kwon, B. S., Suk, K., and Lee, W. H. (2006). Glucocorticoid-induced tumour necrosis factor receptor family related protein (GITR) mediates inflammatory activation of macrophages that can destabilize atherosclerotic plaques. Immunology 119, 421–429.
Koga, N., Suzuki, J., Kosuge, H., Haraguchi, G., Onai, Y., Futamatsu, H., Maejima, Y., Gotoh, R., Saiki, H., Tsushima, F., Azuma, M., and Isobe, M. (2004). Blockade of the interaction between PD-1 and PD-L1 accelerates graft arterial disease in cardiac allografts. Arterioscler. Thromb. Vasc. Biol. 24, 2057–2062.
Krupa, W. M., Dewan, M., Jeon, M. S., Kurtin, P. J., Younge, B. R., Goronzy, J. J., and Weyand, C. M. (2002). Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am. J. Pathol. 161, 1815–1823.
Larsen, C. P., Elwood, E. T., Alexander, D. Z., Ritchie, S. C., Hendrix, R., Tucker-Burden, C., Cho, H. R., Aruffo, A., Hollenbaugh, D., Linsley, P. S., Winn, K. J., and Pearson, T. C. (1996). Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 381, 434–438.
Lee, J., Zhuang, Y., Wei, X., Shang, F., Wang, J., Zhang, Y., Liu, X., Yang, Y., Liu, L., and Zheng, Q. (2009). Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell. Cardiol. 46, 169–176.
Lehman, T. J., Walker, S. M., Mahnovski, V., and McCurdy, D. (1985). Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. 28, 652–659.
Lievens, D., Eijgelaar, W. J., Biessen, E. A., Daemen, M. J., and Lutgens, E. (2009). The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb. Haemost. 102, 206–214.
Liuzzo, G., Biasucci, L. M., Trotta, G., Brugaletta, S., Pinnelli, M., Digianuario, G., Rizzello, V., Rebuzzi, A. G., Rumi, C., Maseri, A., and Crea, F. (2007). Unusual CD4+CD28null T lymphocytes and recurrence of acute coronary events. J. Am. Coll. Cardiol. 50, 1450–1458.
Lutgens, E., Gorelik, L., Daemen, M. J., de Muinck, E. D., Grewal, I. S., Koteliansky, V. E., and Flavell, R. A. (1999). Requirement for CD154 in the progression of atherosclerosis. Nat. Med. 5, 1313–1316.
Mach, F., Schonbeck, U., Sukhova, G. K., Atkinson, E., and Libby, P. (1998). Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature 394, 200–203.
Ma-Krupa, W., Jeon, M.-S., Spoerl, S., Tedder, T. F., Goronzy, J. J., and Weyand, C. M. (2004). Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J. Exp. Med. 199, 173–183.
Maldonado, R. A., and von Andrian, U. H. (2010). How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 108, 111–165.
Miga, A., Masters, S., Gonzalez, M., and Noelle, R. J. (2000). The role of CD40-CD154 interactions in the regulation of cell mediated immunity. Immunol. Invest. 29, 111–114.
Miller, Y. I., Choi, S.-H., Wiesner, P., Fang, L., Harkewicz, R., Hartvigsen, K., Boullier, A., Gonen, A., Diehl, C. J., Que, X., Montano, E., Shaw, P. X., Tsimikas, S., Binder, C. J., and Witztum, J. L. (2011). Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 108, 235–248.
Mitchell, R. N., and Libby, P. (2007). Vascular remodeling in transplant vasculopathy. Circ. Res. 100, 967–978.
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355.
Munroe, M. E. (2009). Functional roles for T cell CD40 in infection and autoimmune disease: the role of CD40 in lymphocyte homeostasis. Semin. Immunol. 21, 283–288.
Murray, P. J., and Wynn, T. A. (2011). Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737.
Nakano, M., Fukumoto, Y., Satoh, K., Ito, Y., Kagaya, Y., Ishii, N., Sugamura, K., and Shimokawa, H. (2010). OX40 ligand plays an important role in the development of atherosclerosis through vasa vasorum neovascularization. Cardiovasc. Res. 88, 539–546.
Nickel, T., Schmauss, D., Hanssen, H., Sicic, Z., Krebs, B., Jankl, S., Summo, C., Fraunberger, P., Walli, A. K., Pfeiler, S., and Weis, M. (2009). oxLDL uptake by dendritic cells induces upregulation of scavenger-receptors, maturation and differentiation. Atherosclerosis 205, 442–450.
Olofsson, P. S., Soderstrom, L. A., Jern, C., Sirsjo, A., Ria, M., Sundler, E., de Faire, U., Wiklund, P. G., Ohrvik, J., Hedin, U., Paulsson-Berne, G., Hamsten, A., Eriksson, P., and Hansson, G. K. (2009). Genetic variants of TNFSF4 and risk for carotid artery disease and stroke. J. Mol. Med. (Berl.) 87, 337–346.
Olofsson, P. S., Soderstrom, L. A., Wagsater, D., Sheikine, Y., Ocaya, P., Lang, F., Rabu, C., Chen, L., Rudling, M., Aukrust, P., Hedin, U., Paulsson-Berne, G., Sirsjo, A., and Hansson, G. K. (2008). CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation 117, 1292–1301.
Onishi, Y., Fehervari, Z., Yamaguchi, T., and Sakaguchi, S. (2008). Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. U.S.A. 105, 10113–10118.
Packard, R. R., Maganto-Garcia, E., Gotsman, I., Tabas, I., Libby, P., and Lichtman, A. H. (2008). CD11c(+) dendritic cells maintain antigen processing, presentation capabilities, and CD4(+) T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ. Res. 103, 965–973.
Paulson, K. E., Zhu, S.-N., Chen, M., Nurmohamed, S., Jongstra-Bilen, J., and Cybulsky, M. I. (2010). Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 106, 383–390.
Randolph, G. J. (2008). Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr. Opin. Lipidol. 19, 462–468.
Rodig, N., Ryan, T., Allen, J. A., Pang, H., Grabie, N., Chernova, T., Greenfield, E. A., Liang, S. C., Sharpe, A. H., Lichtman, A. H., and Freeman, G. J. (2003). Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur. J. Immunol. 33, 3117–3126.
Schmidt, D., Goronzy, J. J., and Weyand, C. M. (1996). CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Invest. 97, 2027–2037.
Schonbeck, U., Sukhova, G. K., Shimizu, K., Mach, F., and Libby, P. (2000). Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 7458–7463.
Schulte, D. J., Yilmaz, A., Shimada, K., Fishbein, M. C., Lowe, E. L., Chen, S., Wong, M., Doherty, T. M., Lehman, T., Crother, T. R., Sorrentino, R., and Arditi, M. (2009). Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking Kawasaki disease. J. Immunol. 183, 5311–5318.
Shamshiev, A. T., Ampenberger, F., Ernst, B., Rohrer, L., Marsland, B. J., and Kopf, M. (2007). Dyslipidemia inhibits Toll-like receptor-induced activation of CD8alpha-negative dendritic cells and protective Th1 type immunity. J. Exp. Med. 204, 441–452.
Shevach, E. M. (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645.
Snyder, M. R., Nakajima, T., Leibson, P. J., Weyand, C. M., and Goronzy, J. J. (2004). Stimulatory killer Ig-like receptors modulate T cell activation through DAP12-dependent and DAP12-independent mechanisms. J. Immunol. 173, 3725–3731.
Stemme, S., Faber, B., Holm, J., Wiklund, O., Witztum, J. L., and Hansson, G. K. (1995). T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 92, 3893–3897.
Tabas, I., Williams, K. J., and Boren, J. (2007). Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116, 1832–1844.
Tivol, E. A., Borriello, F., Schweitzer, A. N., Lynch, W. P., Bluestone, J. A., and Sharpe, A. H. (1995). Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547.
Topalian, S. L., Weiner, G. J., and Pardoll, D. M. (2011). Cancer immunotherapy comes of age. J. Clin. Oncol. 29, 4828–4836.
van Olffen, R. W., de Bruin, A. M., Vos, M., Staniszewska, A. D., Hamann, J., van Lier, R. A., de Vries, C. J., and Nolte, M. A. (2010). CD70-driven chronic immune activation is protective against atherosclerosis. J. Innate Immun. 2, 344–352.
van Wanrooij, E. J., van Puijvelde, G. H., de Vos, P., Yagita, H., van Berkel, T. J., and Kuiper, J. (2007). Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 204–210.
Vinh, A., Chen, W., Blinder, Y., Weiss, D., Taylor, W. R., Goronzy, J. J., Weyand, C. M., Harrison, D. G., and Guzik, T. J. (2010). Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122, 2529–2537.
Wang, X., Ria, M., Kelmenson, P. M., Eriksson, P., Higgins, D. C., Samnegard, A., Petros, C., Rollins, J., Bennet, A. M., Wiman, B., de Faire, U., Wennberg, C., Olsson, P. G., Ishii, N., Sugamura, K., Hamsten, A., Forsman-Semb, K., Lagercrantz, J., and Paigen, B. (2005). Positional identification of TNFSF4, encoding OX40 ligand, as a gene that influences atherosclerosis susceptibility. Nat. Genet. 37, 365–372.
Waterhouse, P., Penninger, J. M., Timms, E., Wakeham, A., Shahinian, A., Lee, K. P., Thompson, C. B., Griesser, H., and Mak, T. W. (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988.
Weber, C., Meiler, S., Doring, Y., Koch, M., Drechsler, M., Megens, R. T., Rowinska, Z., Bidzhekov, K., Fecher, C., Ribechini, E., van Zandvoort, M. A., Binder, C. J., Jelinek, I., Hristov, M., Boon, L., Jung, S., Korn, T., Lutz, M. B., Forster, I., Zenke, M., Hieronymus, T., Junt, T., and Zernecke, A. (2011). CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Invest. 121, 2898–2910.
Wekerle, T., Kurtz, J., Bigenzahn, S., Takeuchi, Y., and Sykes, M. (2002). Mechanisms of transplant tolerance induction using costimulatory blockade. Curr. Opin. Immunol. 14, 592–600.
Wing, K., Yamaguchi, T., and Sakaguchi, S. (2011). Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 32, 428–433.
Yilmaz, A., Rowley, A., Schulte, D. J., Doherty, T. M., Schroder, N. W., Fishbein, M. C., Kalelkar, M., Cicha, I., Schubert, K., Daniel, W. G., Garlichs, C. D., and Arditi, M. (2007). Activated myeloid dendritic cells accumulate and co-localize with CD3+ CD3 T cells in coronary artery lesions in patients with Kawasaki disease. Exp. Mol. Pathol. 83, 93–103.
Keywords: costimulation, coinhibition, T cells, dendritic cells, macrophages vascular disease, atherosclerosis
Citation: Lichtman AH (2012) T cell costimulatory and coinhibitory pathways in vascular inflammatory diseases. Front. Physio. 3:18. doi: 10.3389/fphys.2012.00018
Received: 14 December 2011;
Paper pending published: 06 January 2012;
Accepted: 26 January 2012;
Published online: 16 February 2012.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Ekaterina Koltsova, La Jolla Institute for Allergy and Immunology, USAElena Galkina, Eastern Virginia Medical School, USA
Copyright: © 2012 Lichtman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andrew H. Lichtman, Brigham and Women’s Hospital, NRB Room 752N, 77 Avenue Louis Pasteur, Boston, MA 02115, USA. e-mail: alichtman@rics.bwh.harvard.edu