 Nis Stride1 Steen Larsen1 Jonas Thue Treebak2 Christina Neigaard Hansen1 Martin Hey-Mogensen1 Tobias Speerschneider3 Thomas E. Jensen2 Jacob Jeppesen2 Jørgen F. P. Wojtaszewski2 Erik A. Richter2 Lars Køber4 Flemming Dela1*
Nis Stride1 Steen Larsen1 Jonas Thue Treebak2 Christina Neigaard Hansen1 Martin Hey-Mogensen1 Tobias Speerschneider3 Thomas E. Jensen2 Jacob Jeppesen2 Jørgen F. P. Wojtaszewski2 Erik A. Richter2 Lars Køber4 Flemming Dela1*- 1 Xlab, Faculty of Health Sciences, Department of Biomedical Sciences, Center for Healthy Aging, University of Copenhagen, Copenhagen, Denmark
- 2 Molecular Physiology Group, Faculty of Science, Department of Exercise and Sport Sciences, University of Copenhagen, Copenhagen, Denmark
- 3 Ion-channel group, Faculty of Health Sciences, Department of Biomedical Sciences, Danish Arrythmia Research Centre, University of Copenhagen, Copenhagen, Denmark
- 4 Department of Cardiology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark
5′-adenosine monophosphate-activated protein kinase (AMPK) is considered central in regulation of energy status and substrate utilization within cells. In heart failure the energetic state is compromised and substrate metabolism is altered. We hypothesized that this could be linked to changes in AMPK activity and we therefore investigated mitochondrial oxidative phosphorylation capacity from the oxidation of long- and medium-chain fatty acids (LCFA and MCFA) in cardiomyocytes from young and old mice expressing a dominant negative AMPKα2 (AMPKα2-KD) construct and their wildtype (WT) littermates. We found a 35–45% (P < 0.05) lower mitochondrial capacity for oxidizing MCFA in AMPKα2-KD of both age-groups, compared to WT. This coincided with marked decreases in protein expression (19/29%, P < 0.05) and activity (14/21%, P < 0.05) of 3-hydroxyacyl-CoA-dehydrogenase (HAD), in young and old AMPKα2-KD mice, respectively, compared to WT. Maximal LCFA oxidation capacity was similar in AMPKα2-KD and WT mice independently of age implying that LCFA-transport into the mitochondria was unaffected by loss of AMPK activity or progressing age. Expression of regulatory proteins of glycolysis and glycogen breakdown showed equivocal effects of age and genotype. These results illustrate that AMPK is necessary for normal mitochondrial function in the heart and that decreased AMPK activity may lead to an altered energetic state as a consequence of reduced capacity to oxidize MCFA. We did not identify any clear aging effects on mitochondrial function.
Introduction
The energy supply of the cardiomyocytes in the pumping heart is of vital importance for the sustained delivery of substrates to all other cells in the organism. The hearts own substrate availability must therefore be guaranteed at all times and a tight regulation must exist in order to balance energy mobilization and expenditure. 5′-Adenosine monophosphate-activated protein kinase (AMPK) is considered a key enzyme in this respect, and is classically described as a cellular energy sensor (Hardie et al., 2006). Energetic stress leading to a high AMP:ATP ratio is a potent stimulator of AMPK activity resulting in increased myocellular uptake of glucose and fatty acids (FA) and increased glycolysis (Dolinsky and Dyck, 2006; Viollet et al., 2009).
5′-Adenosine monophosphate-activated protein kinase has primarily been shown to participate in short-term energy regulation of cardiac metabolism and gene expression in healthy organisms whereas less is known about its actions in cellular adaptive responses to disease (Dolinsky and Dyck, 2006; Dyck and Lopaschuk, 2006; Viollet et al., 2009). Transgenic mice are useful tools for elucidating this area and so far it has been shown that loss of function of various AMPK subunits result in different cardiac pathologies (Dolinsky and Dyck, 2006). Mutations in the PRKAG2 gene encoding the AMPK-γ2 regulatory subunit, leads to altered AMPK activity and a cardiac phenotype resembling Wolff–Parkinson–White syndrome, with increased glycogen deposition in the myocardium and potential to induce ventricular pre-excitation (Arad et al., 2002; Gollob and Roberts, 2002; Sidhu et al., 2005). Moreover, a loss of function of the catalytic AMPKα2 subunit is associated with modest left ventricular contractile dysfunction (Russell III et al., 2004; Turdi et al., 2010) and increased susceptibility to ischemic events (Xing et al., 2003; Gundewar et al., 2009; Kim et al., 2011) thereby underlining its role in maintaining cardiac function. We hypothesized that the common effector mechanism of AMPK in these pathologies could be disturbances in mitochondrial substrate utilization and therefore aimed at characterizing mitochondrial substrate utilization in a mouse model over-expressing a dominant negative isoform of the catalytic AMPKα2 subunit as described earlier (Mu et al., 2003). We hypothesized that functioning AMPK is necessary for the orchestration of substrate utilization in the heart and we aimed specifically at investigating mechanisms that could influence the capacity for mitochondrial FA oxidation since this has been shown to be severely affected by various cardiac pathological conditions (Neubauer, 2007). The incidence of cardiac disease is much higher in an aged population and since studies have shown depressed AMPK activity with aging (Ma et al., 2010) we found it relevant to study cardiac metabolic phenotype in both young and old mice.
Materials and Methods
Animals
Protocols were approved by the Danish Animal Experimental Inspectorate and complied with the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes (Council of Europe no. 123, Strasbourg, France, 1985). Male, muscle-specific AMPKα2 kinase dead (AMPKα2-KD) mice (Mu et al., 2003) and wildtype (WT) male littermates on a C57BL/6J background were studied. The mice within each group represented two age-groups: young (Y) (19–22 weeks) and old (O) animals (76–91 weeks). This rendered four groups: WT/Y (n = 12), WT/O (n = 8), AMPKα2-KD/Y (n = 14), and AMPKα2-KD/O (n = 8). Body weight (BW) was lower in young than in old mice (33.1 ± 0.4 vs. 35.8 ± 0.5 g, P < 0.05) with no effect of genotype. All mice were housed in a 10:14-h light-dark cycle and fed standard laboratory chow and water ad libitum. The heart was excised after anesthesia with intraperitoneal pentobarbital/lidocaine injection. The heart was transferred to an ice cold buffer solution (BIOPS, see below), the atria and major vessels were quickly removed, and the ventricles were divided in two portions; one for high resolution respirometry (from apex of left ventricle) and one for clamp freezing in liquid nitrogen and storage at −80°C for later biochemical analyses.
High Resolution Respirometry
Cardiac tissue from the apex of the left ventricle was prepared for measurements of respiratory flux rates by mechanical dissection with sharp forceps in relaxing buffer (BIOPS) on ice. The buffer contained 2.77 mM CaK2EGTA, 7.23 mM K2EGTA, 20 mM imidazole, 20 mM taurine, 6.56 mM MgCl2, 5.77 mM ATP, 15 mM phosphocreatine, 0.5 mM dithiothreitol, and 50 mM K-MES at pH 7.1. Myocardial fibers were permeabilized by gentle agitation for 30 min at 0°C in BIOPS supplemented with 50 μg/ml saponin and then washed in ice cold respiration medium (Mir05) 2 × 10 min during gentle agitation. Mir05 consisted of 110 mM sucrose, 60 mM K-lactobionate, 0.5 mM EGTA, 1 g/l BSA essentially FA free, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM K-HEPES at pH 7.1. The O2 solubility of this medium was taken as 10.5 μM/kPa.
Respiration was measured at 37°C (Oroboros, Oxygraph; Innsbruck, Austria). Data acquisition and analyses were performed using DatLab software (Oroboros). The absolute respiratory rates (oxygen fluxes) were expressed per milligram of wet weight (ww) per second. Two protocols were performed in parallel, diverting with respect to the type of FA added. In protocol A, a medium-chain fatty acid (MCFA) was added whereas a long chain fatty acid (LCFA) was added in protocol B. Protocol A: malate (M) 2 mM, octanoylcarnitine (Oct.car) 0.5 mM, and ADP 5 mM. Protocol B: malate 2 mM, palmitoylcarnitine (Pal.car) 125 μM, and ADP 5 mM. The optimal concentration of octanoylcarnitine and palmitoylcarnitine was chosen on basis of substrate titrations in a pilot study (data not shown) providing maximal state 3 respiration. Cytochrome C (10 mM) was added in four randomly selected separate protocols to test for outer membrane integrity. This did not elicit any additive oxygen consumption during maximally stimulated respiration, verifying the intactness of the outer mitochondrial membranes in the permeabilized muscle fibers (data not shown).
Preparation of Tissue for Western Blotting and PCR Analysis
The frozen hearts were thoroughly pulverized using a steel-morter shaped to accommodate a Cryotube™ and a steel piston shaped to fit the inside of the tube. All utensils were kept on dry-ice in a −20°C environment. Samples were stored at −80°C after processing.
RT-PCR Quantification of Genomic DNA and Mitochondrial DNA
Total DNA was precipitated from 10 mg of the pulverized mouse heart homogenized in DNAzol (Molecular Research Center, Cincinnati, OH, USA). The DNA was dissolved in 200 μl Tris–EDTA pH 8. Five microliters of a 25 times DNA dilution was used for PCR amplification with QuantiTect SYBR Green PCR Master Mix (Qiagen, Hilden, Germany) containing 2.5 pmol of each primer in a total volume of 25 μl. Levels of mitochondrial DNA (mtDNA) and genomic DNA (gDNA) were determined by real-time PCR using a MX3005P QPCR machine (Stratagene, La Jolla, CA, USA) as previously described (Rabol et al., 2009) was quantified using primers for the cytochrome C oxidase Vb (COX-Vb) gene and mtDNA was quantified using primers for the cytochrome C oxidase I (COX-I) gene. Primer sequences are listed in Table 1.
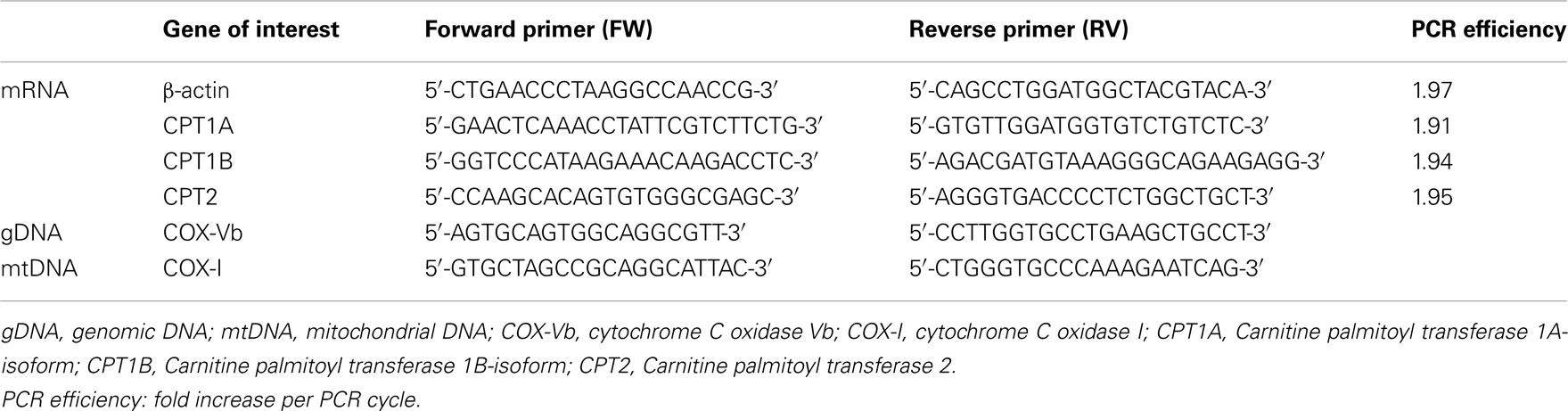
Table 1. Primer sequences used in RT-PCR quantification of mRNA and DNA.
Quantification of MRNA Levels by Real-Time PCR
Total RNA was isolated from 10 mg of pulverized heart tissue by phenol extraction using TRIzol (Invitrogen, San Diego, CA, USA) according to manufacturer’s instructions. One microgram RNA was used for mRNA conversion into 20 μl cDNA, using oligo-dt primers and OmniScript reverse transcriptase (Qiagen, Hilden, Germany). For each target mRNA, 0.25 μl cDNA was mixed with QuantiTect SYBR Green PCR Kit (Qiagen) and 2.5 pmol of each specific primer (Table 1) in a total volume of 25 μl. The PCR amplification was monitored in real-time using the MX3000P QPCR machine (Stratagene, La Jolla, CA, USA) and levels of mRNAs were measured relative to the individual level of β-actin mRNA (2−ΔCt method; Schmittgen and Livak, 2008). Finally the level of specific mRNAs from each group was normalized to the mean of WT/Y.
The primers had less than 10% variation in PCR efficiency (Table 1; Schmittgen and Livak, 2008) and β-actin mRNA was validated as internal reference, via a two-way ANOVA showing similar mRNA expression of β-actin in all groups.
Western Blot Analyses
Approximately 15 mg of frozen pulverized heart biopsies were homogenized in ice cold buffer [25 mM Tris pH 6.8, 5 mM EDTA, 20 mM β-glycerophosphate (Sigma), 10 mM pyrophosphate (Sigma), 2 mM Na Ortovanadate (Sigma), 2.5 mM PMSF and Mini Complete Protease inhibitor tablet according to the manufactures recommendation (Roche)] and added SDS to a final concentration of 3%. From each sample 3–40 μg of total protein were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were probed with antibodies against glycogen-phosphorylase (GP), (AS09 455, Agrisera), 3-hydroxyacyl-CoA-dehydrogenase (HAD), (ab54477, Abcam), hexokinase II (HKII), (#2867, Cell Signaling Technology), malonyl-CoA-decarboxylase (MCD), (15265-1-AP, Protein Tech Group), pyruvate kinase – muscle isoform (PK), (#3106, Cell Signaling Technology), phosphorylated acetyl-CoA-carboxylase (ACCphos), (#07-303, Millipore), phosphorylated AMP-dependent kinase (AMPKphos), (#2531Cell Signaling Technology), or antibody against the AMPK-α2 subunit which was kindly provided by Prof. D. G. Hardie (University of Dundee) followed by incubation with HRP-labeled secondary antibody (Dako, Denmark). The antigen-antibody complexes were visualized by ECL using a CCD system (LAS-4000 Luminescent Image Analyzer, GE Healthcare) and a Kodak Image Station (2000MM, Kodak). Quantification was performed using “Image Quant” analysis software (GE Healthcare) and Kodak ID 3.6 software. Western blotting analyses were performed in two different laboratories. As a result the gels were loaded differently as illustrated by the representative blots shown in Figure 5. Not all blots allowed for direct comparisons between the groups and therefore all protein expression data are reported relative to the same control group, WT/Y, which were included on all gels. All gels were run in duplicates.
Enzyme Activities
Maximal CS and HAD activity was measured in a reaction coupled to conversion of NAD+ to NADH. NADH production was assessed spectrophotometrically, by determination of NADH changes at 340 nm at 37°C, pH 7.0, in myocardial lysates (1 μg/μl protein), using an automatic analyzer (Hitachi automatic analyzer 912; Boehringer Mannheim, Germany). CS activity was measured with acetyl-CoA and oxaloacetate as substrate and HAD measured with acetoacetyl-CoA as substrate.
Statistics
Anthropometric data were compared using an unpaired Student’s t-test. Protein expression data was assessed in a group wise manner by a one-way ANOVA. This allowed us to compare the groups of interest (WT/O/, AMPKα2-KD/Y, and AMPKα2-KD/O) directly to WT/Y but did not allow for detection of interaction effects of either age or genotype as isolated factors. Data on enzyme activities were compared by a two-way ANOVA. In oxygraphic measurements repeated measure two-way ANOVA was used. P values less than 0.05 were considered significant. If the criterion of normal distribution and equal variance was not met, values were log transformed and recalculated. Data are expressed as means ± SE.
Results
Decreased AMPK-α2 Activity in Both Young and Old AMPKα2-KD Mice
The genotypes were confirmed by western blots of the α-p172 AMPK (AMPKphos). The transgenic AMPK-α2 carried a myc tag and migrated slightly slower in the gels (Figure 5). The expression of AMPKphos was expectedly higher in the AMPKα2-KD genotype (data not shown) in line with previous findings (Mu et al., 2003). The decreased functional activity of AMPK in KD mice was confirmed by a 74.0 ± 0.0 and 71.0 ± 0.1% reduction in the protein expression level of ACCphos in young and old AMPKα2-KD mice respectively (Figure 5). ACC is a well known downstream target of AMPK and its use as a marker of AMPK activity has previously been validated (Dyck and Lopaschuk, 2006).
Decrease in Mitochondrial MCFA Respiration in AMPKα2-KD Mice – Irrespective of Age
In protocol A, baseline respiration revealed no significant differences between groups (P > 0.05, data not shown). With the addition of saturating amounts of ADP and MCFA (Oct.car), respiration increased markedly and there was a clear genotype effect, of reduced oxidative capacity in both young and old AMPKα2-KD compared to WT littermates (P < 0.05, Figure 1).
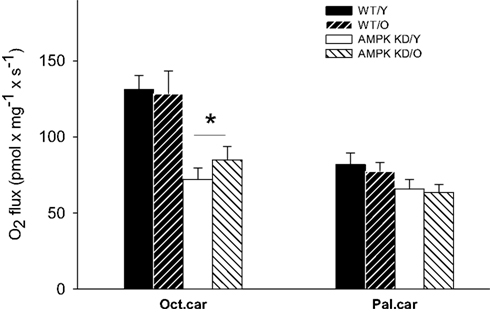
Figure 1. Mitochondrial respiratory capacity in heart muscle samples from wild type mice (WT) and dominant negative AMPKα2 (AMPK KD) mice. Young (Y: 19–22 weeks) and old (O: 76–91 weeks) mice were studied in both groups. State 3 respiration was measured with two different substrate protocols. Malate, ADP, and octanoylcarnitine (Oct.car) or malate, ADP, and palmitoylcarnitine (Pal.car). Values are mean ± SE. *Different from WT/Y (P < 0.05).
Protocol B revealed no differences in LCFA sustained respiration between genotypes or age-groups. Thus, respiration with ADP and LCFA (Pal.car) did not differ between the groups (P > 0.05, Figure 1).
No Change in Mitochondrial Content with Age and/or Genotype
There was no difference in mtDNA content (copy no · mg ww−1) between any groups and the CS activity did also not differ significantly between the groups. (P > 0.05, Figure 2) Thus, we found maintained mitochondrial density irrespective of age and genotype.
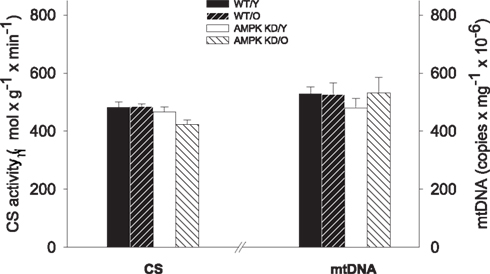
Figure 2. Citrate synthase (CS) enzyme activity (left Y-axis) and mitochondrial DNA content (right Y-axis) in heart muscle samples from wild type mice (WT) and dominant negative AMPKα2 (AMPK KD) mice. Young (Y: 19–22 weeks) and old (O: 76–91 weeks) mice were studied in both groups. *Different from age matched wild type (P < 0.05).
Carnitine Palmitoyl Transferases
There was a high and uniform PCR efficiency between the primers (1.91–1.97, Table 1) and they were thus optimized for the individual genes of interest. The levels of β-actin expression showed no difference between the groups (data not shown) and could therefore be used as reference-gene. We found a modest down-regulation of CPT1B expression in old AMPKα2-KD group compared to both old WT and young AMPKα2-KD (Figure 3). We found no significant differences in gene expression of CPT1A and CPT2.
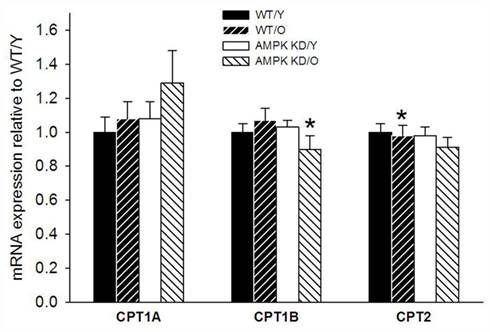
Figure 3. Carnitine palmitoyl transferase isoforms 1A (CPT1A) and 1B (CPT1B) as well as carnitine palmitoyl transferase 2 (CPT2) in heart muscle samples from wild type mice (WT) and dominant negative AMPKα2 (AMPK KD) mice. Young (Y: 19–22 weeks) and old (O: 76–91 weeks) mice were studied in both groups. *Different from WT/Y (P < 0.05).
Myocardial Content and Activity of Metabolic Key Enzymes
3-Hydroxyacyl-CoA-dehydrogenase protein expression showed a clear pattern of down-regulation in the AMPKα2-KD in both age-groups. The expression of HAD was reduced to 0.81 ± 0.05, P < 0.05 in young AMPKα2-KD compared to WT/Y and 0.71 ± 0.04, P < 0.05 in old AMPKα2-KD compared to WT/Y (Figure 4). There was no significant age-effect on HAD expression, indicative of sustained β-oxidative capacity irrespective of old age. Corresponding with the HAD expression, maximal HAD enzyme activity was reduced in both AMPKα2-KD age-groups, compared to their age matched WT littermates (Figure 4).
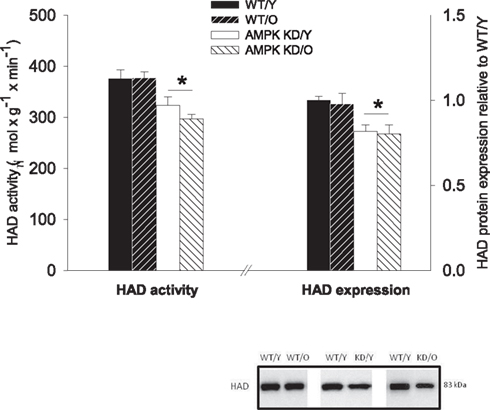
Figure 4. 3-Hydroxyacyl-CoA-dehydrogenase (HAD) enzyme activity (left Y-axis) and protein expression relative to WT/Y (right Y-axis). *Different from WT/Y (P < 0.05).
A considerable decrease in phosphorylated ACCphos was found in both AMPKα2-KD groups being a consequence of reduced AMPK activity, in line with previous studies (Dyck et al., 1999; Zarrinpashneh et al., 2006; Turdi et al., 2010). The expression of malonyl-CoA-decarboxylase (MCD) was reduced in old mice, irrespective of genotype (P < 0.05, Figure 5). GP was up-regulated in WT/O compared to WT/Y (1.20 ± 0.07, P < 0.05, Figure 5). HKII expression was decreased in both young and old AMPKα2-KD (P < 0.05, Figure 5) pointing toward a reduced glycolytic capacity in AMPKα2-KD mice independently of age. In contrast, PK expression was unchanged in both groups of old mice, but surprisingly up-regulated in AMPKα2-KD/Y (Figure 5), indicating a mixed influence of age and genotype on the expression of this enzyme.
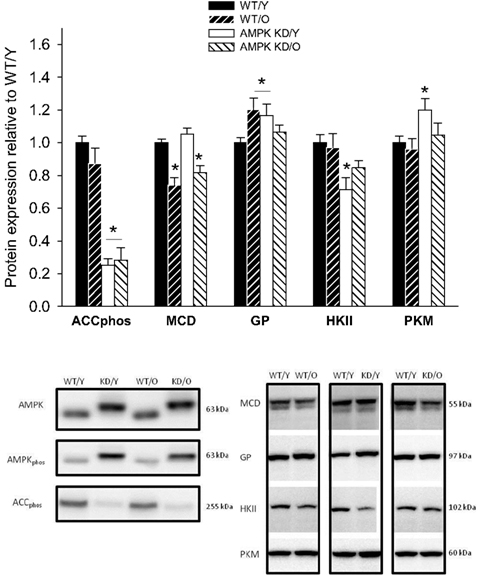
Figure 5. Protein content (Western blot) of pS212 acetyl-CoA-carboxylase (ACCphos) and malonyl-CoA-decarboxylase (MCD), glycogen-phosphorylase (GP), hexokinase II (HKII), and Pyruvatkinase (PKM) in heart muscle samples from wild type mice (WT) and dominant negative AMPKα2 (AMPK KD) mice. Young (Y: 19–22 weeks) and old (O: 76–91 weeks) mice were studied in both groups. Representative blots are shown in the lower panels, which also include blots of AMPK and AMPKphos. *Different from WT/Y (P < 0.05).
Discussion
The major finding of our study is that loss of AMPKα2 activity leads to a reduced mitochondrial capacity for oxidative phosphorylation when the reducing equivalents are provided via β-oxidation of MCFA.
We tested this by assessing mitochondrial FA oxidation capacity, both dependently and independently of the mitochondrial LCFA import apparatus, since palmitoylcarnitine oxidation (protocol B) requires an intact CPT2, whereas this is not the case with octanoylcarnitine (protocol A). The selective reduction in MCFA oxidative capacity within the AMPKα2-KD mice in the present study coincides with a reduction in HAD enzyme activity and protein expression in the AMPKα2-KD mice of both age-groups (Figure 4). This points toward an effect of AMPKα2 in the regulation of β-oxidation capacity modulated by HAD activity and expression and is in line with previous studies showing increased HAD activity and expression in skeletal muscle from rats treated with 5-aminoimidazole-4-carboxamide (AICAR), a pharmacologic activator of AMPK (Putman et al., 2003; Fillmore et al., 2010).
The observed difference between WT and AMPKα2-KD mice in protein expression and HAD activity (∼20%) was slightly exceeded by the reduction in MCFA oxidation capacity (∼35–45%). A linear relationship may not necessarily exist between these variables, but it does leave room for additional factors that may regulate MCFA oxidation. Mitochondrial FA import could be such a factor and CPT1 activity has often been referred to as a key regulator in this respect (Stanley et al., 2005).
The activity of CPT1 is subject to inhibition by malonyl-CoA, the concentration of which is tightly regulated by ACC and MCD. In the present study the phosphorylation of ACC showed an obvious negative effect of the AMPKα2-KD genotype with a 70–74% reduction of ACCphos in these animals.
The decreased ACCphos levels would in theory lead to inhibition of LCFA import via CPTI. However we did not observe any functional difference on LCFA oxidation (protocol B) as palmitoylcarnitine oxidation was unaffected by reduced AMPKα2 activity, arguing against import limitation with this particular FA. It is important to underline that mitochondrial import of palmitoylcarnitine proceeds independently of CPTI and that we were only able to assess a potentially restricting effect exerted via CPTII in this experimental model. MCD protein expression shoved a clear dependence on age with a significant reduction in the old mice of both WT and AMPKα2-KD groups (Figure 5). Low MCD capacity might lead to increased levels of malonyl-CoA in turn inhibiting CPTI in old individuals. Together with the reduced CPT1B mRNA expression, which we observed in the old AMPKα2-KD mice (Figure 3), it could be speculated that CPT1 LCFA import only becomes limiting with advancing age. This could imply that the capacity of CPT1 is sufficient to meet the LCFA import requirements of the young, healthy heart – even in the absence of AMPK stimulation, but that lack of AMPK in the long term could impose a metabolic challenge upon the myocardium – leading to a compromised and vulnerable metabolic phenotype in the old.
Our finding of selectively reduced MCFA oxidative capacity in the heart muscle of the AMPKα2-KD mice may be important in the pathophysiology of heart failure. In a study in dogs with heart failure a reduced medium-chain-acyl-dehydrogenase (MCAD) activity and protein expression was reported (Osorio et al., 2002), whereas in another study, LCFA oxidative capacity was maintained in dogs with compensated hypertrophy (Chandler et al., 2004). Decreased palmitoylcarnitine oxidation in isolated cardiac mitochondria from mice subjected to aortic constriction was only observed after progression from left ventricular hypertrophy to overt heart failure (Faerber et al., 2011). It is therefore conceivable that a compromised capacity for LCFA oxidation is a late event in the metabolic remodeling associated with cardiac pathology and that the metabolic changes induced by a reduction of AMPKα2 function may represent an early, disposing factor in the development of cardiac disease.
In previous studies cardiac phenotype, assessed by echocardiography, did not differ between AMPKα2-KD and WT in young mice (Russell III et al., 2004; Turdi et al., 2010) whereas a compromised LV function was found only in very old AMPKα2-KD mice (aged 24–28 months; Turdi et al., 2010) and a more susceptible phenotype was demonstrated with respect to acute ischemia (Russell III et al., 2004). These observations all indicate that a reduced AMPK activity does not in itself cause cardiac disease, but rather sets the stage for disease development. With the combined negative effects of decreased AMPK activity and aging (limited β-oxidative capacity and compromised LCFA import) a future metabolic challenge, such as acute ischemia, could possibly have negative consequences for the ATP generating capacity within the cardiac mitochondria and lead to development of HF.
In general the present data on protein expression involved in the glycolytic pathway are somewhat complex. The young AMPKα2-KD mice exhibited a significantly decreased expression of HKII compared to WT/Y (Figure 5), previously observed in skeletal muscle of α2-knockout mice (Jorgensen et al., 2007). The increased PK protein expression in AMPKα2-KD/Y compared to WT/Y (Figure 5) is in contrast to the decrease in HKII expression, rendering no clear tendency of the glycolytic capacity depending on age or genotype. The expression of GP was increased in both WT/O and AMPKα2-KD/Y showing no clear effect of age or genotype on the extent of glycogen metabolism either.
Some limitations apply to this study. We have not yet been able to obtain reliable respirometric measurements with LCFA acyl-CoA as substrate in contrast to LCFA acyl-carnitine. This prevents us from assessing the functional capacity of CPT1 via the permeabilized fiber technique. Furthermore, echocardiographic evaluation would have been helpful in the description of aging and AMPK effects on LV phenotype in the present model. Finally, the entire data set applies only to male mice and therefore potential sex differences cannot be addressed.
In conclusion, we report that loss of AMPK activity is characterized by a selectively reduced MCFA oxidative capacity together with reduced protein content and enzyme activity of HAD. We did not find any clear effects of aging on the ability to utilize FA as substrate for mitochondrial respiration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Betina Bolmgren and Irene Bech Nielsen for expert technical assistance. The financial support from The Nordea Foundation, Danish Cardiovascular Research Academy – DaCRA, The Danish Medical Research Council, The Lundbeck Foundation and Ketty, and Ejvind Lyngsbæks foundation is gratefully acknowledged.
References
Arad, M., Benson, D. W., Perez-Atayde, A. R., McKenna, W. J., Sparks, E. A., Kanter, R. J., McGarry, K., Seidman, J. G., and Seidman, C. E. (2002). Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy 1. J. Clin. Invest. 109, 357–362.
Chandler, M. P., Kerner, J., Huang, H., Vazquez, E., Reszko, A., Martini, W. Z., Hoppel, C. L., Imai, M., Rastogi, S., Sabbah, H. N., and Stanley, W. C. (2004). Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am. J. Physiol. Heart Circ. Physiol. 287, H1538–H1543.
Dolinsky, V. W., and Dyck, J. R. (2006). Role of AMP-activated protein kinase in healthy and diseased hearts. Am. J. Physiol. Heart Circ. Physiol. 291, H2557–H2569.
Dyck, J. R., Kudo, N., Barr, A. J., Davies, S. P., Hardie, D. G., and Lopaschuk, G. D. (1999). Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5′-AMP activated protein kinase 1. Eur. J. Biochem. 262, 184–190.
Dyck, J. R., and Lopaschuk, G. D. (2006). AMPK alterations in cardiac physiology and pathology: enemy or ally? J. Physiol. (Lond.) 574, 95–112.
Faerber, G., Barreto-Perreia, F., Schoepe, M., Gilsbach, R., Schrepper, A., Schwarzer, M., Mohr, F. W., Hein, L., and Doenst, T. (2011). Induction of heart failure by minimally invasive aortic constriction in mice: reduced peroxisome proliferator-activated receptor gamma coactivator levels and mitochondrial dysfunction. J. Thorac. Cardiovasc. Surg. 141, 492–500.
Fillmore, N., Jacobs, D. L., Mills, D. B., Winder, W. W., and Hancock, C. R. (2010). Chronic AMP-activated protein kinase activation and a high-fat diet have an additive effect on mitochondria in rat skeletal muscle 1. J. Appl. Physiol. 109, 511–520.
Gollob, M. H., and Roberts, R. (2002). AMP-activated protein kinase and familial Wolff-Parkinson-White syndrome: new perspectives on heart development and arrhythmogenesis. Eur. Heart J. 23, 679–681.
Gundewar, S., Calvert, J. W., Jha, S., Toedt-Pingel, I., Ji, S. Y., Nunez, D., Ramachandran, A., Anaya-Cisneros, M., Tian, R., and Lefer, D. J. (2009). Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 104, 403–411.
Hardie, D. G., Hawley, S. A., and Scott, J. W. (2006). AMP-activated protein kinase – development of the energy sensor concept. J. Physiol. (Lond.) 574, 7–15.
Jorgensen, S. B., Treebak, J. T., Viollet, B., Schjerling, P., Vaulont, S., Wojtaszewski, J. F., and Richter, E. A. (2007). Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am. J. Physiol. Endocrinol. Metab. 292, E331–E339.
Kim, A. S., Miller, E. J., Wright, T. M., Li, J., Qi, D., Atsina, K., Zaha, V., Sakamoto, K., and Young, L. H. (2011). A small molecule AMPK activator protects the heart against ischemia-reperfusion injury 1. J. Mol. Cell. Cardiol. 51, 24–32.
Ma, H., Wang, J., Thomas, D. P., Tong, C., Leng, L., Wang, W., Merk, M., Zierow, S., Bernhagen, J., Ren, J., Bucala, R., and Li, J. (2010). Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart 1. Circulation 122, 282–292.
Mu, J., Barton, E. R., and Birnbaum, M. J. (2003). Selective suppression of AMP-activated protein kinase in skeletal muscle: update on “lazy mice.” Biochem. Soc. Trans. 31, 236–241.
Osorio, J. C., Stanley, W. C., Linke, A., Castellari, M., Diep, Q. N., Panchal, A. R., Hintze, T. H., Lopaschuk, G. D., and Recchia, F. A. (2002). Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation 106, 606–612.
Putman, C. T., Kiricsi, M., Pearcey, J., MacLean, I. M., Bamford, J. A., Murdoch, G. K., Dixon, W. T., and Pette, D. (2003). AMPK activation increases uncoupling protein-3 expression and mitochondrial enzyme activities in rat muscle without fibre type transitions. J. Physiol. (Lond.) 551, 169–178.
Rabol, R., Svendsen, P. F., Skovbro, M., Boushel, R., Haugaard, S. B., Schjerling, P., Schrauwen, P., Hesselink, M. K., Nilas, L., Madsbad, S., and Dela, F. (2009). Reduced skeletal muscle mitochondrial respiration and improved glucose metabolism in nondiabetic obese women during a very low calorie dietary intervention leading to rapid weight loss. Metab. Clin. Exp. 58, 1145–1152.
Russell, R. R. III, Li, J., Coven, D. L., Pypaert, M., Zechner, C., Palmeri, M., Giordano, F. J., Mu, J., Birnbaum, M. J., and Young, L. H. (2004). AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 114, 495–503.
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108.
Sidhu, J. S., Rajawat, Y. S., Rami, T. G., Gollob, M. H., Wang, Z., Yuan, R., Marian, A. J., DeMayo, F. J., Weilbacher, D., Taffet, G. E., Davies, J. K., Carling, D., Khoury, D. S., and Roberts, R. (2005). Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation 111, 21–29.
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129.
Turdi, S., Fan, X., Li, J., Zhao, J., Huff, A. F., Du, M., and Ren, J. (2010). AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 9, 592–606.
Viollet, B., Athea, Y., Mounier, R., Guigas, B., Zarrinpashneh, E., Horman, S., Lantier, L., Hebrard, S., Devin-Leclerc, J., Beauloye, C., Foretz, M., Andreelli, F., Ventura-Clapier, R., and Bertrand, L. (2009). AMPK: lessons from transgenic and knockout animals. Front. Biosci. 14, 19–44.
Xing, Y., Musi, N., Fujii, N., Zou, L., Luptak, I., Hirshman, M. F., Goodyear, L. J., and Tian, R. (2003). Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 278, 28372–28377.
Zarrinpashneh, E., Carjaval, K., Beauloye, C., Ginion, A., Mateo, P., Pouleur, A. C., Horman, S., Vaulont, S., Hoerter, J., Viollet, B., Hue, L., Vanoverschelde, J. L., and Bertrand, L. (2006). Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am. J. Physiol. Heart Circ. Physiol. 291, H2875–H2883.
Keywords: AMPK, metabolic remodeling, mitochondria, oxidative phosphorylation
Citation: Stride N, Larsen S, Treebak JT, Hansen CN, Hey-Mogensen M, Speerschneider T, Jensen TE, Jeppesen J, Wojtaszewski JFP, Richter EA, Køber L and Dela F (2012) 5′-AMP activated protein kinase is involved in the regulation of myocardial β-oxidative capacity in mice. Front. Physio. 3:33. doi: 10.3389/fphys.2012.00033
Received: 12 November 2011;
Accepted: 06 February 2012;
Published online: 22 February 2012.
Edited by:
Jolanda Van Der Velden, VU University Medical Center, NetherlandsReviewed by:
John Konhilas, University of Arizona, USARegis Lamberts, University of Otago, New Zealand
Copyright: © 2012 Stride, Larsen, Treebak, Hansen, Hey-Mogensen, Speerschneider, Jensen, Jeppesen, Wojtaszewski, Richter, Køber and Dela. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Flemming Dela, Faculty of Health Sciences, Department of Biomedical Sciences, Center for Healthy Aging, University of Copenhagen, Blegdamsvej 3b, 2200 Copenhagen N, Denmark. e-mail: fdela@sund.ku.dk