 Roelien A. M. Meijering1 Deli Zhang1 Femke Hoogstra-Berends1,2 Robert H. Henning1 Bianca J. J. M. Brundel1*
Roelien A. M. Meijering1 Deli Zhang1 Femke Hoogstra-Berends1,2 Robert H. Henning1 Bianca J. J. M. Brundel1*- 1 Department of Clinical Pharmacology, Groningen University Institute for Drug Exploration, University Medical Center Groningen, University of Groningen, Groningen, Netherlands
- 2 Nyken BV, Groningen, Netherlands
Atrial fibrillation (AF) is the most common, sustained clinical tachyarrhythmia associated with significant morbidity and mortality. AF is a persistent condition with progressive structural remodeling of the atrial cardiomyocytes due to the AF itself, resulting in cellular changes commonly observed in aging and in other heart diseases. While rhythm control by electrocardioversion or drug treatment is the treatment of choice in symptomatic AF patients, its efficacy is still limited. Current research is directed at preventing first-onset AF by limiting the development of substrates underlying AF progression and resembles mechanism-based therapy. Upstream therapy refers to the use of non-ion channel anti-arrhythmic drugs that modify the atrial substrate- or target-specific mechanisms of AF, with the ultimate aim to prevent the occurrence (primary prevention) or recurrence of the arrhythmia following (spontaneous) conversion (secondary prevention). Heat shock proteins (HSPs) are molecular chaperones and comprise a large family of proteins involved in the protection against various forms of cellular stress. Their classical function is the conservation of proteostasis via prevention of toxic protein aggregation by binding to (partially) unfolded proteins. Our recent data reveal that HSPs prevent electrical, contractile, and structural remodeling of cardiomyocytes, thus attenuating the AF substrate in cellular, Drosophila melanogaster, and animal experimental models. Furthermore, studies in humans suggest a protective role for HSPs against the progression from paroxysmal AF to persistent AF and in recurrence of AF. In this review, we discuss upregulation of the heat shock response system as a novel target for upstream therapy to prevent derailment of proteostasis and consequently progression and recurrence of AF.
Management of Atrial Fibrillation by Upstream Therapy
Atrial fibrillation (AF) is the most common clinical tachyarrhythmia which significantly contributes to cardiovascular morbidity and mortality, mainly through stroke and heart failure (Nattel, 2002). The incidence of AF is escalating due to the increased prevalence of risk factors constituting a substrate for AF, such as obesity (Wang et al., 2004), metabolic syndrome (Watanabe et al., 2008) and increasing age (Feinberg et al., 1995). In addition to the increased first-onset of AF, also the progression of the arrhythmia poses problems, as the longer AF persists the less effective pharmacological and electrical cardioversion therapies are (Van Gelder et al., 1996). In patients with symptomatic AF, rhythm control is the treatment of choice (Camm et al., 2010). However, the effective reversal to sinus rhythm is still inadequate. Therefore, recent research focuses on the identification of mechanisms underlying AF substrate induction and maintenance, which have led to several novel upstream therapeutic approaches, including angiotensin-converting enzyme inhibitors, aldosterone antagonists, statins, polyunsaturated fatty acids, and colchicine (Imazio et al., 2011; Savelieva et al., 2011b). Upstream therapy refers to the use of non-ion channel anti-arrhythmic drugs that modify the atrial substrate- or target-specific mechanisms of AF with the ultimate aim to prevent the occurrence (primary prevention) or recurrence (secondary prevention) of the arrhythmia (Van Gelder et al., 2010; Savelieva et al., 2011a).
It has been recognized that electrical and structural remodeling of cardiomyocytes create a substrate for AF (Nattel et al., 2008). Nevertheless, the exact molecular mechanisms that underlie cardiomyocyte remodeling and AF progression are as yet unidentified. We recently obtained evidence that derailment of proteostasis (i.e., the homeostasis of protein production, breakdown, and function) constitutes an important substrate for induction and progression of AF (Brundel et al., 2001, 2002, 2006b). Proteostasis involves controlling the concentration, conformation, binding interaction, kinetics, and location of individual proteins. Derailment of cellular proteostasis results in many systemic diseases including cardiovascular disorders (Balch et al., 2008). Cells respond to a loss of proteostatic control by inducing the heat shock response (HSR) resulting in the expression of heat shock proteins (HSPs) that facilitate protein folding and function (Westerheide and Morimoto, 2005). Consequently, an emerging target candidate for upstream therapy of AF is the upregulation of HSPs. Indeed, HSP induction alleviates the occurrence and recurrence of AF in various experimental model systems for AF (Brundel et al., 2006a,b, 2008; Sakabe et al., 2008). Furthermore, studies in humans suggest a protective role for HSPs against progression from paroxysmal AF to persistent AF (Brundel et al., 2006a) and the restoration of sinus rhythm in patients with persistent AF (secondary prevention; Cao et al., 2011).
Here we discuss the concept that derailment of cardiomyocyte proteostasis constitutes an important aspect of the substrate for AF. In addition we examine the evidence for induction of the HSR system as a novel target for upstream therapy to prevent the occurrence and the recurrence of AF and address its possible modes of action.
Mechanisms Underlying AF Initiation and Progression: Derailment of Proteostasis
Although the exact molecular mechanism(s) underlying AF initiation, maintenance, and progression has not yet been completely elucidated, an important recognition was that AF induction required a suitable substrate as well as a trigger that acts on the substrate (Nattel et al., 2008). Various clinical conditions, e.g., several heart diseases, hypertension and cardiac surgery, are risk factors for the first-onset of AF, as they create a substrate(s) and/or trigger(s) for the initiation of AF (Figure 1; Benjamin et al., 1998; Kannel et al., 1998; Chelazzi et al., 2011). Key AF-promoting factors have been identified, including inflammation, oxidative stress, active Rho-GTPase, fibrosis, and atrial muscle bundle dissociation (Allessie et al., 2010; de Groot et al., 2010), which induce loss of proteostatic control, creating a substrate for AF. Subsequent triggers will act on the substrate and will induce AF (Sah et al., 1999; Dudley et al., 2005; Engelmann and Svendsen, 2005; Adam et al., 2007; Burstein and Nattel, 2008; Nattel et al., 2008).
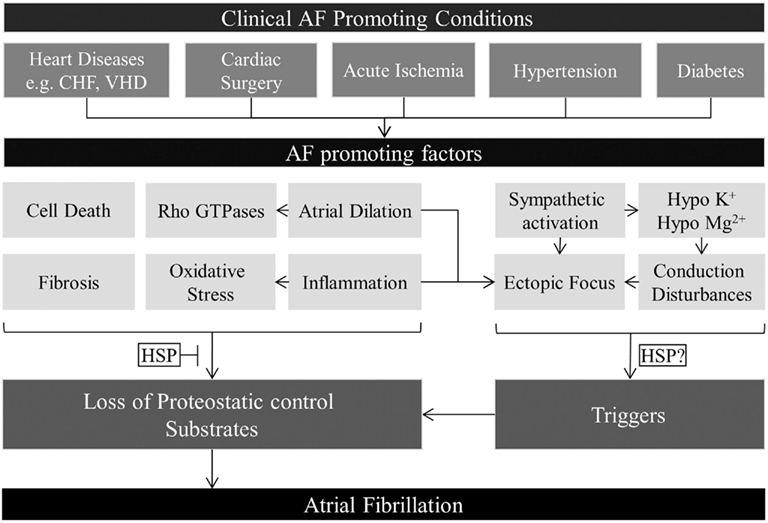
Figure 1. Overview of AF-promoting factors contributing to first-onset AF. Various clinical conditions induce AF-promoting factors. These factors can induce triggers for AF or are responsible for the loss of proteostatic control, thereby inducing remodeling, and creating a substrate for AF. Triggers will act on the vulnerable substrate to induce first-onset AF. Prevention and/or normalization of the cardiomyocyte proteostasis by inducing HSP expression could prevent AF substrate formation and prove an effective approach in preventing first-onset AF in response to various AF-promoting factors.
Once AF is initiated, AF itself will induce further electrical and structural remodeling in a manner that contributes to AF maintenance and progression (Wijffels et al., 1995). A conceptual model is depicted in Figure 2. Electrical remodeling resulting in shortening of action potential duration (APD), slowing of conduction, and abnormal calcium handling will favor AF maintenance (Dobrev and Voigt, 2011). When AF persists, the calcium overload causes irreversible changes in structural remodeling, especially cardiomyocyte hibernation (Ausma et al., 1997b, 2000b; Brundel et al., 2002). Hibernation is characterized by irreversible degradation of the myofibril structure (myolysis) and mitochondrial damage, implying impaired energy production and release of reactive oxygen species (ROS), which leads to contractile dysfunction (Vanoverschelde et al., 1993; Ausma et al., 1997a; Sherman et al., 2000; Bito et al., 2004). Other characteristics are redistribution of nuclear chromatin and pathological gene expression revealing a deficiency in healthy cardiomyocyte proteostasis (Ausma et al., 1995, 1997a,b, 1998; Allessie et al., 2002; Thijssen et al., 2004). While early electrical remodeling is reversible (Schotten et al., 2003), the derailment of proteostasis underlies irreversible structural remodeling and thereby AF progression (Allessie et al., 2002; Brundel et al., 2002; Ausma et al., 2003; Cha et al., 2004; Todd et al., 2004). We and others identified various molecular mechanisms that underlie derailment of proteostasis and AF progression and recurrence (Brundel et al., 2001, 2002, 2006b; Ke et al., 2008, 2011; Zhang et al., 2011a).
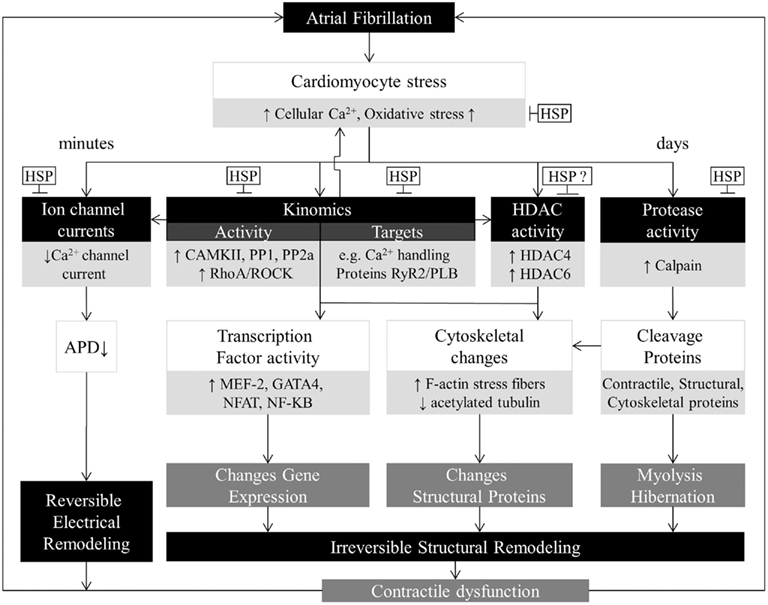
Figure 2. Overview of AF-induced derailment of cardiomyocyte proteostasis. AF induces time-related progressive myocyte remodeling. First, AF causes cellular Ca2+ overload and oxidative stress, which results in a direct inhibition of the L-type Ca2+ channel, shortening of action potential duration, and contractile dysfunction. These changes have an early onset and are reversible. The early processes protect the cardiomyocyte against Ca2+ overload but at the expense of creating a substrate for persistent AF. When AF persists, derailment of proteostasis occurs via activation of calpain, kinases/phosphatases, RhoA-GTPase, HDACs, and exhaustion of protective HSPs. The key modulators also activate each other. Derailment of proteostasis results in irreversible myolysis/hibernation, alterations in structural proteins and pathological gene expression, which are substrates for impaired contractile function and AF persistence. Upstream therapies are directed at modifying the substrate for AF progression. Normalization of the cardiomyocyte proteostasis by inducing HSP expression might represent an effective approach to manage clinical AF.
Ion Channel Currents and Calcium Handling
During AF, atrial cardiomyocytes are subjected to very rapid (400–600 times per minute) and irregular firing, causing a rapid Ca2+ overload resulting in functional downregulation of the L-type Ca2+ channel, which leads to shortening of the APD and contractile dysfunction (hypocontractility), thus providing a further substrate for AF (Figure 2; Goette et al., 1996; Leistad et al., 1996; Ausma et al., 2000a; Brundel et al., 2001; Qi et al., 2008). Decreased ICaL has been consistently found in atrial remodeling, which is believed to significantly contribute to electrical remodeling and AF progression (Van Wagoner et al., 1997; Yue et al., 1997). The functional changes have a rapid onset following initiation of AF and are reversible (Schotten et al., 2003). In addition to the L-type Ca2+ channel, also other channel currents are affected, as reviewed in Dobrev and Voigt (2011). Ion channel currents are regulated by expression level, phosphorylation, and redox status of the channel, all of which are altered during AF (Brundel et al., 1999; Wang, 2003; Dobrev and Voigt, 2011). In dog atrial cardiomyocytes, tachypacing induced the activation of calcineurin via the Ca2+/calmodulin system, which in turn changes cardiomyocyte proteostasis by stimulating nuclear translocation of NFAT, resulting in transcriptional and translational downregulation of the L-type calcium channel resulting in atrial remodeling and AF progression (Qi et al., 2008). Phosphorylation status of ion channels are altered due to changes in activity of various kinases and phosphatases, such as enhanced CaMKII, PP1, and PP2a activity (Anderson, 2004; Dobrev and Voigt, 2011). Further, oxidative stress during AF can contribute to changes in redox status, thereby altering ion channel currents (Wang, 2003; Dobrev and Voigt, 2011).
Kinomics Activity and Target Proteins
Various kinases and phosphatases become activated during AF and contribute to AF-induced remodeling and contractile dysfunction (Anderson, 2004; Christ et al., 2004; El-Armouche et al., 2006; Greiser et al., 2007; Dobrev and Nattel, 2008; Ke et al., 2011). In addition to modification of ion channel currents through (de)phosphorylation, also the function of other downstream target proteins are affected by the altered activity of kinases and phosphatases, including calcium handling proteins, such as RyR2 and PLB (Schwinger et al., 1999; Hagemann and Xiao, 2002; Bers, 2006; Carter et al., 2011). Modification of calcium handling protein function will contribute to calcium overload and subsequent contractile dysfunction (Dobrev and Nattel, 2008). Furthermore, also kinases involved in post-translational modifications of structural proteins, such as actin, have been implicated in AF-induced remodeling. Tachypacing-induced activation of RhoA-GTPase and downstream Rho-kinase (ROCK), induces F-actin stress fiber formation (Figure 2; Ke et al., 2011). Prevention of stress fiber formation by ROCK-inhibition or HSP expression prevented tachypacing-induced remodeling (Ke et al., 2011). These findings imply a key role for alterations in kinomics in derailed proteostasis and progression of AF.
HDAC and Protease Activity
Histone deacetylases (HDACs) have been recently implicated in AF (Backs et al., 2006; Liu et al., 2008; Zhang et al., 2011b). CaMKII was found to phosphorylate class IIa histone deacetylase (HDAC4), resulting in the expression of pathological cardiac genes, contractile dysfunction, and structural remodeling, such as hibernation, all substrates for AF (Backs et al., 2006). HDAC inhibition has been shown to reverse atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy (Liu et al., 2008). Recently we showed that selective inhibition of HDAC6 protected against tachypacing-induced contractile dysfunction and structural remodeling in HL-1 atrial cardiomyocytes and Drosophila (Zhang et al., 2011b), indicating the involvement of HDACs in tachypacing-induced cardiomyocyte remodeling. In addition to HDACs, also the cysteine protease calpain is activated during AF. Calpain is persistently activated by the AF-induced intracellular Ca2+ overload, which results in degradation of contractile and structural proteins (Brundel et al., 2002; Ke et al., 2008), leading to myolysis, thereby further contributing to irreversible structural remodeling and AF progression.
Thus, AF-induced derailment of proteostasis includes changes in ion channel function, kinomics, HDAC and calpain activation and underlies reversible electrical remodeling and irreversible structural remodeling and thereby AF initiation and progression.
Heat Shock Proteins Protect Against AF Initiation and Progression
It has been recognized that heat shock transcription factor 1 (HSF1) is an important regulator of proteostasis by controlling the expression of major HSPs, including HSPB1 (HSP27), HSPA1A (HSP70), and HSPC1 (HSP90), that facilitate protein folding, localization, and function (Balch et al., 2008; Powers et al., 2009). Induction of HSPs provides cytoprotective effects against stress-induced derailment of proteostasis and is beneficial in various cardiac diseases (Table 1; Marber et al., 1995; Plumier et al., 1995; Efthymiou et al., 2004; Kupatt et al., 2004; Brundel et al., 2006b; Hayashi et al., 2006; Inagaki et al., 2006; Kim et al., 2006; Kwon et al., 2007; Lin et al., 2007; Rajasekaran et al., 2007; Ago et al., 2008; Balch et al., 2008; Powers et al., 2009; Fan and Kranias, 2011; Ke et al., 2011). Therefore, recent studies have investigated the cardioprotective potential of HSPs in AF, focusing on AF induction as well as progression.
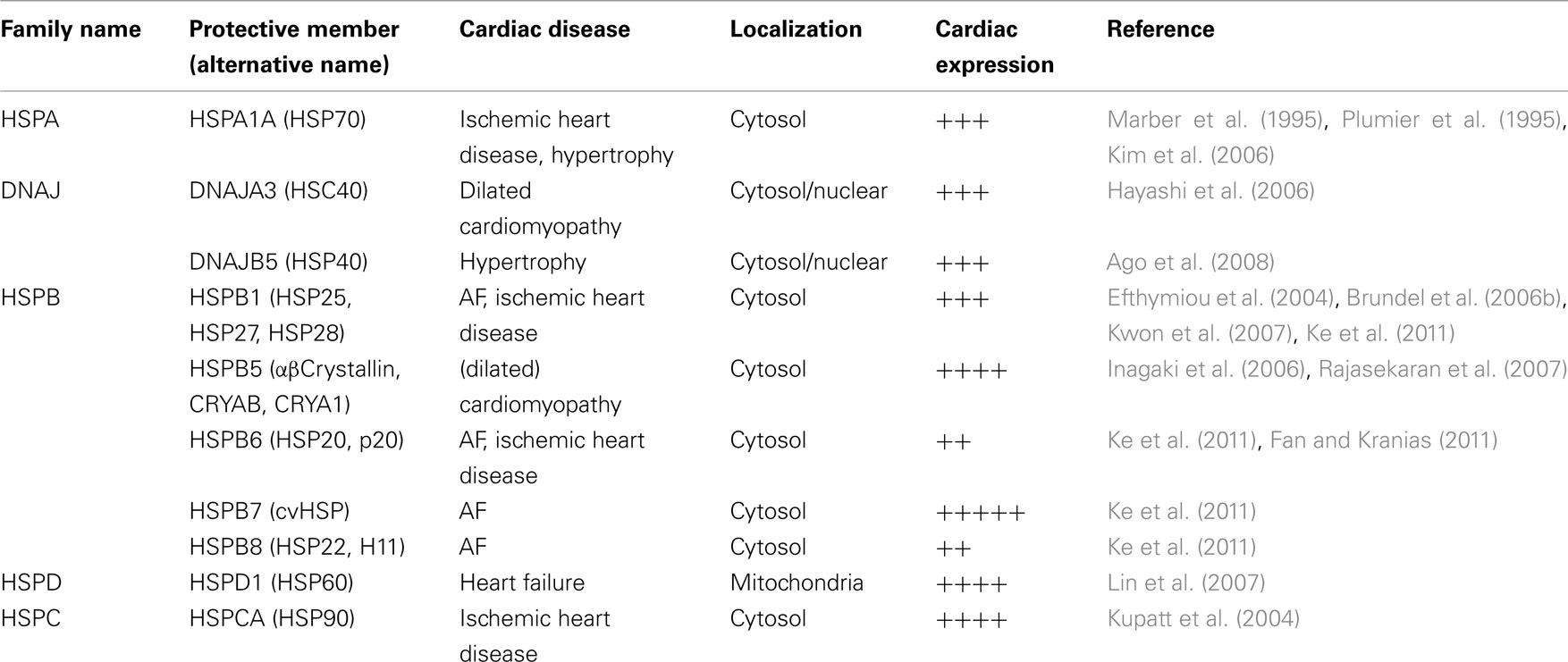
Table 1. Major cardioprotective heat shock proteins, localization, expression, and cardiac disease protective effects.
HSPs in the Prevention of First-Onset AF (Primary Prevention)
Heat shock proteins protect against a variety of AF-promoting factors contributing to first-onset AF (Figure 1). Protective effects of HSPs against cell death, fibrosis, Rho-GTPase activation, oxidative stress, and inflammation have been described, indicating their potential in preventing loss of proteostatic control and formation of AF substrates (Arya et al., 2007; Wakisaka et al., 2007; Jones et al., 2011; Ke et al., 2011). It is unclear if HSPs could affect the formation of triggers. However, since triggers need a vulnerable substrate to act on (Allessie et al., 2010), the prevention of AF substrate formation might be sufficient to protect against first-onset AF. Indeed in various models for first-onset AF, HSPs provide protection against AF substrate formation and hence AF initiation. In a canine model (acute) atrial ischemia related AF, geranylgeranylacetone (GGA) pre-treatment induced HSPA1A expression and revealed protection against prolongation of effective refractory period (ERP) and atrial conduction abnormalities (Brundel et al., 2006b; Sakabe et al., 2008), thereby preventing AF initiation. These observations suggest that HSP induction may protect against some forms of AF in patients with coronary artery disease. Furthermore, a recent study in rats showed that induction of HSPA1A prevents both the angiotensin II mediated atrial fibrosis and increased atrial vulnerability for AF induction (Wakisaka et al., 2007). The findings suggest HSPA1A to play a role in inhibiting the development of a non-cardiomyocyte substrate for AF induction. In two clinical studies, HSPA1A has been implicated as cardioprotective, showing a correlation between HSPA1A atrial expression levels and reduced incidence of post-operative AF in patients in sinus rhythm undergoing cardiac surgery (St Rammos et al., 2002; Mandal et al., 2005).
In addition to HSPA1A, other HSPs might be involved in primary prevention of AF. In patients with AF, increased mitochondrial HSP expression levels, i.e., HSPD1 [37], HSPE1 and mortalin (HSPA9B; Kirmanoglou et al., 2004) have been reported. In addition, increased HSPD1 antibody levels in the serum of patients have been associated with the occurrence of post-operative AF (Oc et al., 2007), suggesting HSPD1 as a marker for mitochondrial and cardiac damage and subsequent increased risk for AF. Increased expression of mitochondrial chaperones could contribute to an increased protection against oxidative stress. Therefore, these HSP family members might contribute to survival of cardiomyocytes by maintaining mitochondrial integrity and capacity for ATP generation. To date, however, several studies showed opposing correlations between the expression of mitochondrial HSPs and AF (Kirmanoglou et al., 2004; Oc et al., 2007; Yang et al., 2007), thereby obscuring their involvement in protection against AF.
HSPs in the Prevention of AF Progression and Recurrence (Secondary Prevention)
Various in vitro and in vivo models for tachypacing-induced AF progression identified HSPs to protect against the derailment of proteostasis and cardiomyocyte remodeling. In tachypaced HL-1 atrial cardiomyocytes and Drosophila melanogaster models for AF, a general HSP induction via a mild heat shock or by a HSP-inducing drug GGA, conserved cardiomyocyte proteostasis during tachypacing and protected against subsequent electrical, contractile, and structural remodeling (Brundel et al., 2006a,b; Zhang et al., 2011a). Furthermore, in canine models for AF progression, GGA pre-treatment induced HSP (HSPA1A and HSPB1) expression and revealed protective effects against shortening of ERP, shortening of APD, reductions in L-type Ca2+ current, and AF progression (Brundel et al., 2006b; Sakabe et al., 2008). Also, in clinical studies, a potent HSR and high HSPB1 levels have been associated with restoration of normal sinus rhythm in patients with permanent AF after mitral valve surgery (Cao et al., 2011). Two other studies comparing paroxysmal vs. persistent AF and sinus rhythm, found an inverse correlation between HSPB1 atrial expression and AF duration and extend of myolysis (Brundel et al., 2006a; Yang et al., 2007). Suggesting, a temporary activation of the HSR during a short duration of AF but exhaustion in time, related to the duration of AF. Consequently, cardiomyocytes lose the ability for proteostatic control, inducing remodeling, which will result in AF progression and recurrence.
Further studies investigated the role of individual HSPs in protection against tachypacing-induced remodeling. HSPB1, and not HSPA1A, was found to play an important role, as its exclusive overexpression appears sufficient to protect against tachypacing-induced remodeling, comparable to GGA pre-treatment (Brundel et al., 2006b). Conversely, the protective effect of a general HSR or GGA pre-treatment on tachypacing-induced changes was annihilated by a selective knockdown of HSPB1. However, in addition to HSPB1, also other HSPB family members (HSPB6, HSPB7, and HSPB8) protect against AF-induced structural remodeling independently from HSPB1 (Ke et al., 2011). Hence, sole overexpression of multiple HSPB family members prevent against AF-induced cardiomyocyte remodeling and AF progression by preserving cell proteostasis, thereby demonstrating their therapeutic potential in AF.
Taken together, there seems to be a strong case for induction of HSPs to prevent AF initiation, recurrences, and progression, by attenuation of electrical, contractile, and structural cardiomyocyte remodeling. There are strong indications that this effect is via normalization of cell proteostasis.
Mode of Action for HSPs to Normalize Proteostasis
It has been recognized that HSPs protect against derailment of proteostasis by preventing cardiomyocyte remodeling at different stages (Figures 1 and 2). The exact mechanisms in prevention of AF initiation (primary prevention) and recurrences (secondary prevention) are not known, but are likely due to HSP regulated protection against various AF-promoting factors that induce the substrate for AF initiation and progression.
Ion Channel Currents
An ion channel current is dependent on the expression level, phosphorylation and redox status of the channel (Hara et al., 2002; Wang, 2003; Dobrev and Voigt, 2011), as well as the stability of the cytoskeleton (Sadoshima et al., 1992) and Rho-GTPase activity (Pochynyuk et al., 2007). The HSP-inducing drug GGA previously showed protective effects against tachypacing-induced reductions in L-type Ca2+ current and shortening of APD (Brundel et al., 2006b). Furthermore, several studies have shown protective effect of HSPs on almost all of these regulating factors. HSPs are known to interact and, in some cases, inhibit kinases and phosphatases, who’s activity is altered during AF (Ding et al., 1998; Lakshmikuttyamma et al., 2004; Dobrev, 2006; Peng et al., 2010; Fan and Kranias, 2011; Qian et al., 2011), thereby potentially preventing or normalizing the phosphorylation status of ion channels, especially L-type Ca2+ channel (Christ et al., 2004). Furthermore, several HSPs (including HSPB1) were shown to provide protection against oxidative stress, thereby potentially preventing or restoring the redox status of the ion channels (Kalmar and Greensmith, 2009). If HSPs can influence the expression levels of ion channels is currently not known. Lastly, also the stability of the actin cytoskeleton and Rho-GTPase activity are regulated by the small HSP family members (see below; Sugiyama et al., 2000; Mounier and Arrigo, 2002; Golenhofen et al., 2004; Brundel et al., 2006b; Salinthone et al., 2008; Ke et al., 2011; Zhang et al., 2011a). The findings reveal a protective role of HSP against AF-induced changes in ion channel current, including reductions in the L-type Ca2+ current.
Kinomics Activity and Target Proteins
Various kinases and phosphatases reveal changed activity during AF, which contributes to cardiomyocyte remodeling depending on their target proteins (Anderson, 2004; Christ et al., 2004; El-Armouche et al., 2006; Greiser et al., 2007; Dobrev and Nattel, 2008; Ke et al., 2011). In addition to ion channels, known targets are transcription factors, various calcium handling proteins and the actin cytoskeleton. Changes in transcription factor phosphorylation, regulates gene expression and hence can induce an altered gene expression profile, possibly contributing to cardiomyocyte hibernation. Interestingly, HSPB1 was shown to interact with certain (downstream) kinases, such as IkappaB kinase and c-Jun N-terminal kinase (JNK), thereby suppressing activation of the transcription factor NF-kappaB (Park et al., 2003; Kammanadiminti and Chadee, 2006). Interestingly, these kinases have also been found to be modulated during AF (Li et al., 2001; Cardin et al., 2003). In addition, HSPB1 is known to interact with other kinases and phosphatases and thereby might prevent the activation of other downstream transcription factors (Ding et al., 1998; Lakshmikuttyamma et al., 2004; Dobrev, 2006; Peng et al., 2010; Fan and Kranias, 2011; Qian et al., 2011).
Changes in phosphorylation status of calcium handling proteins will affect the calcium homeostasis in cardiomyocytes. It is generally accepted that AF-induced abnormalities in intracellular Ca2+ handling leads to atrial cardiomyocyte stress and induces remodeling that contributes to the progression of AF (Qi et al., 2008; Chelu et al., 2009). A calcium overload can be caused by an increase in L-type Ca2+ channel activity, or a changed activity of calcium handling proteins such as RyR2, SR Ca2+ ATPases, or Na+/Ca2+ exchanger. These rapid changes in activity of proteins involved in calcium handling are modulated by kinases and/or phosphatases, including CaMKII and PP1, of which the activities are increased during AF (Christ et al., 2004; Vest et al., 2005). Interestingly, studies showed that HSPs interact with CaMKII (Peng et al., 2010), calcineurin (Lakshmikuttyamma et al., 2004), and PP1 (Dobrev, 2006; Fan and Kranias, 2011). Furthermore, HSPB6 was shown to inhibit PP1 activity (Qian et al., 2011). Also, HSPs increase SR Ca2+ ATPase activity and stimulate both the reuptake of Ca2+ into the SR and the extrusion of Ca2+ out of the cardiomyocyte via Na+/Ca2+ exchanger (Liu et al., 2006; Chen et al., 2010). These findings suggest that HSPs can protect against (tachypacing-induced) changes in calcium handling proteins, resulting in attenuation of AF progression.
Atrial fibrillation is known to activate RhoA-GTPase and ROCK and induces subsequent F-actin stress fiber formation contributing to contractile dysfunction (Ke et al., 2011). Several HSPB family members (HSPB1, HSPB6, and HSPB7) were recently shown to bind to actin and prevent F-actin stress fiber formation downstream of RhoA/ROCK. HSPB1 and HSPB6 even promoted actin stress fiber disassembly. HSPB8 did not directly bind actin, but instead inhibited upstream RhoA-GTPase activation, thereby preventing F-actin stress fiber formation (Ke et al., 2011). As most HSPB family members are known to protect the actin cytoskeleton from remodeling, this action likely represents an important mechanism by which HSPBs attenuate AF-induced derailment of proteostasis and cardiomyocyte remodeling.
HDAC and Protease Activity
Recently, HDAC6 has been implicated in AF-induced cardiomyocyte remodeling (Zhang et al., 2011b). In contrast to the modulation effect of HSPs on kinases and phosphatases, no direct interactions between HSPs and HDACs have yet been described. However, since various kinases and phosphatases regulate the activity of HDACs (de Ruijter et al., 2003) HSPs could theoretically modulate HDAC activity by indirect inhibition of the kinase/phosphatase activity.
In contrast to HDAC activity, HSPs have been shown to attenuate activation of the cysteine protease calpain and subsequently structural remodeling. In a Drosophila model for AF, DmHSP23, presumably the ortholog for human HSPB1, prevents induction of calpain activity, mitochondrial damage, and myolysis (Figure 3). Other studies have also described the modulating effects of HSPB1 on cysteine proteases such as caspase 3 (Garrido et al., 1999; Concannon et al., 2001). Our observation that HSPB protects against AF-induced mitochondrial damage, indicates that it attenuates the impairment of energy production and release of ROS (Figure 3). This finding suggests a protective effect of HSP against induction of early oxidative stress (see below; Figure 2).
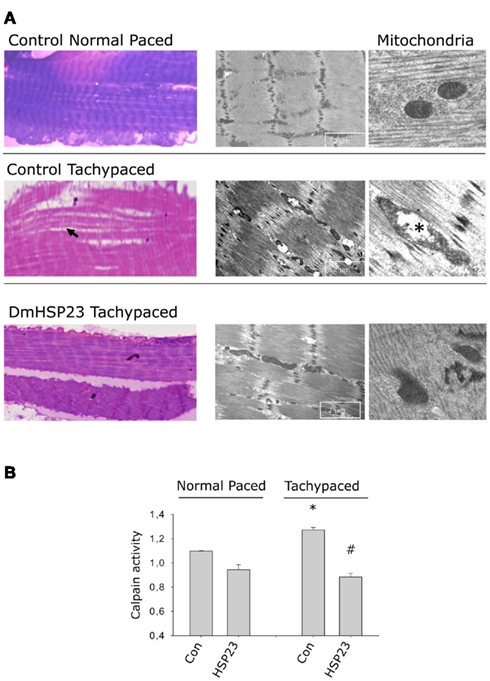
Figure 3. HSP23 overexpressing Drosophila are protected against tachypacing-induced structural remodeling, including loss of sarcomeres (myolysis) and mitochondrial damage. (A) Left panel shows light microscopic structure of the heart wall in normal paced and tachypaced control and DmHSP23 overexpressing strains. Right panel shows electron microscopic details of the cardiomyocytes and mitochondria. Arrowhead shows loss of sarcomeres in the heart wall and asterisk marks mitochondrial damage. (B) Tachypacing induces calpain activity, and DmHSP23 (UAS-actin GAL4-DmHSP23) overexpressing strains were protected. *P < 0.05 vs. control normal paced, #P < 0.05 vs. control tachypaced. Figure adapted from Zhang et al. (2011a).
HSPs and Oxidative Stress
Interestingly, in AF patients, an increase in oxidative stress markers has been observed and anti-inflammatory or anti-oxidant treatment with glucocorticoids and statins (Shiroshita-Takeshita et al., 2004, 2006, 2007; Sanchez-Quinones et al., 2008) suppressed atrial remodeling and have shown some clinical value in prevention of post-surgery AF (primary prevention; Camm et al., 2010) substantiating a role for oxidative stress in AF-induced remodeling. Glucocorticoids and statins have been reported to induce several HSPs (HSPB1, HSPB5, and HSPA1A; Nègre-Aminou et al., 2002; Son et al., 2005), leaving open the possibility that part of their protective pleiotropic effects is due to overexpression of HSPs. HSP induction can provide protection against oxidative stress by several mechanisms. HSPB1 is known to regulate the redox status of cardiomyocytes by maintaining glutathione in its reduced form, thus decreasing the amount of ROS produced in cells exposed to oxidative stress or tumor necrosis factor TNFalpha (Arrigo, 2007). HSPB1 may therefore prevent tachypacing-induced alterations in redox status of cardiomyocytes and thereby preserve cell proteostasis and electrophysiological and contractile function of the cardiomyocyte in AF. In addition to alterations in redox state, oxidative stress can also contribute to actin cytoskeleton instability, resulting in impairment of cardiomyocyte contractile function. Several members of the HSPB family were found to bind the actin filaments and prevent their disruption in response to various stresses, including AF (Sugiyama et al., 2000; Mounier and Arrigo, 2002; Golenhofen et al., 2004; Brundel et al., 2006b; Salinthone et al., 2008; Zhang et al., 2011a). In addition, HSPB family members were also shown protective against AF-induced mitochondrial damage (Zhang et al., 2011a), thereby limiting the release of ROS.
Therapeutic Potential of HSP-Inducing Drugs in AF
Pharmacological and other approaches (e.g., viral gene transfer), preventing the substrate for AF are being studied, with the hope that they might be useful therapeutic agents in treating AF (Amit et al., 2010; Dobrev and Nattel, 2010; Igarashi et al., 2012). Focusing on the current pharmacological options, the efficacy of commonly used drugs, including glucocorticoids and statins, on remodeling is limited (Dobrev and Nattel, 2010) and (serious) adverse effects have been reported, indicating the need for more effective therapeutic agents with less adverse effects. Since derailment of cellular proteostasis results in cardiomyocyte remodeling and AF progression and derailment is counteracted by HSPs, pharmacological induction of the HSR seems to represent a key target for upstream therapy.
Currently, GGA represents the most promising compound for the pharmacological induction of HSPs. Until now it is the most efficacious HSP boosting drug (Brundel et al., 2008). Furthermore, in contrast to other HSP inducers, GGA is a non-toxic compound shown to be capable of inducing HSP expression in various tissues, including gastric mucosa, intestine, liver, myocardium, retina, kidney, and central nervous system. In addition GGA is used clinically in Japan since 1984 as an antiulcer drug (Murakami et al., 1981) and no serious adverse reactions have been reported (Unoshima et al., 2003; Katsuno et al., 2005; Yanaka et al., 2007; Fujimura et al., 2012). GGA rapidly induces HSP expression (HSPB, HSPA1A, and HSPC family members) via activation of the HSF1, in response to a variety of stresses, whereas its effect is weaker under non-stress conditions, providing its main effect when and where needed (Hirakawa et al., 1996; Yamanaka et al., 2003).
The protective effect of GGA-induced HSP expression on early and late remodeling, suggests that it has value in the prevention of clinical AF, although this still needs to be assessed in clinical trials (Brundel et al., 2006a,b; Sakabe et al., 2008). So far, the protective action of GGA has been established regarding electrical, contractile, and structural remodeling in in vitro HL-1 and dog atrial cardiomyocytes and in in vivo Drosophila and dog models for AF. GGA also has beneficial effects in AF of different origin, as observed in AF induced by congestive heart failure and acute ischemia (Shiroshita-Takeshita et al., 2007; Sakabe et al., 2008). The broad protective effects of GGA against AF-related derailment of proteostasis and atrial remodeling suggest that inducers of the HSR have substantial therapeutic value for clinical AF. Other drugs that induce HSP expression, such as bimoclomol, atorvastatin, cyclosporine A, and dexamethasone, still need to be tested for their protective effects against AF-induced remodeling. Nevertheless, their therapeutic potential in other cardiac diseases, such as ischemic heart disease, have already been documented (Sun et al., 2000; Ooie et al., 2001; Chen et al., 2002; Lubbers et al., 2002; Efthymiou et al., 2005; Shinohara et al., 2007).
In summary, AF results in a derailment of cardiomyocyte proteostasis by inducing reversible electrical and irreversible structural remodeling. There is strong evidence that induction of HSPs, in particular HSPB family members, normalizes proteostasis and thereby prevents electrical and structural remodeling. Known upstream targets for HSP protection include L-type Ca2+ channel, calcium handling proteins, calpain, RhoA-GTPase, and F-actin stress fibers. Ultimately, the induction of HSPs, by proteostasis regulators such as GGA, may prevent the occurrence of AF (primary prevention) and may contribute to enhance therapeutic efficacy and treatment options for patient with AF in delaying progression toward persistent AF and/or improve the outcome of cardioversion (secondary prevention).
Conflict of Interest Statement
Femke Hoogstra-Berends is an employee (Technician) of Nyken BV, which holds intellectual property interests in heat shock protein expression as a treatment in atrial fibrillation.
Acknowledgments
We thank the Dutch Organization for Scientific Research (NWO program grant 916. 46. 043), Dutch Heart Foundation (2007B217 and 2009B024), the European Community, European Fund for Regional Development (Operationeel Programma Noord-Nederland 2007-2012, OP-EFRO) and the Province of Groningen, Innovative Action-program Groningen (IAG3). The authors thank Dr. Herman Steen (CEO, Nyken BV) for critically reviewing the manuscript.
References
Adam, O., Frost, G., Custodis, F., Sussman, M. A., Schäfers, H. J., Böhm, M., and Laufs, U. (2007). Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 50, 367.
Ago, T., Liu, T., Zhai, P., Chen, W., Li, H., Molkentin, J. D., Vatner, S. F., and Sadoshima, J. (2008). A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133, 978–993.
Allessie, M., Ausma, J., and Schotten, U. (2002). Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 54, 230–246.
Allessie, M. A., de Groot, N. M., Houben, R. P., Schotten, U., Boersma, E., Smeets, J. L., and Crijns, H. J. (2010). Electropathological substrate of long-standing persistent atrial fibrillation in patients with structural heart disease: longitudinal dissociation. Circ. Arrhythm. Electrophysiol. 3, 606–615.
Amit, G., Kikuchi, K., Greener, I. D., Yang, L., Novack, V., and Donahue, J. K. (2010). Selective molecular potassium channel blockade prevents atrial fibrillation. Circulation 121, 2263–2270.
Anderson, M. E. (2004). Calmodulin kinase and L-type calcium channels; a recipe for arrhythmias? Trends Cardiovasc. Med. 14, 152–161.
Arrigo, A. P. (2007). The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv. Exp. Med. Biol. 594, 14–26.
Arya, R., Mallik, M., and Lakhotia, S. C. (2007). Heat shock genes – integrating cell survival and death. J. Biosci. 32, 595–610.
Ausma, J., Dispersyn, G. D., Duimel, H., Thone, F., Ver Donck, L., Allessie, M., and Borgers, M. (2000a). Changes in ultrastructural calcium distribution in goat atria during atrial fibrillation. J. Mol. Cell. Cardiol. 32, 355–364.
Ausma, J., Duimel, H., Borgers, M., and Allessie, M. (2000b). Recovery of structural remodeling after cardioversion of chronic atrial fibrillation. Circulation 102, II–153.
Ausma, J., Furst, D., Thone, F., Shivalkar, B., Flameng, W., Weber, K., Ramaekers, F., and Borgers, M. (1995). Molecular changes of titin in left ventricular dysfunction as a result of chronic hibernation. J. Mol. Cell. Cardiol. 27, 1203–1212.
Ausma, J., Thon‚, F., Dispersyn, G. D., Flameng, W., Vanoverschelde, J. L., Ramaekers, F. C. S., and Borgers, M. (1998). Dedifferentiated cardiomyocytes from chronic hibernating myocardium are ischemia-tolerant. Mol. Cell. Biochem. 186, 159–168.
Ausma, J., Van Der Velden, H. M. W., Lenders, M. H., Van Ankeren, E. P., Jongsma, H. J., Ramaekers, F., Borgers, M., and Allessie, M. (2003). Reverse structural and gap-junctional remodeling after prolonged atrial fibrillation in the goat. Circulation 107, 2051–2058.
Ausma, J., Wijffels, M., Thone, F., Wouters, L., Allessie, M., and Borgers, M. (1997a). Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation 96, 3157–3163.
Ausma, J., Wijffels, M., Van Eys, G., Koide, M., Ramaekers, F., Allessie, M., and Borgers, M. (1997b). Dedifferentiation of atrial cardiomyocytes as a result of chronic atrial fibrillation. Am. J. Pathol. 151, 985–997.
Backs, J., Song, K., Bezprozvannaya, S., Chang, S., and Olson, E. N. (2006). CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Invest. 116, 1853–1864.
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919.
Benjamin, E. J., Wolf, P. A., D’Agostino, R. B., Silbershatz, H., Kannel, W. B., and Levy, D. (1998). Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 98, 946–952.
Bers, D. M. (2006). Cardiac ryanodine receptor phosphorylation: target sites and functional consequences. Biochem. J. 396, e1–e3.
Bito, V., Heinzel, F. R., Weidemann, F., Dommke, C., Van Der Velden, J., Verbeken, E., Claus, P., Bijnens, B., De Scheerder, I., Stienen, G. J. M., Sutherland, G. R., and Sipido, K. R. (2004). Cellular mechanisms of contractile dysfunction in hibernating myocardium. Circ. Res. 94, 794–801.
Brundel, B., Ausma, J., Van Gelder, I. C., Van Der Want, J. J. L., Van Gilst, W. H., Crijns, H., and Henning, R. H. (2002). Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc. Res. 54, 380–389.
Brundel, B., Henning, R. H., Ke, L., Van Gelder, I. C., Crijns, H., and Kampinga, H. H. (2006a). Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 41, 555–562.
Brundel, B., Shiroshita-Takeshita, A., Qi, X. Y., Yeh, Y. H., Chartier, D., Van Gelder, I. C., Henning, R. H., Kampinga, H. H., and Nattel, S. (2006b). Induction of heat-shock response protects the heart against atrial fibrillation. Circ. Res. 99, 1394–1402.
Brundel, B., Ke, L., Dijkhuis, A. J., Qi, X., Shiroshita-Takeshita, A., Nattel, S., Henning, R. H., and Kampinga, H. H. (2008). Heat shock proteins as molecular targets for intervention in atrial fibrillation. Cardiovasc. Res. 78, 422–428.
Brundel, B., Van Gelder, I. C., Henning, R. H., Tuinenburg, A. E., Deelman, L. E., Tieleman, R. G., Grandjean, J. G., Van Gilst, W. H., and Crijns, H. (1999). Gene expression of proteins influencing the calcium homeostasis in patients with persistent and paroxysmal atrial fibrillation. Cardiovasc. Res. 42, 443–454.
Brundel, B., Van Gelder, I. C., Henning, R. H., Tuinenburg, A. E., Tieleman, R. G., Wietses, M., Grandjean, J. G., Van Gilst, W. H., and Crijns, H. (2001). Ion channel remodeling is related to intra-operative atrial refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation 103, 684–690.
Burstein, B., and Nattel, S. (2008). Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 51, 802–809.
Camm, A. J., Kirchhof, P., Lip, G. Y., Schotten, U., Savelieva, I., Ernst, S., Van Gelder, I. C., Al-Attar, N., Hindricks, G., Prendergast, B., Heidbuchel, H., Alfieri, O., Angelini, A., Atar, D., Colonna, P., De Caterina, R., De Sutter, J., Goette, A., Gorenek, B., Heldal, M., Hohloser, S. H., Kolh, P., Le Heuzey, J. Y., Ponikowski, P., and Rutten, F. H. (2010). Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur. Heart J. 31, 2369–2429.
Cao, H., Xue, L., Xu, X., Wu, Y., Zhu, J., Chen, L., Chen, D., and Chen, Y. (2011). Heat shock proteins in stabilization of spontaneously restored sinus rhythm in permanent atrial fibrillation patients after mitral valve surgery. Cell Stress Chaperones 16, 517–528.
Cardin, S., Li, D., Thorin-Trescases, N., Leung, T. K., Thorin, E., and Nattel, S. (2003). Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways. Cardiovasc. Res. 60, 315–325.
Carter, S., Pitt, S. J., Colyer, J., and Sitsapesan, R. (2011). Ca2+-dependent phosphorylation of RyR2 can uncouple channel gating from direct cytosolic Ca2+ regulation. J. Membr. Biol. 240, 21–33.
Cha, T. J., Ehrlich, J., Zhang, L., Shi, Y. F., Tardif, J. C., Leung, T. K., and Nattel, S. (2004). Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation 109, 412–418.
Chelazzi, C., Villa, G., and De Gaudio, A. R. (2011). Postoperative atrial fibrillation. ISRN Cardiol. PMID: 203179, 10.
Chelu, M. G., Sarma, S., Sood, S., Wang, S., Van Oort, R. J., Skapura, D. G., Li, N., Santonastasi, M., Muller, F. U., Schmitz, W., Schotten, U., Anderson, M. E., Valderrabano, M., Dobrev, D., and Wehrens, X. H. T. (2009). Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951.
Chen, H. W., Chien, C. T., Yu, S. L., Lee, Y. T., and Chen, W. J. (2002). Cyclosporine A regulate oxidative stress-induced apoptosis in cardiomyocytes: mechanisms via ROS generation, iNOS and Hsp70. Br. J. Pharmacol. 137, 771–781.
Chen, Y. C., Kao, Y. H., Huang, C. F., Cheng, C. C., Chen, Y. J., and Chen, S. A. (2010). Heat stress responses modulate calcium regulations and electrophysiological characteristics in atrial myocytes. J. Mol. Cell. Cardiol. 48, 781–788.
Christ, T., Boknik, P., Wöhrl, S., Wettwer, E., Graf, E. M., Bosch, R. F., Knaut, M., Schmitz, W., Ravens, U., and Dobrev, D. (2004). L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110, 2651–2657.
Concannon, C. G., Orrenius, S., and Samali, A. (2001). Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 9, 195–201.
de Groot, N. M., Houben, R. P., Smeets, J. L., Boersma, E., Schotten, U., Schalij, M. J., Crijns, H., and Allessie, M. A. (2010). Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation 122, 1674–1682.
de Ruijter, A. J., Van Gennip, A. H., Caron, H. N., Kemp, S., and Van Kuilenburg, A. B. (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749.
Ding, X. Z., Tsokos, G. C., and Kiang, J. G. (1998). Overexpression of HSP-70 inhibits the phosphorylation of HSF1 by activating protein phosphatase and inhibiting protein kinase C activity. FASEB J. 12, 451–459.
Dobrev, D., and Nattel, S. (2008). Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J. Cardiovasc. Pharmacol. 52, 293–299.
Dobrev, D., and Nattel, S. (2010). New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 375, 1212–1223.
Dobrev, D., and Voigt, N. (2011). Ion channel remodelling in atrial fibrillation. Eur. Cardiol. 7, 7.
Dudley, S. C. Jr., Hoch, N. E., Mccann, L. A., Honeycutt, C., Diamandopoulos, L., Fukai, T., Harrison, D. G., Dikalov, S. I., and Langberg, J. (2005). Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112, 1266–1273.
Efthymiou, C. A., Mocanu, M. M., De Belleroche, J., Wells, D. J., Latchmann, D. S., and Yellon, D. M. (2004). Heat shock protein 27 protects the heart against myocardial infarction. Basic Res. Cardiol. 99, 392–394.
Efthymiou, C. A., Mocanu, M. M., and Yellon, D. M. (2005). Atorvastatin and myocardial reperfusion injury: new pleiotropic effect implicating multiple prosurvival signaling. J. Cardiovasc. Pharmacol. 45, 247–252.
El-Armouche, A., Boknik, P., Eschenhagen, T., Carrier, L., Knaut, M., Ravens, U., and Dobrev, D. (2006). Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 114, 670–680.
Engelmann, M. D., and Svendsen, J. H. (2005). Inflammation in the genesis and perpetuation of atrial fibrillation. Eur. Heart J. 26, 2083–2092.
Fan, G. C., and Kranias, E. G. (2011). Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 51, 574–577.
Feinberg, W. M., Blackshear, J. L., Laupacis, A., Kronmal, R., and Hart, R. G. (1995). Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch. Intern. Med. 155, 469–473.
Fujimura, N., Jitsuiki, D., Maruhashi, T., Mikami, S., Iwamoto, Y., Kajikawa, M., Chayama, K., Kihara, Y., Noma, K., Goto, C., and Higashi, Y. (2012). Geranylgeranylacetone, heat shock protein 90/AMP-activated protein kinase/endothelial nitric oxide synthase/nitric oxide pathway, and endothelial function in humans. Arterioscler. Thromb. Vasc. Biol. 32, 153–160.
Garrido, C., Bruey, J. M., Fromentin, A., Hammann, A., Arrigo, A. P., and Solary, E. (1999). HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 13, 2061–2070.
Goette, A., Honeycutt, C., and Langberg, J. J. (1996). Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation 94, 2968–2974.
Golenhofen, N., Perng, M. D., Quinlan, R. A., and Drenckhahn, D. (2004). Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 122, 415–425.
Greiser, M., Halaszovich, C. R., Frechen, D., Boknik, P., Ravens, U., Dobrev, D., Luckhoff, A., and Schotten, U. (2007). Pharmacological evidence for altered src kinase regulation of I (Ca,L) in patients with chronic atrial fibrillation. Naunyn Schmiedebergs Arch. Pharmacol. 375, 383–392.
Hagemann, D., and Xiao, R. P. (2002). Dual site phospholamban phosphorylation and its physiological relevance in the heart. Trends Cardiovasc. Med. 12, 51–56.
Hara, Y., Wakamori, M., Ishii, M., Maeno, E., Nishida, M., Yoshida, T., Yamada, H., Shimizu, S., Mori, E., Kudoh, J., Shimizu, N., Kurose, H., Okada, Y., Imoto, K., and Mori, Y. (2002). LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 9, 163–173.
Hayashi, M., Imanaka-Yoshida, K., Yoshida, T., Wood, M., Frearns, C., Tatake, R. J., and Lee, J. D. (2006). A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat. Med. 1, 128–132.
Hirakawa, T., Rokutan, K., Nikawa, T., and Kishi, K. (1996). Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 111, 345–357.
Igarashi, T., Finet, J. E., Takeuchi, A., Fujino, Y., Strom, M., Greener, I. D., Rosenbaum, D. S., and Donahue, J. K. (2012). Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 125, 216–225.
Imazio, M., Brucato, A., Ferrazzi, P., Rovere, M. E., Gandino, A., Cemin, R., Ferrua, S., Belli, R., Maestroni, S., Simon, C., Zingarelli, E., Barosi, A., Sansone, F., Patrini, D., Vitali, E., Trinchero, R., Spodick, D. H., and Adler, Y. (2011). Colchicine reduces postoperative atrial fibrillation: results of the colchicine for the prevention of the postpericardiotomy syndrome (COPPS) atrial fibrillation substudy. Circulation 124, 2290–2295.
Inagaki, N., Hayashi, T., Arimura, T., Koga, Y., Takahashi, M., Shibata, H., Teraoka, K., Chikamori, T., Yamashina, A., and Kimura, A. (2006). Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 342, 379–386.
Jones, Q., Voegeli, T. S., Li, G., Chen, Y., and Currie, R. W. (2011). Heat shock proteins protect against ischemia and inflammation through multiple mechanisms. Inflamm. Allergy Drug Targets 10, 247–259.
Kalmar, B., and Greensmith, L. (2009). Induction of heat shock proteins for protection against oxidative stress. Adv. Drug Deliv. Rev. 61, 310–318.
Kammanadiminti, S. J., and Chadee, K. (2006). Suppression of NF-kB activation by Entamoeba histolytica in intestinal epithelial cells is mediated by heat shock protein 27. J. Biol. Chem. 281, 26112–26120.
Kannel, W. B., Wolf, P. A., Benjamin, E. J., and Levy, D. (1998). Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am. J. Cardiol. 82, 2N–9N.
Katsuno, M., Sang, C., Adachi, H., Minamiyama, M., Waza, M., Tanaka, F., Doyu, M., and Sobue, G. (2005). Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl. Acad. Sci. U.S.A. 102, 16801–16806.
Ke, L., Meijering, R. A. M., Hoogstra-Berends, F., Mackovicova, K., Vos, M. J., Van Gelder, I. C., Henning, R. H., Kampinga, H. H., and Brundel, B. (2011). HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE 6, e20395. doi:10.1371/journal.pone.0020395
Ke, L., Qi, X. Y., Dijkhuis, A. J., Chartier, D., Nattel, S., Henning, R. H., Kampinga, H. H., and Brundel, B. (2008). Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 45, 685–693.
Kim, Y. K., Suarez, J., Hu, Y., Mcdonough, P. M., Boer, C., Dix, D. J., and Dillmann, W. H. (2006). Deletion of the inducible 70-kDa heat shock protein genes in mice impairs cardiac contractile function and calcium handling associated with hypertrophy. Circulation 113, 2589–2597.
Kirmanoglou, K., Hannekum, A., and Schafler, A. E. (2004). Expression of mortalin in patients with chronic atrial fibrillation. Basic Res. Cardiol. 99, 404–408.
Kupatt, C., Dessy, C., Hinkel, R., Raake, P., Daneau, G., Bouzin, C., Boekstegers, P., and Feron, O. (2004). Heat shock protein 90 transfection reduces ischemia-reperfusion-induced myocardial dysfunction via reciprocal endothelial NO synthase serine 1177 phosphorylation and threonine 495 dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 24, 1435–1441.
Kwon, J. H., Kim, J. B., Lee, K. H., Kang, S. M., Chung, N., Jang, Y., and Chung, J. H. (2007). Protective effect of heat shock protein 27 using protein transduction domain-mediated delivery on ischemia/reperfusion heart injury. Biochem. Biophys. Res. Commun. 363, 399–404.
Lakshmikuttyamma, A., Selvakumar, P., Anderson, D. H., Datla, R. S., and Sharma, R. K. (2004). Molecular cloning of bovine cardiac muscle heat-shock protein 70 kDa and its phosphorylation by cAMP-dependent protein kinase in vitro. Biochemistry 43, 13340–13347.
Leistad, E., Borgers, M., and Christensen, G. (1996). Atrial contractile dysfunction after short-term atrial fibrillation can be explained by changes in intracellular calcium, but not by atrial ischemia. Circulation 94, I–386–I–387.
Li, D., Shinagawa, K., Pang, L., Leung, T. K., Cardin, S., Wang, Z., and Nattel, S. (2001). Effects of Angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 104, 2608–2614.
Lin, L., Kim, S. C., Wang, Y., Gupta, S., Davis, B., Simon, S. I., Torre-Amione, G., and Knowlton, A. A. (2007). HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 293, H2238–H2247.
Liu, F., Levin, M. D., Petrenko, N. B., Lu, M. M., Wang, T., Yuan, L. J., Stout, A. L., Epstein, J. A., and Patel, V. V. (2008). Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell. Cardiol. 45, 715–723.
Liu, J., Kam, K. W. L., Borchert, G. H., Kravtsov, G. M., Ballard, H. J., and Wong, T. M. (2006). Further study on the role of HSP70 on Ca2+ homeostasis in rat ventricular myocytes subjected to simulated ischemia. Am. J. Physiol. Cell Physiol. 290, C583–C591.
Lubbers, N. L., Polakowski, J. S., Wegner, C. D., Burke, S. E., Diaz, G. J., Daniell, K. M., and Cox, B. F. (2002). Oral bimoclomol elevates heat shock protein 70 and reduces myocardial infarct size in rats. Eur. J. Pharmacol. 435, 79–83.
Mandal, K., Torsney, E., Poloniecki, J., Camm, A. J., Xu, Q., and Jahangiri, M. (2005). Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann. Thorac. Surg. 79, 865–871.
Marber, M. S., Mestril, R., Chi, S. H., Sayen, M. R., Yellon, D. M., and Dillmaann, W. H. (1995). Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Invest. 95, 1446–1456.
Mounier, N., and Arrigo, A. P. (2002). Actin cytoskeleton and small heat shock proteins: how do they interact? Cell Stress Chaperones 7, 167–176.
Murakami, M., Oketani, K., Fujisaki, H., Wakabayashi, T., and Ohgo, T. (1981). Antiulcer effect of geranylgeranylacetone, a new acyclic polyisoprenoid on experimentally induced gastric and duodenal ulcers in rats. Arzneimittelforschung 31, 799–804.
Nattel, S., Burstein, B., and Dobrev, D. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73.
Nègre-Aminou, P., Van Leeuwen, R. E., Van Thiel, G. C., Van Den, I. P., De Jong, W. W., Quinlan, R. A., and Cohen, L. H. (2002). Differential effect of simvastatin on activation of Rac(1) vs. activation of the heat shock protein 27-mediated pathway upon oxidative stress, in human smooth muscle cells. Biochem. Pharmacol. 64, 1483–1491.
Oc, M., Ucar, H. I., Pinar, A., Akbulut, B., Oc, B., Akinci, S. B., Akyon, Y., Kanbak, M., Boke, E., and Dogan, R. (2007). Heat shock protein 60 antibody. A new marker for subsequent atrial fibrillation development. Saudi Med. J. 28, 844–847.
Ooie, T., Takahashi, N., Saikawa, T., Nawata, T., Arikawa, M., Yamanaka, K., Hara, M., Shimada, T., and Sakata, T. (2001). Single oral dose of geranylgeranylacetone induces heat-shock protein 72 and renders protection against ischemia/reperfusion injury in rat heart. Circulation 104, 1837–1843.
Park, K. J., Gaynor, R. B., and Kwak, Y. T. (2003). Heat shock protein 27 association with the I kappa B kinase complex regulates tumor necrosis factor alpha-induced NF-kappa B activation. J. Biol. Chem. 278, 35272–35278.
Peng, W., Zhang, Y., Zheng, M., Cheng, H., Zhu, W., Cao, C. M., and Xiao, R. P. (2010). Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ. Res. 106, 102–110.
Plumier, J. C., Ross, B. M., Currie, R. W., Angelidis, C. E., Kazlaris, H., Kollias, G., and Pagoulatos, G. N. (1995). Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J. Clin. Invest. 95, 1854–1860.
Pochynyuk, O., Stockand, J. D., and Staruschenko, A. (2007). Ion channel regulation by Ras, Rho, and Rab small GTPases. Exp. Biol. Med. (Maywood) 232, 1258–1265.
Powers, E. T., Morimoto, R. I., Dillin, A., Kelly, J. W., and Balch, W. E. (2009). Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991.
Qi, X. Y., Yeh, Y. H., Xiao, L., Burstein, B., Maguy, A., Chartier, D., Villeneuve, L. R., Brundel, B., Dobrev, D., and Nattel, S. (2008). Cellular signalling underlying atrial tachycardia remodeling of L-type calcium-current. Circ. Res. 103, 845–854.
Qian, J., Vafiadaki, E., Florea, S. M., Singh, V. P., Song, W., Lam, C. K., Wang, Y., Yuan, Q., Pritchard, T. J., Cai, W., Haghighi, K., Rodriguez, P., Wang, H. S., Sanoudou, D., Fan, G. C., and Kranias, E. G. (2011). Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ. Res. 108, 1429–1438.
Rajasekaran, N. S., Connell, P., Christians, E. S., Yan, L. J., Taylor, R. P., Orosz, A., Zhang, X. Q., Stevenson, T. J., Peshock, R. M., Leopold, J. A., Barry, W. H., Loscalzo, J., Odelberg, S. J., and Benjamin, I. J. (2007). Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130, 427–439.
Sadoshima, J., Takahashi, T., Jahn, L., and Izumo, S. (1992). Roles of mechano-sensitive ion channels, cytoskeleton, and contractile activity in stretch-induced immediate-early gene expression and hypertrophy of cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 89, 9905–9909.
Sah, V. P., Minamisawa, S., Tam, S. P., Wu, T. H., Dorn, G. W., Ross, J., Chien, K. R., and Brown, J. H. (1999). Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J. Clin. Invest. 103, 1627–1634.
Sakabe, M., Shiroshita-Takeshita, A., Maguy, A., Brundel, B., Fujiki, A., Inoue, H., and Nattel, S. (2008). Effects of heat shock protein induction on atrial fibrillation caused by acute atrial ischemia. Cardiovasc. Res. 78, 63–70.
Salinthone, S., Tyagi, M., and Gerthoffer, W. T. (2008). Small heat shock proteins in smooth muscle. Pharmacol. Ther. 119, 44–54.
Sanchez-Quinones, J., Marin, F., Roldan, V., and Lip, G. Y. (2008). The impact of statin use on atrial fibrillation. QJM 101, 845–861.
Savelieva, I., Kakouros, N., Kourliouros, A., and Camm, A. J. (2011a). Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part I: primary prevention. Europace 13, 308–328.
Savelieva, I., Kakouros, N., Kourliouros, A., and Camm, A. J. (2011b). Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part II: secondary prevention. Europace 13, 610–625.
Schotten, U., Duytschaever, M., Ausma, J., Eijsbouts, S., Neuberger, H. R., and Allessie, M. (2003). Electrical and contractile remodeling during the first days of atrial fibrillation go hand in hand. Circulation 107, 1433–1439.
Schwinger, R. H., Munch, G., Bolck, B., Karczewski, P., Krause, E. G., and Erdmann, E. (1999). Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell. Cardiol. 31, 479–491.
Sherman, A. J., Klocke, F. J., Decker, R. S., Decker, M. L., Kozlowski, K. A., Harris, K. R., Hedjbeli, S., Yaroshenko, Y., Nakamura, S., Parker, M. A., Checchia, P. A., and Evans, D. B. (2000). Myofibrillar disruption in hypocontractile myocardium showing perfusion-contraction matches and mismatches. Am. J. Physiol. Heart Circ. Physiol. 278, 1320–1334.
Shinohara, T., Takahashi, N., Kohno, H., Yamanaka, K., Ooie, T., Wakisaka, O., Murozono, Y., Taniguchi, Y., Torigoe, Y., Hara, M., Shimada, T., Saikawa, T., and Yoshimatsu, H. (2007). Mitochondria are targets for geranylgeranylacetone-induced cardioprotection against ischemia-reperfusion in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 293, H1892–H1899.
Shiroshita-Takeshita, A., Brundel, B., Burnstein, B., Leung, T. K., Mitamura, H., Ogawa, S., and Nattel, S. (2007). Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovasc. Res. 74, 75–84.
Shiroshita-Takeshita, A., Brundel, B., Lavoie, J., and Nattel, S. (2006). Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc. Res. 69, 865–875.
Shiroshita-Takeshita, A., Schram, G., Lavoie, J., and Nattel, S. (2004). Effect of simvastatin and antioxidant vitamins on atrial fibrillation promotion by atrial-tachycardia remodeling in dogs. Circulation 110, 2313–2319.
Son, G. H., Geum, D., Chung, S., Park, E., Lee, K. H., Choi, S., and Kim, K. (2005). A protective role of 27-kDa heat shock protein in glucocorticoid-evoked apoptotic cell death of hippocampal progenitor cells. Biochem. Biophys. Res. Commun. 338, 1751–1758.
St Rammos, K., Koullias, G. J., Hassan, M. O., Argyrakis, N. P., Voucharas, C. G., Scarupa, S. J., and Cowte, T. G. (2002). Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovasc. Surg. 10, 228–232.
Sugiyama, Y., Suzuki, A., Kishikawa, M., Akutsu, R., Hirose, T., Waye, M. M., Tsui, S. K., Yoshida, S., and Ohno, S. (2000). Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J. Biol. Chem. 275, 1095–1104.
Sun, L., Chang, J., Kirchhoff, S. R., and Knowlton, A. A. (2000). Activation of HSF and selective increase in heat-shock proteins by acute dexamethasone treatment. Am. J. Physiol. Heart Circ. Physiol. 278, H1091–H1097.
Thijssen, V. L., Ausma, J., Gorza, L., Van Der Velden, H. M., Allessie, M. A., Van Gelder, I. C., Borgers, M., and Van Eys, G. J. (2004). Troponin I isoform expression in human and experimental atrial fibrillation. Circulation 110, 770–775.
Todd, D. M., Fynn, S. P., Walden, A. P., Hobbs, W. J., Arya, S., and Garratt, C. J. (2004). Repetitive 4-week periods of atrial electrical remodeling promote stability of atrial fibrillation: time course of a second factor involved in the self-perpetuation of atrial fibrillation. Circulation 109 1434–1439.
Unoshima, M., Iwasaka, H., Eto, J., Takita-Sonoda, Y., Noguchi, T., and Nishizono, A. (2003). Antiviral effects of geranylgeranylacetone: enhancement of MxA expression and phosphorylation of PKR during influenza virus infection. Antimicrob. Agents Chemother. 47, 2914–2921.
Van Gelder, I. C., Crijns, H., Tieleman, R. G., De Kam, P. J., Gosseling, A. T. M., Verheugt, F. W. A., and Lie, K. I. (1996). Value and limitation of electrical cardioversion in patients with chronic atrial fibrillation – importance of arrhythmia risk factors and oral anticoagulation. Arch. Intern. Med. 156, 2585–2592.
Van Gelder, I. C., Smit, M. D., Alings, M., and Crijns, H. J. (2010). Upstream therapy in patients with early atrial fibrillation: the relevance of the Routine versus Aggressive upstream rhythm Control for prevention of Early atrial fibrillation in heart failure (RACE 3) study. Neth. Heart J. 18, 522–523.
Van Wagoner, D. R., Lamorgese, M., Kirian, P., Cheng, Y., Efimov, I. R., and Mazgalev, T. N. (1997). Calcium current density is reduced in atrial myocytes isolated from patients in chronic atrial fibrillation. Circulation 96, I–180 (abstract).
Vanoverschelde, J. L. J., Wijns, W., Depr‚, C., Essamri, B., Heyndricks, S., Borgers, M., Bol, A., and Melin, J. (1993). Mechanisms of chronic regional postischemic dysfunction in humans: new insights from the study on non-infarcted collateral dependent myocardium. Circulation 87, 1513–1523.
Vest, J. A., Wehrens, X. H. T., Reiken, S. R., Lehnart, S. E., Dobrev, D., Chandra, P., Danilo, P., Ravens, U., Rosen, M. R., and Marks, A. R. (2005). Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 111, 2025–2032.
Wakisaka, O., Takahashi, N., Shinohara, T., Ooie, T., Nakagawa, M., Yonemochi, H., Hara, M., Shimada, T., Saikawa, T., and Yoshimatsu, H. (2007). Hyperthermia treatment prevents angiotensin II-mediated atrial fibrosis and fibrillation via induction of heat-shock protein 72. J. Mol. Cell Cardiol. 436, 616–626.
Wang, T. J., Parise, H., Levy, D., D’Agostino, R. B. Sr., Wolf, P. A., Vasan, R. S., and Benjamin, E. J. (2004). Obesity and the risk of new-onset atrial fibrillation. JAMA 292, 2471–2477.
Wang, Z. (2003). Role of redox state in modulation of ion channel function by fatty acids and phospholipids. Br. J. Pharmacol. 139, 681–683.
Watanabe, H., Tanabe, N., Watanabe, T., Darbar, D., Roden, D. M., Sasaki, S., and Aizawa, Y. (2008). Metabolic syndrome and risk of development of atrial fibrillation: the Niigata preventive medicine study. Circulation 117, 1255–1260.
Westerheide, S. D., and Morimoto, R. I. (2005). Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 280, 33097–33100.
Wijffels, M. C., Kirchhof, C. J., Dorland, R., and Allessie, M. A. (1995). Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92, 1954–1968.
Yamanaka, K., Takahashi, N., Ooie, T., Kaneda, K., Yoshimatsu, H., and Saikawa, T. (2003). Role of protein kinase C in geranylgeranylacetone-induced expression of heat-shock protein 72 and cardioprotection in the rat heart. J. Mol. Cell. Cardiol. 35, 785–794.
Yanaka, A., Zhang, S., Sato, D., Tauchi, M., Suzuki, H., Shibahara, T., Matsui, H., Nakahara, A., and Hyodo, I. (2007). Geranylgeranylacetone protects the human gastric mucosa from diclofenac-induced injury via induction of heat shock protein 70. Digestion 75, 148–155.
Yang, M., Tan, H., Cheng, L., He, M., Wei, Q., Tanguay, R. M., and Wu, T. (2007). Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones 12, 142–150.
Yue, L., Feng, J., Gaspo, R., Li, G. R., Wang, Z., and Nattel, S. (1997). Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ. Res. 81, 512–525.
Zhang, D., Ke, L., Mackovicova, K., Van Der Want, J. J. L., Sibon, O. C. M., Tanguay, R. M., Morrow, G., Henning, R. H., Kampinga, H. H., and Brundel, B. (2011a). Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 51, 381–389.
Keywords: AF, HSP, Rho-GTPases, remodeling, upstream therapy, myolysis, calpain, geranylgeranylacetone
Citation: Meijering RAM, Zhang D, Hoogstra-Berends F, Henning RH and Brundel BJJM (2012) Loss of proteostatic control as a substrate for atrial fibrillation: a novel target for upstream therapy by heat shock proteins. Front. Physio. 3:36. doi: 10.3389/fphys.2012.00036
Received: 24 December 2011;
Accepted: 09 February 2012;
Published online: 23 February 2012.
Edited by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USAReviewed by:
Yuanfang Xie, University of California at Davis, USAGerard Boink, Academic Medical Center, Netherlands
Copyright: © 2012 Meijering, Zhang, Hoogstra-Berends, Henning and Brundel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Bianca J. J. M. Brundel, Department of Clinical Pharmacology, University Medical Center Groningen, University of Groningen, Deusinglaan 1, 9713 AV Groningen, Netherlands. e-mail: b.j.j.m.brundel@umcg.nl