 Francisco Javier Blanco* Carmelo Bernabéu
Francisco Javier Blanco* Carmelo Bernabéu
- Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas and Centro de Investigación Biomédica en Red de Enfermedades Raras, Madrid, Spain
Aging is the major risk factor per se for the development of cardiovascular diseases. The senescence of the endothelial cells (ECs) that line the lumen of blood vessels is the cellular basis for these age-dependent vascular pathologies, including atherosclerosis and hypertension. During their lifespan, ECs may reach a stage of senescence by two different pathways; a replicative one derived from their preprogrammed finite number of cell divisions; and one induced by stress stimuli. Also, certain physiological stimuli, such as transforming growth factor-β, are able to modulate cellular senescence. Currently, the cellular aging process is being widely studied to identify novel molecular markers whose changes correlate with senescence. This review focuses on the regulation of alternative splicing mediated by the serine–arginine splicing factor 1 (SRSF1, or ASF/SF2) during endothelial senescence, a process that is associated with a differential subcellular localization of SRSF1, which typically exhibits a scattered distribution throughout the cytoplasm. Based on its senescence-dependent involvement in alternative splicing, we postulate that SRSF1 is a key marker of EC senescence, regulating the expression of alternative isoforms of target genes such as endoglin (ENG), vascular endothelial growth factor A (VEGFA), tissue factor (T3), or lamin A (LMNA) that integrate in a common molecular senescence program.
Introduction
It is well established that vascular physiology declines with aging due to the impairment of endothelial functions. This endothelial dysfunction is fairly evidenced by the stiffening of blood vessels that fail to control the dilatory responses associated with hypertension (Versari et al., 2009), and by a pro-inflammatory state of the endothelial microenvironment that favors the development of atherosclerosis (Erusalimsky, 2009). Hence the biology of endothelial cells (ECs) is a key element for elucidating and understanding the cellular and molecular basis of vascular pathologies associated with age.
Endothelial cells form a thin monolayer that lines the blood vessels and is in direct contact with the blood flow. During embryonic development, processes including vasculogenesis and angiogenesis require repeated division of ECs, but in adults these cells are in a quiescent state and divide only occasionally (Conway and Carmeliet, 2004; Semenza, 2007; Carmeliet and Jain, 2011). However, at this latter stage ECs remain highly responsive to mechanical stimuli, such as shear stress and injuries, as well as to chemical signals, including hypoxia, cytokines, and growth factors, among others. Thus, during vascular remodeling or upon angiogenic stimulation, ECs actively proliferate, but progressively lose their capacity to divide, reaching the stage of replicative senescence. This process involves telomere shortening until the cells reach the so-called Hayflick’s limit (Brandes et al., 2005), that renders cell division impossible, constituting a mechanism to prevent genomic instability and the development of cancer (Shay and Wright, 2007). Endothelial senescence can also be triggered by telomere-independent events, including chemotherapy, proto-oncogene-induced senescence, alterations in nuclear lamina, and stress stimuli that usually involve DNA damage. In this sense, oxidative stress and ultraviolet radiation are major stimuli for the induction of this type of senescence; both ultimately generate reactive oxygen species (ROS; Debacq-Chainiaux et al., 2005; Erusalimsky and Skene, 2009; Collins and Tzima, 2011) and activate retinoblastoma protein family pathways, the final effectors of the senescence program (Campisi and d’Adda di Fagagna, 2007; Erusalimsky, 2009).
There is a growing body of evidence that vascular aging and senescence of ECs are the basis of the endothelial dysfunction associated with vascular pathologies. For example, it is well known that aging is associated with decreased angiogenesis (Brandes et al., 2005; Erusalimsky, 2009), and that senescent ECs are unable to form neoangiogenic networks in matrigel assays (Chang et al., 2005). In this regard, certain physiological regulators of angiogenesis, such as transforming growth factor-β (TGF-β), are able to modulate cellular senescence of ECs (Santibanez et al., 2011; Blanco and Bernabeu, 2012; Doetschman et al., 2012; Krieglstein et al., 2012).
Recently, growing interest has focused on the role of alternative processing of certain precursor mRNA transcripts during EC senescence (Meshorer and Soreq, 2002; Harries et al., 2011). Alternative splicing represents an advantageous mechanism by which cells enrich their transcriptome in order to synthesize a specialized proteome for a particular stage or process. An early link between the role of alternative splicing and aging was described in the late 1980s, when it was reported that alternative isoforms from the elastin gene (ELN) could contribute to aging and pathological situations in the cardiovascular and pulmonary systems (Indik et al., 1989). Since then, numerous papers have confirmed this idea, not only in senescent ECs, but also in other cell types (Meshorer and Soreq, 2002).
Currently, active investigations are focused on the molecular components that orchestrate the modulation of alternative splicing events in aging. Briefly, splicing is carried out by the spliceosome, a complex molecular machinery integrated by a core of small nuclear ribonucleoproteins (snRNPs) plus a great variety of essential and accessory splicing factors, or trans-elements. These splicing factors recognize short consensus sequences in a precursor mRNA, or cis-elements, thereby modulating its activity. Thus, it is assumed that the splicing changes observed during aging are part of a genetically programmed mechanism which is not necessarily associated with deleterious mutations, but with the splicing factors that play a critical role affecting the expression of target genes.
Splicing Factors Involved in Endothelial Senescence
Analyses of the splicing factors SNEV and serine–arginine splicing factor 1 (SRSF1) have demonstrated their role in EC senescence. Thus, upon replicative senescence of human umbilical vein endothelial cells (HUVECs), a down-regulation of SNEV was observed (Voglauer et al., 2006). This event takes places concomitantly with down-regulation of the catalytic subunit of the telomerase, hTERT, the classic cellular senescent marker. Conversely, overexpression of SNEV provokes extension of the replicative lifespan in these cells. In addition to being involved in spliceosome assembly and stabilization, SNEV regulates proteasome function due to its U3-ligase activity, and is involved in DNA repair (Grillari et al., 2010). Furthermore, the haploinsufficiency of SNEV has been demonstrated using a heterozygous mouse model, which showed that the proliferative and repopulating capacity of hematopoietic stem cells from these mice was clearly compromised (Schraml et al., 2008).
The splicing factor SRSF1, or ASF/SF2, is the prototypical member of the highly conserved family of serine/arginine-rich proteins (SR proteins). Structurally, these SR proteins are composed of one or two RNA recognition motifs plus a C-terminal serine/arginine-rich domain involved in protein–protein interactions (Figure 1). During the past decade, the role of SR proteins has been extended to include a diverse set of cellular processes, including mRNA nuclear export, mRNA stability and quality control, translation, maintenance of genomic stability, and oncogenic transformation (Manley and Krainer, 2010). Within this family, SRSF1 participates in constitutive and alternative splicing, and in a plethora of biological processes, including viral infections and cancer (Sanford et al., 2005; Biamonti and Caceres, 2009). Recently, the role of SRSF1 in senescence, regulating the expression of the short isoform of endoglin, has been described (Blanco and Bernabeu, 2011). In ECs, SRSF1 promotes retention of the last intron of the endoglin gene (ENG) by binding to an intronic cis-element that overlaps with the branch point consensus sequence, interfering with intron removal (Blanco and Bernabeu, 2011). This retained intron shifts the open reading frame, incorporating an early stop codon that produces an alternate cytoplasmic domain shorter than the one present in the predominantly expressed L-endoglin (Bellon et al., 1993). This process has been associated with aging, contributing to the development of vascular pathologies including hypertension and atherosclerosis (Blanco et al., 2008). The involvement of SRSF1 in senescence has also been studied using mouse embryonic fibroblasts as a cellular model (Verduci et al., 2010). These authors found that two microRNAs, miR-28 and miR-505, are induced during senescence and negatively modulate SRSF1 expression, promoting cell cycle arrest and/or apoptosis.
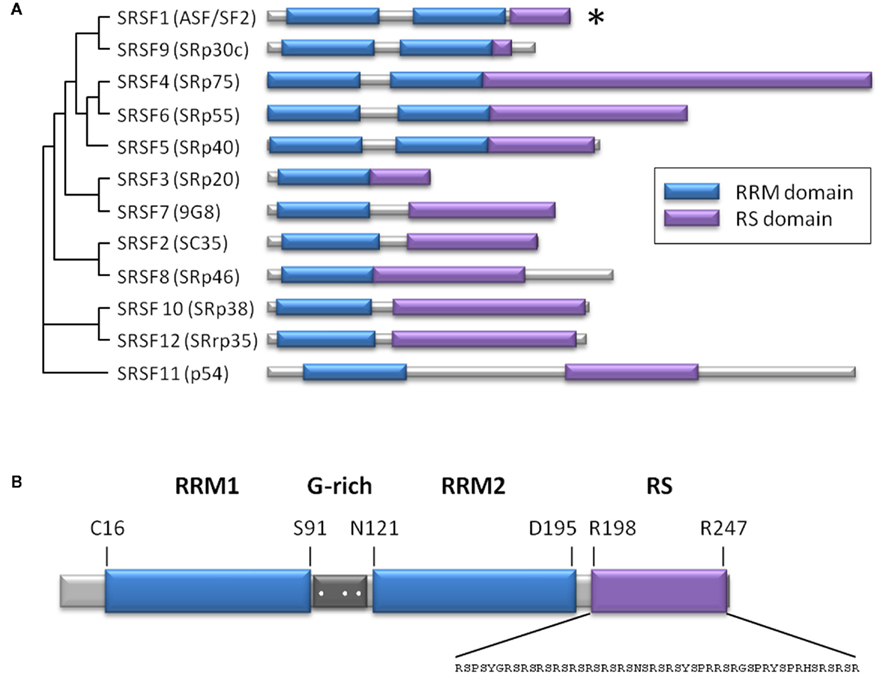
Figure 1. The SR protein family. (A) Scheme representing the level of homology among human SR proteins (Manley and Krainer, 2010), based on the result of a multiple sequence alignment using ClustalW2. Alternate names are in parentheses. Both RS and RRMs are modular domains, which means that RRMs can be exchanged between SR proteins, and may still bind to RNA in the absence of the RS domain. The RS domain can bind to a heterologous RNA-binding domain and be still functional. The asterisk indicates the SRSF1 member. (B) Structural scheme of SRSF1 showing two RRM domains (blue) separated by a glycine-rich sequence (dark gray), where three arginine residues are susceptible to methylation (white dots). The sequence of the RS domain (violet) is indicated. RRM, RNA recognition motif; RS, arginine/serine-rich.
Spliced Genes Regulated by SRSF1 and Vascular Aging
The role of SRSF1 in the endothelial context has been addressed by analyzing the expression of alternative isoforms of the vascular endothelial growth factor A (VEGFA) gene. VEGF is expressed as a disulfide-linked homodimer that specifically acts on ECs. Some of its biological effects include an increase in vascular permeability, induction of growth and migration of ECs (vasculogenesis and angiogenesis), and inhibition of apoptosis. Of note, VEGFA encodes two families of isoforms that are generated by alternate splice-site selection in its terminal exon #8. Thus, selection of the proximal splice-site (PSS) results in pro-angiogenic VEGF isoforms, whereas selection of the distal splice-site (DSS) leads to anti-angiogenic VEGFb isoforms (Neufeld et al., 1999). Interestingly, SRSF1 is a key regulator in the selection of the PSS, an event induced by IGF-1 treatment and abolished by TGF-β stimulus (Nowak et al., 2008; Figure 2A). Also, selection of the pro-angiogenic PSS by SRSF1 is prompted by changes in the cellular microenvironment such as those that occur in solid tumors, including a decrease in pH, among others (Elias and Dias, 2008). Because upon stimulation, SRPK1/2 kinases activate and translocate SRSF1 into the nucleus, it is not surprising to find that inhibition of SRPK1/2 reduced angiogenesis in a mouse model of retinal neovascularization, launching the potential involvement of alternative splicing regulation as a therapeutic target in angiogenic disorders (Nowak et al., 2010; Carter et al., 2011).
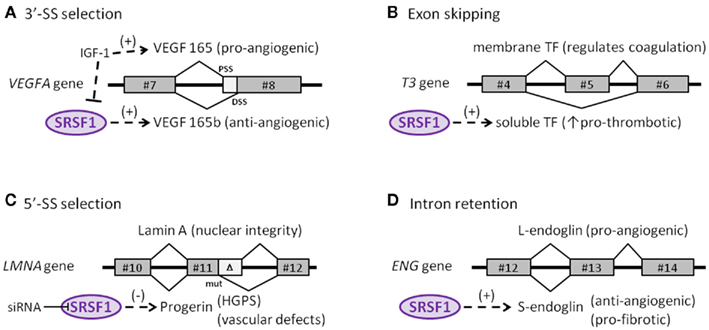
Figure 2. Endothelial target genes of SRSF1. (A) Selection of the proximal (PSS) or distal (DSS) 3′-splice-site determines synthesis of either the pro- or anti-angiogenic isoform of the VEGFA gene, respectively. (B) The skipping of exon #5 in the tissue factor (T3) gene affects the reading frame and generates a soluble, pro-thrombotic isoform of tissue factor. (C) A reinforcing mutation near a cryptic 5′-splice-site in the LMNA gene truncates the final protein (progerin) that accumulates in the nucleus. (D) Retention of the final intron in the ENG gene alters the reading frame and generates the anti-angiogenic, pro-fibrotic isoform S-endoglin.
In addition to being involved in the selection of splice-sites, SRSF1 also participates in the skipping of particular exonic sequences, as in the case of the tissue factor (TF) gene (T3), the primary initiator of blood coagulation. TF is mainly expressed by ECs, usually as a transmembrane glycoprotein, but is also occasionally produced as a soluble form in response to pro-inflammatory cytokines (Szotowski et al., 2005). The activation and overexpression of SRSF1, as well as other SR proteins, in ECs favor skipping exon #5 of TF. Consequently, exon #4 is spliced directly to exon #6, producing a translational frameshift that leads to the generation of a soluble variant that contributes to thrombus growth, thus initiating and propagating the coagulation process (Bogdanov et al., 2003; Figure 2B).
Recently, the involvement of SRSF1 in alternative splicing of the lamin A gene (LMNA) has been described (Lopez-Mejia et al., 2011). LMNA encodes lamins A and C, which are major components of the nuclear lamina in the inner membrane of the nuclear envelope. These proteins are involved in essential functions, including DNA replication, transcription, chromatin organization, and dis/assembly of the nucleus during cell division (Worman et al., 2009). Sporadically, a de novo germline mutation in exon #11 promotes the reinforcement of a weak 5′ splice-site that generates a truncated and pathological version of lamin A named progerin, which accumulates in cell nuclei in a tissue-specific manner (Figure 2C). Interestingly, SRSF1 is critically involved in the control of the lamin A/progerin splicing switch. Thus, specific knock-down of SRSF1 leads to a reduction in progerin expression, resulting in an important impact on the lifespan of the animal (Lopez-Mejia et al., 2011). Mutations in LMNA, that give rise to progerin, are responsible for Hutchinson–Gilford progeria syndrome, a rare disorder characterized by segmental premature aging and death (Gerhard-Herman et al., 2012). Progerin accumulates primarily in vascular cells, suggesting its direct involvement in vascular disease (McClintock et al., 2006), in agreement with the cardiovascular defects, including accelerated atherosclerosis, myocardial infarction, and stroke characteristic of Hutchinson–Gilford progeria syndrome (Trigueros-Motos et al., 2011; Gerhard-Herman et al., 2012).
The role of SRSF1 in intron retention by the endoglin gene (ENG) that leads to formation of the senescence-associated S-endoglin isoform has been demonstrated (Blanco and Bernabeu, 2011). Endoglin is an auxiliary co-receptor for TGF-β expressed in ECs that plays a critical role in vascular remodeling and angiogenesis, as illustrated by the fact that mutations in ENG lead to hereditary hemorrhagic telangiectasia Type 1, a disease characterized by vascular malformations (Lopez-Novoa and Bernabeu, 2010; Shovlin, 2010). While endoglin is predominantly expressed as the long isoform (L-endoglin) which shows a pro-angiogenic role, expression during senescence of the alternatively spliced S-endoglin significantly affects the behavior of ECs, promoting anti-angiogenic and pro-fibrotic phenotypes, and compromising vasodilator responses (Perez-Gomez et al., 2005; Blanco et al., 2008; Velasco et al., 2008). In this context, S-endoglin tilts the TGF-β signaling pathway toward its pro-fibrotic side, via Smad3 signaling, inducing expression of plasminogen activator inhibitor (PAI)-1, a key regulator of synthesis and deposition of the extracellular matrix in tissue homeostasis (Ghosh and Vaughan, 2012). Moreover, the TGF-β/S-endoglin signaling pathway represses the expression of Id1 factor, which is essential for cellular proliferation (Tang et al., 2002; Blanco et al., 2008). Compatible with this finding, elevated levels of TGF-β have been reported in aging varicose veins; these likely favor the fibrotic process and subsequent venous insufficiency (Pascual et al., 2007). Because TGF-β-induced Smad3 is able to interact with transcription factor c-myc, thus repressing the promoter of the hTERT gene that encodes the catalytic subunit of telomerase, up-regulation of S-endoglin would be predicted to contribute to endothelial senescence by compromising telomere integrity and arresting the cell cycle (Li and Liu, 2007). It has been postulated that the S-endoglin mRNA, which bears the last intron, may escape from the activity of the major spliceosome in the nucleus. Under normal conditions, this intron would typically be removed by the minor spliceosome in the cytoplasm, giving rise to the predominant isoform L-endoglin. However, during senescence, SRSF1 translocates to the cytoplasm, binding to the consensus SRSF1 cis-element within the branch point of the S-endoglin mRNA, interfering with the activity of the minor spliceosome and leading to an increase in the level of S-endoglin (Figure 2D).
Effect of Senescence-Associated Stress Signals on the Role of SRSF1
Precursor mRNA splicing is one target of several environmental stress factors, including oxidative stress and ultraviolet radiation, that frequently cause DNA damage (Biamonti and Caceres, 2009). Depending on the intensity of the stress signal, cells may react by activating the check-point pathway mediated by p53 and entering into cell cycle arrest, which, in turn, prompts senescence (Borras et al., 2011). Many of these stress stimuli induce the cytoplasmic accumulation of several splicing factors, including SRSF1 (Biamonti and Caceres, 2009), hnRNP A1 (Guil et al., 2006), Slu7 (Shomron et al., 2005), and PTB (Xie et al., 2003). Furthermore, SRSF1 translocates to the cytoplasm during replicative or stress-induced senescence, suggesting that SRSF1 is a new senescence target (Blanco and Bernabeu, 2011).
Serine–arginine splicing factor 1 is a shuttling factor that localizes either in the nucleus or the cytoplasm, depending on the phosphorylation state of its RS domain (Cazalla et al., 2002; Sanford et al., 2005) and/or the methylation of three arginine residues between the RRM domains (Sinha et al., 2010; Figure 1B). The kinase SRPK1 is a major contributor to SRSF1 phosphorylation and translocation. Under normal conditions, SRPK1 is located in the cytoplasm where this kinase continuously phosphorylates SRSF1, favoring its transport to the nucleus. However, some stress factors may alter the subcellular distribution of SRPK1, triggering the nuclear import of the kinase (Giannakouros et al., 2011). Consequently, the cytoplasmic pool of SRSF1 is augmented by the absence of SRPK1 in the cytoplasm, as well as by the nuclear phosphatase activity that facilitates the cytoplasmic export of SRSF1. Thus, it can be postulated that the cytoplasmic localization of SRSF1 that occurs during endothelial senescence, whether replicative or induced by stress stimuli, may be due, at least in part, to nuclear translocation of SRPK1.
Importantly, cytoplasmic SRSF1 is able to modulate the alternative splicing of specific precursor mRNAs, as shown for ENG in a process that involves the minor spliceosome (m-Sp) machinery (Blanco and Bernabeu, 2011). This m-Sp, also known to be U12-dependent, co-exists with the U2-dependent major spliceosome (m-Sp) in most organisms (Patel and Steitz, 2003). First described as responsible for the removal of a rare class of introns (U12-type), it is currently recognized that m-Sp is also involved in the processing of standard introns (Sheth et al., 2006) and may be localized in the cytoplasm (Konig et al., 2007). The m-Sp contains four unique small nuclear ribonucleic acids (snRNAs): U11, U12, U4atac, and U6atac, which are paralogs of the U1, U2, U4, and U6 snRNAs of the m-Sp, respectively, while U5 snRNA is shared between the two spliceosomes. While m-Sp is able to splice and remove the intron encoded in S-endoglin, the cytoplasmic localization of SRSF1 during endothelial senescence interferes with this splicing, promoting intron retention (Blanco and Bernabeu, 2011). This is in agreement with the findings that: (i) depletion of protein components essential for m-Sp activity leads to cellular growth arrest (Will et al., 2004); and (ii) during cell senescence, changes in gene expression levels affect the composition and activity of m-Sp (Meshorer and Soreq, 2002; Harries et al., 2011).
Conclusion
Numerous and increasing efforts are focused on aging research, to try to elucidate its underlying molecular mechanisms. Aging is one of the major risk factors for the development of cardiovascular diseases, and is associated with cellular senescence of the vascular endothelium. In this context, there is growing interest in the molecular components that orchestrate the modulation of alternative splicing events in senescent ECs. Due to its senescence-induced cytoplasmic localization, the splicing factor SRSF1 can be considered as a new marker for endothelial senescence. In addition, the cytoplasmic SRSF1 regulates alternative splicing of the endoglin pre-mRNA, leading to the senescence-associated S-endoglin isoform, which contributes to a senescent environment in the vessels. The data summarized in this review support the link between SRSF1 and senescence, and also suggest the existence of a common genetic program involving alternative splicing of a cluster of genes, including ENG, VEGFA, T3, and LMNA, whose resulting variants orchestrate vascular functions (Figure 3). Deciphering this senescent program will shed light on new therapeutic targets for cardiovascular diseases.
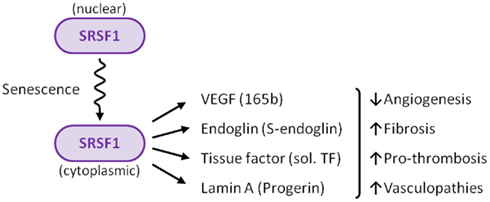
Figure 3. Summary of the role of SRSF1 in ECs. Several stimuli, including senescence, induce translocation of SRSF1 to the cytoplasm. SRSF1 is a key factor that modulates the expression of critical regulators of vascular physiology, including VEGF, endoglin, tissue factor (TF), and lamin A.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the Ministerio de Ciencia e Innovación of Spain (SAF2010-19222) and Genoma España (MEICA) to Carmelo Bernabéu. The Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) is an initiative of the Instituto de Salud Carlos III (ISCIII) of Spain.
References
Bellon, T., Corbi, A., Lastres, P., Cales, C., Cebrian, M., Vera, S., Cheifetz, S., Massague, J., Letarte, M., and Bernabeu, C. (1993). Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 23, 2340–2345.
Biamonti, G., and Caceres, J. F. (2009). Cellular stress and RNA splicing. Trends Biochem. Sci. 34, 146–153.
Blanco, F. J., and Bernabeu, C. (2011). Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell 10, 896–907.
Blanco, F. J., and Bernabeu, C. (2012). “Alternative splicing in endothelial senescence. Role of the TGF-β co-receptor endoglin,” in Senescence, ed. T. Nagata (InTech). Available from: http://www.intechopen.com/articles/show/title/alternative-splicing-in-endothelial-senescence-role-of-the-tgf-beta-co-receptor-endoglin
Blanco, F. J., Grande, M. T., Langa, C., Oujo, B., Velasco, S., Rodriguez-Barbero, A., Perez-Gomez, E., Quintanilla, M., Lopez-Novoa, J. M., and Bernabeu, C. (2008). S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ. Res. 103, 1383–1392.
Bogdanov, V. Y., Balasubramanian, V., Hathcock, J., Vele, O., Lieb, M., and Nemerson, Y. (2003). Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat. Med. 9, 458–462.
Borras, C., Gomez-Cabrera, M. C., and Vina, J. (2011). The dual role of p53: DNA protection and antioxidant. Free Radic. Res. 45, 643–652.
Campisi, J., and d’Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740.
Carmeliet, P., and Jain, R. K. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307.
Carter, J. G., Cherry, J., Williams, K., Turner, S., Bates, D. O., and Churchill, A. J. (2011). Splicing factor polymorphisms, the control of VEGF isoforms and association with angiogenic eye disease. Curr. Eye Res. 36, 328–335.
Cazalla, D., Zhu, J., Manche, L., Huber, E., Krainer, A. R., and Caceres, J. F. (2002). Nuclear export and retention signals in the RS domain of SR proteins. Mol. Cell. Biol. 22, 6871–6882.
Chang, M. W., Grillari, J., Mayrhofer, C., Fortschegger, K., Allmaier, G., Marzban, G., Katinger, H., and Voglauer, R. (2005). Comparison of early passage, senescent and hTERT immortalized endothelial cells. Exp. Cell Res. 309, 121–136.
Collins, C., and Tzima, E. (2011). Hemodynamic forces in endothelial dysfunction and vascular aging. Exp. Gerontol. 46, 185–188.
Conway, E. M., and Carmeliet, P. (2004). The diversity of endothelial cells: a challenge for therapeutic angiogenesis. Genome Biol. 5, 207.
Debacq-Chainiaux, F., Borlon, C., Pascal, T., Royer, V., Eliaers, F., Ninane, N., Carrard, G., Friguet, B., De Longueville, F., Boffe, S., Remacle, J., and Toussaint, O. (2005). Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-beta1 signaling pathway. J. Cell Sci. 118, 743–758.
Doetschman, T., Barnett, J. V., Runyan, R. B., Camenisch, T. D., Heimark, R. L., Granzier, H. L., Conway, S. J., and Azhar, M. (2012). Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 347, 203–223.
Elias, A. P., and Dias, S. (2008). Microenvironment changes (in pH) affect VEGF alternative splicing. Cancer Microenviron. 1, 131–139.
Erusalimsky, J. D. (2009). Vascular endothelial senescence: from mechanisms to pathophysiology. J. Appl. Physiol. 106, 326–332.
Erusalimsky, J. D., and Skene, C. (2009). Mechanisms of endothelial senescence. Exp. Physiol. 94, 299–304.
Gerhard-Herman, M., Smoot, L. B., Wake, N., Kieran, M. W., Kleinman, M. E., Miller, D. T., Schwartzman, A., Giobbie-Hurder, A., Neuberg, D., and Gordon, L. B. (2012). Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 59, 92–97.
Giannakouros, T., Nikolakaki, E., Mylonis, I., and Georgatsou, E. (2011). Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 278, 570–586.
Grillari, J., Grillari-Voglauer, R., and Jansen-Durr, P. (2010). Post-translational modification of cellular proteins by ubiquitin and ubiquitin-like molecules: role in cellular senescence and aging. Adv. Exp. Med. Biol. 694, 172–196.
Guil, S., Long, J. C., and Caceres, J. F. (2006). hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 26, 5744–5758.
Harries, L. W., Hernandez, D., Henley, W., Wood, A. R., Holly, A. C., Bradley-Smith, R. M., Yaghootkar, H., Dutta, A., Murray, A., Frayling, T. M., Guralnik, J. M., Bandinelli, S., Singleton, A., Ferrucci, L., and Melzer, D. (2011). Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10, 868–878.
Indik, Z., Yeh, H., Ornstein-Goldstein, N., Kucich, U., Abrams, W., Rosenbloom, J. C., and Rosenbloom, J. (1989). Structure of the elastin gene and alternative splicing of elastin mRNA: implications for human disease. Am. J. Med. Genet. 34, 81–90.
Konig, H., Matter, N., Bader, R., Thiele, W., and Muller, F. (2007). Splicing segregation: the minor spliceosome acts outside the nucleus and controls cell proliferation. Cell 131, 718–729.
Krieglstein, K., Miyazono, K., Ten Dijke, P., and Unsicker, K. (2012). TGF-beta in aging and disease. Cell Tissue Res. 347, 5–9.
Li, H., and Liu, J. P. (2007). Mechanisms of action of TGF-beta in cancer: evidence for Smad3 as a repressor of the hTERT gene. Ann. N. Y. Acad. Sci. 1114, 56–68.
Lopez-Mejia, I. C., Vautrot, V., De Toledo, M., Behm-Ansmant, I., Bourgeois, C. F., Navarro, C. L., Osorio, F. G., Freije, J. M., Stevenin, J., De Sandre-Giovannoli, A., Lopez-Otin, C., Levy, N., Branlant, C., and Tazi, J. (2011). A conserved splicing mechanism of the LMNA gene controls premature aging. Hum. Mol. Genet. 20, 4540–4555.
Lopez-Novoa, J. M., and Bernabeu, C. (2010). The physiological role of endoglin in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 299, H959–H974.
Manley, J. L., and Krainer, A. R. (2010). A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 24, 1073–1074.
McClintock, D., Gordon, L. B., and Djabali, K. (2006). Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proc. Natl. Acad. Sci. U.S.A. 103, 2154–2159.
Meshorer, E., and Soreq, H. (2002). Pre-mRNA splicing modulations in senescence. Aging Cell 1, 10–16.
Neufeld, G., Cohen, T., Gengrinovitch, S., and Poltorak, Z. (1999). Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22.
Nowak, D. G., Amin, E. M., Rennel, E. S., Hoareau-Aveilla, C., Gammons, M., Damodoran, G., Hagiwara, M., Harper, S. J., Woolard, J., Ladomery, M. R., and Bates, D. O. (2010). Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J. Biol. Chem. 285, 5532–5540.
Nowak, D. G., Woolard, J., Amin, E. M., Konopatskaya, O., Saleem, M. A., Churchill, A. J., Ladomery, M. R., Harper, S. J., and Bates, D. O. (2008). Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 121, 3487–3495.
Pascual, G., Mendieta, C., Garcia-Honduvilla, N., Corrales, C., Bellon, J. M., and Bujan, J. (2007). TGF-beta1 upregulation in the aging varicose vein. J. Vasc. Res. 44, 192–201.
Patel, A. A., and Steitz, J. A. (2003). Splicing double: insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 4, 960–970.
Perez-Gomez, E., Eleno, N., Lopez-Novoa, J. M., Ramirez, J. R., Velasco, B., Letarte, M., Bernabeu, C., and Quintanilla, M. (2005). Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene 24, 4450–4461.
Sanford, J. R., Ellis, J. D., Cazalla, D., and Caceres, J. F. (2005). Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc. Natl. Acad. Sci. U.S.A. 102, 15042–15047.
Santibanez, J. F., Quintanilla, M., and Bernabeu, C. (2011). TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin. Sci. 121, 233–251.
Schraml, E., Voglauer, R., Fortschegger, K., Sibilia, M., Stelzer, I., Grillari, J., and Schauenstein, K. (2008). Haploinsufficiency of senescence evasion factor causes defects of hematopoietic stem cells functions. Stem Cells Dev. 17, 355–366.
Semenza, G. L. (2007). Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J. Cell. Biochem. 102, 840–847.
Shay, J. W., and Wright, W. E. (2007). Hallmarks of telomeres in ageing research. J. Pathol. 211, 114–123.
Sheth, N., Roca, X., Hastings, M. L., Roeder, T., Krainer, A. R., and Sachidanandam, R. (2006). Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 34, 3955–3967.
Shomron, N., Alberstein, M., Reznik, M., and Ast, G. (2005). Stress alters the subcellular distribution of hSlu7 and thus modulates alternative splicing. J. Cell Sci. 118, 1151–1159.
Shovlin, C. L. (2010). Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 24, 203–219.
Sinha, R., Allemand, E., Zhang, Z., Karni, R., Myers, M. P., and Krainer, A. R. (2010). Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF. Mol. Cell. Biol. 30, 2762–2774.
Szotowski, B., Antoniak, S., Poller, W., Schultheiss, H. P., and Rauch, U. (2005). Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ. Res. 96, 1233–1239.
Tang, J., Gordon, G. M., Nickoloff, B. J., and Foreman, K. E. (2002). The helix-loop-helix protein id-1 delays onset of replicative senescence in human endothelial cells. Lab. Invest. 82, 1073–1079.
Trigueros-Motos, L., Gonzalez, J. M., Rivera, J., and Andres, V. (2011). Hutchinson-Gilford progeria syndrome, cardiovascular disease and oxidative stress. Front. Biosci. (Schol. Ed.) 3, 1285–1297.
Velasco, S., Alvarez-Munoz, P., Pericacho, M., Dijke, P. T., Bernabeu, C., Lopez-Novoa, J. M., and Rodriguez-Barbero, A. (2008). L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 121, 913–919.
Verduci, L., Simili, M., Rizzo, M., Mercatanti, A., Evangelista, M., Mariani, L., Rainaldi, G., and Pitto, L. (2010). MicroRNA (miRNA)-mediated interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J. Biol. Chem. 285, 39551–39563.
Versari, D., Daghini, E., Virdis, A., Ghiadoni, L., and Taddei, S. (2009). Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br. J. Pharmacol. 157, 527–536.
Voglauer, R., Chang, M. W., Dampier, B., Wieser, M., Baumann, K., Sterovsky, T., Schreiber, M., Katinger, H., and Grillari, J. (2006). SNEV overexpression extends the life span of human endothelial cells. Exp. Cell Res. 312, 746–759.
Will, C. L., Schneider, C., Hossbach, M., Urlaub, H., Rauhut, R., Elbashir, S., Tuschl, T., and Luhrmann, R. (2004). The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. RNA 10, 929–941.
Worman, H. J., Fong, L. G., Muchir, A., and Young, S. G. (2009). Laminopathies and the long strange trip from basic cell biology to therapy. J. Clin. Invest. 119, 1825–1836.
Keywords: alternative splicing, endothelial senescence, SRSF1, endoglin, progerin, VEGF, tissue factor
Citation: Blanco FJ and Bernabéu C (2012) The splicing factor SRSF1 as a marker for endothelial senescence. Front. Physio. 3:54. doi: 10.3389/fphys.2012.00054
Received: 16 December 2011; Accepted: 26 February 2012;
Published online: 28 March 2012.
Edited by:
Francesc Jiménez-Altayó, Universitat Aut‘noma de Barcelona, SpainReviewed by:
Michael Ladomery, University of the West of England Bristol, UKXiu-Fen Ming, University of Fribourg, Switzerland
Gerardo Ferbeyre, Université de Montréal, Canada
Copyright: © 2012 Blanco and Bernabéu. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Francisco Javier Blanco, Department of Cellular and Molecular Medicine, Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas, C/Ramiro de Maeztu 9, Madrid 28040, Spain. e-mail: fjblanco@cib.csic.es