
- Department of Experimental Biology, University of Jaen, Jaen, Spain,
Pitx2 is a homeobox transcription factor involved in left–right signaling during embryogenesis. Disruption of left–right signaling in mice within its core nodal/lefty cascade, results in impaired expression of the last effector of the left–right cascade, Pitx2, leading in many cases to absence or bilateral expression of Pitx2 in lateral plate mesoderm (LPM). Loss of Pitx2 expression in LPM results in severe cardiac malformations, including right cardiac isomerism. Pitx2 is firstly expressed asymmetrically in the left but not right LPM, before the cardiac crescent forms, and subsequently, as the heart develops, becomes confined to the left side of the linear heart tube. Expression of Pitx2 is remodeled during cardiac looping, becoming localized to the ventral portion of the developing ventricular chambers, while maintaining a distinct left-sided atrial expression. The importance of Pitx2 during cardiogenesis has been illustrated by the complex and robust cardiac defects observed on systemic deletion of Pitx2 in mice. Lack of Pitx2 expression leads to embryonic lethality at mid-term, and Pitx2-deficient embryos display isomeric hearts with incomplete closure of the body wall. However, whereas the pivotal role of Pitx2 during cardiogenesis is well sustained, its putative role in the fetal and adult heart is largely unexplored. Recent genome-wide association studies have identified several genetic variants highly associated with atrial fibrillation (AF). Among them are genetic variants located on chromosome 4q25 adjacent to PITX2. Since then several transgenic approaches have provided evidences of the role of the homeobox transcription factor PITX2 and atrial arrhythmias. Here, we review new insights into the cellular and molecular links between PITX2 and AF.
Cardiac arrhythmia can be defined as a deviation from the normal heart rate and/or rhythm under physiological circumstances which are thus reflected on specific ECG patterns (Bellet, 1971). Atrial arrhythmias may be regular, as in the case of flutter or monomorphic tachycardia, or irregular, as in the case of fibrillation or polymorphic tachycardia (Gussak and Antzelevitch, 2008). We focus herein on atrial fibrillation (AF), the most prevalent type of arrhythmia in humans. AF is characterized by uncoordinated atrial activation with consequent weakening of atrial mechanical function with intact AV conduction (Gussak and Antzelevitch, 2008). AF prevalence ranges from 1% in young adults to >10% of those over 80 years old (Fuster et al., 2001) and is frequently associated with hypertension, coronary disease, valvular diseases, and cardiomyopathies (Kannel et al., 1998), yet in 10–20% of AF patients, no underlying disease is found (Brand et al., 1985), i.e., idiopathic or lone AF. Great insights have been gained concerning the cellular and molecular mechanisms that underlie AF (Schotten et al., 2011; Wakili et al., 2011). Enhanced automaticity in one or several rapidly depolarizing foci leads to AF (Haissaguerre et al., 1998), being frequently located within the superior pulmonary veins, and more rarely within the superior vena cava or coronary sinus (Moe and Abildskov, 1959). Alternatively, multiple wave fronts propagate within the atria, resulting in derived wavelets that self-perpetuate (Arnar et al., 2006). However, the molecular substrates of AF remain obscure.
Although AF is the most prevalent type of arrhythmia, its genetic etiology remains elusive. The first AF genetic locus was reported by Brugada et al. (1997) identifying linkage to chromosome 10q22–q24 and soon thereafter Chen et al. (2003) identify a mutation in KCNQ1. Mutations in KCNQ1 have been subsequently confirmed (Ellinor et al., 2007) as well as in other ion channels including KCNE2, KCNE3, KCNJ2, KCNA5, and SCN5A (Ellinor et al., 2004; Mc Nair et al., 2004; Yang et al., 2004; Xia et al., 2005). Surprisingly, mutations in ion channels would underlie less 1% of AF cases.
The advent of new genetic strategies, in particular genome-wide association studies (GWAS), has shed new light on the genetic bases of arrhythmogenic syndromes and the identification of novel disease-causing genes (Larson et al., 2007). GWAS have identified several genetic variants highly associated with AF, which are located at 9p21, 1q21, 16q22, and 4q25, respectively. On chromosome 9p21, no candidate gene has been linked to AF, and thus its relevance awaits further experimental and functional evidence (Larson et al., 2007). Distinct genetic variants at 1q21 are linked to KCNN3, a potassium channel involved in atrial repolarization (Ellinor et al., 2011) and IL6R, a gene involved in inflammatory response (Schnabel et al., 2011), respectively. Genetic variants at 16q22 are linked to ZFHX3, a zinc finger homeobox transcription factor involved in liver gene expression (Gudbjartsson et al., 2009). However, in both cases 1q21 and 16q22 risk variants, their role in AF remains obscure. Finally, genetic variants on chromosome 4q25 which are strongly associated with lone AF in three distinct populations of European descent (Gudbjartsson et al., 2007) map adjacent to PITX2, which is known to have a critical function in establishing left–right asymmetry (Gage et al., 1999; Lin et al., 1999) and plays a critical role in cardiac development (Campione et al., 1999, 2001; Franco and Campione, 2003) and thus supporting the notion that impaired function of PITX2 might underlie AF (Gudbjartsson et al., 2009).
Pitx2 is a homeobox transcription factor involved in left–right signaling during embryogenesis (Gage et al., 1999; Lin et al., 1999). Disruption of left–right signaling in mice within its core nodal/lefty cascade (Blum et al., 1999; Hamada et al., 2001; Bamforth et al., 2004) results in impaired expression of Pitx2, the last effector of the left–right cascade, being absent or bilateral expressed in lateral plate mesoderm (LPM). Loss of Pitx2 expression in LPM results in severe cardiac malformations, including transposition of the great arteries, double-outlet right ventricle, and right cardiac isomerism (Gage et al., 1999; Lin et al., 1999). However, whereas the pivotal role of Pitx2 during cardiogenesis is well sustained, its putative role in the fetal and adult heart is largely unexplored.
The Pitx2 gene encodes three distinct isoforms, Pitx2a, Pitx2b, and Pitx2c, through alternative splicing and distinct promoter usage (Schweickert et al., 2000). Pitx2c is the most prominent isoform in the developing mouse heart and only weak and transient expression of Pitx2a is observed during early embryonic (E9.5–E12.5) stages, whereas Pitx2b displays progressively decreasing expression in embryonic and fetal stages (D. Franco and A. Aranega, unpublished data). In man, a fourth isoform, PITX2D is also expressed, which acts as a dominant negative isoform (Cox et al., 2002). PITX2 mutations are linked to Axenfeld–Rieger syndrome, which is characterized by abnormal morphogenesis of the anterior segment of the eye, variable degree of maxillary hypoplasia, skeletal abnormalities, and abdominal defects (Hjalt and Semina, 2005). In rodents and avian, Pitx2 displays a highly dynamic expression profile during cardiogenesis. Pitx2 is initially expressed asymmetrically in the left but not right LPM, before the cardiac crescent forms, and subsequently, as the heart develops, becomes confined to the left side of the linear heart tube (Campione et al., 1999, 2001; Franco and Campione, 2003). Pitx2 expression is later remodeled during cardiac looping, becoming confined to the ventral portion of the forming ventricular chambers, while keeping left-sided atrial expression (Campione et al., 2001). With further development, Pitx2 is down-regulated in the ventricular chambers, while high and robust expression of Pitx2 is maintained in the atrial chambers as well as in discrete components of the inflow tract, including the left atrial chamber, left superior caval vein, the pulmonary veins, and the interatrial septum (Franco et al., 2000; Kahr et al., 2011).
The significance of Pitx2 during heart development is illustrated by the complex and robust cardiac defects observed on Pitx2 defective mouse models. Systemic deficiency of Pitx2 leads to embryonic lethality at mid-term, leading to severe cardiovascular defects such as double-outlet right ventricle and right atrial isomerism as well as incomplete closure of the body wall (Gage et al., 1999; Lin et al., 1999; Liu et al., 2001, 2002). Isoform-specific Pitx2 mutants revealed that Pitx2c is the most relevant isoform during cardiogenesis, since lack of Pitx2c expression, but not Pitx2a or Pitx2b, recapitulate the morphogenetic defects similar to systemic Pitx2-deficient mice (Liu et al., 2001). More recently, with the advent of tissue-specific conditional mutants, our understanding of the role of Pitx2 during cardiogenesis has greatly expanded. Ai et al. (2006) revealed that lack of Pitx2 function in the secondary heart field, a subpopulation of cardiovascular progenitor cells emanating from the medio-lateral cardiogenic mesoderm (Kelly et al., 2001), leads to developmental defects reminiscent of systemic Pitx2c null mutants. In addition, further evidence on the role of Pitx2 within the secondary heart field has been reported at the arterial (Nowotschin et al., 2006; Yashiro et al., 2007) and venous (Galli et al., 2008) poles of the heart. Importantly, Pitx2 function is not exclusive related to cardiac precursor cells but is also relevant in differentiated cardiomyocytes as reported by Tessari et al. (2008). Using αMHC-Cre driver mice, these authors demonstrated that functional impairment of Pitx2 in cardiomyocytes leads to cytoarchitectural disarray in ventricular myocytes and to abnormal left–right expression of Bmp10 in the atria. Unexpectedly, αMHCCrePitx2 mutants do not display atrial isomerism, suggesting that left/right atrial morphological identity is acquired before αMHC-Cre activation. Overall, these data demonstrate a developmental role of Pitx2 with precursor cells as well as in cardiomyocytes, including importantly, morphological structures prone to trigger atrial arrhythmias, yet no evidence of such electrophysiological defects was reported in these studies. Moreover, although it is clearly established that Pitx2 plays a role in pulmonary vein development (Mommersteeg et al., 2007), a frequent AF foci, Pitx2 driven alterations occur in the setting of complex cardiac abnormalities such as atrial isomerism, a condition which is not linked to atrial arrhythmias in men. Therefore, additional evidence is required to fill the gap between Pitx2 function in the embryo and the onset of AF.
Genetic variants at 4q25, close to the Pitx2 locus, are highly associated with AF (Gudbjartsson et al., 2007), providing the conjecture thus that Pitx2 dysfunction might be to AF. Linkage of 4q25 AF risk variants have been consistently reported within distinct cohorts, highlighting thus the reproducibility of these findings (Chinchilla et al., 2011; Henningsen et al., 2011; Kiliszek et al., 2011; Schnabel et al., 2011; Delaney et al., 2012; Liu et al., 2012; Olesen et al., 2012). A twofold question arises from this hypothesis, first is whether the 4q25 genetic variants can modulate Pitx2 expression, a question that remains unanswered, and secondly, whether Pitx2 impairment can lead to AF. Reports in human AF right and left atrial biopsies demonstrated that PITX2 expression is severely diminished in AF patients as compared to patients with no clinical history of AF (Chinchilla et al., 2011), validating thus the hypothesis that PITX2 is impaired in AF patients. In addition to the human data, several independent experimental studies have provided evidences for this second issue, using subtle distinct experimental models. Wang et al. (2010) have demonstrated that Pitx2 haploinsufficiency predisposes to AF in electrically stimulated adult mice with structurally normal hearts. These authors reported a developmental link in that Pitx2 inhibits the sinoatrial node gene program in the embryonic left atrium, by regulating Shox2 and Nkx2.5 expression. When Pitx2 function is partially (heterozygous) or completely (null) impaired in the embryonic (E12.5) mouse heart, Shox2, as well as several other sinoatrial node markers such as Hcn4 and Tbx3, are ectopically expressed in the forming inflow tract of the heart. These findings are further underscored by the recent study of Ammirabile et al. (2012) using a myocardiac specific conditional Pitx2 mouse model (cardiac troponin T Cre deletor mice). Thus, these observations provide a link between the embryonic function of Pitx2 and AF.
Kirchhof et al. (2011) have recently described impaired gap and tight junction expression in microarray analysis of the left atria of adult Pitx2c heterozygous mutant mice which display no overt structural and/or morphological alterations. These data provide a putative link to AF since mutations in GJA5 (Cx40) are associated with AF (Gollob et al., 2006) and further implicate impaired Pitx2 function at fetal and/or adult stages in predisposition to AF. In line with these findings, recent work in our laboratory using a Pitx2 over-expression model of embryonic stem cell-derived cardiomyocytes (Lozano-Velasco et al., 2011) has shown that Pitx2 does not modify the expression of contractile proteins such as actin and myosin heavy and light-chain isoforms, thus suggesting that Pitx2 does not impair the contractile properties of cardiomyocytes, but importantly, Pitx2 mis-expression impairs connexin-40 expression.
In addition, our recent work using atrial-specific conditional Pitx2 mouse mutants, in which right-sided sinoatrial node formation is intact, also supports a role of Pitx2 impairment on the onset of left-sided electrophysiological defects (Chinchilla et al., 2011) which are prone substrates for AF. Voltage-gated sodium and inward rectifying K+ (Kir2.1, Kir2.2, and Kir2.3) ion channels are abnormally expressed in the atrial myocardium both in fetal and adult stages of atrial-specific conditional Pitx2 mouse mutants, while cardiac morphogenesis is not compromised. Importantly, inward rectifying K+ channel de-regulation, but not sodium, is mediated by impaired miR-1 expression in atrial-specific conditional Pitx2 mutant mice. As a consequence, in these mice, the action potential amplitude is decreased, while the resting membrane potential is more depolarized. Further evidence on the de-regulation of ion channels by Pitx2 impairment has been recently reported also by Kirchhof et al. (2011) using mRNA microarray analyses in Pitx2c heterozygous adult mutant mice. Thus, these data reveal a role for Pitx2 controlling ion channel expression, and thus a link to atrial arrhythmogenesis, as summarized in Figure 1.
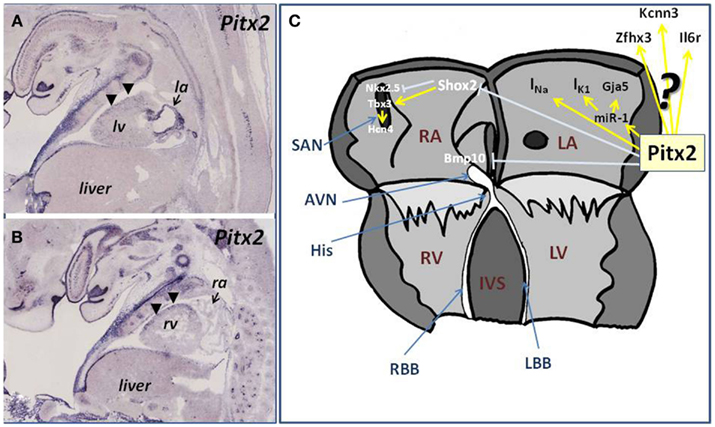
Figure 1. (A,B) Illustrations of Pitx2 expression in the developing fetal embryo (E14.5) in longitudinal histological sections. (A) Illustrate the expression of Pitx2 at the level of the left ventricle (lv) and left atria (la), showing the robust expression of Pitx2 in the developing left atrial chamber, whereas (B) illustrates the expression of Pitx2 at the level of the right ventricle (rv) and right atrium (ra). Observe the marked contrast of expression between the right atrium (low) and the left atrium (high) as revealed by the arrows. Note also the dorso-ventral difference of Pitx2 expression at the ventricular level [arrowheads, (A,B)]. (C) Schematic representation of the current state-of-the-art role of Pitx2 in the developing and adult atrial chambers linked to atrial arrhythmogenesis. Pitx2 controls miR-1 expression in the left atrial chamber, providing thus a regulatory mechanism for IK1 and Gja1 function. In addition Pitx2 regulates INa while inhibits the expression right atrial (Bmp10) and sinoatrial (Shox2) markers in the left atrium, which in turn play key regulatory roles on cardiomyocyte cell cycle and pacemaker activity, providing additional links to atrial arrhythmias.
Concluding Remarks and Perspective
We have highlighted herein our current knowledge on the role of Pitx2 in the developing and adult heart as well as on the state-of-the-art link between PITX2 and AF. Several studies have shown that Pitx2 is essential for pulmonary vein development (Mommersteeg et al., 2007), a highly recurrent pro-arrhythmogenic region in humans. Experimental evidence has shown that Pitx2 is important for transcriptional regulation in the secondary heart field (Ai et al., 2006), derivatives of which will form the venous pole of the heart. Similarly, additional evidence has revealed a role of Pitx2 in the regulation of cell-to-cell communication proteins and essential ion channels, which modulate critical aspects of the cardiac action potential, as well as a role repressing the sinoatrial node gene program (Wang et al., 2010; Chinchilla et al., 2011; Kirchhof et al., 2011). Overall, these data strongly implicate Pitx2 as an upstream regulator of pro-arrhythmogenic events. However, there are several missing links. The first one is how the identified genetic variants alter Pitx2 expression and to date, this point remains unanswered. Secondly, it remains elusive how altered Pitx2 expression would modify conductive elements leading to the onset of AF (although new insights have progressively being gained; Figure 1), in particular whether the impaired conductive elements might be deregulated already from embryonic stages, as suggested by Wang et al. (2010) and Ammirabile et al. (2012) or exclusively in the fetal/adult heart as reported by Kirchhof et al. (2011) and Chinchilla et al. (2011). On the basis of the developmental role of Pitx2, it might be postulated that impaired Pitx2 in the developing heart might be a predisposing factor for AF. However, an early defect in Pitx2 function would be expected to have severe consequences for cardiac embryogenesis, and thus it is more likely that fetal or adult dysfunction might underlie Pitx2 predisposition to AF. In addition, it is unclear if Pitx2 deficiency might also lead to impaired formation and/or function of other components of the ventricular cardiac conduction system such as the AV node and the left and right AV bundle branches. Morphological analysis of the ventricular conduction system in the left–right mutant mouse model iv/iv suggests that left/right cues can influence AV node formation (Franco and Icardo, 2001). Nonetheless, further studies are required to demonstrate a putative role of Pitx2 in ventricular conduction system deployment.
Interestingly, in addition to reports linking 4q25 risk variants to AF, new GWAS have identified several other genetic variants highly associated with AF, which are located at 9p21, 1q21, and 16q22, respectively. On chromosome 9p21, no candidate gene has been linked to AF, and thus its relevance awaits further experimental and functional evidence (Larson et al., 2007). However, genetic variants at 1q21 are linked to KCNN3 (Ellinor et al., 2011) and IL6R (Schnabel et al., 2011), respectively whereas risk genetic variants at 16q22 are linked to ZFHX3 (Gudbjartsson et al., 2009). To date it remains elusive the putative involvement of these genes in the pathophysiology of AF. A putative scenario might be that PITX2 might modulate these genes. Thus, further insights are required to explore both the links between all GWAS associated genes as well as the cellular and molecular mechanisms that might underlie a functional role between KCNN3, IL6R and ZFHX3, and AF, yet this aspect remains to be elucidated. Thus, new exciting dates are ahead right now.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is partially supported by the VI EU Integrated Project “Heart Failure and Cardiac Repair” LSHM-CT-2005-018630 to Diego Franco, a grant from the Junta de Andalucía Regional Council to Diego Franco (CTS-1614), a grant from the Junta de Andalucía Regional Council to Amelia E. Aránega (CTS-03878), and grants from the Ministry of Science and Innovation of the Spanish Government to Diego Franco (MICINN BFU2009-11566) and to Amelia E. Aránega (MICINN BFU-2008-01217). This work is partially supported by a translational CNIC grant (2009-08) to Diego Franco.
References
Ai, D., Liu, W., Ma, L., Dong, F., Lu, M. F., Wang, D., Verzi, M. P., Cai, C., Gage, P. J., Evans, S., Black, B. L., Brown, N. A., and Martin, J. F. (2006). Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev. Biol. 296, 437–449.
Ammirabile, G., Tessari, A., Pignataro, V., Szumska, D., Sutera Sardo, F., Benes, J. Jr., Balistreri, M., Bhattacharya, S., Sedmera, D., and Campione, M. (2012). Pitx2 confers left morphological, molecular, and functional identity to the sinus venosus myocardium. Cardiovasc. Res. 93, 291–301.
Arnar, D. O., Thorvaldsson, S., Manolio, T. A., Thorgeirsson, G., Kristjansson, K., Hakonarson, H., and Stefansson, K. (2006). Familial aggregation of atrial fabrillation in Iceland. Eur. Heart J. 27, 708–712.
Bamforth, S. D., Bragança, J., Farthing, C. R., Schneider, J. E., Broadbent, C., Michell, A. C., Clarke, K., Neubauer, S., Norris, D., Brown, N. A., Anderson, R. H., and Bhattacharya, S. (2004). Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 36, 1189–1196.
Blum, M., Steinbeisser, H., Campione, M., and Schweickert, A. (1999). Vertebrate left-right asymmetry: old studies and new insights. Cell. Mol. Biol. 45, 505–516.
Brand, F. N., Abbott, R. D., Kannel, W. B., and Wolf, P. A. (1985). Characteristics and prognosis of lone atrial fibrillation. 30-year follow-up in the Framingham Study. JAMA 254, 3449–3453.
Brugada, R., Tapscott, T., Czernuszewicz, G. Z., Marian, A. J., Iglesias, A., Mont, L., Brugada, J., Girona, J., Domingo, A., Bachinski, L. L., and Roberts, R. (1997). Identification of a genetic locus for familial atrial fibrillation. N. Engl. J. Med. 336, 905–911.
Campione, M., Ros, M. A., Icardo, J. M., Piedra, E., Christoffels, V. M., Schweickert, A., Blum, M., Franco, D., and Moorman, A. F. (2001). Pitx2 expression defines a left cardiac lineage of cells: evidence for atrial and ventricular molecular isomerism in the iv/iv mice. Dev. Biol. 231, 252–264.
Campione, M., Steinbeisser, H., Schweickert, A., Deissler, K., van Bebber, F., Lowe, L. A., Nowotschin, S., Viebahn, C., Haffter, P., Kuehn, M. R., and Blum, M. (1999). The homeobox gene Pitx2: mediator of asymmetric left-right signaling in vertebrate heart and gut looping. Development 126, 1225–1234.
Chen, Y. H., Xu, S. J., Bendahhou, S., Wang, X. L., Wang, Y., Xu, W. Y., Jin, H.-W., Sun, H., Su1, X.-Y., Zhuang, Q. N., Yang, Y.-Q., Li, Y.-B., Liu, Y., Xu, H. J., Li, X. F., Ma, N., Mou, C. P., Chen, Z., Barhanin, J., and Huang, W. (2003). KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299, 251–254.
Chinchilla, A., Daimi, H., Lozano-Velasco, E., Dominguez, J. N., Caballero, R., Delpon, E., Tamargo, J., Cinca, J., Hove-Madsen, L., Aranega, A. E., and Franco, D. (2011). Pitx2 insufficiency leads to atrial electrical and structural remodelling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 4, 269–279.
Cox, C. J., Espinoza, H. M., McWilliams, B., Chappell, K., Morton, L., Hjalt, T. A., Semina, E. V., and Amendt, B. A. (2002). Differential regulation of gene expression by PITX2 isoforms. J. Biol. Chem. 277, 25001–25010.
Delaney, J. T., Jeff, J. M., Brown, N. J., Pretorius, M., Okafor, H. E., Darbar, D., Roden, D. M., and Crawford, D. C. (2012). Characterization of genome-wide association-identified variants for atrial fibrillation in African Americans. PLoS ONE 7, e32338. doi:10.1371/journal.pone.0032338
Ellinor, P. T., Lunetta, K. L., Glazer, N. L., Pfeufer, A., Alonso, A., Chung, M. K., Sinner, M. F., de Bakker, P. I., Mueller, M., Lubitz, S. A., Fox, E., Darbar, D., Smith, N. L., Smith, J. D., Schnabel, R. B., Soliman, E. Z., Rice, K. M., Van Wagoner, D. R., Beckmann, B. M., van Noord, C., Wang, K., Ehret, G. B., Rotter, J. I., Hazen, S. L., Steinbeck, G., Smith, A. V., Launer, L. J., Harris, T. B., Makino, S., Nelis, M., Milan, D. J., Perz, S., Esko, T., Köttgen, A., Moebus, S., Newton-Cheh, C., Li, M., Möhlenkamp, S., Wang, T. J., Kao, W. H., Vasan, R. S., Nöthen, M. M., MacRae, C. A., Stricker, B. H., Hofman, A., Uitterlinden, A. G., Levy, D., Boerwinkle, E., Metspalu, A., Topol, E. J., Chakravarti, A., Gudnason, V., Psaty, B. M., Roden, D. M., Meitinger, T., Wichmann, H. E., Witteman, J. C., Barnard, J., Arking, D. E., Benjamin, E. J., Heckbert, S. R., and Kääb, S. (2011). Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 42, 240–244.
Ellinor, P. T., Moore, R. K., Patton, K. K., Ruskin, J. N., Pollak, M. R., and Macrae, C. A. (2004). Mutations in the long QT gene, KCNQ1, are an uncommon cause of atrial fibrillation. Heart 90, 1487–1488.
Ellinor, P. T., Nam, E. G., Shea, M. A., Milan, D. J., Ruskin, J. N., and MacRae, C. A. (2007). Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 5, 99–105.
Franco, D., and Campione, M. (2003). The role of Pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc. Med. 13, 157–163.
Franco, D., Campione, M., Kelly, R., Zammit, P. S., Buckingham, M., Lamers, W. H., and Moorman, A. F. (2000). Multiple transcriptional domains, with distinct left and right components, in the atrial chambers of the developing heart. Circ. Res. 87, 984–991.
Franco, D., and Icardo, J. M. (2001). Molecular characterization of the ventricular conduction system in the developing mouse heart: topographical correlation in normal and congenitally malformed hearts. Cardiovasc. Res. 49, 417–429.
Fuster, V., Rydean, L. E., Asinger, R. W., Cannom, D. S., Crijns, H. J., Frye, R. L., Halperin, J. L., Kay, G. N., Klein, W. W., Lévy, S., McNamara, R. L., Prystowsky, E. N., Wann, L. S., Wyse, D. G., and Gibbons, R. J. (2001). ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 104, 2118–2150.
Gage, P. J., Suh, H., and Camper, S. A. (1999). Dosage requirement of Pitx2 for development of multiple organs. Development 126, 4643–4465.
Galli, D., Domínguez, J. N., Zaffran, S., Munk, A., Brown, N. A., and Buckingham, M. E. (2008). Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as Pitx2c is expressed. Development 135, 1157–1167.
Gollob, M. H., Jones, D. L., Krahn, A. D., Danis, L., Gong, X. Q., Shao, Q., Liu, X., Veinot, J. P., Tang, A. S., Stewart, A. F., Tesson, F., Klein, G. J., Yee, R., Skanes, A. C., Guiraudon, G. M., Ebihara, L., and Bai, D. (2006). Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677–2688.
Gudbjartsson, D. F., Arnar, D. O., Helgadottir, A., Gretarsdottir, S., Holm, H., Sigurdsson, A., Jonasdottir, A., Baker, A., Thorleifsson, G., Kristjansson, K., Palsson, A., Blondal, T., Sulem, P., Backman, V. M., Hardarson, G. A., Palsdottir, E., Helgason, A., Sigurjonsdottir, R., Sverrisson, J. T., Kostulas, K., Ng, M. C., Baum, L., So, W. Y., Wong, K. S., Chan, J. C., Furie, K. L., Greenberg, S. M., Sale, M., Kelly, P., MacRae, C. A., Smith, E. E., Rosand, J., Hillert, J., Ma, R. C., Ellinor, P. T., Thorgeirsson, G., Gulcher, J. R., Kong, A., Thorsteinsdottir, U., and Stefansson, K. (2007). Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448, 353–357.
Gudbjartsson, D. F., Holm, H., Gretarsdottir, S., Thorleifsson, G., Walters, G. B., Thorgeirsson, G., Walters, G. B., Thorgeirsson, G., Gulcher, J., Mathiesen, E. B., Njølstad, I., Nyrnes, A., Wilsgaard, T., Hald, E. M., Hveem, K., Stoltenberg, C., Kucera, G., Stubblefield, T., Carter, S., Roden, D., Ng, M. C., Baum, L., So, W. Y., Wong, K. S., Chan, J. C., Gieger, C., Wichmann, H. E., Gschwendtner, A., Dichgans, M., Kuhlenbäumer, G., Berger, K., Ringelstein, E. B., Bevan, S., Markus, H. S., Kostulas, K., Hillert, J., Sveinbjörnsdóttir, S., Valdimarsson, E. M., Løchen, M. L., Ma, R. C., Darbar, D., Kong, A., Arnar, D. O., Thorsteinsdottir, U., and Stefansson, K. (2009). A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat. Genet. 41, 876–878.
Gussak, I., and Antzelevitch, C. (2008). Electrical Diseases of the Heart: Genetics, mechanisms, Treatment, Prevention. London: Springer-Verlag.
Haissaguerre, M., Jais, P., Shah, D. C., Takahashi, A., Hocini, M., Quiniou, G., Garrigue, S., Le Mouroux, A., Le Métayer, P., and Clémenty, J. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666.
Hamada, H., Meno, C., Saijoh, Y., Adachi, H., Yashiro, K., Sakuma, R., and Shiratori, H. (2001). Role of asymmetric signals in left-right patterning in the mouse. Am. J. Med. Genet. 101, 324–327.
Henningsen, K. M., Olesen, M. S., Haunsoe, S., and Svendsen, J. H. (2011). Association of rs2200733 at 4q25 with early onset of lone atrial fibrillation in young patients. Scand. Cardiovasc. J. 45, 324–326.
Hjalt, T. A., and Semina, E. V. (2005). Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev. Mol. Med. 7, 1–17.
Kahr, P. C., Piccini, I., Fabritz, L., Greber, B., Schöler, H., Scheld, H. H., Hoffmeier, A., Brown, N. A., and Kirchhof, P. (2011). Systematic analysis of gene expression differences between left and right atria in different mouse strains and in human atrial tissue. PLoS ONE 6, e26389. doi:10.1371/journal.pone.0026389
Kannel, W. B., Wolf, P. A., Benjamin, E. J., and Levy, D. (1998). Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am. J. Cardiol. 82, 2N–9N.
Kelly, R. G., Brown, N. A., and Buckingham, M. E. (2001). The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 1, 435–440.
Kiliszek, M., Franaszczyk, M., Kozluk, E., Lodzinski, P., Piatkowska, A., Broda, G., Ploski, R., and Opolski, G. (2011). Association between variants on chromosome 4q25, 16q22 and 1q21 and atrial fibrillation in the Polish population. PLoS ONE 6, e21790. doi:10.1371/journal.pone.0021790
Kirchhof, P., Kahr, P. C., Kaese, S., Piccini, I., Vokshi, I., Scheld, H. H., Rotering, H., Fortmueller, L., Laakmann, S., Verheule, S., Schotten, U., Fabritz, L., and Brown, N. A. (2011). PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ. Cardiovasc. Genet. 4, 123–133.
Larson, M. G., Atwood, L. D., Benjamin, E. J., Cupples, L. A., D’Agostino, R. B. Sr., Fox, C. S., Govindaraju, D. R., Guo, C. Y., Heard-Costa, N. L., Hwang, S. J., Murabito, J. M., Newton-Cheh, C., O’Donnell, C. J., Seshadri, S., Vasan, R. S., Wang, T. J., Wolf, P. A., and Levy, D. (2007). Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med. Genet. 8(Suppl. 1), S5. doi:10.1186/1471-2350-8-S1-S5
Lin, C. R., Kioussi, C., O’Connell, S., Briata, P., Szeto, D., Liu, F., Izpisúa-Belmonte, J. C., and Rosenfeld, M. G. (1999). Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature 401, 279–282.
Liu, C., Liu, W., Lu, M. F., Brown, N. A., and Martin, J. F. (2001). Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development 128, 2039–2048.
Liu, C., Liu, W., Palie, J., Lu, M. F., Brown, N. A., and Martin, J. F. (2002). Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development 129, 5081–5091.
Liu, X., Wang, F., Knight, A. C., Zhao, J., and Xiao, J. (2012). Common variants for atrial fibrillation: results from genome-wide association studies. Hum. Genet. 131, 33–39.
Lozano-Velasco, E., Chinchilla, A., Martinez-Fernandez, S., Hernandez-Torres, F., Navarro, F., Lyons, G., Franco, D., and Aránega, A. E. (2011). Pitx2c modulates cardiac specific transcription factor networks in differentiating cardiomyocytes from murine embryonic stem cells. Cells Tissues Organs (Print) 194, 349–362.
Mc Nair, W. P., Ku, L., Taylor, M. R., Fain, P. R., Dao, D., Wolfel, E., Mestroni, L., and The Familial Cardiomyopathy Registry Research Group. (2004). SCN5A mutation associated with dilated car-diomyopathy, conduction disorder, and arrhythmia. Circulation 110, 2163–2167.
Moe, G. K., and Abildskov, J. A. (1959). Atrial fibrillation as a self sustaining arrhythmia independent of focal discharge. Am. Heart J. 58, 59–70.
Mommersteeg, M. T., Brown, N. A., Prall, O. W., de Gier-de Vries, C., Harvey, R. P., Moorman, A. F., and Christoffels, V. M. (2007). Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ. Res. 101, 902–909.
Nowotschin, S., Liao, J., Gage, P. J., Epstein, J. A., Campione, M., and Morrow, B. E. (2006). Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development 133, 1565–1573.
Olesen, M. S., Holst, A. G., Jabbari, J., Nielsen, J. B., Christophersen, I. E., Sajadieh, A., Haunsø, S., and Svendsen, J. H. (2012). Genetic loci on chromosomes 4q25, 7p31, and 12p12 are associated with onset of lone atrial fibrillation before the age of 40 years. Can. J. Cardiol. 28, 191–195.
Schnabel, R. B., Kerr, K. F., Lubitz, S. A., Alkylbekova, E. L., Marcus, G. M., Sinner, M. F., Magnani, J. W., Wolf, P. A., Deo, R., Lloyd-Jones, D. M., Lunetta, K. L., Mehra, R., Levy, D., Fox, E. R., Arking, D. E., Mosley, T. H., Müller-Nurasyid, M., Young, T. R., Wichmann, H. E., Seshadri, S., Farlow, D. N., Rotter, J. I., Soliman, E. Z., Glazer, N. L., Wilson, J. G., Breteler, M. M., Sotoodehnia, N., Newton-Cheh, C., Kääb, S., Ellinor, P. T., Alonso, A., Benjamin, E. J., Heckbert, S. R., and Candidate Gene Association Resource (CARe) Atrial Fibrillation/Electrocardiography Working Group (2011). Large-scale candidate gene analysis in whites and African Americans identifies IL6R polymorphism in relation to atrial fibrillation: the National Heart, Lung, and Blood Institute’s Candidate Gene Association Resource (CARe) project. Circ. Cardiovasc. Genet. 4, 557–564.
Schotten, U., Verheule, S., Kirchhof, P., and Goette, A. (2011). Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol. Rev. 91, 265–325.
Schweickert, A., Campione, M., Steinbeisser, H., and Blum, M. (2000). Pitx2 isoforms:involvement of Pitx2c but not Pitx2a or Pitx2b in vertebrate left-right asymmetry. Mech. Dev. 90, 41–51.
Tessari, A., Pietrobon, M., Notte, A., Cifelli, G., Gage, P. J., Schneider, M. D., Lembo, G., and Campione, M. (2008). Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ. Res. 102, 813–822.
Wakili, R., Voigt, N., Kääb, S., Dobrev, D., and Nattel, S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968.
Wang, J., Klysik, E., Sood, S., Johnson, R. L., Wehrens, X. H., and Martin, J. F. (2010). Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl. Acad. Sci. U.S.A. 107, 9753–9758.
Xia, M., Jin, Q., Bendahhou, S., He, Y., Larroque, M. M., Chen, Y., Zhou, Q., Yang, Y., Liu, Y., Liu, B., Zhu, Q., Zhou, Y., Lin, J., Liang, B., Li, L., Dong, X., Pan, Z., Wang, R., Wan, H., Qiu, W., Xu, W., Eurlings, P., Barhanin, J., and Chen, Y. (2005). A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 332, 1012–1019.
Yang, Y., Xia, M., Jin, Q., Bendahhou, S., Shi, J., Chen, Y., Liang, B., Lin, J., Liu, Y., Liu, B., Zhou, Q., Zhang, D., Wang, R., Ma, N., Su, X., Niu, K., Pei, Y., Xu, W., Chen, Z., Wan, H., Cui, J., Barhanin, J., and Chen, Y. (2004). Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 75, 899–905.
Keywords: Pitx2, atrial fibrillation, microRNAs, ion channel, electrophysiology
Citation: Franco D, Chinchilla A and Aránega AE (2012) Transgenic insights linking Pitx2 and atrial arrhythmias. Front. Physio. 3:206. doi: 10.3389/fphys.2012.00206
Received: 15 March 2012; Paper pending published: 05 April 2012;
Accepted: 23 May 2012; Published online: 12 June 2012.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsReviewed by:
Larissa Fabritz, Hospital of the University of Münster, GermanyBas Boukens, Academic Medical Center, Netherlands
Copyright: © 2012 Franco, Chinchilla and Aránega. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Diego Franco, Department of Experimental Biology, University of Jaen, 23071 Jaen, Spain. e-mail: dfranco@ujaen.es