 Mickael Derangeon1,2,3
Mickael Derangeon1,2,3- 1 INSERM, UMR 1087, l’Institut du Thorax, Nantes, France
- 2 CNRS, UMR 6291, l’Institut du Thorax, Nantes, France
- 3 Université de Nantes, Nantes, France
Mutations of SCN5A gene, which encodes the α-subunit of the voltage-gated Na+ channel NaV1.5, underlie hereditary cardiac arrhythmic syndromes such as the type 3 long QT syndrome, cardiac conduction diseases, the Brugada syndrome, the sick sinus syndrome, a trial standstill, and numerous overlap syndromes. Patch-clamp studies in heterologous expression systems have provided important information to understand the genotype-phenotype relationships of these diseases. However, they could not clarify how SCN5A mutations can be responsible for such a large spectrum of diseases, for the late age of onset or the progressiveness of some of these diseases and for the overlapping syndromes. Genetically modified mice rapidly appeared as promising tools for understanding the pathophysiological mechanisms of cardiac SCN5A-related arrhythmic syndromes and several mouse models have been established. This review presents the results obtained on these models that, for most of them, recapitulate the clinical phenotypes of the patients. This includes two models knocked out for Nav1.5 β1 and β3 auxiliary subunits that are also discussed. Despite their own limitations that we point out, the mouse models still appear as powerful tools to elucidate the pathophysiological mechanisms of SCN5A-related diseases and offer the opportunity to investigate the secondary cellular consequences of SCN5A mutations such as the expression remodeling of other genes. This points out the potential role of these genes in the overall human phenotype. Finally, they constitute useful tools for addressing the role of genetic and environmental modifiers on cardiac electrical activity.
Mutations in the SCN5A gene, which encodes the α-subunit of the cardiac voltage-gated Na+ channel NaV1.5, underlie hereditary arrhythmic syndromes (so-called cardiac channelopathies). On one hand, gain-of-function mutations, that increase the late component of the Na+ current (INa) and thus prolong the ventricular action potential, are responsible for the type 3 long QT syndrome (LQT3; Moss and Kass, 2005). On the other hand, loss-of-function mutations decrease INa and are responsible for cardiac conduction diseases (Schott et al., 1999; Tan et al., 2001; Probst et al., 2003), Brugada syndrome (Gussak et al., 1999), sick sinus syndrome (Benson et al., 2003), and atrial standstill (Groenewegen et al., 2003). To complicate matters further, some SCN5A mutations can lead to more complex diseases associating different phenotypic traits such as, for instance, bradycardia, conduction disease, LQT3, and Brugada syndrome (so-called overlap syndromes; Bezzina et al., 1999; Kyndt et al., 2001; Grant et al., 2002; Rossenbacker et al., 2004; Smits et al., 2005; for review, see Remme et al., 2008). Finally, there is also an association between SCN5A genetic defects and susceptibility to dilated cardiomyopathy (DCM; McNair et al., 2004) and atrial fibrillation (Laitinen-Forsblom et al., 2006; Ellinor et al., 2008).
Although patch-clamp studies in heterologous expression systems have provided a great deal of information to understand the genotype-phenotype relationships of these diseases, these models could not clarify how loss-of-function-mutations can be responsible for such a large spectrum of diseases and the late age of onset or the progressiveness of some of them. They are even less adapted to overlap syndromes.
Genetically modified mice thus appeared as promising tools for understanding the pathophysiological sequence of cardiac SCN5A-related channelopathies and several mouse models have been established (Table 1). Fortunately, whereas some genetically modified mice related to K+ channel dysfunction did not reach investigators’ expectations because of major electrophysiological differences between mice and men (Nerbonne et al., 2001; Charpentier et al., 2004), mouse models of inherited SCN5A channelopathies, on the contrary, appeared to be informative.
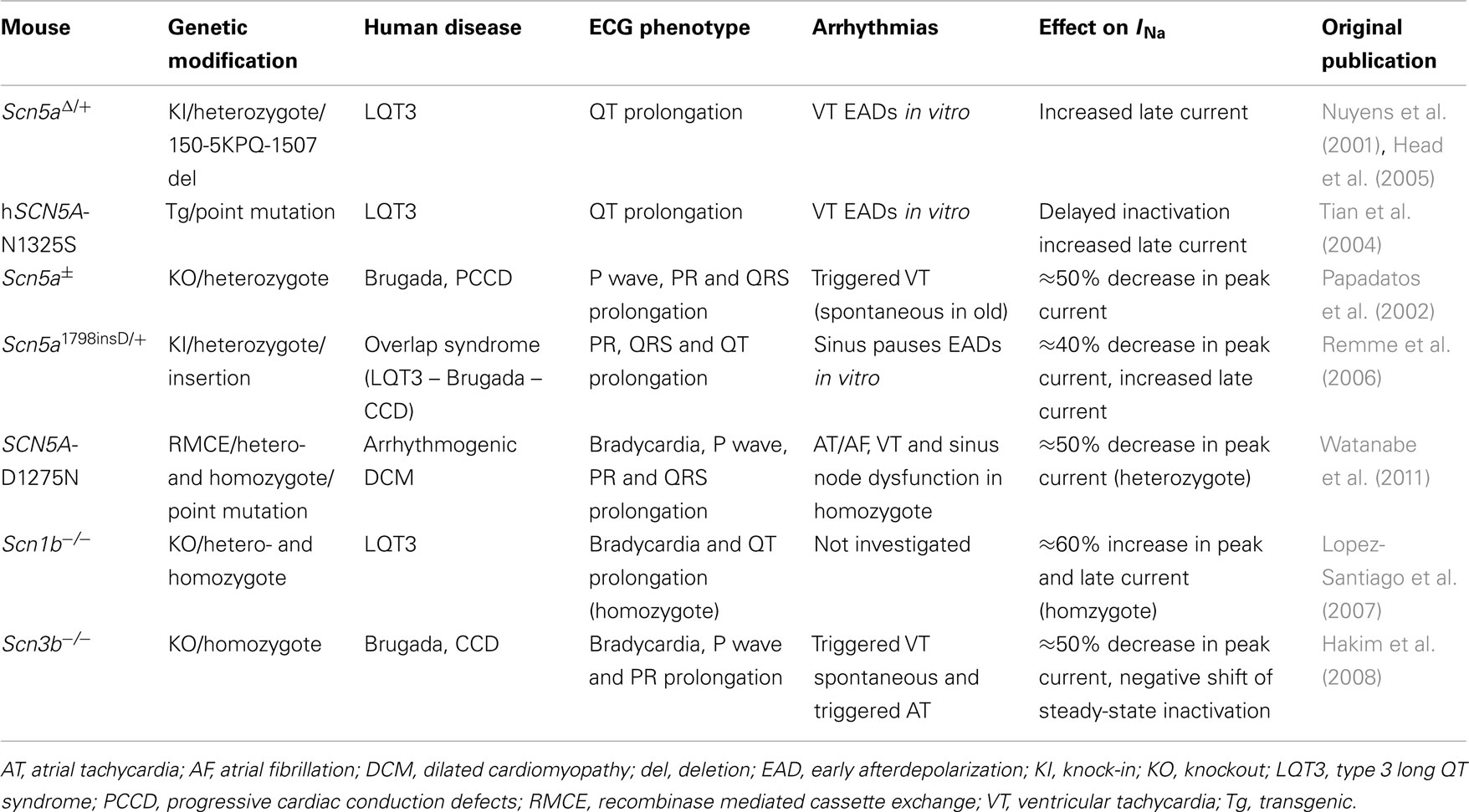
Table 1. Mouse models of SCN5A-related cardiac genetic diseases.
Models for the LQT3 Syndrome
SCN5A-related long QT syndrome (LQT3; Wang et al., 1995) is less frequent but often more lethal than LQT1 and LQT2 (Priori et al., 2003). Like in other LQT syndromes, the abnormal prolongation of ventricular repolarization is associated with a susceptibility to a specific polymorphic ventricular tachycardia called torsades de pointes and ventricular fibrillation, leading to syncope and sudden death. Clinically, LQT3 is characterized by an increased duration of the ST segment with a late appearance of the T-wave (Moss, 2002). Bradycardia and pauses occurring at rest, and more particularly during sleep, are often at the origin of the arrhythmias. However, fatal tachycardia-induced arrhythmias have also been reported for a third of the patients (Schwartz et al., 2001). Two thirds of the SCN5A mutations found in LQT3 alter the fast inactivation process of the channel (Zimmer and Surber, 2008). For example, the first identified mutation, which leads to the deletion of three amino acids (1505-KPQ-1507) in the inactivation domain of NaV1.5, results in a persistent inward Na+ current (Wang et al., 1996) and prolongation of the action potential plateau phase (Moss and Kass, 2005).
Scn 5aΔ1505-1507KPQ/+ knock-in mouse
Nuyens et al. (2001) generated a knock-in mouse model lacking the same KPQ residues. Mice heterozygous for this mutation (Scn5aΔ/+) showed typical features of the LQT3 syndrome and spontaneously developed ventricular arrhythmias. Compared with wild-type mice, transmembrane action potentials recorded in ventricular preparations were markedly prolonged in Scn5aΔ/+ mice and showed a steeper “heart rate” versus “action potential duration” relationship (Nuyens et al., 2001). Action potential amplitude and upstroke velocity were not altered. Whole-cell patch-clamp recordings performed on cardiomyocytes confirmed the increase in late inward Na+ current previously observed in heterologous expression system, which explains the action potential prolongation (Wang et al., 1996). Surprisingly, there was also a two-fold increase in the peak Na+ current density, which could not be explained by an over-expression of NaV1.5 or changes in the steady-state activation and inactivation of the current. However, this finding was not confirmed in a more recent study on the same model (Fredj et al., 2006) and the reason for this discrepancy remains to be clarified. Nuyens et al.’s (2001) study also revealed an acceleration of the recovery from inactivation, a property that might favor re-entrant arrhythmias. Indeed, intracardiac pacing with extra stimuli, short-long-short pacing sequences and abrupt accelerations of heart rate reproducibly induced polymorphic ventricular arrhythmias in Scn5aΔ/+ mice but not in wild-type mice. The Scn5aΔ/+ mice were also characterized by a paradoxical transient action potential prolongation upon abrupt acceleration of pacing rate that favored the occurrence of early after depolarizations. This prolongation was blocked by the Na+ channel blocker mexiletine and the adrenergic agonist isoproterenol. It can be explained by the increase of both fast and slow components of the Na+ current which causes a sudden increase of Na+ load, and secondarily of Ca2+ load. As a consequence, during each action potential, the increased Ca2+ release from the sarcoplasmic reticulum activates a larger Na/Ca exchanger current which generates early after depolarizations. The autonomic modulation of cardiac electrical activity and arrhythmias of Scn5aΔ/+ mice has been recently investigated in more details both in vivo in freely moving mice and ex vivo (Fabritz et al., 2010). As expected from clinical investigations, cholinergic stimulation was shown to favor arrhythmias by inducing bradycardia. In contrast, β-adrenergic stimulation suppressed arrhythmias by shortening repolarization and increasing cardiac rate, while β-blockers had no effect on their own.
A second Scn5aΔ/+ mouse line, exhibiting the same phenotype as the previous line, was created (Head et al., 2005) and submitted to an extended pharmacological analysis. Electrophysiological studies performed on Langendorff-perfused whole hearts confirmed the anti-arrhythmic properties of mexiletine (Fabritz et al., 2003; Head et al., 2005) and showed that another Na+ channel inhibitor, flecainide, could also prevent arrhythmias by preferentially inhibiting the late Na+ current (Fredj et al., 2006), whereas β-blockers were inefficient (Head et al., 2005; Stokoe et al., 2007b; Sabir et al., 2008). These results have been confirmed in freely moving mice (Fabritz et al., 2010). More recently, it was shown that a specific inhibition of the L-type Ca2+ channels with nifedipine was also efficient in preventing arrhythmias through the suppression of early after depolarizations (Thomas et al., 2007). Finally, nicorandil, an activator of the ATP-sensitive K+ channel, also reduces arrhythmogenicity in Scn5aΔ/+ mice (Hothi et al., 2008), confirming previous results obtained with the canine wedge preparation pharmacological model of LQT3 (Shimizu and Antzelevitch, 2000). In contrast, quinidine was proarrhythmic (Sabir et al., 2008). Ex vivo studies also confirmed the key role played by bradycardia and increased dispersion of repolarization on the occurrence of spontaneous ventricular arrhythmias (Fabritz et al., 2003). Sudden rate accelerations initially and transiently increased the dispersion of repolarization due to early after depolarizations and action potential alternans (Fabritz et al., 2003; Hothi et al., 2008), as previously observed in vivo (Nuyens et al., 2001), and then secondarily suppressed and prevented ventricular tachycardia by decreasing dispersion of repolarization and suppressing early after depolarizations. Arrhythmogenesis, as assessed by programmed electrical stimulation on Langendorff-perfused hearts, has also been associated with abnormal patterns of myocardial activation and abnormally reduced transmural gradients of repolarization resulting from a larger increase in action potential duration in subepicardium than in subendocardium (Stokoe et al., 2007b). In addition, Scn5aΔ/+ mice exhibit larger differences between action potential duration and refractory period, so-called local critical interval (APD-ERP), in subepicardial regions than in wild-type mice. Those anomalies may support re-entrant events following premature excitation. More recently, Sabir et al. (2008) proposed that the increases of the slopes of restitution curves (plotting action potential duration versus the preceding diastolic interval; Gilmour, 2003) to values greater than one, rather than increased dispersion of repolarization, was associated with arrhythmogenicity. All these anomalies, including action potential alternans are corrected by nicorandil (Hothi et al., 2008).
Altogether, Scn5aΔ/+ mice recapitulate most of the LQT3 associated clinical symptoms. They confirmed previous studies suggesting that arrhythmias in LQT3 syndrome may result from reentries provoked by early after depolarizations and enabled to unveil the mechanisms of tachycardia-induced arrhythmias. Studies of Scn5aΔ/+ mice have also provided a rationale for the use of Na+ channel blockers and pacing to prevent cardiac arrhythmias and sudden death in the context of LQT3, for which β-blockers have shown low efficacy. Finally, they suggest that Ca2+ channels inhibitors and ATP-sensitive K+ channel activators might be interesting pharmacological approaches to prevent arrhythmias in this context.
In addition to early after depolarizations, the occurrence of delayed after depolarizations, resulting from the transient inward current (Iti), was also clearly demonstrated in myocytes of Scn5aΔ/+ mice (Fredj et al., 2006). Most recently, Lindegger et al. (2009) showed that Iti induction occurs under conditions of elevated sarcoplasmic reticulum Ca2+ content that is most probably related to the mutation-induced increase in late Na+ current. Indeed, both Iti and its underlying Ca2+ wave were suppressed by ranolazine, an antianginal agent well known to block the late Na+ current (Hale et al., 2008). Whether delayed after depolarizations participate to spontaneous arrhythmic events in Scn5aΔ/+ mice, and of course in LQT3 patients, remains to be determined. Investigating the potential anti-arrhythmic effects of ranolazine in this model also remains to be performed.
Patients with LQT syndrome (El Yaman et al., 2008; Johnson et al., 2008; Zellerhoff et al., 2009), including LQT3 (Benito et al., 2008), may be at increased risk for atrial fibrillation. Therefore, the atrial phenotype of Scn5aΔ/+ mice was characterized. Using Langendorff-perfused hearts, Blana et al. (2010) showed that atrial action potential duration and effective refractory period were prolonged in Scn5aΔ/+ mice. Short runs of rapid pacing-induced a larger increase in post-pacing spontaneous atrial cycle length and action potential duration in Scn5aΔ/+ mice than in wild-type mice, leading to the occurrence of atrial tachycardia in Scn5aΔ/+ mice but not in wild-type mice. Extra stimuli triggered arrhythmias similarly in wild-type and Scn5aΔ/+ mice (in 30–40% of the mice). Contrasting results had been previously observed with the second mouse line suggesting that Scn5aΔ/+ mice are protected against pacing-induced atrial tachyarrhythmias when young (3 months; Dautova et al., 2009), whereas they exhibit an increased atrial arrhythmogenicity when old (1 year; Guzadhur et al., 2010). Without excluding the influence of variable experimental conditions, those discrepancies might also rely on the fact that the two lines have different genetic backgrounds, as we will discuss later for the Scn5a1798insD/+ knock-in mouse. Both mouse lines, however, exhibit decreased heart rate and atrioventricular conduction (prolonged PR interval) versus wild-type mice.
Sinus node dysfunction has been reported in patients with SCN5A-related long QT syndrome whether these mutations only lead to an increased late Na+ current (Moss et al., 1995; Wei et al., 1999) or lead to more complex biophysical alterations of the channel (Bezzina et al., 1999; van den Berg et al., 2001; Grant et al., 2002). Because QT prolongation is more pronounced at lower heart rates, bradycardia represents an important indirect factor in predisposition to lethal arrhythmias in LQT3 families (Schwartz et al., 1995). The mechanisms for LQT3-related bradycardia were investigated for the 1795insD mutation of NaV1.5 (Veldkamp et al., 2003), a mutation inducing an overlap syndrome with bradycardia, conduction disease, LQT3 and Brugada syndrome (Bezzina et al., 1999). This mutation induces both a decrease in Na+ peak current and the occurrence of a late Na+ current (see dedicated section below for details). By combining current clamp studies and computational modeling, Veldkamp et al. (2003) suggested that both the decreased availability of Na+ channels and the increase of late current accounted for bradycardia. However, the effect of action potential prolongation secondary to the increased late current appeared more prominent. The contribution of the late Na+ current to sinus node dysfunction has been further confirmed on Scn5aΔ/+ mice (Wu et al., 2012). Scn5aΔ/+ mice exhibit sinus node dysfunction with episodes of bradycardia, sinus pauses, and longer sinus node recovery times. Sino-atrial preparations from Scn5aΔ/+ mice had lower intrinsic rate from the sinus node to the surrounding atrium than wild-type preparations. Computational studies confirmed that the decrease in sinus rate could be attributed to the increased late current. Ex vivo experiments on sino-atrial preparations also showed that conduction through the sinus node to the surrounding atrium was decreased, a result that computer modeling could only explain by a decrease in Na+ peak current amplitude, which has not been experimentally confirmed.
hSCN5A-N1325S transgenic mouse model
The N1325S mutation of NaV1.5, located in the intracellular region between segments 4 and 5 of domain III, also causes LQT3 syndrome (Wang et al., 1995). This mutation, like many similar mutations, leads to an increased late inward Na+ current (Dumaine et al., 1996; Wang et al., 1996). A transgenic mouse with cardiac selective expression of human SCN5A-N1325S mutation under the control of the mouse α-myosin heavy chain promoter was established (Tian et al., 2004). Transgenic mice showed some of the typical features of the LQT3 syndrome such as prolonged QT interval, ventricular arrhythmias, and a high rate of sudden deaths due to documented polymorphic ventricular tachycardia and ventricular fibrillation. However, transgenic mice were also characterized by increased heart rate and shorter PR intervals that are not observed in LQT3 patients. Some of these features might result from over-expression of the channel rather than from the mutation itself since short PR interval was also observed in the control mouse over-expressing wild-type hSCN5A gene (Zhang et al., 2007).
Voltage-clamp experiments on cardiomyocytes isolated from wild-type and hSCN5A-N1325S mice confirmed that the N1325S mutation does not modify the peak current but delays the inactivation process and generates a late persistent current (Tian et al., 2004), which decreases with increasing pacing rates (Yong et al., 2007). Action potentials from hSCN5A-N1325S ventricular myocytes were longer than wild-type ones and exhibited early after depolarizations. As in Scn5aΔ/+ mice, mexiletine shortened ventricular action potential duration and prevented arrhythmias (Tian et al., 2004). Interestingly, action potential duration of transgenic but not wild-type myocytes paradoxically increased with pacing rate (Tian et al., 2004) and became highly variable with occurrence of repolarization alternans (Yong et al., 2007). Whether this phenomenon was only transient, as in Scn5aΔ/+ mice, or persisted during long periods of increased pacing rate is unclear. The Ca2+ channel blocker verapamil reduced these alternans, suggesting an involvement of intracellular Ca2+ deregulation in this phenomenon. Previous studies had shown that patients with long QT syndrome can exhibit T-wave alternans during sinus tachycardia (Cruz Filho et al., 2000; Viskin et al., 2004).
Like the Scn5aΔ/+ mouse model, the hSCN5A-N1325S mouse provided key information to our understanding of arrhythmias in LQT3 syndrome. For instance, it showed that the N1325S mutant dysfunction can explain both bradycardia- and non-bradycardia-related arrhythmias observed in patients carrying this mutation (Yong et al., 2007). However, the model might be taken with caution because of the limitations linked to the over-expression of the channel. Indeed, there is a clear relationship between the level of hSCN5A-N1325S expression and the extent of QT prolongation, i.e., by 23% with 1–2 copies and by 91% with 10 copies. Most recently, this model was shown to exhibit cardiomyocyte apoptosis, cardiac fibrosis, and contractile dysfunction during aging (Zhang et al., 2011). But signs of heart failure were markedly larger in the mouse line with 10 copies. Although heart failure is probably not related to SCN5A over-expression per se, because old mice over-expressing 10 times the wild-type channel remained normal, one should better consider the model over-expressing the least the mutant channel to extrapolate the results to the patients. In this second line, left ventricular fractional shortening was less decreased and only 30% of the mice showed features of DCM, a result more consistent with the scarcity of heart failure in LQT3 patients (Nguyen et al., 2008; Shi et al., 2008).
A model for the Brugada Syndrome and Conduction Disorders: The Scn5a Knockout Mouse
The Brugada syndrome is a genetic disease which associates ST segment elevation in the right precordial leads V1 to V3 of the ECG, often with signs of conduction slowing, with a high risk of sudden cardiac death secondary to ventricular tachycardia or fibrillation (Gussak et al., 1999; Antzelevitch et al., 2005). In 20–30% of the patients, the disease is related to mutations in SCN5A leading to a complete or partial loss-of-function of NaV1.5 (Tan et al., 2003). Loss-of-function mutations are also responsible for inherited Progressive Cardiac Conduction Defects (PCCD), also called Lev-Lenègre disease (Schott et al., 1999), or non-progressive conduction defects (Tan et al., 2001). More recently, PCCD has also been linked to gain-of-function mutations in TRPM4 gene (Transient Receptor Potential cation channel, subfamily M, member 4), which encodes a Ca2+-activated non-selective channel (Kruse et al., 2009; Liu et al., 2010). PCCD is a slowly evolving disease which progressively affects cardiac conduction leading ultimately to pacemaker implantation to prevent the risk of complete atrioventricular block and Stokes–Adams syncope. Inherited PCCD associates haploinsufficiency of SCN5A together with an additional yet unknown mechanism related to aging (Probst et al., 2003). Despite their major clinical differences, Brugada syndrome, and inherited conduction diseases share common features. First, both diseases can be caused by SCN5A haploinsufficiency. Second, Brugada patients with SCN5A mutations exhibit altered conduction whereas Brugada patients without SCN5A mutations do not (Smits et al., 2002). Finally, both Brugada syndrome and PCCD can be found in the same pedigree (Kyndt et al., 2001). In other families, patients carrying loss-of-function mutations can exhibit symptoms of Brugada syndrome, conduction disease, and sick sinus syndrome, either combined or not (Smits et al., 2005). Severe forms of sick sinus syndrome have also been found in patients with double inheritance of SCN5A loss-of-function mutations (Benson et al., 2003).
A mouse model with targeted disruption of Scn5a gene has been established (Papadatos et al., 2002). Homozygous knockout mouse embryos die during mid-gestation due to severe defects in ventricular morphogenesis whereas heterozygous (Scn5a±) mice show normal survival. However, 8- to 10-week-old Scn5a± mice have several cardiac electrical defects such as decreased atrial and atrioventricular conduction and increased susceptibility to pacing-induced ventricular arrhythmias (Papadatos et al., 2002). Following this initial report, the model was further analyzed to investigate the potential pathophysiological mechanisms involved in the progressive evolution of SCN5A-related PCCD (Royer et al., 2005; van Veen et al., 2005). It was found that in addition to atrial and atrioventricular conduction, ventricular conduction was also decreased. Moreover, ECG studies performed on mice ranging from 4 to 71 weeks of age showed that the ventricular phenotype of Scn5a± mice worsened with age. This feature was confirmed by activation mapping studies performed on Langendorff-perfused hearts. In young Scn5a± mice, conduction velocity was only affected in the right ventricle. In old mice, right ventricular conduction defect worsened and was in addition associated with conduction velocity defect in the left ventricle. This age-dependent deterioration of ventricular conduction was associated with the occurrence of fibrosis in ventricular myocardium. In some aspects, this phenotype resembles PCCD, although in the later fibrosis was found to be limited to the conduction system area (Lenègre and Moreau, 1963; Lev, 1964). However, it is worth noting that fibrosis has also been observed in patients with the Brugada syndrome (Coronel et al., 2005; Frustaci et al., 2005). More recently, it was shown that Scn5a± mice display variable degrees of conduction defects when young (10 weeks old): some mice have only mild prolongation of the QRS interval while others have more severe QRS prolongation associated with RR′ or SS′ patterns (Leoni et al., 2010). The extent of fibrotic remodeling was dependent on the severity of the initial conduction disorders: mice with larger QRS prolongation when young have the most severe fibrotic remodeling when they get older, suggesting that the ventricular activation pattern is a key element for triggering remodeling. Whatever their triggering mechanism, fibrosis, and redistribution of connexin 43 expression most likely contribute to the age-dependent degradation of ventricular conduction in the mouse model. Whether SCN5A-related inherited PCCD in human also results from a primary decrease in Na+ current and a secondary progressive fibrosis with aging remains to be clarified. In this context, Scn5a± mice could be used for testing preventive therapies as an alternative to pacemaker implantation.
In their initial report on the Scn5a± mouse model, Papadatos et al. (2002) showed that programmed ventricular electrical pacing of Langendorff-perfused hearts induced ventricular tachyarrhythmias in two thirds of Scn5a± mice but not in wild-type mice and proposed that the model could be used for elucidating the mechanisms of arrhythmias in the context of SCN5A-related Brugada syndrome. Pathophysiological findings have implicated the right ventricular outflow tract as the primary location for electrical disorders in Brugada syndrome. Two hypotheses have been proposed to explain both ST segment elevation and occurrence of arrhythmias (Wilde et al., 2010). The first one, mainly based on experimental data obtained in arterially perfused canine right ventricular wedges, implicates alterations in transmural and regional repolarization gradients leading to localized losses of the subepicardial action potential dome and phase 2 reentries as the arrhythmogenic mechanism. The second one, mainly based on clinical studies, implicates abnormal conduction. To elucidate the arrhythmogenic mechanisms in Scn5a± mice programmed electrical stimulation, monophasic action potential (MAP) recording, and bipolar electrogram recording was performed on Langendorff-perfused hearts. These experiments showed that Scn5a± mice were characterized by increased electrogram duration with shortening extrasystoles intervals, especially in the right ventricular outflow tract, and increased transmural and regional heterogeneity of MAP duration and refractory periods in the right ventricle with the shortest in the right ventricular outflow tract (Stokoe et al., 2007a; Martin et al., 2010, 2011a,b). Scn5a± mice also exhibited increased incidence of MAP alternans. As a consequence, ventricular tachycardia occurred earlier in the right ventricular outflow tract (Martin et al., 2011b). All these mechanisms were potentiated by flecainide, a Na+ channel inhibitor which is well described to favor ST segment elevation and arrhythmias in patients with the Brugada syndrome. Ventricular tachycardia also occurred spontaneously both in vivo (Leoni et al., 2010; Martin et al., 2011c) and ex vivo (Martin et al., 2011d), but with a much higher incidence under flecainide treatment (Martin et al., 2011c,d). Multielectrode array studies showed that the initiation of spontaneous arrhythmias resulted from a combination of conduction slowing and repolarization heterogeneities leading to lines of conduction blocks and unidirectional conduction (Martin et al., 2011d). Altogether these results seem to contradict the hypothesis that arrhythmias in the Brugada syndrome result from phase 2 re-entrant mechanisms. However, the main limitation of the mouse model is that the ventricular action potential lacks a plateau phase, and for the subepicardial action potential, a spike, and dome morphology, present in larger mammals and prerequisite for phase 2 reentries.
In addition, Scn5a± mice turned out to be useful for understanding the role of NaV1.5 in sinus node normal function and the pathophysiology of sinus node dysfunction associated to SCN5A loss-of-function mutations (Benson et al., 2003; Smits et al., 2005). Indeed, Scn5a± mice also display moderate bradycardia that can be associated with occasional sino-atrial blocks in older animals (Lei et al., 2005). Lei et al. showed that small central sino-atrial node cells, which do not express NaV1.5, exhibited normal intrinsic pacemaker rate. In contrast, larger peripheral sino-atrial node cells, which normally express NaV1.5, exhibited reduced intrinsic pacemaker rate associated to reduced expression of NaV1.5. These data were consistent with the slower sino-atrial conduction and frequent sino-atrial conduction blocks observed in Scn5a± sino-atrial node preparations compared to wild-type although they exhibited normal activation patterns. A computer model successfully reproduced these findings and implicated INa in action potential propagation through the sino-atrial node, from sino-atrial node to atria, and in modifying heart rate through a coupling of sino-atrial node and atrial cells. More recently, this same team has shown that both aging and Scn5a disruption affect sino-atrial node function through electrical remodeling and TGF-β1-mediated fibrosis, with the most severe phenotype observed in old Scn5a± mice (Hao et al., 2011). Interestingly, this in vivo sino-atrial node dysfunction appears less severe than expected from ex vivo experiments and computer simulations seems to be due to an increase in sympathetic regulation of sino-atrial node with age, partly explained by an over-expression of β1-adrenoceptors (Hao et al., 2011).
An Overlap Syndrome Model: The Scn5a1798insD/+ Knock-in Mouse
In 1999, a SCN5A mutation (1795insD) associated with an overlap syndrome of cardiac Na+ channelopathies was described in a large Dutch family. The patients’ ECG presented features of bradycardia, conduction disease, LQT3, and Brugada syndrome (Bezzina et al., 1999). The analysis of the 1795insD mutation biophysical properties led to contrasting results depending on the heterologous expression system used. In Xenopus oocytes, the mutation reduced the Na+ current through a negative shift in voltage-dependence of steady-state inactivation but did not produce the persistent current commonly observed in LQT3 (Bezzina et al., 1999). In contrast, in HEK293 cells, 1795insD mutation disrupted fast inactivation leading to sustained inward Na+ current that could prolong repolarization at slow heart rates and, at the same time, increased slow inactivation, delaying recovery of Na+ channel availability between stimuli and reducing the Na+ current under conditions mimicking fast heart rates (Veldkamp et al., 2000). Computer modeling showed that this dual mechanism could indeed explain the overlap syndrome, depending on the heart rate (Clancy and Rudy, 2002). Because of these discrepancies and because the co-inheritance of other genetic variants besides the 1795insD mutation as a cause of the phenotype complexity in the family could not be ruled out, Remme et al. (2006) generated a knock-in mouse carrying the mouse equivalent (1798insD) of the human SCN5A-1795insD mutation. Homozygous mice are not viable. Heterozygous Scn5a1798insD/+ mice recapitulate many of the pathological features of the patients including sinus node dysfunction, conduction slowing, and QT prolongation at slow rates. Patch-clamp experiments performed on ventricular Scn5a1798insD/+ cardiomyocytes have shown that action potential duration was longer than in wild-type cardiomyocytes and that this prolongation was more pronounced at slow pacing rates. Also, at fast pacing rates, action potential upstroke velocity, which reflects Na+ channel availability, was reduced in Scn5a1798insD/+ mice compared to wild-type. However, the potential biophysical mechanisms thought to explain decreased availability of Na+ channels in heterologous expression systems were not confirmed in Scn5a1798insD/+ mice. Indeed, rather than changes in the voltage-dependence of activation and inactivation or slow inactivation properties, dysfunctional mutant channels at the membrane or ineffective trafficking of mutant channels to the membrane more likely explained the marked decrease of Na+ current in Scn5a1798insD/+ mice. In addition, a small persistent inward Na+ current was found in Scn5a1798insD/+ cardiomyocytes, explaining the prolongation of action potential duration and QT interval. These studies constitute another example of the limitations of heterologous expression systems and illustrate the benefits of genetically engineered mouse models. In summary, the phenotypic characterization of this mouse model has demonstrated that a single SCN5A mutation is sufficient to cause an overlap syndrome of cardiac Na+ channel diseases.
This unique model also represented a useful tool to investigate the impact of genetic and environmental modifiers on cardiac conduction and repolarization. Indeed, also genetic background has often been proposed to affect the phenotypic consequences of ion channel mutations, the identification of genetic modifiers of disease severity in genetically inherited arrhythmias is rare (Scicluna et al., 2008). In order to identify genetic modifiers of conduction disease, Remme et al. (2009) studied the effect of the Scn5a-1798insD mutation in mice of two distinct inbred genetic backgrounds. They showed that the phenotype severity of mice carrying the Scn5a1798insD/+ mutation varies depending on the genetic background. This variability is associated with differential expression of a large number of genes including some encoding ion channels. In particular, ventricular expression levels of NaV1.5 regulatory β4-subunit (encoded by Scn4b gene) were markedly reduced in the mouse strain with the most severe conduction defects, leading to a shift of NaV1.5 channel activation curve toward more positive voltages and consequently to a further decrease of INa. Most recently, these authors went a step further and applied linkage analysis to a F2 cross resulting from the two strains carrying the Scn5a-1798insD mutation to identify quantitative trait loci (QTL) for heart rate, ECG parameters, and susceptibility to arrhythmias (Scicluna et al., 2011). This first genetic mapping analysis for cardiac electrical traits in mice identified 3 QTL linked with heart rate (chromosome 4), PR interval (chromosome 3), and QRS interval (chromosome 7) at baseline. Additional significant linkage to chromosome 4 locus was observed for post-flecainide heart rate and ventricular arrhythmias. Unexpectedly, numerous sex-specific QTLs were also identified. Finally, QTL for ECG parameters and arrhythmias coincided at five chromosomal locations suggesting pleiotropic effects at these loci. Interestingly, identified QTLs coincided with chromosomal locations of genes found differentially expressed between the two parental strains in the prior study (Remme et al., 2009). This considerably reduces the number of candidate genes responsible for these QTLs. Nevertheless, additional experiments are still needed to reduce linkage intervals and identify the causal genes. In spite of their success, genome-wide association studies (GWAS) performed in large human populations identified only a very small fraction of heritability of ECG traits and often failed to identify the causal genes. In this context, QTL analysis studies performed in mice could provide independent information pointing to the right genes (Dina, 2011).
A Model for SCN5A-Related Dilated Cardiomyopathy: The Scn5a-D1275N Mouse
The D1275N SCN5A mutation has been related to different cardiac diseases. It was first reported in a family affected by atrial standstill, mild conduction disease, and atrial enlargement. However, all affected members also carried a variant in the connexin 40 promoter (Groenewegen et al., 2003). Subsequently, D1275N mutation was reported in a family affected by DCM, sinus node dysfunction, atrial and ventricular tachyarrhythmias and conduction disorders (McNair et al., 2004; Olson et al., 2005; Ge et al., 2008). More recently, it was also reported in a family with atrial tachyarrhythmias, conduction disease, and ventricular enlargement, but without impaired contractility, as opposed to the previous family (Laitinen-Forsblom et al., 2006). Finally, this mutation was also identified in a patient with atrial flutter, atrial standstill, conduction disease, and sinus node dysfunction (Watanabe et al., 2011). Despite these phenotypic differences, patch-clamp studies that have compared wild-type and D1275N channels in heterologous expression systems have not shown major differences in the biophysical properties of the mutant channel (Groenewegen et al., 2003; Gui et al., 2010; Watanabe et al., 2011).
To address this discrepancy, Watanabe et al. (2011) used recombinase mediated cassette exchange to engineer a mouse model expressing the human D1275N mutant channel and compare it to the mouse model expressing the human wild-type channel (Watanabe et al., 2011). The D1275 allele was associated with slow heart rate and prolonged atrial, atrioventricular and ventricular conduction in a gene-dose-dependent manner, i.e., D1275N homozygous mice had a more severe phenotype. Interestingly, conduction disease increased with age. In addition, sinus node dysfunction, second degree atrioventricular block, atrial tachycardia/fibrillation, and polymorphic ventricular tachycardia occurred in homozygous D1275N mice. Patch-clamp studies of isolated cardiomyocytes demonstrated that D1275N mutation causes a severe decrease in peak INa by 54% in heterozygous mice and 78% in homozygous mice in relation to a decreased sarcolemmal expression of NaV1.5. Moreover, homozygous D1275N mice exhibited an increased late Na+ current, leading to prolonged action potential duration. Finally, the mutant was also associated with a gene-dose-dependent reduction of contractile function and DCM. These results are in line with the clinical findings described in the mutation carriers.
Scn1b Knockout Mouse
The cardiac Na+ channel is a heterotrimer composed of Nav1.5, the pore-forming α-subunit, and two auxiliary β-subunits (Meadows and Isom, 2005): a non-covalently linked β1-subunit (NaVβ1 or β3) and a disulfide-linked subunit (NaVβ2 or β4). In a genetic screening of 282 probands with Brugada syndrome and 44 with conduction disease, none of whom had mutations in SCN5A gene, three mutations in the gene encoding NaVβ1 (SCN1B) were identified in three kindred (Watanabe et al., 2008). Two mutations were located in a newly described alternately spliced transcript, NaVβ1B (Qin et al., 2003). Both transcripts are expressed in human heart and to a greater extent in Purkinje fibers than in ventricular myocardium (Watanabe et al., 2008). The E87Q mutation, located in the extracellular immunoglobulin loop of both β1- and β1B-subunits, was identified in a family affected by conduction disease. The two other mutations, which resulted in a β1B-subunit truncated after the amino-acid tryptophan-179, were identified in a family affected with both Brugada syndrome and conduction disease and in a kindred affected with conduction disease. Electrophysiological studies showed that peak INa amplitude was lower when Nav1.5 was co-expressed with mutant NaVβ1- and NaVβ1B-subunits versus wild-type subunits. This result was consistent with the patients’ phenotypes.
The cardiac electrophysiological phenotype of a mouse model invalidated for Scn1b markedly differed from the phenotype of the patients presented above (Lopez-Santiago et al., 2007). Compared to wild-type mice, Scn1b knockout mice exhibited lower heart rates and prolonged QT intervals, without any signs of conduction disease. Surprisingly, loss of NaVβ1 expression resulted in an increase in both peak and persistent Na+ current while channel gating and kinetics were unaffected. This increase in current was most likely due to the increased expression of Nav1.5 observed in these mice. Immunostaining studies revealed no alterations in the localization of Na+ channel α- or β-subunits. Action potentials were prolonged, supporting the increased QT interval. These results are consistent with the observation that the D1790G mutation of SCN5A, which affects the interaction of NaV1.5 with NaVβ1, also leads to a long QT phenotype (Benhorin et al., 1998; An et al., 1998). Once again, studies performed in heterologous expression systems led to contrasting results. Indeed, the D1790G mutation was first found to shift negatively steady-state inactivation of the Na+ current without increasing its late component (An et al., 1998). The action potential prolongation was attributed to alterations of calcium-sensitive exchange and ion channel currents (Wehrens et al., 2000). By contrast, a second study showed that the mutation-induced a marked prolongation of the late Na+ current, a result more consistent with the long QT phenotype (Baroudi and Chahine, 2000). Interestingly, the phenotype of Scn1b knockout mice resembles that found in a family of patients carrying a mutation on SCN4B, the gene encoding the β4-subunit. These patients exhibit a markedly prolonged QT interval (LQT10; Medeiros-Domingo et al., 2007) due to a large increase of the late Na+ current amplitude, as in LQT3 patients.
Scn3b Knockout Mouse
In order to understand better the role of NaVβ3 subunit on NaV1.5 function, Hakim and co-workers generated a mouse model lacking the Scn3b gene (Scn3b−/− mouse; Hakim et al., 2008). They showed that the lack of NaVβ3 induced a ≈30% decrease in cardiac Na+ peak current amplitude associated with a shift of the steady-state inactivation curve toward negative potentials while other INa biophysical parameters were not altered, thus confirming previous studies in mammalian heterologous expression system (Ko et al., 2005). As a consequence, Scn3b] mice exhibited decreased ventricular conduction, shortened ventricular action potential duration, and refractory period and increased incidence of arrhythmias under programmed electrical stimulation in ex vivo Langendorff-perfused hearts (Hakim et al., 2008). Arrhythmogenic incidence was reduced by flecainide and quinidine (Hakim et al., 2010b), in contrast to what had been observed previously for flecainide in Scn5a± mice (Martin et al., 2011c,d). Like Scn5a± mice, Scn3b−/− mice also exhibited sinus node dysfunction and decreased atrial and atrioventricular conduction, sometimes leading to atrioventricular block (Hakim et al., 2010a). Finally, Scn3b−/− mice were more prone than wild-type mice to develop atrial tachycardia and fibrillation upon burst pacing in ex vivo Langendorff-perfused hearts. Altogether, these studies suggested that Scn3b gene could also be involved in cardiac channelopathies in human.
This hypothesis has been confirmed. Indeed, SCN3B mutations have been identified in patients with either Brugada syndrome (Hu et al., 2009), idiopathic ventricular fibrillation (Valdivia et al., 2010) or lone atrial fibrillation (Olesen et al., 2011). All the mutations lead to a loss-of-function of NaVβ3 and consequently a decrease in cardiac Na+ current.
Conclusion
Most mouse models of SCN5A-related cardiac channelopathies recapitulate most of the diverse clinical phenotypes observed in patients. However mouse models also have their own limitation as a phenotype observed in mice may not be encountered in patients. For example, the Scn1b knockout mouse shows prolonged ventricular repolarization, a feature that is not seen in patients with SCN1B loss-of-function mutations. These discrepancies relate to either species differences or differences between a heterozygous point mutation and complete invalidation of a gene on one allele.
Mouse models also turned out to be powerful tools to elucidate the pathophysiological mechanisms of SCN5A-related diseases. For instance, the Scn5a1798insD/+ mouse clearly demonstrated that a single SCN5A mutation is sufficient to cause an overlap syndrome. This mouse also illustrated the limitations of heterologous expression systems in the elucidation of the biophysical consequences of the mutation on the channel function.
Finally, mouse models of NaV1.5 (or NaV1.5-associated proteins) dysfunction offer the unique opportunity to investigate the secondary cellular consequences of SCN5A mutations such as the expression remodeling of other genes, which is hardly accessible in the hearts of patients, but that might participate to the overall phenotype and explain some of the differences among patients. The most exiting benefit of mouse models is that they raise new working hypotheses such as the putative link between NaV1.5 loss-of-function and occurrence of fibrosis in the context of PCCD, thereby providing new potential therapeutic approaches. One limitation, though, is that remodeling might be partly species dependent. They also constitute useful tools for future studies addressing as yet unanswered questions, such as the role of genetic and environmental modifiers on cardiac conduction and repolarization.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the seventh framework program of the European Union (EUTrigTreat collaborative project; grant agreement #241526) for financial support.
References
An, R. H., Wang, X. L., Kerem, B., Benhorin, J., Medina, A., Goldmit, M., and Kass, R. S. (1998). Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta1-subunits. Circ. Res. 83, 141–146.
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D., Gussak, I., Le Marec, H., Nademanee, K., Perez Riera, A. R., Shimizu, W., Schulze-Bahr, E., Tan, H., and Wilde, A. (2005). Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670.
Baroudi, G., and Chahine, M. (2000). Biophysical phenotypes of SCN5A mutations causing long QT and Brugada syndromes. FEBS Lett. 487, 224–228.
Benhorin, J., Goldmit, M., MacCluer, J. W., Blangero, J., Goffen, R., Leibovitch, A., Rahat, A., Wang, Q., Medina, A., Towbin, J., and Kerem, B. (1998). Identification of a new SCN5A mutation associated with the long QT syndrome. Hum. Mutat. 12, 72–74.
Benito, B., Brugada, R., Perich, R. M., Lizotte, E., Cinca, J., Mont, L., Berruezo, A., Tolosana, J. M., Freixa, X., Brugada, P., and Brugada, J. (2008). A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 5, 1434–1440.
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., Rhodes, T. H., and George, A. L. Jr. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028.
Bezzina, C., Veldkamp, M. W., van den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J. W., van Langen, I. M., Tan-Sindhunata, G., Bink-Boelkens, M. T., van Der Hout, A. H., Mannens, M. M., and Wilde, A. A. (1999). A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213.
Blana, A., Kaese, S., Fortmüller, L., Laakmann, S., Damke, D., van Bragt, K., Eckstein, J., Piccini, I., Kirchhefer, U., Nattel, S., Breithardt, G., Carmeliet, P., Carmeliet, E., Schotten, U., Verheule, S., Kirchhof, P., and Fabritz, L. (2010). Knock-in gain-of-function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm 7, 1862–1869.
Charpentier, F., Demolombe, S., and Escande, D. (2004). Cardiac channelopathies: from men to mice. Ann. Med. 36(Suppl. 1), 28–34.
Clancy, C. E., and Rudy, Y. (2002). Na+ channel mutation that causes both Brugada and long-QT syndrome phenotypes: a simulation study of mechanism. Circulation 105, 1208–1213.
Coronel, R., Casini, S., Koopmann, T. T., Wilms-Schopman, F. J., Verkerk, A. O., de Groot, J. R., Bhuiyan, Z., Bezzina, C. R., Veldkamp, M. W., Linnenbank, A. C., van der Wal, A. C., Tan, H. L., Brugada, P., Wilde, A. A., and de Bakker, J. M. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777.
Cruz Filho, F. E., Maia, I. G., Fagundes, M. L., Barbosa, R. C., Alves, P. A., Sá, R. M., Boghossian, S. H., and Ribeiro, J. C. (2000). Electrical behavior of T-wave polarity alternans in patients with congenital long QT syndrome. J. Am. Coll. Cardiol. 36, 167–173.
Dautova, Y., Zhang, Y., Sabir, I., Grace, A. A., and Huang, C. L. (2009). Atrial arrhythmogenesis in wild-type and Scn5a+/delta murine hearts modelling LQT3 syndrome. Pflugers Arch. 458, 443–457.
Dumaine, R., Wang, Q., Keating, M. T., Hartmann, H. A., Schwartz, P. J., Brown, A. M., and Kirsch, E. (1996). Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ. Res. 78, 916–924.
El Yaman, M., Perry, J., Makielski, J. C., and Ackerman, M. J. (2008). Suppression of atrial fibrillation with mexiletine pharmacotherapy in a young woman with type 1 long QT syndrome. Heart Rhythm 5, 472–474.
Ellinor, P. T., Nam, E. G., Shea, M. A., Milan, D. J., Ruskin, J. N., and MacRae, C. A. (2008). Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 5, 99–105.
Fabritz, L., Damke, D., Emmerich, M., Kaufmann, S. G., Theis, K., Blana, A., Fortmüller, L., Laakmann, S., Hermann, S., Aleynichenko, E., Steinfurt, J., Volkery, D., Riemann, B., Kirchhefer, U., Franz, M. R., Breithardt, G., Carmeliet, E., Schäfers, M., Maier, S. K., Carmeliet, P., and Kirchhof, P. (2010). Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc. Res. 87, 60–72.
Fabritz, L., Kirchhof, P., Franz, M. R., Nuyens, D., Rossenbacker, T., Ottenhof, A., Haverkamp, W., Breithardt, G., Carmeliet, E., and Carmeliet, P. (2003). Effect of pacing and mexiletine on dispersion of repolarisation and arrhythmias in DeltaKPQ SCN5A (long QT3) mice. Cardiovasc. Res. 57, 1085–1093.
Fredj, S., Lindegger, N., Sampson, K. J., Carmeliet, P., and Kass, R. S. (2006). Altered Na+ channels promote pause-induced spontaneous diastolic activity in long QT syndrome type 3 myocytes. Circ. Res. 99, 1225–1232.
Frustaci, A., Priori, S. G., Pieroni, M., Chimenti, C., Napolitano, C., Rivolta, I., Sanna, T., Bellocci, F., and Russo, M. A. (2005). Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 112, 3680–3687.
Gussak, I., Antzelevitch, C., Bjerregaard, P., Towbin, J. A., and Chaitman, B. R. (1999). The Brugada syndrome: clinical, electrophysiologic and genetic aspects. J. Am. Coll. Cardiol. 33, 5–15.
Ge, J., Sun, A., Paajanen, V., Wang, S., Su, C., Yang, Z., Li, Y., Wang, S., Jia, J., Wang, K., Zou, Y., Gao, L., Wang, K., and Fan, Z. (2008). Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ. Arrhythm. Electrophysiol. 1, 83–92.
Gilmour, R. F. Jr. (2003). A novel approach to identifying antiarrhythmic drug targets. Drug Discov. Today 8, 162–167.
Grant, A. O., Carboni, M. P., Neplioueva, V., Starmer, C. F., Memmi, M., Napolitano, C., and Priori, S. (2002). Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110, 1201–1209.
Groenewegen, W. A., Firouzi, M., Bezzina, C. R., Vliex, S., van Langen, I. M., Sandkuijl, L., Smits, J. P., Hulsbeek, M., Rook, M. B., Jongsma, H. J., and Wilde, A. A. (2003). A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 92, 14–22.
Gui, J., Wang, T., Jones, R. P., Trump, D., Zimmer, T., and Lei, M. (2010). Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS ONE 5, e10985. doi:10.1371/journal.pone.0010985
Guzadhur, L., Pearcey, S. M., Duehmke, R. M., Jeevaratnam, K., Hohmann, A. F., Zhang, Y., Grace, A. A., Lei, M., and Huang, C. L. (2010). Atrial arrhythmogenicity in aged Scn5a+/DeltaKPQ mice modeling long QT type 3 syndrome and its relationship to Na+ channel expression and cardiac conduction. Pflugers Arch. 460, 593–601.
Hakim, P., Brice, N., Thresher, R., Lawrence, J., Zhang, Y., Jackson, A. P., Grace, A. A., and Huang, C. L. (2010a). Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta Physiol. (Oxf.) 198, 47–59.
Hakim, P., Thresher, R., Grace, A. A., and Huang, C. L. (2010b). Effects of flecainide and quinidine on action potential and ventricular arrhythmogenic properties in Scn3b knockout mice. Clin. Exp. Pharmacol. Physiol. 37, 782–789.
Hakim, P., Gurung, I. S., Pedersen, T. H., Thresher, R., Brice, N., Lawrence, J., Grace, A. A., and Huang, C. L. (2008). Scn3b knockout mice exhibit abnormal ventricular electrophysiological properties. Prog. Biophys. Mol. Biol. 98, 251–266.
Hale, S. L., Shryock, J. C., Belardinelli, L., Sweeney, M., and Kloner, R. A. (2008). Late sodium current inhibition as a new cardioprotective approach. J. Mol. Cell. Cardiol. 44, 954–967.
Hao, X., Zhang, Y., Zhang, X., Nirmalan, M., Davies, L., Konstantinou, D., Yin, F., Dobrzynski, H., Wang, X., Grace, A. A., Zhang, H., Boyett, M., Huang, C. L., and Lei, M. (2011). TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythm. Electrophysiol. 4, 397–406.
Head, C. E., Balasubramaniam, R., Thomas, G., Goddard, C. A., Lei, M., Colledge, W. H., Grace, A. A., and Huang, C. L. (2005). Paced electrogram fractionation analysis of arrhythmogenic tendency in DeltaKPQ Scn5a mice. J. Cardiovasc. Electrophysiol. 16, 1329–1340.
Hothi, S. S., Booth, S. W., Sabir, I. N., Killeen, M. J., Simpson, F., Zhang, Y., Grace, A. A., and Huang, C. L. (2008). Arrhythmogenic substrate and its modification by nicorandil in a murine model of long QT type 3 syndrome. Prog. Biophys. Mol. Biol. 98, 267–280.
Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A., Pfeiffer, R., Koopmann, T. T., Cordeiro, J. M., Guerchicoff, A., Pollevick, G. D., and Antzelevitch, C. (2009). A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270–278.
Johnson, J. N., Tester, D. J., Perry, J., Salisbury, B. A., Reed, C. R., and Ackerman, M. J. (2008). Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 5, 704–709.
Ko, S. H., Lenkowski, P. W., Lee, H. C., Mounsey, J. P., and Patel, M. K. (2005). Modulation of Na(v)1.5 by beta1 – and beta3-subunit co-expression in mammalian cells. Pflugers Arch. 449, 403–412.
Kruse, M., Schulze-Bahr, E., Corfield, V., Beckmann, A., Stallmeyer, B., Kurtbay, G., Ohmert, I., Schulze-Bahr, E., Brink, P., and Pongs, O. (2009). Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Invest. 119, 2737–2744.
Kyndt, F., Probst, V., Potet, F., Demolombe, S., Chevallier, J. C., Baró, I., Moisan, J. P., Boisseau, P., Schott, J. J., Escande, D., and Le Marec, H. (2001). Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 104, 3081–3086.
Laitinen-Forsblom, P. J., Mäkynen, P., Mäkynen, H., Yli-Mäyry, S., Virtanen, V., Kontula, K., and Aalto-Setälä, K. (2006). SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J. Cardiovasc. Electrophysiol. 17, 480–485.
Lei, M., Goddard, C., Liu, J., Leoni, AL., Royer, A., Fung, S. S., Xiao, G., Ma, A., Zhang, H., Charpentier, F., Vandenberg, J. I., Colledge, W. H., Grace, A. A., and Huang, C. L. (2005). Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. J. Physiol. (Lond.) 567, 387–400.
Lenègre, J., and Moreau, P. (1963). Le bloc auriculo-ventriculaire chronique. Etude anatomique, clinique et histologique. Arch. Mal. Coeur. Vaiss. 56, 867–888.
Leoni, A. L., Gavillet, B., Rougier, J. S., Marionneau, C., Probst, V., Le Scouarnec, S., Schott, J. J., Demolombe, S., Bruneval, P., Huang, C. L., Colledge, W. H., Grace, A. A., Le Marec, H., Wilde, A. A., Mohler, P. J., Escande, D., Abriel, H., and Charpentier, F. (2010). Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(±) mouse model. PLoS ONE 5, e9298. doi:10.1371/journal.pone.0009298
Lindegger, N., Hagen, B. M., Marks, A. R., Lederer, W. J., and Kass, R. S. (2009). Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J. Mol. Cell. Cardiol. 47, 326–334.
Liu, H., El Zein, L., Kruse, M., Guinamard, R., Beckmann, A., Bozio, A., Kurtbay, G., Mégarbané, A., Ohmert, I., Blaysat, G., Villain, E., Pongs, O., and Bouvagnet, P. (2010). Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 3, 374–385.
Lopez-Santiago, L. F., Meadows, L. S., Ernst, S. J., Chen, C., Malhotra, J. D., McEwen, D. P., Speelman, A., Noebels, J. L., Maier, S. K., Lopatin, A. N., and Isom, L. L. (2007). Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J. Mol. Cell. Cardiol. 43, 636–647.
Martin, C. A., Grace, A. A., and Huang, C. L. (2011a). Refractory dispersion promotes conduction disturbance and arrhythmias in a Scn5a (±) mouse model. Pflugers Arch. 462, 495–504.
Martin, C. A., Grace, A. A., and Huang, C. L. (2011b). Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. Am. J. Physiol. Heart Circ. Physiol. 300, H605–H616.
Martin, C. A., Zhang, Y., Grace, A. A., and Huang, C. L. (2011c). In vivo studies of Scn5a± mice modeling Brugada syndrome demonstrate both conduction and repolarization abnormalities. J. Electrocardiol. 43, 433–439.
Martin, C. A., Guzadhur, L., Grace, A. A., Lei, M., and Huang, C. L. (2011d). Mapping of re-entrant spontaneous polymorphic ventricular tachycardia in a Scn5a± mouse model. Am. J. Physiol. Heart Circ. Physiol. 300, H1853–H1862.
Martin, C. A., Zhang, Y., Grace, A. A., and Huang, C. L. (2010). Increased right ventricular repolarization gradients promote arrhythmogenesis in a murine model of Brugada syndrome. J. Cardiovasc. Electrophysiol. 21, 1153–1159.
McNair, W. P., Ku, L., Taylor, M. R., Fain, P. R., Dao, D., Wolfel, E., Mestroni, L., and Familial Cardiomyopathy Registry Research Group. (2004). SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 110, 2163–2167.
Meadows, L. S., and Isom, L. L. (2005). Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc. Res. 67, 448–458.
Medeiros-Domingo, A., Kaku, T., Tester, D. J., Iturralde-Torres, P., Itty, A., Ye, B., Valdivia, C., Ueda, K., Canizales-Quinteros, S., Tusié-Luna, M. T., Makielski, J. C., and Ackerman, M. J. (2007). SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134–142.
Moss, A. J. (2002). T-wave patterns associated with the hereditary long QT syndrome. Card. Electrophysiol. Rev. 6, 311–315.
Moss, A. J., and Kass, R. S. (2005). Long QT syndrome: from channels to cardiac arrhythmias. J. Clin. Invest. 115, 2018–2024.
Moss, A. J., Zareba, W., Benhorin, J., Locati, E. H., Hall, W. J., Robinson, J. L., Schwartz, P. J., Towbin, J. A., Vincent, G. M., Lehmann, M. H., Keating, M. T., MacCluer, J. W., and Timothy, K. W. (1995). ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 92, 2929–2934.
Nerbonne, J. M., Nichols, C. G., Schwarz, T. L., and Escande, D. (2001). Genetic manipulation of cardiac K+ channel function in mice: what have we learned, and where do we go from here? Circ. Res. 89, 944–956.
Nguyen, T. P., Wang, D. W., Rhodes, T. H., and George, A. L. Jr. (2008). Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ. Res. 102, 364–371.
Nuyens, D., Stengl, M., Dugarmaa, S., Rossenbacker, T., Compernolle, V., Rudy, Y., Smits, J. F., Flameng, W., Clancy, C. E., Moons, L., Vos, M. A., Dewerchin, M., Benndorf, K., Collen, D., Carmeliet, E., and Carmeliet, P. (2001). Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat. Med. 7, 1021–1027.
Olesen, M. S., Jespersen, T., Nielsen, J. B., Liang, B., Møller, D. V., Hedley, P., Christiansen, M., Varró, A., Olesen, S. P., Haunsø, S., Schmitt, N., and Svendsen, J. H. (2011). Mutations in sodium channel β-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 89, 786–793.
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., Horton, S. C., Rodeheffer, R. J., and Anderson, J. L. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454.
Papadatos, G. A., Wallerstein, P. M., Head, C. E., Ratcliff, R., Brady, P. A., Benndorf, K., Saumarez, R. C., Trezise, A. E., Huang, C. L., Vandenberg, J. I., Colledge, W. H., and Grace, A. A. (2002). Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. U.S.A. 99, 6210–6215.
Priori, S. G., Schwartz, P. J., Napolitano, C., Bloise, R., Ronchetti, E., Grillo, M., Vicentini, A., Spazzolini, C., Nastoli, J., Bottelli, G., Folli, R., and Cappelletti, D. (2003). Risk stratification in the long-QT syndrome. N. Engl. J. Med. 348, 1866–1874.
Probst, V., Kyndt, F., Potet, F., Trochu, J. N., Mialet, G., Demolombe, S., Schott, J. J., Baró, I., Escande, D., and Le Marec, H. (2003). Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenègre disease. J. Am. Coll. Cardiol. 41, 643–652.
Qin, N., D’Andrea, M. R., Lubin, M. L., Shafaee, N., Codd, E. E., and Correa, A. M. (2003). Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur. J. Biochem. 270, 4762–4770.
Remme, C. A., Scicluna, B. P., Verkerk, A. O., Amin, A. S., van Brunschot, S., Beekman, L., Deneer, V. H., Chevalier, C., Oyama, F., Miyazaki, H., Nukina, N., Wilders, R., Escande, D., Houlgatte, R., Wilde, A. A., Tan, H. L., Veldkamp, M. W., de Bakker, J. M., and Bezzina, C. R. (2009). Genetically determined differences in sodium current characteristics modulate conduction disease severity in mice with cardiac sodium channelopathy. Circ. Res. 104, 1283–1292.
Remme, C. A., Verkerk, A. O., Nuyens, D., van Ginneken, A. C., van Brunschot, S., Belterman, C. N., Wilders, R., van Roon, M. A., Tan, H. L., Wilde, A. A., Carmeliet, P., de Bakker, J. M., Veldkamp, M. W., and Bezzina, C. R. (2006). Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114, 2584–2594.
Remme, C. A., Wilde, A. A., and Bezzina, C. R. (2008). Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc. Med. 18, 78–87.
Rossenbacker, T., Carroll, S. J., Liu, H., Kuipéri, C., de Ravel, T. J. L., Devriendt, K., Carmeliet, P., Kass, R. S., and Heidbüchel, H. (2004). Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Heart Rhythm 1, 610–615.
Royer, A., van Veen, T. A., Le Bouter, S., Marionneau, C., Griol-Charhbili, V., Leoni, A. L., Steenman, M., van Rijen, H. V., Demolombe, S., Goddard, C. A., Richer, C., Escoubet, B., Jarry-Guichard, T., Colledge, W. H., Gros, D., de Bakker, J. M., Grace, A. A., Escande, D., and Charpentier, F. (2005). Mouse model of SCN5A-linked hereditary Lenegre’s disease: age-related conduction slowing and myocardial fibrosis. Circulation 111, 1738–1746.
Sabir, I. N., Li, L. M., Jones, V. J., Goddard, C. A., Grace, A. A., and Huang, C. L. (2008). Criteria for arrhythmogenicity in genetically-modified Langendorff-perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflugers Arch. 455, 637–651.
Schott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M., Wilde, A. A., Escande, D., Mannens, M. M., and Le Marec, H. (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21.
Schwartz, P. J., Priori, S. G., Locati, E. H., Napolitano, C., Cantu, F., Towbin, J. A., Keating, M. T., Hammoude, H., Brown, A. M., Chen, L. K., and Colatsky, T. J. (1995). Long-QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate: implications for gene-specific therapy. Circulation 92, 3381–3386.
Schwartz, P. J., Priori, S. G., Spazzolini, C., Moss, A. J., Vincent, G. M., Napolitano, C., Denjoy, I., Guicheney, P., Breithardt, G., Keating, M. T., Towbin, J. A., Beggs, A. H., Brink, P., Wilde, A. A., Toivonen, L., Zareba, W., Robinson, J. L., Timothy, K. W., Corfield, V., Wattanasirichaigoon, D., Corbett, C., Haverkamp, W., Schulze-Bahr, E., Lehmann, M. H., Schwartz, K., Coumel, P., and Bloise, R. (2001). Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95.
Scicluna, B. P., Tanck, M. W., Remme, C. A., Beekman, L., Coronel, R., Wilde, A. A., and Bezzina, C. R. (2011). Quantitative trait loci for electrocardiographic parameters and arrhythmia in the mouse. J. Mol. Cell. Cardiol. 50, 380–389.
Scicluna, B. P., Wilde, A. A., and Bezzina, C. R. (2008). The primary arrhythmia syndromes: same mutation, different manifestations. Are we starting to understand why? J. Cardiovasc. Electrophysiol. 19, 445–452.
Shi, R., Zhang, Y., Yang, C., Huang, C., Zhou, X., Qiang, H., Grace, A. A., Huang, C. L., and Ma, A. (2008). The cardiac sodium channel mutation delQKP 1507-1509 is associated with the expanding phenotypic spectrum of LQT3, conduction disorder, dilated cardiomyopathy, and high incidence of youth sudden death. Europace 10, 1329–1335.
Shimizu, W., and Antzelevitch, C. (2000). Effects of a K(+) channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation 102, 706–712.
Smits, J. P., Eckardt, L., Probst, V., Bezzina, C. R., Schott, J. J., Remme, C. A., Haverkamp, W., Breithardt, G., Escande, D., Schulze-Bahr, E., LeMarec, H., and Wilde, A. A. (2002). Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J. Am. Coll. Cardiol. 40, 350–356.
Smits, J. P., Koopmann, T. T., Wilders, R., Veldkamp, M. W., Opthof, T., Bhuiyan, Z. A., Mannens, M. M., Balser, J. R., Tan, H. L., Bezzina, C. R., and Wilde, A. A. (2005). A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell. Cardiol. 38, 969–981.
Stokoe, K. S., Balasubramaniam, R., Goddard, C. A., Colledge, W. H., Grace, A. A., and Huang, C. L. (2007a). Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a± murine hearts modelling the Brugada syndrome. J. Physiol. (Lond.) 581, 255–275.
Stokoe, K. S., Thomas, G., Goddard, C. A., Colledge, W. H., Grace, A. A., and Huang, C. L. (2007b). Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Delta murine hearts modelling long QT syndrome 3. J. Physiol. (Lond.) 578, 69–84.
Tan, H. L., Bezzina, C. R., Smits, J. P., Verkerk, A. O., and Wilde, A. A. (2003). Genetic control of sodium channel function. Cardiovasc. Res. 57, 961–973.
Tan, H. L., Bink-Boelkens, M. T., Bezzina, C. R., Viswanathan, P. C., Beaufort-Krol, G. C., van Tintelen, P. J., van den Berg, M. P., Wilde, A. A., and Balser, J. R. (2001). A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047.
Thomas, G., Gurung, I. S., Killeen, M. J., Hakim, P., Goddard, C. A., Mahaut-Smith, M. P., Colledge, W. H., Grace, A. A., and Huang, C. L. (2007). Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J. Physiol. (Lond.) 578, 85–97.
Tian, X. L., Yong, S. L., Wan, X., Wu, L., Chung, M. K., Tchou, P. J., Rosenbaum, D. S., Van Wagoner, D. R., Kirsch, G. E., and Wang, Q. (2004). Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc. Res. 61, 256–267.
Valdivia, C. R., Medeiros-Domingo, A., Ye, B., Shen, W. K., Algiers, T. J., Ackerman, M. J., and Makielski, J. C. (2010). Loss-of-function mutation of the SCN3B-encoded sodium channel {beta}3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc. Res. 86, 392–400.
van den Berg, M. P., Wilde, A. A. M., Viersma, J. W., Brouwer, J., Haaksma, J., van der Hout, A. H., Stolte-Dijkstra, I., Bezzina, C. R., van Langen, I. M., Beaufort-Krol, G. C. M., Cornel, J. H., and Crijns, H. J. G. M. (2001). Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J. Cardiovasc. Electrophysiol. 12, 630–636.
van Veen, T. A., Stein, M., Royer, A., Le Quang, K., Charpentier, F., Colledge, W. H., Huang, C. L., Wilders, R., Grace, A. A., Escande, D., de Bakker, J. M., and van Rijen, H. V. (2005). Impaired impulse propagation in Scn5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation 112, 1927–1235.
Veldkamp, M. W., Viswanathan, P. C., Bezzina, C., Baartscheer, A., Wilde, A. A., and Balser, J. R. (2000). Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ. Res. 86, E91–E97.
Veldkamp, M. W., Wilders, R., Baartscheer, A., Zegers, J. G., Bezzina, C. R., and Wilde, A. A. (2003). Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circ. Res. 92, 976–983.
Viskin, S., Rosso, R., Rogowski, O., Belhassen, B., Levitas, A., Wagshal, A., Katz, A., Fourey, D., Zeltser, D., Oliva, A., Pollevick, G. D., Antzelevitch, C., and Rozovski, U. (2004). Provocation of sudden heart rate oscillation with adenosine exposes abnormal QT responses in patients with long QT syndrome: a bedside test for diagnosing long QT syndrome. Eur. Heart J. 27, 469–475.
Wang, D. W., Yazawa, K., George, A. L. Jr., and Bennett, P. B. (1996). Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 93, 13200–13205.
Wang, Q., Shen, J., Li, Z., Timothy, K., Vincent, G. M., Priori, S. G., Schwartz, P. J., and Keating, M. T. (1995). Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum. Mol. Genet. 4, 1603–1607.
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J. J., Demolombe, S., Probst, V., Anselme, F., Escande, D., Wiesfeld, A. C., Pfeufer, A., Kääb, S., Wichmann, H. E., Hasdemir, C., Aizawa, Y., Wilde, A. A., Roden, D. M., and Bezzina, C. R. (2008). Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268.
Watanabe, H., Yang, T., Stroud, D. M., Lowe, J. S., Harris, L., Atack, T. C., Wang, D. W., Hipkens, S. B., Leake, B., Hall, L., Kupershmidt, S., Chopra, N., Magnuson, M. A., Tanabe, N., Knollmann, B. C., George, A. L. Jr., and Roden, D. M. (2011). Striking In vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 124, 1001–1011.
Wehrens, X. H., Abriel, H., Cabo, C., Benhorin, J., and Kass, R. S. (2000). Arrhythmogenic mechanism of an LQT-3 mutation of the human heart Na(+) channel alpha-subunit: a computational analysis. Circulation 102, 584–590.
Wei, J., Wang, D. W., Alings, M., Fish, F., Wathen, M., Roden, D. M., and George, A. L. (1999). Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation 99, 3165–3171.
Wilde, A. A., Postema, P. G., Di Diego, J. M., Viskin, S., Morita, H., Fish, J. M., and Antzelevitch, C. (2010). The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J. Mol. Cell. Cardiol. 49, 543–553.
Wu, J., Zhang, Y., Zhang, X., Cheng, L., Lammers, W. J., Grace, A. A., Fraser, J. A., Zhang, H., Huang, C. L., and Lei, M. (2012). Altered sino atrial node function and intra-atrial conduction in murine gain-of-function Scn5a+/{Delta}KPQ hearts suggest an overlap syndrome. Am. J. Physiol. Heart Circ. Physiol. 302, H1510–H1523.
Yong, S. L., Ni, Y., Zhang, T., Tester, D. J., Ackerman, M. J., and Wang, Q. K. (2007). Characterization of the cardiac sodium channel SCN5A mutation, N1325S, in single murine ventricular myocytes. Biochem. Biophys. Res. Commun. 352, 378–383.
Zellerhoff, S., Pistulli, R., Mönnig, G., Hinterseer, M., Beckmann, B. M., Köbe, J., Steinbeck, G., Kääb, S., Haverkamp, W., Fabritz, L., Gradaus, R., Breithardt, G., Schulze-Bahr, E., Böcker, D., and Kirchhof, P. (2009). Atrial Arrhythmias in long-QT syndrome under daily life conditions: a nested case control study. J. Cardiovasc. Electrophysiol. 20, 401–407.
Zhang, T., Yong, S. L., Drinko, J. K., Popovic, Z. B., Shryock, J. C., Belardinelli, L., and Wang, Q. K. (2011). LQTS mutation N1325S in cardiac sodium channel gene SCN5A causes cardiomyocyte apoptosis, cardiac fibrosis and contractile dysfunction in mice. Int. J. Cardiol. 147, 239–245.
Zhang, T., Yong, S. L., Tian, X. L., and Wang, Q. K. (2007). Cardiac-specific overexpression of SCN5A gene leads to shorter P wave duration and PR interval in transgenic mice. Biochem. Biophys. Res. Commun. 355, 444–450.
Keywords: NaV1.5, arrhythmia, conduction disease, long QT syndrome, Brugada syndrome, sinus node dysfunction
Citation: Derangeon M Montnach J Baró I and Charpentier F (2012) Mouse models of SCN5A-related cardiac arrhythmias. Front. Physio. 3:210. doi: 10.3389/fphys.2012.00210
Received: 12 March 2012; Paper pending published: 05 April 2012;
Accepted: 29 May 2012; Published online: 22 June 2012.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsReviewed by:
Toon Van Veen, University Medical Center Utrecht, NetherlandsMing Lei, University of Manchester, UK
Copyright: © 2012 Derangeon, Montnach, Baró and Charpentier. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Flavien Charpentier, INSERM, UMR1087, CNRS UMR6291, l’Institut du Thorax, IRT-UN, 8 quai Moncousu, BP 70721, 44007 Nantes Cedex 1, France. e-mail: flavien.charpentier@inserm.fr