
- 1Inserm U769, IFR141, Labex Lermit, University of Paris-Sud 11, Châtenay-Malabry, France
- 2Unidad de Hipertensión, Hospital Universitario 12 de Octubre, Instituto de Investigación Sanitaria i+12, and Instituto Pluridisciplinar, Universidad Complutense, Madrid, Spain
Ca2+ is probably the most versatile signal transduction element used by all cell types. In the heart, it is essential to activate cellular contraction in each heartbeat. Nevertheless Ca2+ is not only a key element in excitation-contraction coupling (EC coupling), but it is also a pivotal second messenger in cardiac signal transduction, being able to control processes such as excitability, metabolism, and transcriptional regulation. Regarding the latter, Ca2+ activates Ca2+-dependent transcription factors by a process called excitation-transcription coupling (ET coupling). ET coupling is an integrated process by which the common signaling pathways that regulate EC coupling activate transcription factors. Although ET coupling has been extensively studied in neurons and other cell types, less is known in cardiac muscle. Some hints have been found in studies on the development of cardiac hypertrophy, where two Ca2+-dependent enzymes are key actors: Ca2+/Calmodulin kinase II (CaMKII) and phosphatase calcineurin, both of which are activated by the complex Ca2+/Calmodulin. The question now is how ET coupling occurs in cardiomyocytes, where intracellular Ca2+ is continuously oscillating. In this focused review, we will draw attention to location of Ca2+ signaling: intranuclear ([Ca2+]n) or cytoplasmic ([Ca2+]c), and the specific ionic channels involved in the activation of cardiac ET coupling. Specifically, we will highlight the role of the 1,4,5 inositol triphosphate receptors (IP3Rs) in the elevation of [Ca2+]n levels, which are important to locally activate CaMKII, and the role of transient receptor potential channels canonical (TRPCs) in [Ca2+]c, needed to activate calcineurin (Cn).
Ca2+ has evolved as the most versatile signal transduction pathway used by all cells (Berridge et al., 2000), but perhaps no other cell type uses Ca2+ in such different ways as cardiac myocytes do, in normal physiology and as a major contributor to heart disease. First evidenced by Ringer as the signal carrier initiating contraction (Ringer, 1883), Ca2+ is known to control other key cardiac cell processes (Berridge et al., 1998) including initiation of pacemaker activity, action potential (AP) shape, regulation of cell–cell communication, arrhythmogenesis, metabolism, and transcriptional regulation. All these processes use Ca2+ as a nexus, which auto controls its own cellular fluxes, as illustrated by the Ca2+-induced Ca2+ release mechanism (Fabiato, 1983) underlying excitation-contraction (EC) coupling, as well as the Ca2+-induced Ca2+-entry (Richard et al., 2006) participating in excitation-transcription (ET) coupling. ET coupling is the process by which signaling molecules that regulate EC-coupling activate Ca2+-dependent transcription factors (Anderson, 2000). In the adult heart, neurohormonal/mechanical stress enhances ET coupling, resulting in cell growth (hypertrophy), reexpression of the fetal gene program, and alteration of ionic channels and transporter expression (Chevalier et al., 1989; Marbán and Koretsune, 1990; Chien et al., 1991; Moalic et al., 1993; Gidh-Jain et al., 1995; Nass et al., 2008). The transcription factors involved in cardiac hypertrophy have been reviewed by Heineke and Molkentin (Heineke and Molkentin, 2006). Among them, myocyte enhancer factor 2 (MEF2) and GATA4 are initiated by a cascade activated by Ca2+/Calmodulin (CaM): CaM Kinase II (CaMKII) for MEF2 (Passier et al., 2000) and calcineurin (Cn) for GATA4 (Molkentin et al., 1998; Houser and Molkentin, 2008).
Thus, Ca2+ activates contraction in the heart in a beat-to-beat fashion, while it is also able to activate hypertrophy by ET coupling at a longer time scale (Maier and Bers, 2002). The mechanisms by which the heart differentiates between Ca2+ signals are only beginning to be elucidated. In this review, we will focus on the implication of local pools of Ca2+ in activating gene transcription in adult ventricular cardiomyocytes, as during hypertrophy development.
Ca2+ Signaling in Ventricular Myocytes
Ca2+ is a key element in cardiac EC coupling. In each heartbeat, membrane depolarization during an AP activates L-type Ca2+ channels (LTCCs) located in the sarcolemma. Ca2+ entry activates intracellular Ca2+ release channels, named ryanodine receptors (RyRs), located in the membrane of the sarcoplasmic reticulum (SR). RyRs amplify the initial Ca2+ signal, providing enough Ca2+ to activate contractile myofibrils. Relaxation occurs when cytosolic Ca2+ concentration ([Ca2+]c) returns to diastolic values, due mainly to Ca2+ pumped back into the SR by the Ca2+-ATPase (SERCA) and extrusion from the cell via the Na+/Ca2+ exchanger (Bers, 2002). New roles for intracellular Ca2+ ([Ca2+]i) are being elucidated (Bers, 2008). For instance, prohypertrophic transcription factors are activated by nuclear/perinuclear activation of CaMKII promoted by local elevation of nuclear [Ca2+] ([Ca2+]n) (Wu et al., 2006): CaMKII phosphorylates histone deacetylases (HDAC) 4 and 5, resulting in their translocation out of the nucleus, derepriming the transcription factor MEF2. Cytoplasmic Ca2+ elevations are also involved in ET coupling by activating Cn, which dephosphorylates the nuclear factor of activated T cells (NFAT), which is imported into the nucleus where it activates the transcription factor GATA (Molkentin et al., 1998).
However, it is still not fully understood whether [Ca2+]n variations can be dissociated from bulk [Ca2+]i oscillations during contraction-relaxation cycles. The proposed mechanisms are the location and the specificity of the channels. Thus, rapid elevations in cytoplasmic Ca2+ activate contraction, while [Ca2+]n activates Ca2+-dependent transcription factors. Regarding the channels and oversimplifying the situation: if Ca2+ comes from SR, the channel involved in contractile activity is the RyR, while the one involved in transcription is the inositol 1,4,5 triphosphate receptor (IP3R). The location, RyR in the SR and IP3R in the nuclear envelope (NE) and perinuclear area, preferentially affects cytosolic and [Ca2+]n, respectively. When Ca2+ enters through the sarcolemma, the specific channel involved may also help to differentiate contractile vs. transcriptional Ca2+. LTCCs are mainly involved in contraction, while other less known Ca2+ permeating channels in the cardiomyocyte, such as TRPCs, play an important role in hypertrophy development (Wu et al., 2010). However, LTCCs may also be involved in transcription activation. It has been shown that the C-terminal part of LTCCs may travel from the membrane to the nucleus, activating transcription. The T-type Ca2+ channels have been shown to be involved in cell growth. However, in the adult myocyte this channel is not or is only very weakly expressed. At late stages of Ca2+ hypertrophy, the T-type Ca2+ channels are reexpressed (Nuss and Houser, 1993; Martinez et al., 1999), but their implication in the initiation of hypertrophy has not been demonstrated.
Below we summarize some of the known aspects of transcription induction by [Ca2+]n, focusing on the role of IP3R, and by [Ca2+]c, focusing on the role of TRPCs. The involvement of two Ca2+-dependent enzymes, Cn and CaMKII, has been established. Their involvement in cardiac hypertrophy-ET coupling is reviewed in Bers (2008) and Molkentin (2000), among others.
Nuclear Ca2+ in ET Coupling
The question of how ET coupling can co-exist in cardiac myocytes in which [Ca2+]c continuously oscillates within each heartbeat remains a matter of debate. Localization of the Ca2+ signal restricted to microdomains may be the answer. It has thus been postulated that intranuclear/perinuclear Ca2+ is involved in ET coupling, whereas [Ca2+]c is responsible for EC coupling. While there is no doubt on the second, whether or not [Ca2+]n signaling is independently regulated from cytosolic Ca2+ is not that clear. In fact, the NE [which also acts as a Ca2+ reservoir, continuously to the SR (Wu and Bers, 2006)] has pores permeable to Ca2+ (Bootman et al., 2009). Thus, [Ca2+]c can passively diffuse into the nucleus, challenging the possibility of an independence of [Ca2+]n from cytosolic [Ca2+]c. This important question is still not answered. However, the hypothesis of separately controlled domains is supported by the following: (1) the location of Ca2+ release channels is different in SR and NE; (2) some molecules preferentially affect [Ca2+]n; and (3) [Ca2+]n signal decay is slower, due mainly to the lack of SERCA in the inner membrane of the NE (Bootman et al., 2009), and thus under conditions of fast pacing Ca2+ can be accumulated in the nucleoplasm initiating the hypertrophic signaling.
1. The location of RyRs on the junctional SR, facing LTCCs (located on the T-tubules), is crucial for EC coupling in ventricular myocytes. Other Ca2+ release channels expressed in cardiac myocytes are the IP3Rs, which are concentrated on the NE/perinuclear area (Escobar et al., 2011). After activation of Gq-coupled protein receptors, phospholipase C (PLC) is activated, producing IP3. Activation of IP3Rs provide Ca2+ to the intranuclear or perinuclear region where activate local CaMKII, which phosphorylates class II HDAC, prompting their translocation out of the nucleus and derepressing the prohypertrophic transcription factor MEF2 (McKinsey et al., 2000; Zhang et al., 2002). IP3Rs are also expressed at the junctional SR of hypertrophied hearts, where they may play a role in EC coupling (Harzheim et al., 2009) under this pathological condition. Furthermore, RyRs may also be expressed in the NE (Bootman et al., 2009), although its role there is not known.
2. Some prohypertrophic molecules have shown an action elevating [Ca2+]n more than [Ca2+]c. For instance, endothelin, which activates Gq and PLC producing IP3, increases [Ca2+]n in both atrial (Kockskamper et al., 2008a,b) and ventricular myocytes (Wu et al., 2006) independently of [Ca2+]c. Recently, we analyzed the effects on [Ca2+]n of Epac (De Rooij et al., 1998), a protein with prohypertrophic actions in cardiac myocytes (Morel et al., 2005; Metrich et al., 2008). This protein is directly activated by cAMP and contributes to β-adrenergic-induced cardiac hypertrophy (Metrich et al., 2008). Epac induces IP3 production (Metrich et al., 2010; Pereira et al., 2012) and a significant increase in [Ca2+]n, correlating with the perinuclear expression pattern of Epac (Pereira et al., 2012). Moreover, sustained Epac activation (from 30 min) drives the HDAC5 nuclear export in a manner that is CaMKII- and IP3Rs-dependent, with the consequent activation of MEF2 (Metrich et al., 2010; Pereira et al., 2012).
3. Oscillating Ca2+ may also be an important contributor to the activation of gene transcription. Increasing the frequency of [Ca2+]i transients (as in tachycardia) induces cardiac hypertrophy and heart failure (HF). It is not known whether the cell is stimulated by an increase in the time-average [Ca2+]i or if, because [Ca2+]n dynamics are slower than cytoplasmic ones, there is an accumulation of Ca2+ in the nucleoplasm at higher frequencies.
Cytoplasmic Ca2+ in ET Coupling
Although nuclear localization is involved in ET coupling, mathematical models have predicted that separate compartments may not be necessary in vitro (Cooling et al., 2009). Without disregarding the relevance of [Ca2+]n in ET coupling, [Ca2+]c may also play a role. In fact, Ca2+/CaM activates Cn, found in the cytosol, which is involved in hypertrophy (Molkentin et al., 1998). When activated, Cn dephosphorylates NFAT in the cytoplasm, permitting its translocation to the nucleus where it participates in the hypertrophic gene expression (Heineke and Molkentin, 2006). Moreover, the plasma membrane Ca2+ ATPase antagonizes Ca2+ hypertrophy, suggesting that extruding Ca2+ from the cytosol, probably close to Cn, prevents its activation (Wu et al., 2009).
The Ca2+ entry pathways which may activate Cn are being elucidated. LTCCs located in lipid rafts could form a Ca2+ signaling microdomain (Houser and Molkentin, 2008). But other Ca2+-permeable channels may be located on these microdomains to activate Cn. Ca2+ entry through TRPC channels is necessary to induce hypertrophy (Wu et al., 2010). Most of the TRPC studies have been conducted in non-excitable cells, and thus their role in ventricular myocytes is not yet completely clear, although the proof that they are needed for cardiac hypertrophy has highlighted an important role in the heart (Wu et al., 2010). Ca2+ influxes through LTCCs and TRPCs are thus the proximal sources of Ca2+ influx that regulate cardiac gene expression in adult ventricular cells. These Ca2+ influxes might influence gene expression by several mechanisms. Ca2+ can diffuse to the nucleus and activate nuclear calcium-dependent transcription factors and coregulators (Hardingham et al., 2001) or Ca2+ can activate calcium-dependent signaling proteins around the mouth of the channel, which propagate the signal to the nucleus (Deisseroth et al., 1998; Dolmetsch et al., 2001). Another mechanism was recently observed in neurons (Gomez-Ospina et al., 2006) and cardiac myocytes (Schroder et al., 2009). The C-terminal domain of the LTCC pore-forming subunit, Cav1.2, might be truncated as a result of post-translational processing. The cleaved fragment, in a Ca2+-dependent manner, translocates to the nucleus and acts as a transcription factor to control the transcription of a variety of genes, including Cav1.2.
L-Type Ca2+ Channels (LTCCs)
Treating myocardial cultures with high potassium to inhibit spontaneous contractions (and LTCCs) results in decreased myosin and ribosomal RNA expression (McDermott et al., 1985, 1991; Samarel and Engelmann, 1991). In neonatal rat ventricular cell cultures, LTCC activators stimulate atrial natriuretic factor (ANF) expression (Sei et al., 1991), and ANF expression induced by electrical stimulation of contractions was inhibited by nifedipine, an LTCC blocker (McDonough and Glembotski, 1992). Moreover, Zn2+ influx via voltage-dependent Ca2+ channels can turn on gene expression (Atar et al., 1995). Similarly to what was previously described in skeletal muscle cells (Taouis et al., 1991; Duff et al., 1992), treatment with verapamil, a Ca2+ channel blocker, increases the Na+ channel α-subunit mRNA levels in neonatal rat cardiac myocytes, while treatment with A23187, a Ca2+ ionophore, leads to a decrease in the mRNA levels (Chiamvimonvat et al., 1995). In adult ventricular myocytes, transient changes in [Ca2+]i can modulate Cav1.2 mRNA and protein abundance, producing a corresponding change in functional Ca2+ channels (Davidoff et al., 1997). Surprisingly, whereas early studies in mammalian heart muscle were unable to detect an increased number of channels (Nishiyama et al., 1986; Gengo et al., 1988), an LTCC block by in vivo pharmacological treatment might result in up-regulation of L-type Ca2+ current (ICa,L), CaV1.2 protein, and mRNA (Chapados et al., 1992; Chiappe De Cingolani et al., 1994; De Cingolani et al., 1996; Morgan et al., 1999; Schroder et al., 2007). We found some lines of evidence supporting this hypothesis. We saw that aldosterone, a neurohormone involved in HF, (1) activates LTCC expression (Bénitah and Vassort, 1999), (2) increases diastolic Ca2+ release by decreasing the expression of the RyR accessory proteins FKBP12 and 12.6 (Gomez et al., 2009), and (3) decreases the expression of the channel responsible for the transient outward potassium current (Ito) secondarily to an increase in [Ca2+]i and activation of Cn (Bénitah et al., 2003; Perrier et al., 2004), thereby recapitulating some of the outcomes of HF (Bénitah et al., 1993, 2002; Gómez et al., 1997; Marx et al., 2000). Interestingly, the increase in LTCC expression precedes cell hypertrophy (Perrier et al., 2003).
There is evidence that physiopathological perturbations in CaV1.2 Ca2+ influx regulate K+ channel expression. We have seen that aldosterone increases LTCC expression (Bénitah and Vassort, 1999), which secondarily decreases the expression of the channel responsible for Ito (Bénitah et al., 2003). Consistently, we have reported that increased Ca2+ influx results in decreased Ito density, as a result of down-regulation of Kv4.2 transcript expression mediated by Cn (Perrier et al., 2004). Although it has been reported that expression of a constitutively active form of Cn increases Ito densities through the up-regulation of Kv4.2 transcript expression in neonatal rat ventricular myocytes (Gong et al., 2006), the transcriptional down-regulation of Kv4.2 across the ventricular wall (Rossow et al., 2006), as well as following myocardial infarction (Rossow et al., 2004), results from differences in [Ca2+]i that appear to underlie a differential activation of Cn and NFAT. In addition, it has been reported that increased CaMKII activity down-regulates Kv4.3 transcript expression, resulting in decreased Ito densities in isolated canine ventricular myocytes (Xiao et al., 2008).
Thus in cardiac myocytes, although not as broadly illustrated in other cell types (Barbado et al., 2009), it clearly appears that Ca2+ itself, or even other divalent cations like Zn2+ influx through LTCCs, is involved in transcriptional regulation and/or post-transcriptional events in response to membrane depolarization. This is of particular importance but it is not always taken into account in acquired or inherited cardiac diseases, during which AP duration is altered.
Although LTCCs have been the focus of the majority of the studies with regard to non-cardiac and cardiac gene regulation, some studies also suggest the implication of Ca2+ entry through non-L-type channels in ET coupling, notably TRPC channels.
TRPC Channels
TRPC channels provide Ca2+ entry pathways, modulate the driving force for Ca2+ entry, and also likely provide intracellular pathways for Ca2+ release from cellular organelles. Preferentially localized to the peripheral plasma membrane in cardiomyocytes (Kuwahara et al., 2006; Seth et al., 2009; Wu et al., 2010), they are cation-selective channels that initiate cardiac hypertrophy by Ca2+ influx and subsequent Cn activation (Bush et al., 2006; Kuwahara et al., 2006; Nakayama et al., 2006; Onohara et al., 2006).
The TRPC family includes 7 isoforms (TRPC1–7) divided into 2 general subfamilies based on structural and functional similarities: TRPC1/4/5 and TRPC3/6/7. TRPC2 is not expressed in humans (Lof et al., 2011). TRPC channels can be homomeric or heteromeric assemblies between 4 TRPC subunits. Each TRPC subunit has a transmembrane region flanked by functionally important intracellular N and C termini (Clapham, 2003). TRPC3/6/7 are activated by diacylglycerol (DAG) generated by G-protein coupled receptors Gαq/PLC signaling. TRPC1/4/5 can be activated by depletion of intracellular Ca2+ stores or by stretch (Nilius et al., 2007; Abramowitz and Birnbaumer, 2009). Once activated, these channels induce signal transduction through elevations in [Ca2+]i and Na+ or through refilling of ER Ca2+ stores to ensure prolonged signaling events (Nilius et al., 2007; Abramowitz and Birnbaumer, 2009).
One controversy surrounding TRPC channels concerns their participation in store-operated Ca2+ entry (SOCE) versus receptor-operated Ca2+ entry (ROCE) (Figure 1). TRPC1/4/5 channels are proposed candidate subunits of store-operated channels (SOCs). These types of channels are activated by IP3-dependent mechanisms (Nishida et al., 2006). TRPC3/6/7 are directly activated by DAG, independently of the stores (Hofmann et al., 1999) linked to PLC activation. TRPC channels might also sense and transduce mechanical stress (stretch-activated Ca2+ channels, Figure 1). Another study suggested that TRPC3/6 are activated by DAG causing membrane depolarization with effects on LTCCs and Ca2+ oscillations (Onohara et al., 2006) (Figure 1).
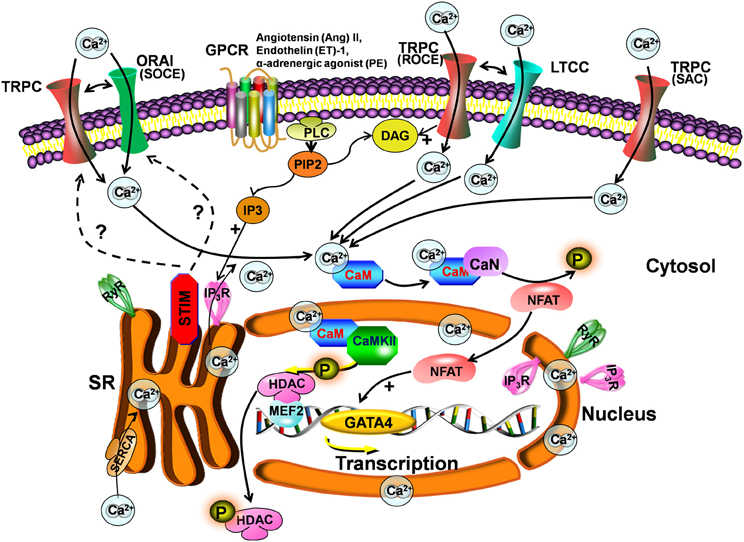
Figure 1. Scheme for the TRPC signaling pathway in hypertrophy. Stimulation of Gq-protein coupled receptors (GPCR) and subsequent activation of PLC leads to IP3 and DAG generation. DAG directly activates TRPCs and induces receptor-operated Ca2+ entry (ROCE), causing membrane depolarization and secondarily activating LTCCs. IP3 activates IP3Rs, which induce depletion of stores and activation of STIM. STIM1 provokes store-operated Ca2+ entry (SOCE) through Orai and/or TRPCs. TRPC channels can also be activated by stretch (SAC). The increase in intracellular Ca2+ following TRPC activation is involved in hypertrophy development via activation of the calcineurin-NFAT pathway.
The role of TRPC channels in SOCE is less clear since the discovery of stromal interaction molecule 1 (STIM1) and Orai1 as mediators of SOCE. STIM1 serves as a Ca2+ sensor in the endoplasmic reticulum/SR, which, when is Ca2+ depleted, clusters proximal to the plasma membrane to activate Orai1, the pore-forming subunit of the Ca2+ release-activated channel (Frischauf et al., 2008) but possibly also to activate TRPC channels (Figure 1). Indeed, it has been shown that TRPC1/4/5 can directly bind STIM1, activating SOCE (Yuan et al., 2007). STIM1 can also indirectly activate TRPC3/6, but not TRPC7 (Liao et al., 2009). Interestingly, TRPC channels can also colocalize with STIM1 and Orai in lipid raft domains (Pani et al., 2008). One study even suggests that Orai and TRPC form complexes that participate in SOCE and ROCE (Liao et al., 2009). However, other investigators have not observed a role for TRPC channels in the Orai/STIM1 complex, suggesting a model whereby these 2 mechanisms of Ca2+ entry are distinct and not coregulated (Dehaven et al., 2009). Interestingly, STIM1 amplifies agonist-induced hypertrophy via activation of the Cn-NFAT pathway (Luo et al., 2012). Figure 1 summarizes some of the TRPC pathways involved in ET coupling.
In conclusion, [Ca2+]i, besides its major role in EC coupling, is an important messenger in signal transduction regulating cardiac hypertrophy by activation of Ca2+-dependent transcription factors. Here we have attempted to present some of the pathways by which cardiac Ca2+ signaling is involved in ET coupling, notably during cardiac hypertrophy development. Although the profound influence of Ca2+ signaling on gene expression has been recognized mainly in neurons (Dolmetsch, 2003), the notion of cardiac ET coupling has recently emerged (Atar et al., 1995; Anderson, 2000; Richard et al., 2006). Evidence is growing that intracellular signaling pathways are laid down in a very sophisticated manner to enable cardiac cells to distinguish between Ca2+ signals. This is particularly important during cardiac hypertrophy, which occurs in response to a variety of stimuli (neurohumoral stimulation, stretch, and pacing) but is initiated in many cases by an elevation in [Ca2+]i. New discoveries are expected in the near future on cardiac Ca2+ regulation to further enrich our understanding in this fascinating research field.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Inserm, ANR grants (Geno-09-012 and Geno-09-034), and CODDIM (COD 100256). Fondation pour la Recherche Médicale (FRM, programme cardiovasculaire) and ESAC (European Society of Aldosterone Council, R12075LL). Inserm U-769 is a member of the Laboratory of Excellence LERMIT, which is supported by an ANR grant, “Investissements d'avenir”. Gema Ruiz-Hurtado was funded by the Ministerio de Economía y Competitividad in the Juan de la Cierva postdoctoral program, Spain.
References
Abramowitz, J., and Birnbaumer, L. (2009). Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 23, 297–328.
Anderson, M. E. (2000). Connections count: excitation-contraction meets excitation-transcription coupling. Circ. Res. 86, 717–719.
Atar, D., Backx, P. H., Appel, M. M., Gao, W. D., and Marbán, E. (1995). Excitation-transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J. Biol. Chem. 270, 2473–2477.
Barbado, M., Fablet, K., Ronjat, M., and De Waard, M. (2009). Gene regulation by voltage-dependent calcium channels. Biochim. Biophys. Acta 1793, 1096–1104.
Bénitah, J. P., Gómez, A. M., Bailly, P., Da Ponte, J. P., Berson, G., Delgado, C., et al. (1993). Heterogeneity of the early outward current in ventricular cells isolated from normal and hypertrophied rat hearts. J. Physiol. (Lond.) 469, 111–138.
Bénitah, J. P., Gómez, A. M., Virsolvy, A., and Richard, S. (2003). New perspectives on the key role of calcium in the progression of heart disease. J. Muscle Res. Cell Motil. 24, 275–283.
Bénitah, J. P., Kerfant, B. G., Vassort, G., Richard, S., and Gómez, A. M. (2002). Altered communication between L-type calcium channels and ryanodine receptors in heart failure. Front. Biosci. 7:e263–e275.
Bénitah, J. P., and Vassort, G. (1999). Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circ. Res. 85, 1139–1145.
Berridge, M. J., Bootman, M. D., and Lipp, P. (1998). Calcium–a life and death signal. Nature 395, 645–648.
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21.
Bers, D. M. (2008). Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49.
Bootman, M. D., Fearnley, C., Smyrnias, I., Macdonald, F., and Roderick, H. L. (2009). An update on nuclear calcium signalling. J. Cell Sci. 122, 2337–2350.
Bush, E. W., Hood, D. B., Papst, P. J., Chapo, J. A., Minobe, W., Bristow, M. R., et al. (2006). Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 281, 33487–33496.
Chapados, R. A., Gruver, E. J., Ingwall, J. S., Marsh, J. D., and Gwathmey, J. K. (1992). Chronic administration of cardiovascular drugs: altered energetics and transmembrane signaling. Am. J. Physiol. 263, H1576–H1586.
Chevalier, B., Callens, F., Charlemagne, D., Delcayre, C., Lompre, A. M., Lelievre, L., et al. (1989). Signal and adaptational changes in gene expression during cardiac overload. J. Mol. Cell. Cardiol. 21(Suppl. 5), 71–77.
Chiamvimonvat, N., Kargacin, M. E., Clark, R. B., and Duff, H. J. (1995). Effects of intracellular calcium on sodium current density in cultured neonatal rat cardiac myocytes. J. Physiol. 483, 307–318.
Chiappe De Cingolani, G. E., Mosca, S. M., Vila Petroff, M., and Cingolani, H. E. (1994). Chronic administration of nifedipine induces upregulation of dihydropyridine receptors in rabbit heart. Am. J. Physiol. 267, H1222–H1226.
Chien, K. R., Knowlton, K. U., Zhu, H., and Chien, S. (1991). Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. Faseb. J. 5, 3037–3046.
Cooling, M. T., Hunter, P., and Crampin, E. J. (2009). Sensitivity of NFAT cycling to cytosolic calcium concentration: implications for hypertrophic signals in cardiac myocytes. Biophys. J. 96, 2095–2104.
Davidoff, A. J., Maki, T. M., Ellingsen, O., and Marsh, J. D. (1997). Expression of calcium channels in adult cardiac myocytes is regulated by calcium. J. Mol. Cell. Cardiol. 29, 1791–1803.
De Cingolani, G. E., Mosca, S. M., Moreyra, A. E., and Cingolani, H. E. (1996). Chronic nifedipine treatment diminishes cardiac inotropic response to nifedifine: functional upregulation of dihydropyridine receptors. J. Cardiovasc. Pharmacol. 27, 240–246.
Dehaven, W. I., Jones, B. F., Petranka, J. G., Smyth, J. T., Tomita, T., Bird, G. S., et al. (2009). TRPC channels function independently of STIM1 and Orai1. J. Physiol. 587, 2275–2298.
Deisseroth, K., Heist, E. K., and Tsien, R. W. (1998). Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392, 198–202.
De Rooij, J., Zwartkruis, F. J., Verheijen, M. H., Cool, R. H., Nijman, S. M., Wittinghofer, A., et al. (1998). Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477.
Dolmetsch, R. (2003). Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci. STKE 2003,PE4.
Dolmetsch, R. E., Pajvani, U., Fife, K., Spotts, J. M., and Greenberg, M. E. (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339.
Duff, H. J., Offord, J., West, J., and Catterall, W. A. (1992). Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel alpha subunit in rat cardiac muscle. Mol. Pharmacol. 42, 570–574.
Escobar, M., Cardenas, C., Colavita, K., Petrenko, N. B., and Franzini-Armstrong, C. (2011). Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J. Mol. Cell. Cardiol. 50, 451–459.
Fabiato, A. (1983). Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1–C14.
Frischauf, I., Schindl, R., Derler, I., Bergsmann, J., Fahrner, M., and Romanin, C. (2008). The STIM/Orai coupling machinery. Channels (Austin) 2, 261–268.
Gengo, P., Skattebol, A., Moran, J. F., Gallant, S., Hawthorn, M., and Triggle, D. J. (1988). Regulation by chronic drug administration of neuronal and cardiac calcium channel, beta-adrenoceptor and muscarinic receptor levels. Biochem. Pharmacol. 37, 627–633.
Gidh-Jain, M., Huang, B., Jain, P., Battula, V., and El-Sherif, N. (1995). Reemergence of the fetal pattern of L-type calcium channel gene expression in non infarcted myocardium during left ventricular remodeling. Biochem. Biophys. Res. Commun. 216, 892–897.
Gómez, A. M., Bénitah, J. P., Henzel, D., Vinet, A., Lorente, P., and Delgado, C. (1997). Modulation of electrical heterogeneity by compensated hypertrophy in rat left ventricle. Am. J. Physiol. 272, H1078–H1086.
Gomez, A. M., Rueda, A., Sainte-Marie, Y., Pereira, L., Zissimopoulos, S., Zhu, X., et al. (2009). Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation 119, U2179–U2189.
Gomez-Ospina, N., Tsuruta, F., Barreto-Chang, O., Hu, L., and Dolmetsch, R. (2006). The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell 127, 591–606.
Gong, N., Bodi, I., Zobel, C., Schwartz, A., Molkentin, J. D., and Backx, P. H. (2006). Calcineurin increases cardiac transient outward K+ currents via transcriptional up-regulation of Kv4.2 channel subunits. J. Biol. Chem. 281, 38498–38506.
Hardingham, G. E., Arnold, F. J., and Bading, H. (2001). Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 4, 261–267.
Harzheim, D., Movassagh, M., Foo, R. S., Ritter, O., Tashfeen, A., Conway, S. J., et al. (2009). Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 106, 11406–11411.
Heineke, J., and Molkentin, J. D. (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600.
Hofmann, T., Obukhov, A. G., Schaefer, M., Harteneck, C., Gudermann, T., and Schultz, G. (1999). Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397, 259–263.
Houser, S. R., and Molkentin, J. D. (2008). Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci. Signal. 1, pe31.
Kockskamper, J., Seidlmayer, L., Walther, S., Hellenkamp, K., Maier, L. S., and Pieske, B. (2008a). Endothelin-1 enhances nuclear Ca2+ transients in atrial myocytes through Ins(1, 4, 5)P3-dependent Ca2+ release from perinuclear Ca2+ stores. J. Cell Sci. 121, 186–195.
Kockskamper, J., Zima, A. V., Roderick, H. L., Pieske, B., Blatter, L. A., and Bootman, M. D. (2008b). Emerging roles of inositol 1, 4, 5-trisphosphate signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 45, 128–147.
Kuwahara, K., Wang, Y., McAnally, J., Richardson, J. A., Bassel-Duby, R., Hill, J. A., et al. (2006). TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Invest. 116, 3114–3126.
Liao, Y., Plummer, N. W., George, M. D., Abramowitz, J., Zhu, M. X., and Birnbaumer, L. (2009). A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC: Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. U.S.A. 106, 3202–3206.
Lof, C., Viitanen, T., Sukumaran, P., and Tornquist, K. (2011). TRPC2: of mice but not men. Adv. Exp. Med. Biol. 704, 125–134.
Luo, X., Hojayev, B., Jiang, N., Wang, Z. V., Tandan, S., Rakalin, A., et al. (2012). STIM1-dependent store-operated Ca(2) entry is required for pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 52, 136–147.
Maier, L. S., and Bers, D. M. (2002). Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J. Mol. Cell. Cardiol. 34, 919–939.
Marbán, E., and Koretsune, Y. (1990). Cell calcium, oncogenes, and hypertrophy. Hypertension 15, 652–658.
Martinez, M. L., Heredia, M. P., and Delgado, C. (1999). Expression of T-type Ca(2+) channels in ventricular cells from hypertrophied rat hearts. J. Mol. Cell. Cardiol. 31, 1617–1625.
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376.
McDermott, P., Daood, M., and Klein, I. (1985). Contraction regulates myosin synthesis and myosin content of cultured heart cells. Am. J. Physiol. 249, H763–H769.
McDermott, P. J., Carl, L. L., Conner, K. J., and Allo, S. N. (1991). Transcriptional regulation of ribosomal RNA synthesis during growth of cardiac myocytes in culture. J. Biol. Chem. 266, 4409–4416.
McDonough, P. M., and Glembotski, C. C. (1992). Induction of atrial natriuretic factor and myosin light chain-2 gene expression in cultured ventricular myocytes by electrical stimulation of contraction. J. Biol. Chem. 267, 11665–11668.
McKinsey, T. A., Zhang, C. L., Lu, J., and Olson, E. N. (2000). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111.
Metrich, M., Laurent, A. C., Breckler, M., Duquesnes, N., Hmitou, I., Courillau, D., et al. (2010). Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell. Signal. 22, 1459–1468.
Metrich, M., Lucas, A., Gastineau, M., Samuel, J. L., Heymes, C., Morel, E., et al. (2008). Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 102, 959–965.
Moalic, J. M., Charlemagne, D., Mansier, P., Chevalier, B., and Swynghedauw, B. (1993). Cardiac hypertrophy and failure—a disease of adaptation. Modifications in membrane proteins provide a molecular basis for arrhythmogenicity. Circulation 87, IV21–IV26.
Molkentin, J. D. (2000). Calcineurin and beyond: cardiac hypertrophic signaling. Circ. Res. 87, 731–738.
Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Robbins, J., et al. (1998). A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228.
Morel, E., Marcantoni, A., Gastineau, M., Birkedal, R., Rochais, F., Garnier, A., et al. (2005). cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 97, 1296–1304.
Morgan, P. E., Aiello, E. A., Chiappe De Cingolani, G. E., Mattiazzi, A. R., and Cingolani, H. E. (1999). Chronic administration of nifedipine induces up-regulation of functional calcium channels in rat myocardium. J. Mol. Cell. Cardiol. 31, 1873–1883.
Nakayama, H., Wilkin, B. J., Bodi, I., and Molkentin, J. D. (2006). Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 20, 1660–1670.
Nass, R. D., Aiba, T., Tomaselli, G. F., and Akar, F. G. (2008). Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat. Clin. Pract. Cardiovasc. Med. 5, 196–207.
Nilius, B., Owsianik, G., Voets, T., and Peters, J. A. (2007). Transient receptor potential cation channels in disease. Physiol. Rev. 87, 165–217.
Nishida, M., Hara, Y., Yoshida, T., Inoue, R., and Mori, Y. (2006). TRP channels: molecular diversity and physiological function. Microcirculation 13, 535–550.
Nishiyama, T., Kobayashi, A., Haga, T., and Yamazaki, N. (1986). Chronic treatment with nifedipine does not change the number of [3H]nitrendipine and [3H]dihydroalprenolol binding sites. Eur. J. Pharmacol. 121, 167–172.
Nuss, H. B., and Houser, S. R. (1993). T-type Ca2+ current is expressed in hypertrophied adult feline left ventricular myocytes. Circ. Res. 73, 777–782.
Onohara, N., Nishida, M., Inoue, R., Kobayashi, H., Sumimoto, H., Sato, Y., et al. (2006). TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 25, 5305–5316.
Pani, B., Ong, H. L., Liu, X., Rauser, K., Ambudkar, I. S., and Singh, B. B. (2008). Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ entry (SOCE). J. Biol. Chem. 283, 17333–17340.
Passier, R., Zeng, H., Frey, N., Naya, F. J., Nicol, R. L., Mckinsey, T. A., et al. (2000). CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 105, 1395–1406.
Pereira, L., Ruiz-Hurtado, G., Morel, E., Laurent, A. C., Metrich, M., Dominguez-Rodriguez, A., et al. (2012). Epac enhances excitation–transcription coupling in cardiac myocytes. J. Mol. Cell. Cardiol. 52, 283–291.
Perrier, E., Kerfant, B., Vassort, G., Rossier, M., Gomez, A. M., and Benitah, J. P. (2003). Aldosterone-antagonist prevents ionic remodelling that precedes cardiac hypertrophy. Biophys. J. 84, 423A.
Perrier, E., Perrier, R., Richard, S., and Bénitah, J. P. (2004). Ca2+ controls functional expression of the cardiac K+ transient outward current via the calcineurin pathway. J. Biol. Chem. 279, 40634–40639.
Richard, S., Perrier, E., Fauconnier, J., Perrier, R., Pereira, L., Gómez, A. M., et al. (2006). ‘Ca(2+)-induced Ca(2+) entry’ or how the L-type Ca(2+) channel remodels its own signalling pathway in cardiac cells. Prog. Biophys. Mol. Biol. 90, 118–135.
Ringer, S. (1883). A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J. Physiol. 4, 29–42.3.
Rossow, C. F., Dilly, K. W., and Santana, L. F. (2006). Differential calcineurin/NFATc3 activity contributes to the Ito transmural gradient in the mouse heart. Circ. Res. 98, 1306–1313.
Rossow, C. F., Minami, E., Chase, E. G., Murry, C. E., and Santana, L. F. (2004). NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ. Res. 94, 1340–1350.
Samarel, A. M., and Engelmann, G. L. (1991). Contractile activity modulates myosin heavy chain-beta expression in neonatal rat heart cells. Am. J. Physiol. 261, H1067–H1077.
Schroder, E., Byse, M., and Satin, J. (2009). L-type calcium channel C terminus autoregulates transcription. Circ. Res. 104, 1373–1381.
Schroder, E., Magyar, J., Burgess, D., Andres, D., and Satin, J. (2007). Chronic verapamil treatment remodels ICa, L in mouse ventricle. Am. J. Physiol. Heart Circ. Physiol. 292, H1906–H1916.
Sei, C. A., Irons, C. E., Sprenkle, A. B., McDonough, P. M., Brown, J. H., and Glembotski, C. C. (1991). The alpha-adrenergic stimulation of atrial natriuretic factor expression in cardiac myocytes requires calcium influx, protein kinase C, and calmodulin-regulated pathways. J. Biol. Chem. 266, 15910–15916.
Seth, M., Zhang, Z. S., Mao, L., Graham, V., Burch, J., Stiber, J., et al. (2009). TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 105, 1023–1030.
Taouis, M., Sheldon, R. S., and Duff, H. J. (1991). Upregulation of the rat cardiac sodium channel by in vivo treatment with a class I antiarrhythmic drug. J. Clin. Invest. 88, 375–378.
Wu, X., and Bers, D. M. (2006). Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ. Res. 99, 283–291.
Wu, X., Chang, B., Blair, N. S., Sargent, M., York, A. J., Robbins, J., et al. (2009). Plasma membrane Ca2+-ATPase isoform 4 antagonizes cardiac hypertrophy in association with calcineurin inhibition in rodents. J. Clin. Invest. 119, 976–985.
Wu, X., Eder, P., Chang, B., and Molkentin, J. D. (2010). TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 107, 7000–7005.
Wu, X., Zhang, T., Bossuyt, J., Li, X., McKinsey, T. A., Dedman, J. R., et al. (2006). Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Invest. 116, 675–682.
Xiao, L., Coutu, P., Villeneuve, L. R., Tadevosyan, A., Maguy, A., Le Bouter, S., et al. (2008). Mechanisms underlying rate-dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ. Res. 103, 733–742.
Yuan, J. P., Zeng, W., Huang, G. N., Worley, P. F., and Muallem, S. (2007). STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 9, 636–645.
Keywords: heart, calcium, excitation-transcription coupling, TRPC, nuclear calcium
Citation: Domínguez-Rodríguez A, Ruiz-Hurtado G, Benitah J-P and Gómez AM (2012) The other side of cardiac Ca2+ signaling: transcriptional control. Front. Physio. 3:452. doi: 10.3389/fphys.2012.00452
Received: 13 April 2012; Accepted: 12 November 2012;
Published online: 28 November 2012.
Edited by:
Eric A. Sobie, Mount Sinai School of Medicine, USAReviewed by:
Jeffrey Varner, Cornell University, USARavi Radhakrishnan, University of Pennsylvania, USA
Copyright © 2012 Domínguez-Rodríguez, Ruiz-Hurtado, Benitah and Gómez. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Ana M. Gómez, Inserm U769, Faculté de Pharmacie, Tour D4, 5ème étage, 5 Rue Jean Baptiste Clément, 92296 Châtenay-Malabry, France. e-mail: ana-maria.gomez@inserm.fr