
- Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado School of Medicine, Denver, CO, USA
Chronic Obstructive Pulmonary Disease (COPD) is one of the foremost causes of death worldwide. It is primarily caused by tobacco smoke, making it an easily preventable disease, but facilitated by genetic α-1 antitrypsin deficiency. In addition to active smokers, health problems also occur in people involuntarily exposed to second hand smoke (SHS). Currently, the relationship between SHS and COPD is not well established. Knowledge of pathogenic mechanisms is limited, thereby halting the advancement of new treatments for this socially and economically detrimental disease. Here, we attempt to summarize tobacco smoke studies undertaken in animal models, applying both mainstream (direct, nose only) and side stream (indirect, whole body) smoke exposures. This overview of 155 studies compares cellular and molecular mechanisms as well as proteolytic, inflammatory, and vasoreactive responses underlying COPD development. This is a difficult task, as listing of exposure parameters is limited for most experiments. We show that both mainstream and SHS studies largely present similar inflammatory cell populations dominated by macrophages as well as elevated chemokine/cytokine levels, such as TNF-α. Additionally, SHS, like mainstream smoke, has been shown to cause vascular remodeling and neutrophil elastase-mediated proteolytic matrix breakdown with failure to repair. Disease mechanisms and therapeutic interventions appear to coincide in both exposure scenarios. One of the more widely applied interventions, the anti-oxidant therapy, is successful for both mainstream and SHS. The comparison of direct with indirect smoke exposure studies in this review emphasizes that, even though there are many overlapping pathways, it is not conclusive that SHS is using exactly the same mechanisms as direct smoke in COPD pathogenesis, but should be considered a preventable health risk. Some characteristics and therapeutic alternatives uniquely exist in SHS-related COPD.
Introduction
Cigarette smoke is the main preventable cause for chronic obstructive pulmonary disease (COPD), resulting in progressive proteolytic, inflammatory, and vasoactive responses that lead to emphysema, small airway obstruction, and pulmonary hypertension. COPD in itself is a serious burden throughout the world, both economically and socially, costing $193 billion in the United States alone (CDC, 2008). The disease is the third largest cause of death in the United States and the fourth worldwide (Pauwels et al., 2001; Minino, 2010). An estimated 95% of COPD cases are attributed to smoking (Barnes et al., 2003), while only a relatively small margin of smokers is susceptible (Fletcher and Peto, 1977). The United States Centers for Disease Control and Prevention considers tobacco smoke to be “the single most important preventable risk to human health in developed countries and an important cause of premature death worldwide,” not only to smokers, but also to those involuntary exposed to second hand smoke (SHS or environmental tobacco smoke, known as ETS) (Oberg et al., 2011).
There are three kinds of smoke that humans are exposed to: mainstream or first hand smoke that is directly inhaled through a person's mouth after taking a puff on a lit cigarette; Side stream smoke, which goes into the air directly from a burning cigarette, cigar, or smoking pipe; and SHS, which is a combination of both, side stream smoke being the main component of SHS, also known as ETS. Cigarette smoke thereby not only affects smokers, but also contributes to health problems in non-smokers. While a smoker voluntarily inhales the first hand smoke, the non-smoker is inadvertently exposed to SHS that comes from the burning end of a cigarette and the smoke exhaled by the smoker. SHS is therefore also a potential risk factor for COPD and entails symptomatic disease in individuals who are not actually smokers. At present, the dose-response relationship between SHS exposure and COPD is not well established and there is limited understanding of the mechanisms responsible for its pathogenesis, thus halting the development of new advanced treatments for this detrimental disease.
This review is an attempt to sort out the cigarette smoke exposure studies according to levels (low/high) and manner (mainstream or whole body) of the exposure, focusing largely on newer studies applying the rodent model.
Second Hand Cigarette Smoke Exposure
While the qualitative composition of the components is nearly identical in mainstream smoke, side stream smoke, and SHS, the quantitative composition of each is different. In the enclosed environment, due to relatively low ventilation rates (Lofroth, 1989; Jinot and Bayard, 1994), some compounds are emitted at levels up to more than 10 times greater in side stream smoke and SHS when compared with mainstream smoke (Moritsugu, 2007). Side stream smoking has therefore been classified as a Class A carcinogen by the US Environmental Protection Agency. Still, data regarding the biological evidence linking SHS exposure and COPD are scarce. A 2006 US Surgeon General Report on the health consequences of SHS concluded that the evidence linking SHS and COPD is suggestive (www.surgeongeneral.gov/library/secondhandsmoke), but not sufficient, to infer a causal relationship. The conclusion of the report was drawn primarily from dated epidemiologic evidence that did not establish a biological link (Hirayama, 1981; Kalandidi et al., 1987; Sandler et al., 1989; Robbins et al., 1993; Dayal et al., 1994; Leuenberger et al., 1994; Piitulainen et al., 1998; Berglund et al., 1999). A very recent retrospective analysis of data from 192 countries was published in 2011 and shows that worldwide 40% of children, 33% of male non-smokers, and 35% of female non-smokers are exposed to SHS (Oberg et al., 2011). In 2004 alone, 603,000 deaths were attributable to SHS, accounting for approximately 1% of worldwide mortality. For this reason, the US Surgeon General Report's conclusion should be challenged by providing evidence that SHS exposure can in fact cause COPD by utilizing and comparing animal models as well as assessing patient exposure to SHS.
The Animal Model and Exposure System
In order to gain insight into the mechanisms of disease development during cigarette smoke exposure, both mainstream and second hand, animal models have been of exceptional use. Animals are exposed to cigarette smoke in a smoking apparatus for either mainstream (nose or head only) or side stream (whole body) applications. In addition, smoke can either be filtered or not, which either depends on the cigarette used (with a filter or without) or the set-up of the apparatus. Another diversity factor is the selection of different animal species with varying susceptibilities, which can be particular to each strain within a species. Early studies of SHS used self-constructed chambers for laboratory animals, allowing environmental smoke to diffuse within a confined space in which the animals were kept. These chambers, unique to each group, did not have the capabilities to measure the parameters essential for assessing cigarette smoke dose and composition. Also, the number and type of cigarettes as well as the length of exposure varied in each study as much as they do today, adding to the difficulty of comparing results. Generally, standardized research-grade cigarettes should be used to easily define a specified dose of total suspended particles (TSP) or total particulate matter (TPM), including nicotine and carbon monoxide levels. Standardized cigarettes became available for worldwide use in 1969 (Roemer et al., 2012) and are most commonly from the University of Kentucky (http://www.ca.uky.edu/refcig/), although there are still often publications using non-reference cigarettes. The introduction of the Teague chamber in 1994 (Teague et al., 1994) has revolutionized the field by allowing maintenance of consistent levels of TSP/TPM that can be set at a variety of concentrations for the exposure of animals to SHS that can mimic human exposures.
In human exposure studies, only the distribution of fine particles (PM2.5, which are under 2.5 μm in size and are able to reach the alveoli of the lung) is measured. This accounts for only a small fraction (about 0.1%) of the TSP measurements that are usually reported in animal studies. Data from 66 US casinos with smoking in California, Delaware, Nevada, New Jersey, and Pennsylvania, developed PM2.5 frequency distributions, comparing them with three non-smoking casinos. Geometric means for PM2.5 were 53.8 μg/m3 (range 18.5–205 μg/m3) inside smoking casinos, 4.3 μg/m3 (range 0.26–29.7 μg/m3) outside those casinos, and 3.1 μg/m3 (range 0.6–9 μg/m3) inside the three non-smoking casinos (Jiang et al., 2010; Lu et al., 2011; Repace et al., 2011; Cochran et al., 2012).
Most commonly for SHS studies listed in Table 2, the Teague chamber was set to exposures between 70 and 150 mg/m3. Concurrently, the mainstream smoke exposures listed in Table 1 were as low as 75 mg/m3 and as high as 600 mg/m3 TSP/TPM (Hodge-Bell et al., 2007). Most of the mainstream smoke studies were performed at 140 mg/m3 TSP/TPM levels (Table 1). Ideally, more recent protocols should thereby produce results that can compare the assessed proteolytic, inflammatory, and vasoactive reaction based on similar exposure methods, duration, and cigarette content. This can only be achieved if researchers pertain to the standard procedures available today, such as the use of reference cigarettes in a chamber with defined settings for exposure. These conformities are essential, since a majority of studies are performed in rodents (mice, rats, and guinea pigs), where there is an overwhelming assortment of strains with varying susceptibility, especially when considering mice.
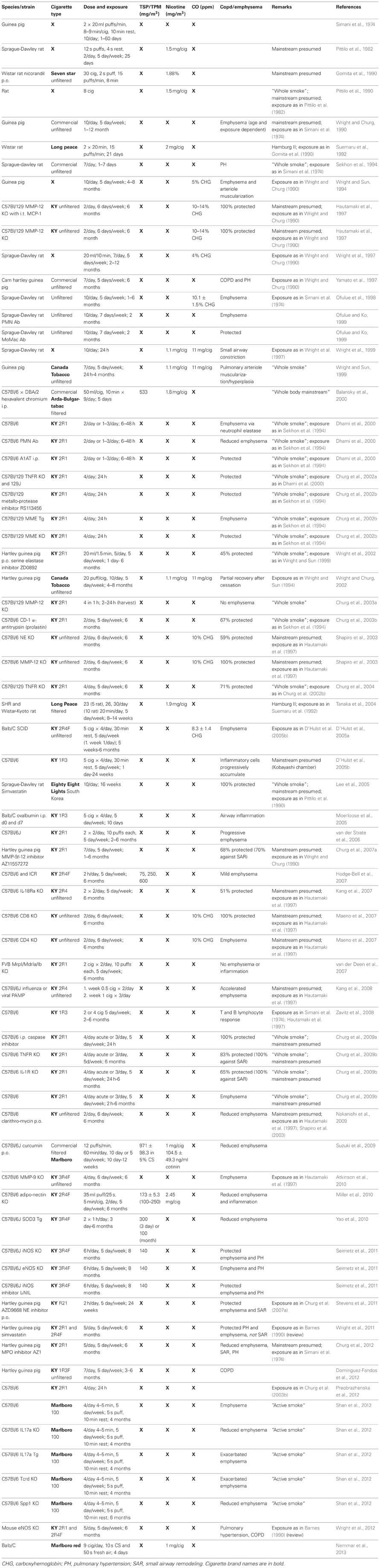
Table 1. Mainstream cigarette smoke exposure modeled in animals.
Today, rodents are the most commonly used models. While mice are surely favored for their wide variety of applicable gene expression manipulations, it remains difficult to standardize measurements of pulmonary function to assess disease parameters. The guinea pig model is also occasionally applied, mainly by one group of investigators (Simani et al., 1974; Wright and Churg, 1990, 2002; Wright and Sun, 1994, 1999; Wright et al., 2002, 2011) though increased inflammatory cells and muscularization of pulmonary vessels was recently documented (Dominguez-Fandos et al., 2012). The rat is a favorable model, since measurable emphysematous changes which further progress can be detected after only 2 months of smoke exposure (Kratzer et al., 2013).
Mainstream smoke and SHS exposure studies are summarized in Tables 1, 2, respectively. Animal studies with an unidentified smoke exposure are presented in Table 3. The cigarette brands used are listed in Table 4.
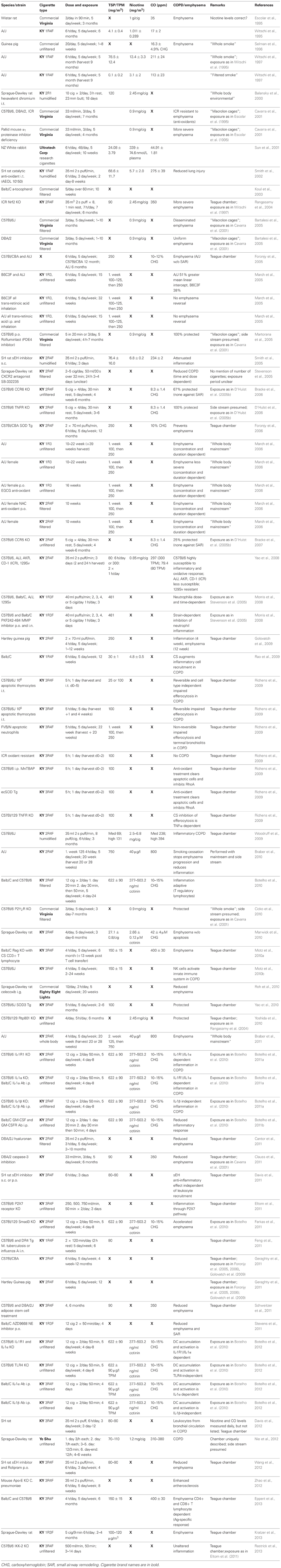
Table 2. Second hand cigarette smoke exposure modeled in animals.
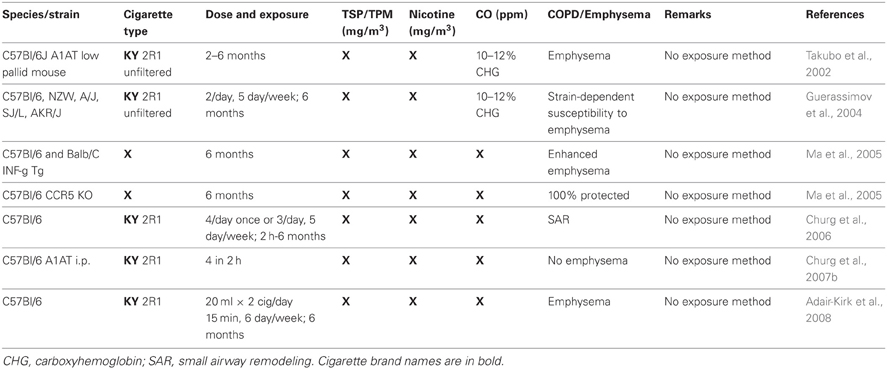
Table 3. Animal studies where the exposure (mainstream or second hand smoke) is not known.
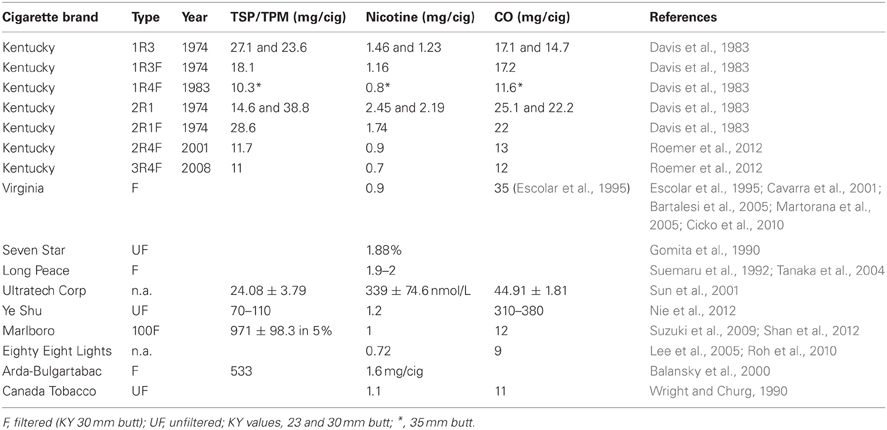
Table 4. Cigarette types used for the summarized experiments.
Chronic Cigarette Smoke Exposure
It is important to differentiate acute versus chronic smoke exposure in respect to lung structure and function (Martin and Tamaoka, 2006). Though both models induce airway narrowing to a certain extent, the inflammation seen initially diminishes during the time-course of exposure, along the lines of early repair and late-stage failure to repair (Churg and Wright, 2009; Kratzer et al., 2013). Mechanisms of repair seem to be sufficient for the first month of cigarette smoke exposure, but since lesions change over the exposure time, it is predicted that the pathological mechanisms differ in acute and chronic cases. Acute models can be as short as 24 h or last up to as many as 2 weeks, while chronic models should be standardized to 6 months (24 weeks) of exposure to induce the anatomical changes characterizing COPD (Churg and Wright, 2009). Both acute and chronic cigarette smoke exposure lead to oxidant stress, which pathologically affects airway cells to promote remodeling (Martin and Tamaoka, 2006).
Since COPD can only manifest the pathological characteristics seen in humans after long-term cigarette smoke exposure, animals are often utilized in chronic studies to relate to the human disease. Emphysema, airway remodeling and pulmonary hypertension, which progress over time, can only be induced in chronic models where structural alterations occur. Irreversible matrix destruction, fibrosis, airway wall thickening, and hyperplasia of smooth muscle and goblet cells as well as fibroblasts can only be seen in chronic cigarette exposure. Since COPD in smoking patients is typically GOLD (Global Initiative for Chronic Obstructive Lung Disease) stage III or IV and less commonly I and II (Retamales et al., 2001), animal models must attempt to mimic this by developing emphysema, small airway remodeling (SAR), or pulmonary hypertension. The excess mucus production that defines chronic bronchitis is also thought to be important in the pathogenesis of acute exacerbations of COPD (Burge and Wedzicha, 2003). This was previously considered difficult to reproduce in animal models due to the fact that, in contrast to humans, the anatomical localization of bronchial glands in rats and mice are concentrated in the proximal trachea (Churg et al., 2008). Recently, the mucus secretion was demonstrated in two rat models (Nie et al., 2012). Authors showed significantly increased mucus production in epithelium of trachea, bronchi, and bronchiole in a 6 week cigarette smoke model and in a chronic bronchitis model with 6 weeks cigarette smoke exposure combined with a single intratracheal injection of LPS on day 39. There was also a significant increase in MUC5A protein levels in broncheoalveolar lavage in both models (Nie et al., 2012).
The Anatomical Manifestations of COPD
For chronic cigarette smoke exposure studies, many months are required to model the lung pathology, as acute models do not have this effect on lung structure and therefore do not allow the prediction of chronic outcomes. In order to evaluate, for example, drug efficacies, a chronic exposure most reliably modeling the human disease is essential. Only chronic models can present the lesions of COPD defined as emphysema, SAR, and pulmonary hypertension, though still more mildly than in human COPD (Wright and Churg, 2008; Churg and Wright, 2009). This is especially important when noting that, because rodents develop a mild form of COPD, drug tests should be performed as late as possible to mimic a treatment versus a prophylaxis (Churg et al., 2011).
Emphysema
The pathogenesis of emphysema is studied by inducing the disease with various methods not necessarily restricted to cigarette smoke. The protease/anti-protease hypothesis has been the main focus in elucidating the cause of emphysema. In addition, oxidative stress has been shown to cause lung cell apoptosis in an emphysema model using a vascular endothelial growth factor (VEGF) receptor blocker (Kasahara et al., 2000; Tuder et al., 2003). Cigarette smoke induces both oxidative stress and the infiltration of inflammatory cells, which release proteases that overwhelm the anti-proteolytic defenses. Inflammatory cell and consequently endothelial cell proteases lead to the breakdown of pulmonary matrix and alveolar walls following particle inhalation, creating airspace enlargement and minimizing surface area for gas exchange (March et al., 2000). This protease theory is based on the 1963 finding that patients deficient in α-1 antitrypsin, a major neutrophil elastase inhibitor, have accelerated emphysema development (Laurell and Eriksson, 1963). The capability of proteases to cause emphysema was then verified by instilling them intratracheally (Gross et al., 1965; Janoff et al., 1977; Snider et al., 1984). Protease inhibition has since been tested in the animal model with varying efficacies, depending on the protease depleted. Serine proteases from neutrophils (59% protection in neutrophil elastase deficient mice) (Shapiro et al., 2003) as well as macrophage metalloproteases (Hautamaki et al., 1997) have been considered most important in a cigarette smoke induced disease model, but the more broad application of anti-inflammatory interventions has proven useful in smoked rodents as well (Churg et al., 2008). This is especially of interest, as sampling of human COPD specimens noted T lymphocytes in alveolar destruction and airflow obstruction, though smoking history was not assessed in the patient population (Hogg et al., 2004). This has shed light on the participation of auto-immunity contributing to the overall pathogenesis of emphysema through anti-endothelial cell antibodies and pathogenic CD4+ T lymphocytes (Taraseviciene-Stewart et al., 2005). Because emphysema was initially believed to occur through the same mechanism as SAR and to be the underlying cause of pulmonary hypertension, both in COPD as well as following cigarette smoke exposure, many studies have focused only on the emphysematous pathology, especially on the proteolytic aspect, neglecting further complex contributing factors.
Small Airway Remodeling (SAR)
Emphysema is not the only manifestation of cigarette smoke induced COPD. SAR is also a major contributor to the disease as it limits airflow (Hogg et al., 2004) and is characterized by continuous repair for the duration of smoke exposure (Churg and Wright, 2009). While emphysema is the breakdown of extracellular matrix in the lung parenchyma, SAR pathogenesis involves a fibrotic process leading to airway narrowing due to airway wall thickening. Therefore, it is not surprising that SAR appears to develop through a mechanism independent of the one that drives emphysema. This correlates well with the fact that methods for emphysema prevention cannot be uniformly applied to treat SAR. Only TNF-α and IL-1 receptor knockouts (Churg et al., 2009b) as well as mice treated with an MMP-9/MMP-12 inhibitor (Churg et al., 2007a) were protected against the development of both emphysema and SAR when exposed to cigarette smoke, while other interventions such as chemokine receptor 5 (CCR5) (Bracke et al., 2007) and CCR6 (Bracke et al., 2006) knockouts are merely protected from emphysema following cigarette smoke.
Pulmonary Hypertension
COPD morbidity and mortality are significantly increased by the not uncommon occurrence of pulmonary hypertension associated with cigarette smoke (Chaouat et al., 2008; Elwing and Panos, 2008), which correlates with a poor prognosis (Weitzenblum et al., 1981). Originally, it was believed that the increase in pulmonary artery pressure is secondary to the lung pathology associated with COPD, as a result of hypoxia, emphysema, and loss of the vascular bed (Wright et al., 2005). With the realization that smoke and its mediators effect vasculature to induce remodeling, and that pulmonary hypertension is an early symptom that manifests long before airflow obstruction, emphysema, and SAR, the independent mechanism became evident (Wright et al., 2005). It has recently been shown that inducible nitric oxide synthase (iNOS) is important in the development of pulmonary hypertension following cigarette smoke exposure (Gielis et al., 2011; Kratzer et al., 2013). Mice lacking iNOS or treated with an iNOS inhibitor are protected against the cigarette smoke induced development of emphysema and pulmonary hypertension (Nathan, 2011; Seimetz et al., 2011). Both prostacyclin and endothelial NOS are protective against pulmonary hypertension induced by hypoxia, while endothelin-1 and VEGF contribute to the pathogenesis (Wright et al., 2004, 2005; Voelkel, 2008; Wright and Churg, 2008). The oxidative damage to the vasculature results from reactive nitrogen species that are not of endothelial source, but more likely contained in the cigarette smoke itself (Wright et al., 2012). The result is altered vasoconstriction and vasodilation as well as vascular cell proliferation leading to the pathological thickening of vessels. In the intima, elastin and collagen are deposited, while smooth muscle cells proliferate (muscularization), and adventitia harbors increased numbers of CD8+ T lymphocytes (Wright et al., 1983, 1992; Barbera et al., 2003).
Smoking is not only the main risk factor for COPD and pulmonary hypertension, but also for coronary artery disease summarized in a recent review (Ghoorah et al., 2012).
The Proteolytic and Inflammatory Response in COPD
COPD, like asthma, is characterized by chronic and abnormal inflammation of the distal airways resulting in airflow limitation, but differs from asthma in that it is progressive and largely irreversible. This chronic disease encompasses bronchiolitis and fibrosis with obstruction of small airways, vascular remodeling leading to pulmonary hypertension, destruction of lung parenchyma with loss of the alveolar wall and subsequent airspace enlargement defined as emphysema, as well as loss of lung elasticity. The underlying mechanisms involve protease/anti-protease imbalance following inflammation, with subsequent proteolytic matrix destruction (Churg et al., 2008), oxidant damage that leads to apoptosis of resident lung structure cells (Yoshida and Tuder, 2007; Churg et al., 2011). accelerated aging (Csiszar et al., 2009), a failure to repair, and the contribution of autoimmunity (Taraseviciene-Stewart et al., 2006; Maeno et al., 2007; Stampfli and Anderson, 2009). The most prominent and frequent etiology of COPD remains cigarette smoke and its onset is mid-life. For COPD, the inflammatory response is slowly progressive and leads to the actual destruction of lung parenchyma where alveolar walls disappear and the distal airspaces become pathologically and permanently enlarged, resulting in emphysema (Barnes, 2004). The contributing inflammatory cells in COPD are neutrophilic granulocytes, alveolar macrophages, and CD8+ T lymphocytes (Taraseviciene-Stewart and Voelkel, 2008; Stampfli and Anderson, 2009). The accumulation of both T and B lymphocytes following apoptosis of resident lung cells has been described to create follicles within the lung parenchyma, adjacent to airways, in COPD (Hogg et al., 2004; Taraseviciene-Stewart et al., 2006). In a rat model, neutrophil depletion did not prevent smoke induced emphysema, while treatment with anti-monocyte/macrophage antibody did. These findings implicate macrophages rather than neutrophils as the critical pathogenic factor in cigarette smoke induced emphysema (Ofulue and Ko, 1999). Macrophages are known to secrete the cytokines interleukin 8 (IL-8) and tumor necrosis factor alpha (TNF-α) as well as leukotriene B4 (LTB4), all found to be increased in COPD patients (Keatings et al., 1996). Additionally, macrophages also generate reactive oxygen species (ROS), monocyte chemotactic protein 1 (MCP-1) and elastolytic enzymes such as matrix metalloproteinase (MMP)-2, MMP-9, MMP-12, and cathepsins K, L, and S thus contributing to the lung destruction (Barnes et al., 2003).
Neutrophils are found to be increased in bronchial biopsies and sputum of patients during COPD, correlating with severity (Keatings et al., 1996; Di Stefano et al., 1998; Pesci et al., 1998; Retamales et al., 2001). Neutrophils apparently contribute to the disease pathogenesis by secreting serine proteases (neutrophil elastase, cathepsin G, proteinase) and metalloelastases MMP-8 and MMP-9 (Barnes et al., 2003).
The major pathogenetic factor of neutrophil and macrophage accumulation in emphysema is their secretion of proteases and inflammatory mediators. Cleaved fragments of elastin, collagen, and fibronectin are believed to possibly act as auto-antibodies, which would explain the accumulation of T and B lymphocytes in COPD patients (Hogg et al., 2004; Taraseviciene-Stewart and Voelkel, 2008), It has been shown that antibodies against endothelial cells correlate with alveolar septal cell apoptosis as well as the activation of MMP-2 and MMP-9, thereby initiating the proteolytic cascade and inducing emphysema independent of cigarette smoke (Taraseviciene-Stewart et al., 2005). This specific mechanism of anti-endothelial cell auto-immunity is dependent on CD4+ T lymphocytes believed to be pathogenic (Voelkel and Taraseviciene-Stewart, 2005; Taraseviciene-Stewart et al., 2007). Anti-endothelial cell antibodies were also detected in a rat model of cigarette induced emphysema (Taraseviciene-Stewart, unpublished observation).
Genetic Predispositions for COPD Development
Interestingly, only 15–20% of smokers are susceptible to developing COPD, underlining the contribution of genetic factors (Fletcher and Peto, 1977). This susceptibility is mirrored in the animal model, where the development of cigarette smoke induced emphysema is strain-dependent (Guerassimov et al., 2004; Bartalesi et al., 2005). To this date, though, the only human genetic predisposition identified is the rare hereditary deficiency in α-1 antitrypsin (Laurell and Eriksson, 1963). Transgenic mice over-expressing genes of interest or knockout mice lacking a specific gene are exposed to mainstream or SHS elucidating how a specific gene modifies the pathogenesis of this disease. Though mice are so small that the assessment of disease progression and COPD symptoms is hindered, the application of transgenic and knockout mice to define the effects of a gene insertion or expression increase as well as that of a gene deletion, can answer many questions. Additionally, 80% of the mouse genome contains genes that have direct orthologues in the human genome and less than 300 genes (1%) are unique to the murine species (Waterston et al., 2002; Pennacchio, 2003). Interestingly, 89–90% of rat genes contain single orthologues in the human genome and 76% of well-characterized human disease genes are found in the rat genome (Gibbs et al., 2004). Both rats (Pauwels et al., 1985; Martin et al., 1992) and mice (Guerassimov et al., 2004; Bartalesi et al., 2005) have strain-dependent susceptibilities to excessive airway inflammation and smoke-induced emphysema, respectively, just as humans have genetic susceptibility to developing COPD.
Preventative Therapies
The most prominent interventions studied for COPD are anti-proteolytic, anti-inflammatory, as well as anti-oxidant and are directed against the development of emphysema, often not even being tested for SAR or pulmonary hypertension efficacies (Churg et al., 2011). Proteases are released by both accumulating inflammatory as well as resident cells, causing matrix destruction in the alveolar wall and therefore emphysema. This hypothesis is summarized in the term protease/anti-protease imbalance, because both an excessive protease release as well as a hindered anti-protease activity is necessary to contribute to the pathogenesis. The inhibition of various proteases during cigarette smoke exposure has been tested: serine proteases (neutrophil elastase) (Cavarra et al., 2001; Takubo et al., 2002; Wright et al., 2002; Churg et al., 2003b; Shapiro et al., 2003; Pemberton et al., 2006), different metalloproteases (MMP-9 and MMP-12) (Hautamaki et al., 1997; Selman et al., 2003; Mahadeva and Shapiro, 2005; Pemberton et al., 2005; Churg et al., 2007a), and cysteine proteases (cathepsins B and S) (Kang et al., 2007). The preventative success depends on the specific protease inhibited.
The immune reaction to chronic smoke exposure has been investigated by blocking certain inflammatory responses using knockout mice or simply applying an anti-inflammatory treatment. Both anti-TNF-α receptor (Churg et al., 2004; D'Hulst et al., 2006) and anti-PDE4 (Rofluminlast) (Martorana et al., 2005) therapies are protective anti-inflammatory measures in the animal model, though these results could not be as successfully reproduced for the chronic human disease (Barnes, 2007). Significant protection against SHS induced emphysema in the animal model was also achieved using a hyaluronan aerosol (Cantor et al., 2005) and by blocking interferon γ (INF-γ) or CCR5 (Ma et al., 2005; Bracke et al., 2007) or CCR6 (Bracke et al., 2006), After it was shown that SCID (severe combined immuno-deficient) and Rag (recombinase activating gene) knockout mice, which both lack functional T and B lymphocytes, are not protected from mainstream smoke induced emphysema (D'Hulst et al., 2005a), CD4+ and CD8+ T lymphocyte-deficient mice were tested individually. While CD4+ T lymphocyte-defecient mice are not at all protected, CD8+ T lymphocyte-deficient mice are 100% protected against emphysema following mainstream exposure (Maeno et al., 2007). Toll-like receptor 4 (TLR4) knockout mice were also not protected (Maes et al., 2006). Statins, however, have proven to be protective against cigarette smoke induced emphysema in rats (Lee et al., 2005) and appear to be promising therapy in humans as well (Soyseth et al., 2007). Simvastatin additionally is effective against pulmonary hypertension, but unfortunately does not prevent SAR (Wright et al., 2011), unless administered before disease onset (Lee et al., 2005; Ou et al., 2009).
ROS are both contained in cigarette smoke and released by the resultant infiltrating cells, such as neutrophils and macrophages (Pryor and Stone, 1993; Rahman and MacNee, 1996; MacNee, 2001). This, again, leads to an imbalance, that of oxidants and anti-oxidants, so that the lung is exposed to oxidative stress (Petrache et al., 2006). Susceptibility to oxidative stress is, like the genetic predisposition for cigarette smoke induced emphysema, specific to a certain strain of mice (Cavarra et al., 2001; Bartalesi et al., 2005). Oxidant sensitive mice show less anti-oxidant capacity when exposed to cigarette smoke, developing emphysema. This effect can be reduced when applying an oral anti-oxidant (Koul et al., 2003), while elimination of the redox sensitive transcription gene Nuclear factor 2 (Nrf2) enhanced the cigarette smoke induced oxidative stress and emphysema in the otherwise resistant ICR mouse strain (Rangasamy et al., 2004).
Concisely, emphysema induced by cigarette smoke is a multi-factorial disease and therapeutic approaches should be undertaken to repair the lung structure by preventing marix degradation by reducing proteases, inflammation, oxidative stress, cell death, and/or autoimmune-mediated destruction.
Study Parameters thus Far
Here we have compiled 114 publications containing 155 studies addressing cigarette smoke exposure in the animal model. Approximately half of the studies (71 experiments) are mainstream exposure (Table 1). Side stream smoke exposures (Table 2) make up 77 experiments of the studies listed herein and a small portion (7 studies) is unclear about the exposure method utilized (Table 3). Of the studies listed here, a large portion (115 studies) utilizes quite a strain diversity of mice (C57Bl/6, DBA/2, Balb/C, A/J, ICR, FVB, or a strain not described), although C57Bl/6 mice are dominant (83 experiments). C57Bl/6 mice are a common strain for knockout studies and generally a popular model for cigarette smoke induced emphysema (Takubo et al., 2002; Guerassimov et al., 2004; Bartalesi et al., 2005; Yao et al., 2008; Botelho et al., 2010) due to their deficiency in anti-elastase (Gardi et al., 1994; Cavarra et al., 2001). Balb/C mice with higher levels of the cytokine granulocyte-macrophage colony stimulating factor (GM-CSF) (Morris et al., 2008; Botelho et al., 2010, 2011b) and the oxidant sensitive strain DBA/2 (Cavarra et al., 2001; Bartalesi et al., 2005) have also been shown to have severe emphysema following cigarette smoke exposure. ICR mice are considered oxidant resistant and are of interest when attempting to induce disease (Cavarra et al., 2001; Rangasamy et al., 2004; Hodge-Bell et al., 2007; Yao et al., 2008). A/J express less severe pulmonary complications (Guerassimov et al., 2004; Yao et al., 2008; Braber et al., 2010) and have found use in drug tests, but have been used as a model for chronic cigarette smoke exposure as well (Foronjy et al., 2005; March et al., 2005, 2006). Apart from mice, one study addresses rabbits, 15 utilize guinea pigs (either Hartley strain or not named), while 24 studies gravitate toward using rats. These are almost exclusively Sprague-Dawley, while spontaneously hypertensive rats (SH) as well as Wistar Kyoto are also popular.
Originally, only studies from 1997 onwards were added to Tables 1–3, but with such consistency of certain researchers to repeatedly reference earlier publications, the original publishers of certain exposure procedures were included as far back as 1974. A number of authors stumble when it comes to actually stating clearly whether their exposure method was mainstream or side stream. This is most likely due to the assumption that this would be known according to the exposure system more or less detailed in the Materials and Methods section or merely mentioned in the abstract. The Materials and Methods section, though, frequently references a publication that, in turn, references the original design. Such a basic fact as mainstream or side stream exposure must be pointed out, as must the fact that there was a cigarette smoke exposure at all, also something that can be quite hidden in a less specific manuscript. Fortunately, many publications do announce a head/nose only or a whole body exposure in the Materials and Methods section, if mainstream or side stream (environmental or second hand) has not already been mentioned in the abstract or introduction. There are some that allow you to look up their chamber of choice, although this leads to lengthy product guidelines tediously researched online (i.e., the long outdated Hamburg II chamber). There are only a few groups that generally use the same procedure and one can at least assume that they are utilizing the same method, simply using their latest publication as the current reference (which then must be referenced back to the original procedure). It is a relatively safe assumption that the method always remains mainstream or side stream within the same research group, unless otherwise noted, but it is left for the reader to assume, nonetheless. In this summary, only two research groups resort to this form of documentation.
Seven publications segregated in Table 3 leave it completely unknown what exposure they have used, and one uses a “whole body mainstream” exposure, apparently employing a high concentration of cigarette particulates into a chamber to mimic mainstream smoking (Braber et al., 2011), but that is, again, left to be assumed. Generally, one can still assess the exposure method in most cases, it becomes less clear at the level of nicotine, carbon monoxide, and particulate levels. Some researches are very diligent here, while others may not have measured these parameters and therefore do not address them. There is also somewhat variability to whether nicotine and TSP/TPM are measured as mg/m3 or μg/l (which can easily be calculated to mg/m3). Some quote mg/cig for the nicotine levels (Pittilo et al., 1982, 1990; Suemaru et al., 1992; Escolar et al., 1995; Wright and Sun, 1999; Wright et al., 1999; Balansky et al., 2000; Cavarra et al., 2001; Wright and Churg, 2002; Rangasamy et al., 2004; Tanaka et al., 2004; Bartalesi et al., 2005; Martorana et al., 2005; Yao et al., 2008; Woodruff et al., 2009; Cicko et al., 2010; Miller et al., 2010; Yoshida et al., 2010; Nie et al., 2012; Nemmar et al., 2013), meaning this was likely looked up through the manufacturer. The same is true for carbon monoxide, which is either measured in ppm or percentage of carboxyhemoglobin in the serum. It is clear that the vast majority of studies has, at bare minimum, one of these parameters missing, as only 24 (15.5%) exemplary studies listed all three. Usually, the levels named are an average kept during the entire exposure period, which is of importance when considering SHS exposure. The TSP/TPM levels in SHS exposures is as low as 24 mg/m3 using a unique rabbit model (Sun et al., 2001), but as high as 250 mg/m3 in standard Teague chamber mouse models using Kentucky reference cigarettes (Foronjy et al., 2005, 2006; Golovatch et al., 2009; Richens et al., 2009). The Stampfli lab (Botelho et al., 2010, 2011a,b, 2012; Farkas et al., 2011) and one other group (Braber et al., 2010) go beyond 700 mg/m3 for their mouse models that do not utilize the Teague chamber.
There is still diversity in the use of cigarettes (Table 4), despite the fact that reference cigarettes, specifically designed for this type of research, have been available for approximately four decades now. The vast majority of studies (111 studies making up 71.6%) do use the reference cigarettes provided by the University of Kentucky's Tobacco Research and Development Center (formerly the Tobacco and Health Research Institute). Two groups have listed all necessary parameters with the unique cigarettes (Ultratech Corp and Ye Shu) they have chosen (Sun et al., 2001; Nie et al., 2012). Unfortunately, it appears that commercial Virginia cigarettes are popular, without specifying which kind, though apparently generally a type with 0.9 mg nicotine per cigarette, except for one study that claims to use Virginia cigarettes with 1 g nicotine per cigarette, presumably a typo (Escolar et al., 1995). A total of 17 studies do not list the cigarettes used (either commercial unfiltered, simply unfiltered, or no reference at all), while five publications compromising eight studies do opt for Kentucky reference cigarettes, but then neglected to list which kind, let alone the parameters (Hautamaki et al., 1997; Shapiro et al., 2003; Maeno et al., 2007; Nakanishi et al., 2009; Clauss et al., 2011).
Other cigarette brands listed in Tables 1–3 stated only some or none of the parameters for TSP/TPM, nicotine, and carbon monoxide levels. The eight remaining brands of cigarettes are Marlboro 100 and Marlboro red (for Marlboro Medium sort, the nicotine level is 1 mg/cig and carbon monoxide is 12 mg/cig), Long Peace and Seven Star (Japan), Eighty Eight Lights (South Korea), Arda-Bulgartabac (Bulgaria), Canada Tobacco, and Virginia brand. For these cigarettes, the parameters could not be found to supplement the information available in the original publications themselves, as the actual type of cigarette was not stated in any one of them. Therefore, data regarding reference cigarettes used in the studies summarized here have been left incomplete in Table 4.
Kentucky reference cigarettes, on the other hand, are very well documented, but sometimes it is unclear in a publication whether a mix of two types was used for smoke exposure. At times, the filtered version (named with an “F” by the manufacturer) is used, but the filter is removed. There are also those who list using 2R4, which presumably means that the filter was removed for the study. The first line of reference cigarettes from the University of Kentucky were from 1974 and included 1R3, 1R3F, and 2R1, of which the filtered kind (1R3F) had 30 mm butts and the unfiltered cigarettes (1R3 and 2R1) could be purchased with either 23 or 30 mm butt lengths. In 1983, the new generation of reference cigarettes was 1R4F, only available in a 35 mm butt length. In 2001, the next generation, named 2R4F, was launched and as of 2008 3R4F has been produced. Studies presumably reflect the reference cigarette of the time and the well-documented levels of Kentucky reference cigarettes can be found in their entirety in Table 4.
Comparing Mainstream and Second Hand Exposures
Although parameters appear to rarely be listed to the extent they should be, we have attempted to compare the direct exposure of smoke in these animal studies to those of animals exposed via a whole body method, mimicking SHS. One must remember that while mainstream smoke is inhaled into the lungs from the cigarette directly, SHS is the inhalation of suspended particles from the smoker's exhale and the burning end of the cigarette, thereby concentrations of particles and individual components are not necessarily reduced, as one would hope. What becomes clear is, that SHS exposure does indeed lead to emphysema and other COPD symptoms in animal models. A multitude of studies elucidate this fact and they are summarized in Table 2, which also highlights what effects (whether emphysema, SAR, or pulmonary hypertension) were assessed.
Studies to unravel the pathogenic mechanisms of cigarette smoke exposure were undertaken in the knockout models, where the effect can easily be seen based on only one factor. The central role of TNF-α has been documented for both exposure applications. This was made evident by inhibition of TNF-α using a receptor knockout model (TNFR KO), which attenuated the development of emphysema by 71–83% and of SAR by 100% in mainstream cigarette smoke exposure (Churg et al., 2002a, 2004, 2009b). TNFR KO were 100% protected against emphysema using side stream smoke (D'Hulst et al., 2006). Additionally, inhibition of efferocytosis is TNF-α dependent in the side stream knockout model (Richens et al., 2009).
C–C CCR5 knockout mice were 100% protected against emphysema in an exposure method that was not defined, where INF-γ over-expression enhanced emphysema (Ma et al., 2005). In a side stream exposure study (Bracke et al., 2007), the effect of CCR5 knockout was only protective to 25%, whereas CCR6 knockout was more effective with 67% protection against emphysema (Bracke et al., 2006). It is possible that the same manipulation would be more protective in a mainstream versus a second hand exposure, although the exposure in the first study, which assessed a number of pathways leading to apoptosis, is not known.
Another well-documented knockout model for the mechanism of smoke induced pathology is that of macrophage elastases, specifically MMP-12 (Hautamaki et al., 1997; Churg et al., 2002a, 2003a; Shapiro et al., 2003) and MMP-9 (Atkinson et al., 2010). The elastinolytic activity central to matrix breakdown and alveolar enlargement had already been attributed to macrophages (Ofulue et al., 1998; Ofulue and Ko, 1999) and these two enzymes were pinpointed via inhibition (Churg et al., 2007a). Here, MMP-12 (also termed macrophage metalloelastase MME) has been identified to be of foremost importance in lung destruction, since MMP-12 knockout mice are resistant to smoke induced emphysema, while emphysema can occur independent of MMP-9 (Atkinson et al., 2010). Mice deficient in monocytes and macrophages do not develop enlarged airspaces upon cigarette smoke exposure, while those deficient in polymorphonuclear cells (PMN) do (Ofulue et al., 1998; Ofulue and Ko, 1999; Dhami et al., 2000). Unfortunately, all studies regarding MMP contributions, which are considerable, have solely been tested in the mainstream smoke model.
The neutrophil-derived serine elastase has been implicated in both a mainstream and a side stream exposure model (Dhami et al., 2000). In a direct smoke exposure model, the neutrophil elastase knockout mouse is 59% protected against emphysema development (Shapiro et al., 2003), while specific neutrophil elastase inhibitors are similarly protective in the guinea pig model (Wright et al., 2002; Stevens et al., 2011). In a SHS exposure model of the mouse, one group (Stevens et al., 2011) was also able to detect a marked decrease in SAR development in both their mainstream and side stream exposure models upon treatment with neutrophil elastase inhibitor AZD9668.
The only thus far defined true genetic factor described in man predisposing to COPD is α-1 antitrypsin deficiency. This deficiency leads to accelerated development of emphysema following cigarette smoke exposure. Although the importance of α-1 antitrypsin has been documented in the animal model of direct cigarette smoke (Dhami et al., 2000; Churg et al., 2003b), there are two studies where it is not clear whether an indirect exposure was utilized (Takubo et al., 2002; Churg et al., 2007b). Nevertheless, α-1 antitrypsin is indeed protective against emphysema in a dose-dependent manner, indicating that this is likely also true for indirect cigarette smoke exposure (Dhami et al., 2000; Churg et al., 2003b).
In addition to genetic manipulations elucidating molecular mechanisms of cigarette smoke pathogenesis, there are also models for drug testing, more specific to an intervention protocol. These go beyond simply attenuating the proteolytic breakdown via inhibition of just one protease and encompass mainly statins and anti-oxidants. Simvastatin, as mentioned above, has a protective effect against emphysema and pulmonary hypertension, but not SAR, and has been tested in both the guinea pig (Wright et al., 2011) and the rat (Lee et al., 2005), but only using a mainstream exposure method. Therefore, a homologous mechanism for side stream smoke is yet to be described.
Beyond being influenced by mouse strain (Guerassimov et al., 2004; Bartalesi et al., 2005; Foronjy et al., 2005), emphysema development is also dependent on the airspace content of anti-oxidants. This was demonstrated by comparing bronchoalveolar lavage (BAL) levels of endogenous anti-oxidants from the oxidant resistant mouse strain ICR with that from other strains (Cavarra et al., 2001; Richens et al., 2009). Successful anti-oxidant therapy is seen for mainstream (March et al., 2006; Suzuki et al., 2009; Churg et al., 2012) and side stream exposure models (Smith et al., 2002; Richens et al., 2009), as well as in a Rtp801 knockout model for side stream smoke (Yoshida et al., 2010). This is reflected in the success of superoxide dismutase (SOD) transgenic mice (Foronjy et al., 2006; Richens et al., 2009), specifically those with enhanced SOD3 expression (Yao et al., 2010), in protecting against emphysema in both exposure scenarios.
What is Missing
Unfortunately, even with chronic exposure studies, the animal model has limitations to how well it can mimic COPD, as seen by the inadequacy of interventions that protect mice against cigarette smoke induced emphysema, but not humans (Barnes, 2007). A procedure that is applied in the same way for each study could greatly contribute to our understanding of pathogenesis and how the animal model relates to the human disease. For example, with a standard 6-month exposure period for mouse models, research groups have been able to establish a disease time frame that is utilized for most chronic studies. In rats, this chronic period starts at 2 months of exposure (Ofulue et al., 1998; Kratzer et al., 2013) and in guinea pigs after 3 months of exposure (Wright et al., 2002). In order to truly compare results, though, it is of importance to standardize exposure methods and levels as well.
Generally, there is favoritism toward the somewhat more recent application of chambers, which was greatly improved so as to be utilized as a standard method for SHS exposure in 1994 (Teague et al., 1994). A chamber may contain any number of animals and is often applied to produce a cigarette smoke mixture consisting of 89% side stream and 11% mainstream smoke, exposing animals to environmental smoke versus attaching a nose or head piece to introduce smoke first hand. Often, though, even mainstream or side stream smoke exposure are neglected to be pointed out, as it is assumed that using a chamber method should suffice as information, even though it can require extensive back-referencing to discover the actual original procedure. Beyond the smoke chamber, which wasn't developed until 1994 (Teague et al., 1994), few standardized procedures were possible. Since then, though, there is still very little homogeneity in the applications used for research in this field. We could greatly improve our understanding of the consequences of inhaled cigarette smoke if we were to apply a similar and comparable model in every case. This would require selecting conform research cigarettes, whether filtered or not, preferably from the same source, so that contents and levels of important toxins and mediators can be compared.
If a variety of cigarettes are necessary to accommodate research across the globe, it is imperative that at least cigarette parameters are assessed and noted. Reference cigarettes were specifically designed from 1969 onwards for the purpose homogenous and comparable studies around the world. To this day, commercial cigarettes with varying or unknown parameters are applied.
There is still an abundance of publications that do not mention or may even not determine nicotine or carbon monoxide amounts or the contributing particles in cigarettes (whether as TSP or TPM), though it could easily be evaluated from online supplements for major cigarettes in use. For randomly selected commercial cigarettes, of course, there is not even this option, as only nicotine and in very few cases carbon monoxide levels are available. Some studies do not even list the cigarette type or brand used, so there is practically no information concerning the exposure. These factors complicate matters even further, as there are already numerous ways that cigarette smoke is administered, using as few as two cigarettes a day in mice (Hautamaki et al., 1997; Shapiro et al., 2003) or as many as 12 cigarettes a day in rats (Nie et al., 2012).
Additionally, many studies use an initial acclimation period for their cigarette smoke exposure, employing an increasing number of cigarettes or longer smoke periods after beginning the study, thereby also allowing variability, but acclimation is usually necessary. Inconsistencies can be avoided by assessing the most important parameters, such as nicotine, carbon monoxide, and particle levels. If the scientific community could agree on a protocol to establish similar exposures both for mainstream and side stream cigarette smoke, results would be much more comparable. At a bare minimum, simply correctly documenting the method used would add a great deal to the knowledge that could have already been gained. In this way, cigarette smoke researchers all around the world would be able to collectively assess and compare their data.
Conclusion
Cigarette smoke exposure is the most important risk factor for developing COPD, both in smokers and in non-smokers who are involuntarily exposed to the toxic and carcinogenic contents of environmental cigarette smoke. Studies of long-term cigarette smoke exposure effects are very costly, time-consuming, and require an abundance of resources. For this reason, tests should be optimized so that experimental models are comparable and provide as much knowledge as possible. It is evident from the literature that there is, to this day, enormous unnecessary diversity in study designs despite improvements to our experimental exposure methods (chambers and reference cigarettes). There is a great need for a standard protocol defining parameters to be evaluated and exposure procedures to be followed. With this quite simple documentation of easily assessed facts and we would be able to gain much more knowledge with each study. It should be possible to determine molecular mechanisms, oxidative stress, protease participation, inflammatory cytokine levels, and cellular death following mainstream and side stream cigarette smoke exposure, comparing them. These insights would greatly improve and expand today's comprehension of cellular and molecular mechanisms implicated in the pathology of COPD. A more defining knowledge of the pathogenesis of mainstream and second hand cigarette smoke induced emphysema, SAR, and pulmonary hypertension could have been achieved with the undertaken studies if a unified procedural method had been applied.
Despite the shortcomings in terms of documentation in a large amount of experiments, they have nonetheless led to great advances through gene knockout models or transgenic mice as well as inhibitory tactics and drug treatments. When combining these animal studies in one overview, it becomes highly evident that second hand cigarette smoke has many of the same mechanisms and detrimental effects that mainstream smoke does. For this reason, we wish to point out that the US Surgeon General's assessment of a missing link should be revisited, so that the public becomes more aware of the health risks to innocent bystanders, where SHS exposure causes COPD/emphysema in non-smokers.
Funding
Funded by AHA 0735388N, 11GRNT7520020, FAMRI CIA 072053, Emphysema Research Fund and Bixler Family Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adair-Kirk, T. L., Atkinson, J. J., Griffin, G. L., Watson, M. A., Kelley, D. G., DeMello, D., et al. (2008). Distal airways in mice exposed to cigarette smoke: Nrf2-regulated genes are increased in Clara cells. Am. J. Respir. Cell Mol. Biol. 39, 400–411.
Atkinson, J. J., Lutey, B. A., Suzuki, Y., Toennies, H. M., Kelley, D. G., Kobayashi, D. K., et al. (2010). The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am. J. Respir. Crit. Care Med. 183, 876–884.
Balansky, R. M., D'Agostini, F., Izzotti, A., and De Flora, S. (2000). Less than additive interaction between cigarette smoke and chromium(VI) in inducing clastogenic damage in rodents. Carcinogenesis 21, 1677–1682.
Barbera, J. A., Peinado, V. I., and Santos, S. (2003). Pulmonary hypertension in chronic obstructive pulmonary disease. Eur. Respir. J. 21, 892–905.
Barnes, P. J. (1990). Reactive oxygen species and airway inflammation. Free Radic. Biol. Med. 9, 235–243.
Barnes, P. J. (2004). Mediators of chronic obstructive pulmonary disease. Pharmacol. Rev. 56, 515–548.
Barnes, P. J. (2007). Unexpected failure of anti-tumor necrosis factor therapy in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 175, 866–867.
Barnes, P. J., Shapiro, S. D., and Pauwels, R. A. (2003). Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur. Respir. J. 22, 672–688.
Bartalesi, B., Cavarra, E., Fineschi, S., Lucattelli, M., Lunghi, B., Martorana, P. A., et al. (2005). Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur. Respir. J. 25, 15–22.
Berglund, D. J., Abbey, D. E., Lebowitz, M. D., Knutsen, S. F., and McDonnell, W. F. (1999). Respiratory symptoms and pulmonary function in an elderly nonsmoking population. Chest 115, 49–59.
Botelho, F. M., Bauer, C. M., Finch, D., Nikota, J. K., Zavitz, C. C., Kelly, A., et al. (2011a). IL-1alpha/IL-1R1 expression in chronic obstructive pulmonary disease and mechanistic relevance to smoke-induced neutrophilia in mice. PLoS ONE 6:e28457. doi: 10.1371/journal.pone.0028457
Botelho, F. M., Nikota, J. K., Bauer, C., Davis, N. H., Cohen, E. S., Anderson, I. K., et al. (2011b). A mouse GM-CSF receptor antibody attenuates neutrophilia in mice exposed to cigarette smoke. Eur. Respir. J. 38, 285–294.
Botelho, F. M., Gaschler, G. J., Kianpour, S., Zavitz, C. C., Trimble, N. J., Nikota, J. K., et al. (2010). Innate immune processes are sufficient for driving cigarette smoke-induced inflammation in mice. Am. J. Respir. Cell Mol. Biol. 42, 394–403.
Botelho, F. M., Nikota, J. K., Bauer, C. M., Morissette, M. C., Iwakura, Y., Kolbeck, R., et al. (2012). Cigarette smoke-induced accumulation of lung dendritic cells is interleukin-1alpha-dependent in mice. Respir. Res. 13:81. doi: 10.1186/1465-9921-13-81
Braber, S., Henricks, P. A., Nijkamp, F. P., Kraneveld, A. D., and Folkerts, G. (2010). Inflammatory changes in the airways of mice caused by cigarette smoke exposure are only partially reversed after smoking cessation. Respir. Res. 11:99. doi: 10.1186/1465-9921-11-99
Braber, S., Koelink, P. J., Henricks, P. A., Jackson, P. L., Nijkamp, F. P., Garssen, J., et al. (2011). Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L255–L265.
Bracke, K. R., D'Hulst, A. I., Maes, T., Demedts, I. K., Moerloose, K. B., Kuziel, W. A., et al. (2007). Cigarette smoke-induced pulmonary inflammation, but not airway remodelling, is attenuated in chemokine receptor 5-deficient mice. Clin. Exp. Allergy 37, 1467–1479.
Bracke, K. R., D'Hulst, A. I., Maes, T., Moerloose, K. B., Demedts, I. K., Lebecque, S., et al. (2006). Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J. Immunol. 177, 4350–4359.
Burge, S., and Wedzicha, J. A. (2003). COPD exacerbations: definitions and classifications. Eur. Respir. J. Suppl. 41, 46s–53s.
Cantor, J. O., Cerreta, J. M., Ochoa, M., Ma, S., Chow, T., Grunig, G., et al. (2005). Aerosolized hyaluronan limits airspace enlargement in a mouse model of cigarette smoke-induced pulmonary emphysema. Exp. Lung Res. 31, 417–430.
Cantor, J. O., Cerreta, J. M., Ochoa, M., Ma, S., Liu, M., and Turino, G. M. (2011). Therapeutic effects of hyaluronan on smoke-induced elastic fiber injury: does delayed treatment affect efficacy? Lung 189, 51–56.
Cavarra, E., Bartalesi, B., Lucattelli, M., Fineschi, S., Lunghi, B., Gambelli, F., et al. (2001). Effects of cigarette smoke in mice with different levels of alpha(1)-proteinase inhibitor and sensitivity to oxidants. Am. J. Respir. Crit. Care Med. 164, 886–890.
Centers for Disease Control (CDC), Centers for Disease Control and Prevention (C. f. D. C. a. P). (2008). Smoking-attributable mortality, years of potential life lost, and productivity losses – United State. Morb. Mortal. Wkly Rep. 57, 1226–1228.
Chaouat, A., Naeije, R., and Weitzenblum, E. (2008). Pulmonary hypertension in COPD. Eur. Respir. J. 32, 1371–1385.
Churg, A., Cosio, M., and Wright, J. L. (2008). Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L612–L631.
Churg, A., Dai, J., Tai, H., Xie, C., and Wright, J. L. (2002a). Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am. J. Respir. Crit. Care Med. 166, 849–854.
Churg, A., Zay, K., Shay, S., Xie, C., Shapiro, S. D., Hendricks, R., et al. (2002b). Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am. J. Respir. Cell Mol. Biol. 27, 368–374.
Churg, A., Marshall, C. V., Sin, D. D., Bolton, S., Zhou, S., Thain, K., et al. (2012). Late intervention with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 185, 34–43.
Churg, A., Sin, D. D., and Wright, J. L. (2011). Everything prevents emphysema: are animal models of cigarette smoke-induced chronic obstructive pulmonary disease any use? Am. J. Respir. Cell Mol. Biol. 45, 1111–1115.
Churg, A., Tai, H., Coulthard, T., Wang, R., and Wright, J. L. (2006). Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wall. Am. J. Respir. Crit. Care Med. 174, 1327–1334.
Churg, A., Wang, R. D., Tai, H., Wang, X., Xie, C., Dai, J., et al. (2003a). Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am. J. Respir. Crit. Care Med. 167, 1083–1089.
Churg, A., Wang, R. D., Xie, C., and Wright, J. L. (2003b). alpha-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 168, 199–207.
Churg, A., Wang, R. D., Tai, H., Wang, X., Xie, C., and Wright, J. L. (2004). Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 170, 492–498.
Churg, A., Wang, R., Wang, X., Onnervik, P. O., Thim, K., and Wright, J. L. (2007a). Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 62, 706–713.
Churg, A., Wang, X., Wang, R. D., Meixner, S. C., Pryzdial, E. L., and Wright, J. L. (2007b). Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am. J. Respir. Cell Mol. Biol. 37, 144–151.
Churg, A., and Wright, J. L. (2009). Testing drugs in animal models of cigarette smoke-induced chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 6, 550–552.
Churg, A., Zhou, S., Preobrazhenska, O., Tai, H., Wang, R., and Wright, J. L. (2009a). Expression of profibrotic mediators in small airways versus parenchyma after cigarette smoke exposure. Am. J. Respir. Cell Mol. Biol. 40, 268–276.
Churg, A., Zhou, S., Wang, X., Wang, R., and Wright, J. L. (2009b). The role of interleukin-1beta in murine cigarette smoke-induced emphysema and small airway remodeling. Am. J. Respir. Cell Mol. Biol. 40, 482–490.
Cicko, S., Lucattelli, M., Muller, T., Lommatzsch, M., De Cunto, G., Cardini, S., et al. (2010). Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J. Immunol. 185, 688–697.
Clauss, M., Voswinckel, R., Rajashekhar, G., Sigua, N. L., Fehrenbach, H., Rush, N. I., et al. (2011). Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. J. Clin. Invest. 121, 2470–2479.
Cochran, C., Henriques, D., York, N., and Lee, K. (2012). Risk of exposure to second hand smoke for adolescents in Las Vegas casinos: an evaluation of the Nevada Clean Indoor Air Act. J. Health Hum. Serv. Adm. 35, 231–252.
Csiszar, A., Podlutsky, A., Wolin, M. S., Losonczy, G., Pacher, P., and Ungvari, Z. (2009). Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Front. Biosci. 14, 3128–3144.
D'Hulst, A. I., Bracke, K. R., Maes, T., De Bleecker, J. L., Pauwels, R. A., Joos, G. F., et al. (2006). Role of tumour necrosis factor-alpha receptor p75 in cigarette smoke-induced pulmonary inflammation and emphysema. Eur. Respir. J. 28, 102–112.
D'Hulst, A. I., Maes, T., Bracke, K. R., Demedts, I. K., Tournoy, K. G., Joos, G. F., et al. (2005a). Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respir. Res. 6:147. doi: 10.1186/1465-9921-6-147
D'Hulst, A. I., Vermaelen, K. Y., Brusselle, G. G., Joos, G. F., and Pauwels, R. A. (2005b). Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur. Respir. J. 26, 204–213.
Davis, B. B., Liu, J. Y., Tancredi, D. J., Wang, L., Simon, S. I., Hammock, B. D., et al. (2011). The anti-inflammatory effects of soluble epoxide hydrolase inhibitors are independent of leukocyte recruitment. Biochem. Biophys. Res. Commun. 410, 494–500.
Davis, B. B., Shen, Y. H., Tancredi, D. J., Flores, V., Davis, R. P., and Pinkerton, K. E. (2012). Leukocytes are recruited through the bronchial circulation to the lung in a spontaneously hypertensive rat model of COPD. PLoS ONE 7:e33304. doi: 10.1371/journal.pone.0033304
Davis, D. L., Vaught, A., Tso, T. C., and Bush, L. P. (1983). Analyses of a New Lower Yield Research Cigarette. Lexington, KY: Tobacco and Health Research Institute.
Dayal, H. H., Khuder, S., Sharrar, R., and Trieff, N. (1994). Passive smoking in obstructive respiratory disease in an industrialized urban population. Environ. Res. 65, 161–171.
Dhami, R., Zay, K., Gilks, B., Porter, S., Wright, J. L., and Churg, A. (2000). Pulmonary epithelial expression of human alpha1-antitrypsin in transgenic mice results in delivery of alpha1-antitrypsin protein to the interstitium. J. Mol. Med. (Berl.) 77, 377–385.
Di Stefano, A., Capelli, A., Lusuardi, M., Balbo, P., Vecchio, C., Maestrelli, P., et al. (1998). Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 158, 1277–1285.
Dominguez-Fandos, D., Peinado, V. I., Puig-Pey, R., Ferrer, E., Musri, M. M., Ramirez, J., et al. (2012). Pulmonary inflammatory reaction and structural changes induced by cigarette smoke exposure in the guinea pig. COPD. 9, 473–484.
Eltom, S., Stevenson, C. S., Rastrick, J., Dale, N., Raemdonck, K., Wong, S., et al. (2011). P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS ONE 6:e24097. doi: 10.1371/journal.pone.0024097
Elwing, J., and Panos, R. J. (2008). Pulmonary hypertension associated with COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 3, 55–70.
Eppert, B. L., Wortham, B. W., Flury, J. L., and Borchers, M. T. (2013). Functional characterization of T cell populations in a mouse model of chronic obstructive pulmonary disease. J. Immunol. 190, 1331–1340.
Escolar, J. D., Martinez, M. N., Rodriguez, F. J., Gonzalo, C., Escolar, M. A., and Roche, P. A. (1995). Emphysema as a result of involuntary exposure to tobacco smoke: morphometrical study of the rat. Exp. Lung Res. 21, 255–273.
Farkas, L., Farkas, D., Warburton, D., Gauldie, J., Shi, W., Stampfli, M. R., et al. (2011). Cigarette smoke exposure aggravates air space enlargement and alveolar cell apoptosis in Smad3 knockout mice. Am. J. Physiol. Lung. Cell Mol. Physiol. 301, L391–L401.
Feng, Y., Kong, Y., Barnes, P. F., Huang, F. F., Klucar, P., Wang, X., et al. (2011). Exposure to cigarette smoke inhibits the pulmonary T-cell response to influenza virus and Mycobacterium tuberculosis. Infect. Immun. 79, 229–237.
Fletcher, C., and Peto, R. (1977). The natural history of chronic airflow obstruction. Br. Med. J. 1, 1645–1648.
Foronjy, R. F., Mercer, B. A., Maxfield, M. W., Powell, C. A., D'Armiento, J., and Okada, Y. (2005). Structural emphysema does not correlate with lung compliance: lessons from the mouse smoking model. Exp. Lung Res. 31, 547–562.
Foronjy, R. F., Mirochnitchenko, O., Propokenko, O., Lemaitre, V., Jia, Y., Inouye, M., et al. (2006). Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am. J. Respir. Crit. Care Med. 173, 623–631.
Gardi, C., Cavarra, E., Calzoni, P., Marcolongo, P., de Santi, M., Martorana, P. A., et al. (1994). Neutrophil lysosomal dysfunctions in mutant C57 Bl/6J mice: interstrain variations in content of lysosomal elastase, cathepsin G and their inhibitors. Biochem. J. 299(Pt. 1), 237–245.
Geraghty, P., Wallace, A., and D'Armiento, J. M. (2011). Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4-C/EBP homologous protein mediated process. Int. J. Chron. Obstruct. Pulmon. Dis. 6, 309–319.
Ghoorah, K., De Soyza, A., and Kunadian, V. (2012). Increased cardiovascular risk in patients with chronic obstructive pulmonary disease and the potential mechanisms linking the two conditions: a review. Cardiol. Rev. doi: 10.1097/CRD.0b013e318279e907. [Epub ahead of print].
Gibbs, R. A., Weinstock, G. M., Metzker, M. L., Muzny, D. M., Sodergren, E. J., Scherer, S., et al. (2004). Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521.
Gielis, J. F., Lin, J. Y., Wingler, K., Van Schil, P. E., Schmidt, H. H., and Moens, A. L. (2011). Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic. Biol. Med. 50, 765–776.
Golovatch, P., Mercer, B. A., Lemaitre, V., Wallace, A., Foronjy, R. F., and D'Armiento, J. (2009). Role for cathepsin K in emphysema in smoke-exposed guinea pigs. Exp. Lung Res. 35, 631–645.
Gomita, Y., Eto, K., Furuno, K., and Araki, Y. (1990). Effects of standard cigarette and nicotine-less cigarette smoke inhalings on nicorandil plasma levels in rats. Pharmacology 40, 312–317.
Gross, P., Pfitzer, E. A., Tolker, E., Babyak, M. A., and Kaschak, M. (1965). Experimental emphysema: its production with papain in normal and silicotic rats. Arch. Environ. Health 11, 50–58.
Guerassimov, A., Hoshino, Y., Takubo, Y., Turcotte, A., Yamamoto, M., Ghezzo, H., et al. (2004). The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am. J. Respir. Crit. Care Med. 170, 974–980.
Hautamaki, R. D., Kobayashi, D. K., Senior, R. M., and Shapiro, S. D. (1997). Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277, 2002–2004.
Hirayama, T. (1981). Non-smoking wives of heavy smokers have a higher risk of lung cancer: a study from Japan. Br. Med. J. (Clin. Res. Ed.) 282, 183–185.
Hodge-Bell, K. C., Lee, K. M., Renne, R. A., Gideon, K. M., Harbo, S. J., and McKinney, W. J. (2007). Pulmonary inflammation in mice exposed to mainstream cigarette smoke. Inhal. Toxicol. 19, 361–376.
Hogg, J. C., Chu, F., Utokaparch, S., Woods, R., Elliott, W. M., Buzatu, L., et al. (2004). The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl. J. Med. 350, 2645–2653.
Janoff, A., Sloan, B., Weinbaum, G., Damiano, V., Sandhaus, R. A., Elias, J., et al. (1977). Experimental emphysema induced with purified human neutrophil elastase: tissue localization of the instilled protease. Am. Rev. Respir. Dis. 115, 461–478.
Jiang, R. T., Cheng, K. C., Acevedo-Bolton, V., Klepeis, N. E., Repace, J. L., Ott, W. R., et al. (2010). Measurement of fine particles and smoking activity in a statewide survey of 36 California Indian casinos. J. Expo. Sci. Environ. Epidemiol. 21, 31–41.
Jinot, J., and Bayard, S. (1994). Respiratory health effects of passive smoking: EPA's weight-of-evidence analysis. J. Clin. Epidemiol. 47, 339–349. discussion 351–353.
Kalandidi, A., Trichopoulos, D., Hatzakis, A., Tzannes, S., and Saracci, R. (1987). Passive smoking and chronic obstructive lung disease. Lancet 2, 1325–1326.
Kang, M. J., Homer, R. J., Gallo, A., Lee, C. G., Crothers, K. A., Cho, S. J., et al. (2007). IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J. Immunol. 178, 1948–1959.
Kang, M. J., Lee, C. G., Lee, J. Y., Dela Cruz, C. S., Chen, Z. J., Enelow, R., et al. (2008). Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J. Clin. Invest. 118, 2771–2784.
Kasahara, Y., Tuder, R. M., Taraseviciene-Stewart, L., Le Cras, T. D., Abman, S., Hirth, P. K., et al. (2000). Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest. 106, 1311–1319.
Keatings, V. M., Collins, P. D., Scott, D. M., and Barnes, P. J. (1996). Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 153, 530–534.
Koul, A., Singh, A., and Sandhir, R. (2003). Effect of alpha-tocopherol on the cardiac antioxidant defense system and atherogenic lipids in cigarette smoke-inhaling mice. Inhal. Toxicol. 15, 513–522.
Kratzer, A., Salys, J., Nold-Petry, C., Cool, C., Zamora, M., Bowler, R., et al. (2013). Role of IL-18 in second hand smoke-induced emphysema. Am. J. Respir. Cell Mol. Biol. doi: 10.1165/rcmb.2012-0173OC. [Epub ahead of print].
Laurell, C., and Eriksson, S. (1963). The electrophoretic pattern alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. Scand. J. Clin. Lab. Invest. 15, 132–140.
Lee, J. H., Lee, D. S., Kim, E. K., Choe, K. H., Oh, Y. M., Shim, T. S., et al. (2005). Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am. J. Respir. Crit. Care Med. 172, 987–993.
Leuenberger, P., Schwartz, J., Ackermann-Liebrich, U., Blaser, K., Bolognini, G., Bongard, J. P., et al. (1994). Passive smoking exposure in adults and chronic respiratory symptoms (SAPALDIA Study). Swiss study on air pollution and lung diseases in adults, SAPALDIA team. Am. J. Respir. Crit. Care Med. 150(5 Pt. 1), 1222–1228.
Lofroth, G. (1989). Environmental tobacco smoke: overview of chemical composition and genotoxic components. Mutat. Res. 222, 73–80.
Lu, S. Q., Fielding, R., Hedley, A. J., Wong, L. C., Lai, H. K., Wong, C. M., et al. (2011). Secondhand smoke (SHS) exposures: workplace exposures, related perceptions of SHS risk, and reactions to smoking in catering workers in smoking and nonsmoking premises. Nicotine Tob. Res. 13, 344–352.
Ma, B., Kang, M. J., Lee, C. G., Chapoval, S., Liu, W., Chen, Q., et al. (2005). Role of CCR5 in IFN-gamma-induced and cigarette smoke-induced emphysema. J. Clin. Invest. 115, 3460–3472.
MacNee, W. (2001). Oxidative stress and lung inflammation in airways disease. Eur. J. Pharmacol. 429, 195–207.
Maeno, T., Houghton, A. M., Quintero, P. A., Grumelli, S., Owen, C. A., and Shapiro, S. D. (2007). CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J. Immunol. 178, 8090–8096.
Maes, T., Bracke, K. R., Vermaelen, K. Y., Demedts, I. K., Joos, G. F., Pauwels, R. A., et al. (2006). Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammation. Int. Arch. Allergy Immunol. 141, 354–368.
Mahadeva, R., and Shapiro, S. D. (2005). Animal models of pulmonary emphysema. Curr. Drug Targets Inflamm. Allergy 4, 665–673.
March, T. H., Bowen, L. E., Finch, G. L., Nikula, K. J., Wayne, B. J., and Hobbs, C. H. (2005). Effects of strain and treatment with inhaled aII-trans-retinoic acid on cigarette smoke-induced pulmonary emphysema in mice. COPD 2, 289–302.
March, T. H., Green, F. H., Hahn, F. F., and Nikula, K. J. (2000). Animal models of emphysema and their relevance to studies of particle-induced disease. Inhal. Toxicol. 12(Suppl 4), 155–187.
March, T. H., Wilder, J. A., Esparza, D. C., Cossey, P. Y., Blair, L. F., Herrera, L. K., et al. (2006). Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. Toxicol. Sci. 92, 545–559.
Martin, J. G., Opazo-Saez, A., Du, T., Tepper, R., and Eidelman, D. H. (1992). In vivo airway reactivity: predictive value of morphological estimates of airway smooth muscle. Can. J. Physiol. Pharmacol. 70, 597–601.
Martin, J. G., and Tamaoka, M. (2006). Rat models of asthma and chronic obstructive lung disease. Pulm. Pharmacol. Ther. 19, 377–385.
Martorana, P. A., Beume, R., Lucattelli, M., Wollin, L., and Lungarella, G. (2005). Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am. J. Respir. Crit. Care Med. 172, 848–853.
Marwick, J. A., Edirisinghe, I., Arunachalam, G., Stevenson, C. S., Macnee, W., Kirkham, P. A., et al. (2010). Cigarette smoke regulates VEGFR2-mediated survival signaling in rat lungs. J. Inflamm. (Lond.) 7:11. doi: 10.1186/1476-9255-7-11
Miller, M., Pham, A., Cho, J. Y., Rosenthal, P., and Broide, D. H. (2010). Adiponectin-deficient mice are protected against tobacco-induced inflammation and increased emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 299, L834–L842.
Minino, A. (2010). Mortality among teenagers aged 12–19 years: United States, 1999–2006. NCHS Data Brief 37, 1–8.
Moerloose, K. B., Pauwels, R. A., and Joos, G. F. (2005). Short-term cigarette smoke exposure enhances allergic airway inflammation in mice. Am. J. Respir. Crit. Care Med. 172, 168–172.
Moritsugu, K. P. (2007). The 2006 report of the surgeon general: the health consequences of involuntary exposure to tobacco smoke. Am. J. Prev. Med. 32, 542–543.
Morris, A., Kinnear, G., Wan, W. Y., Wyss, D., Bahra, P., and Stevenson, C. S. (2008). Comparison of cigarette smoke-induced acute inflammation in multiple strains of mice and the effect of a matrix metalloproteinase inhibitor on these responses. J. Pharmacol. Exp. Ther. 327, 851–862.
Motz, G. T., Eppert, B. L., Wesselkamper, S. C., Flury, J. L., and Borchers, M. T. (2010a). Chronic cigarette smoke exposure generates pathogenic T cells capable of driving COPD-like disease in Rag2-/- mice. Am. J. Respir. Crit. Care Med. 181, 1223–1233.
Motz, G. T., Eppert, B. L., Wortham, B. W., Amos-Kroohs, R. M., Flury, J. L., Wesselkamper, S. C., et al. (2010b). Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease. J. Immunol. 184, 4460–4469.
Nakanishi, Y., Kobayashi, D., Asano, Y., Sakurai, T., Kashimura, M., Okuyama, S., et al. (2009). Clarithromycin prevents smoke-induced emphysema in mice. Am. J. Respir. Crit. Care Med. 179, 271–278.
Nemmar, A., Raza, H., Subramaniyan, D., Yasin, J., John, A., Ali, B. H., et al. (2013). Short-term systemic effects of nose-only cigarette smoke exposure in mice: role of oxidative stress. Cell Physiol. Biochem. 31, 15–24.
Nie, Y. C., Wu, H., Li, P. B., Luo, Y. L., Zhang, C. C., Shen, J. G., et al. (2012). Characteristic comparison of three rat models induced by cigarette smoke or combined with LPS: to establish a suitable model for study of airway mucus hypersecretion in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 25, 349–356.
Oberg, M., Jaakkola, M. S., Woodward, A., Peruga, A., and Pruss-Ustun, A. (2011). Worldwide burden of disease from exposure to second-hand smoke: a retrospective analysis of data from 192 countries. Lancet 377, 139–146.
Ofulue, A. F., and Ko, M. (1999). Effects of depletion of neutrophils or macrophages on development of cigarette smoke-induced emphysema. Am. J. Physiol. 277(1 Pt. 1), L97–L105.
Ofulue, A. F., Ko, M., and Abboud, R. T. (1998). Time course of neutrophil and macrophage elastinolytic activities in cigarette smoke-induced emphysema. Am. J. Physiol. 275(6 Pt. 1), L1134–L1144.
Ou, X. M., Wen, F. Q., Uhal, B. D., Feng, Y. L., Huang, X. Y., Wang, T., et al. (2009). Simvastatin attenuates experimental small airway remodelling in rats. Respirology 14, 734–745.
Pauwels, R., Van Der Straeten, M., Weyne, J., and Bazin, H. (1985). Genetic factors in non-specific bronchial reactivity in rats. Eur. J. Respir. Dis. 66, 98–104.
Pauwels, R. A., Buist, A. S., Ma, P., Jenkins, C. R., and Hurd, S. S. (2001). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: national heart, lung, and blood institute and world health organization global initiative for chronic obstructive lung disease (GOLD): executive summary. Respir. Care 46, 798–825.
Pemberton, P. A., Cantwell, J. S., Kim, K. M., Sundin, D. J., Kobayashi, D., Fink, J. B., et al. (2005). An inhaled matrix metalloprotease inhibitor prevents cigarette smoke-induced emphysema in the mouse. COPD 2, 303–310.
Pemberton, P. A., Kobayashi, D., Wilk, B. J., Henstrand, J. M., Shapiro, S. D., and Barr, P. J. (2006). Inhaled recombinant alpha 1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. COPD 3, 101–108.
Pesci, A., Balbi, B., Majori, M., Cacciani, G., Bertacco, S., Alciato, P., et al. (1998). Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur. Respir. J. 12, 380–386.
Petrache, I., Fijalkowska, I., Zhen, L., Medler, T. R., Brown, E., Cruz, P., et al. (2006). A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am. J. Respir. Crit. Care Med. 173, 1222–1228.
Piitulainen, E., Tornling, G., and Eriksson, S. (1998). Environmental correlates of impaired lung function in non-smokers with severe alpha 1-antitrypsin deficiency (PiZZ). Thorax 53, 939–943.
Pittilo, R. M., Bull, H. A., Gulati, S., Rowles, P. M., Blow, C. M., Machin, S. J., et al. (1990). Nicotine and cigarette smoking: effects on the ultrastructure of aortic endothelium. Int. J. Exp. Pathol. 71, 573–586.
Pittilo, R. M., Mackie, I. J., Rowles, P. M., Machin, S. J., and Woolf, N. (1982). Effects of cigarette smoking on the ultrastructure of rat thoracic aorta and its ability to produce prostacyclin. Thromb. Haemost. 48, 173–176.
Preobrazhenska, O., Wright, J. L., and Churg, A. (2012). Regional heterogeneity in murine lung fibroblasts from normal mice or mice exposed once to cigarette smoke. PLoS ONE 7:e39761. doi: 10.1371/journal.pone.0039761
Pryor, W. A., and Stone, K. (1993). Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 686, 12–27. discussion 27–28.
Rahman, I., and MacNee, W. (1996). Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic. Biol. Med. 21, 669–681.
Rangasamy, T., Cho, C. Y., Thimmulappa, R. K., Zhen, L., Srisuma, S. S., Kensler, T. W., et al. (2004). Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 114, 1248–1259.
Rao, S. P., Sikora, L., Hosseinkhani, M. R., Pinkerton, K. E., and Sriramarao, P. (2009). Exposure to environmental tobacco smoke induces angiogenesis and leukocyte trafficking in lung microvessels. Exp. Lung Res. 35, 119–135.
Rastrick, J. M., Stevenson, C. S., Eltom, S., Grace, M., Davies, M., Kilty, I., et al. (2013). Cigarette smoke induced airway inflammation is independent of NF-kappaB signalling. PLoS ONE 8:e54128. doi: 10.1371/journal.pone.0054128
Repace, J. L., Jiang, R. T., Acevedo-Bolton, V., Cheng, K. C., Klepeis, N. E., Ott, W. R., et al. (2011). Fine particle air pollution and secondhand smoke exposures and risks inside 66 US casinos. Environ. Res. 111, 473–484.
Retamales, I., Elliott, W. M., Meshi, B., Coxson, H. O., Pare, P. D., Sciurba, F. C., et al. (2001). Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am. J. Respir. Crit. Care Med. 164, 469–473.
Richens, T. R., Linderman, D. J., Horstmann, S. A., Lambert, C., Xiao, Y. Q., Keith, R. L., et al. (2009). Cigarette smoke impairs clearance of apoptotic cells through oxidant-dependent activation of RhoA. Am. J. Respir. Crit. Care Med. 179, 1011–1021.
Robbins, A. S., Abbey, D. E., and Lebowitz, M. D. (1993). Passive smoking and chronic respiratory disease symptoms in non-smoking adults. Int. J. Epidemiol. 22, 809–817.
Roemer, E., Schramke, H., Weiler, H., Buettner, A., Kausche, S., Weber, S., et al. (2012). Mainstream smoke chemistry and in vitro and in vivo toxicity of the reference cigarettes 3R4F and 2R4F. Contribute. Tobacco Res. 25, 316–335.
Roh, G. S., Yi, C. O., Cho, Y. J., Jeon, B. T., Nizamudtinova, I. T., Kim, H. J., et al. (2010). Anti-inflammatory effects of celecoxib in rat lungs with smoke-induced emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 299, L184–L191.
Sandler, D. P., Comstock, G. W., Helsing, K. J., and Shore, D. L. (1989). Deaths from all causes in non-smokers who lived with smokers. Am. J. Public Health 79, 163–167.
Schweitzer, K. S., Johnstone, B. H., Garrison, J., Rush, N. I., Cooper, S., Traktuev, D. O., et al. (2011). Adipose stem cell treatment in mice attenuates lung and systemic injury induced by cigarette smoking. Am. J. Respir. Crit. Care Med. 183, 215–225.
Seimetz, M., Parajuli, N., Pichl, A., Veit, F., Kwapiszewska, G., Weisel, F. C., et al. (2011). Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 147, 293–305.
Sekhon, H. S., Wright, J. L., and Churg, A. (1994). Cigarette smoke causes rapid cell proliferation in small airways and associated pulmonary arteries. Am. J. Physiol. 267(5 Pt. 1), L557–L563.
Selman, M., Cisneros-Lira, J., Gaxiola, M., Ramirez, R., Kudlacz, E. M., Mitchell, P. G., et al. (2003). Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in guinea pigs. Chest 123, 1633–1641.
Selman, M., Montano, M., Ramos, C., Vanda, B., Becerril, C., Delgado, J., et al. (1996). Tobacco smoke-induced lung emphysema in guinea pigs is associated with increased interstitial collagenase. Am. J. Physiol. 271(5 Pt. 1), L734–L743.
Shan, M., Yuan, X., Song, L. Z., Roberts, L., Zarinkamar, N., Seryshev, A., et al. (2012). Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci. Transl. Med. 4:117ra9. doi: 10.1126/scitranslmed.3003041
Shapiro, S. D., Goldstein, N. M., Houghton, A. M., Kobayashi, D. K., Kelley, D., and Belaaouaj, A. (2003). Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol. 163, 2329–2335.
Simani, A. S., Inoue, S., and Hogg, J. C. (1974). Penetration of the respiratory epithelium of guinea pigs following exposure to cigarette smoke. Lab. Invest. 31, 75–81.
Smith, K. R., Pinkerton, K. E., Watanabe, T., Pedersen, T. L., Ma, S. J., and Hammock, B. D. (2005). Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 102, 2186–2191.
Smith, K. R., Uyeminami, D. L., Kodavanti, U. P., Crapo, J. D., Chang, L. Y., and Pinkerton, K. E. (2002). Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic. Biol. Med. 33, 1106–1114.
Snider, G. L., Lucey, E. C., Christensen, T. G., Stone, P. J., Calore, J. D., Catanese, A., et al. (1984). Emphysema and bronchial secretory cell metaplasia induced in hamsters by human neutrophil products. Am. Rev. Respir. Dis. 129, 155–160.
Soyseth, V., Brekke, P. H., Smith, P., and Omland, T. (2007). Statin use is associated with reduced mortality in COPD. Eur. Respir. J. 29, 279–283.
Stampfli, M. R., and Anderson, G. P. (2009). How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 9, 377–384.
Stevens, T., Ekholm, K., Granse, M., Lindahl, M., Kozma, V., Jungar, C., et al. (2011). AZD9668: pharmacological characterization of a novel oral inhibitor of neutrophil elastase. J. Pharmacol. Exp. Ther. 339, 313–320.
Stevenson, C. S., Coote, K., Webster, R., Johnston, H., Atherton, H. C., Nicholls, A., et al. (2005). Characterization of cigarette smoke-induced inflammatory and mucus hypersecretory changes in rat lung and the role of CXCR2 ligands in mediating this effect. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L514–L522.
Suemaru, K., Oishi, R., Gomita, Y., Saeki, K., and Araki, Y. (1992). Effect of long-term cigarette smoke exposure on locomotor activity and brain monoamine levels in rats. Pharmacol. Biochem. Behav. 41, 655–658.
Sun, Y. P., Zhu, B. Q., Browne, A. E., Sievers, R. E., Bekker, J. M., Chatterjee, K., et al. (2001). Nicotine does not influence arterial lipid deposits in rabbits exposed to second-hand smoke. Circulation 104, 810–814.
Suzuki, M., Betsuyaku, T., Ito, Y., Nagai, K., Odajima, N., Moriyama, C., et al. (2009). Curcumin attenuates elastase- and cigarette smoke-induced pulmonary emphysema in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L614–L623.
Takubo, Y., Guerassimov, A., Ghezzo, H., Triantafillopoulos, A., Bates, J. H., Hoidal, J. R., et al. (2002). Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am. J. Respir. Crit. Care Med. 166(12 Pt. 1), 1596–1603.
Tanaka, T., Ohno, N., Kita, T., Kubo, K., Yonetani, Y., and Nakashima, T. (2004). Pharmacodynamic effects of chronic cigarette smoke exposure in spontaneously hypertensive rats. Methods Find. Exp. Clin. Pharmacol. 26, 9–18.
Taraseviciene-Stewart, L., Douglas, I. S., Nana-Sinkam, P. S., Lee, J. D., Tuder, R. M., Nicolls, M. R., et al. (2006). Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc. Am. Thorac. Soc. 3, 687–690.
Taraseviciene-Stewart, L., Scerbavicius, R., Choe, K. H., Moore, M., Sullivan, A., Nicolls, M. R., et al. (2005). An animal model of autoimmune emphysema. Am. J. Respir. Crit. Care Med. 171, 734–742.
Taraseviciene-Stewart, L., and Voelkel, N. F. (2008). Molecular pathogenesis of emphysema. J. Clin. Invest. 118, 394–402.
Taraseviciene-Stewart, L. K., Burns, N., and Voelkel, N. F. (2007). “Anti-endothelial cell antibodies in patients with COPD,” in European Respiratory Society Annual Congress, (Stockholm, Sweden) September 15–19, E496.
Teague, S. V., Pinkerton, K. E., Goldsmith, M., Gebremichael, A., Chang, S., Jenkins, R. A., et al. (1994). Sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhal. Toxicol. 6, 79–93.
Tuder, R. M., Zhen, L., Cho, C. Y., Taraseviciene-Stewart, L., Kasahara, Y., Salvemini, D., et al. (2003). Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am. J. Respir. Cell Mol. Biol. 29, 88–97.
van der Deen, M., Timens, W., Timmer-Bosscha, H., van der Strate, B. W., Scheper, R. J., Postma, D. S., et al. (2007). Reduced inflammatory response in cigarette smoke exposed Mrp1/Mdr1a/1b deficient mice. Respir. Res. 8:49. doi: 10.1186/1465-9921-8-49
van der Strate, B. W., Postma, D. S., Brandsma, C. A., Melgert, B. N., Luinge, M. A., Geerlings, M., et al. (2006). Cigarette smoke-induced emphysema: a role for the B cell? Am. J. Respir. Crit. Care Med. 173, 751–758.
Voelkel, N. (2008). Vascular Endothelial Growth Factor and its Role in Emphysema and Asthma in Chronic Obstructive Lung Diseases 2. Hamilton: BC Decker Inc., 77–83.
Voelkel, N., and Taraseviciene-Stewart, L. (2005). Emphysema: an autoimmune vascular disease? Proc. Am. Thorac. Soc. 2, 23–25.
Wang, L., Yang, J., Guo, L., Uyeminami, D., Dong, H., Hammock, B. D., et al. (2012). Use of a soluble epoxide hydrolase inhibitor in smoke-induced chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 46, 614–622.
Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562.
Weitzenblum, E., Hirth, C., Ducolone, A., Mirhom, R., Rasaholinjanahary, J., and Ehrhart, M. (1981). Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax 36, 752–758.
Witschi, H., Espiritu, I., Maronpot, R. R., Pinkerton, K. E., and Jones, A. D. (1997). The carcinogenic potential of the gas phase of environmental tobacco smoke. Carcinogenesis 18, 2035–2042.
Witschi, H., Oreffo, V. I., and Pinkerton, K. E. (1995). Six-month exposure of strain A/J mice to cigarette sidestream smoke: cell kinetics and lung tumor data. Fundam. Appl. Toxicol. 26, 32–40.
Woodruff, P. G., Ellwanger, A., Solon, M., Cambier, C. J., Pinkerton, K. E., and Koth, L. L. (2009). Alveolar macrophage recruitment and activation by chronic second hand smoke exposure in mice. COPD 6, 86–94.
Wright, J. L., and Churg, A. (1990). Cigarette smoke causes physiologic and morphologic changes of emphysema in the guinea pig. Am. Rev. Respir. Dis. 142(6 Pt. 1), 1422–1428.
Wright, J. L., and Churg, A. (2002). Smoking cessation decreases the number of metaplastic secretory cells in the small airways of the Guinea pig. Inhal. Toxicol. 14, 1153–1159.
Wright, J. L., and Churg, A. (2008). Animal models of COPD: barriers, successes, and challenges. Pulm. Pharmacol. Ther. 21, 696–698.
Wright, J. L., Farmer, S. G., and Churg, A. (2002). Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigs. Am. J. Respir. Crit. Care Med. 166, 954–960.
Wright, J. L., Lawson, L., Pare, P. D., Hooper, R. O., Peretz, D. I., Nelems, J. M., et al. (1983). The structure and function of the pulmonary vasculature in mild chronic obstructive pulmonary disease. The effect of oxygen and exercise. Am. Rev. Respir. Dis. 128, 702–707.
Wright, J. L., Levy, R. D., and Churg, A. (2005). Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax 60, 605–609.
Wright, J. L., Petty, T., and Thurlbeck, W. M. (1992). Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: national institutes of health nocturnal oxygen therapy trial. Lung 170, 109–124.
Wright, J. L., and Sun, J. P. (1994). Effect of smoking cessation on pulmonary and cardiovascular function and structure: analysis of guinea pig model. J. Appl. Physiol. 76, 2163–2168.
Wright, J. L., and Sun, J. P. (1999). Dissociation of chronic vascular cell proliferation and vascular contractility after chronic cigarette smoke exposure. Eur. Respir. J. 14, 832–838.
Wright, J. L., Sun, J. P., and Churg, A. (1999). Cigarette smoke exposure causes constriction of rat lung. Eur. Respir. J. 14, 1095–1099.
Wright, J. L., Sun, J. P., and Vedal, S. (1997). A longitudinal analysis of pulmonary function in rats during a 12 month cigarette smoke exposure. Eur. Respir. J. 10, 1115–1119.
Wright, J. L., Tai, H., and Churg, A. (2004). Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries. Am. J. Respir. Cell Mol. Biol. 31, 501–509.
Wright, J. L., Zhou, S., and Churg, A. (2012). Pulmonary hypertension and vascular oxidative damage in cigarette smoke exposed eNOS(−/−) mice and human smokers. Inhal. Toxicol. 24, 732–740.
Wright, J. L., Zhou, S., Preobrazhenska, O., Marshall, C., Sin, D. D., Laher, I., et al. (2011). Statin reverses smoke-induced pulmonary hypertension and prevents emphysema but not airway remodeling. Am. J. Respir. Crit. Care Med. 183, 50–58.
Yamato, H., Sun, J. P., Churg, A., and Wright, J. L. (1997). Guinea pig pulmonary hypertension caused by cigarette smoke cannot be explained by capillary bed destruction. J. Appl. Physiol. 82, 1644–1653.
Yao, H., Arunachalam, G., Hwang, J. W., Chung, S., Sundar, I. K., Kinnula, V. L., et al. (2010). Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. U.S.A. 107, 15571–15576.
Yao, H., Edirisinghe, I., Rajendrasozhan, S., Yang, S. R., Caito, S., Adenuga, D., et al. (2008). Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am. J. Physiol. Lung. Cell Mol. Physiol. 294, L1174–L1186.
Yoshida, T., Mett, I., Bhunia, A. K., Bowman, J., Perez, M., Zhang, L., et al. (2010). Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat. Med. 16, 767–773.
Yoshida, T., and Tuder, R. M. (2007). Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol. Rev. 87, 1047–1082.
Zavitz, C. C., Gaschler, G. J., Robbins, C. S., Botelho, F. M., Cox, P. G., and Stampfli, M. R. (2008). Impact of cigarette smoke on T and B cell responsiveness. Cell. Immunol. 253, 38–44.
Keywords: second hand cigarette smoke, COPD, emphysema, inflammation, animal model, chamber, pulmonary hypertension, matrix degradation
Citation: Leberl M, Kratzer A and Taraseviciene-Stewart L (2013) Tobacco smoke induced COPD/emphysema in the animal model—are we all on the same page? Front. Physiol. 4:91. doi: 10.3389/fphys.2013.00091
Received: 28 December 2012; Accepted: 10 April 2013;
Published online: 15 May 2013.
Edited by:
Michael Borchers, University of Cincinnati College of Medicine, USAReviewed by:
Michael Borchers, University of Cincinnati College of Medicine, USAPeter Di, University of Pittsburgh, USA
Copyright © 2013 Leberl, Kratzer and Taraseviciene-Stewart. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Laimute Taraseviciene-Stewart, Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado School of Medicine, Anschutz Medical Campus, Denver, CO 80262, USA. e-mail: Laima.Taraseviciene@ucdenver.edu
†Present address: Adelheid Kratzer, Institute of Physiology/Cardiovascular Research, University of Zurich, Zurich, Switzerland.